

Журнал аналитической химии, 2020, T. 75, № 8, стр. 721-729
Определение двадцати протеиногенных аминокислот и добавок в культуральной жидкости методом высокоэффективной жидкостной хроматографии
А. Д. Аскретков a, *, А. А. Клишин b, Д. И. Зыбин a, Н. В. Орлова c, А. В. Холодова c, Н. В. Лобанова c, Ю. А. Серегин b
a МИРЭА – Российский технологический университет
119454 Москва, просп. Вернадского, 78, Россия
b Государственный научно-исследовательский институт генетики и селекции промышленных микроорганизмов Национального исследовательского центра “Курчатовский институт”
1117545 Москва, 1-й Дорожный проезд, Россия
c ООО “Фармапарк”
117246 Москва, Научный проезд, 8, стр. 1, Россия
* E-mail: askretkov.a.d@gmail.com
Поступила в редакцию 07.08.2019
После доработки 30.09.2019
Принята к публикации 12.02.2020
Аннотация
Разработана ВЭЖХ-методика определения двадцати протеиногенных аминокислот, а также аммония, цистина и дипептида аланил-глутамина в культуральной жидкости. Для дериватизации аминокислот использовали флуоресцентную метку 6-аминохинолилгидроксисукцимидил карбамат с последующим ВЭЖХ-разделением на обращенно-фазовой колонке. В ходе валидации методики оценили специфичность, линейность, правильность, прецизионность, диапазон определяемых содержаний и предел количественного определения для каждого компонента. Диапазон определяемых содержаний аминокислот составил 10–800 мкМ (20–800 мкМ для триптофана и цистина), а предел количественного определения – не более 5 мкМ (15 мкМ для триптофана и 8 мкМ для цистина).
За последние 30 лет развитие производства рекомбинантных белков в клетках млекопитающих позволило многократно улучшить показатели производимых белков, а также повысить выход продукта [1]. Продуктивности для моноклональных антител за данный период увеличилась с 50 мг/л культуральной жидкости (КЖ) до более чем 5 г/л [2], причем важную роль сыграла оптимизация состава питательных сред с целью улучшения ростовых свойств и минимизации производства культурой токсичных метаболитов [3–5]. Аминокислоты являются одними из наиболее важных и незаменимых питательных веществ в средах, используемых при культивировании продуцентов рекомбинантных белков. В многочисленных исследованиях показано, что контроль содержания аминокислот в культуральной жидкости в процессе культивирования и, как следствие, своевременное введение подпиток быстро метаболизируемых аминокислот приводит к значительному увеличению продуктивности по целевому белку, что позволяет снизить стоимость процесса и повысить его эффективность [4, 5].
На данный момент имеется множество способов определения содержания ряда аминокислот в таких сложных матрицах, как КЖ, основанных на различных типах хроматографии [6], капиллярного электрофореза [7, 8], ферментативных реакций [9] и т.д.
Капиллярный электрофорез является достаточно мощным инструментом для определения многих аналитов, в том числе аминокислот, однако основными недостатком данного метода являются низкая чувствительность, неудовлетворительное разделение некоторых аминокислот и, в ряде случаев, недоступность оборудования [7]. Ферментативные реакции находят широкое применение в биотехнологическом производстве для мониторинга содержания основных питательных веществ и продуктов метаболизма клеток, однако спектр аминокислот, определяемых данными методами, как правило, ограничен глутамином и глутаматом [9, 10], при этом определение каждой аминокислоты требует своего набора реактивов и отдельной постановки анализа.
Для хроматографического определения аминокислот применяют различные виды хроматографии. Классическое разделение проводят при помощи ионообменной хроматографии с последующим импульсным амперометрическим [11], а также спектрофотометрическим и флуориметрическим детектированием производных нингидрина и ортофталевого альдегида [6, 12]. Так как до ввода в хроматограф проба подвергается минимуму воздействий, возможная деструкция или сорбция аминокислот в процессе пробоподготовки сведена к минимуму. Следует отметить, однако, что анализ по такой схеме требует специфического и дорогостоящего оборудования и сложных систем элюентов.
Определение аминокислот обращенно-фазовой ВЭЖХ получает все большее распространение ввиду простоты, надежности, высокой чувствительности и доступности данного метода для многих лабораторий. Дериватизацию с последующим спектрофотометрическим и флуориметрическим детектированием осуществляют орто-фталевым альдегидом [13], флуоренилметоксикарбонил хлоридом либо их комбинациями [12, 14]. Недостаток данных реагентов – низкая стабильность производных аминокислот, возможное неполное протекание реакции или образование ряда производных аминокислот [15, 16]. Применение флуоресцентной метки 6-аминохинолил-N-гидроксисукцимидил карбамата [17] позволило решить данные проблемы. При анализе гидролизатов было достаточно разделить 17 основных аминокислот [18], однако в случае образцов, содержащих аспаргин, глутамин и триптофан, наблюдалось со-элюирование серина с аспарагином и гистидина с глутамином. Для преодоления данной проблемы использовали сложную градиентную четырехкомпонентную систему элюентов [19, 20], применяли масс-спектрометрическое детектирование [12, 21] или системы для ультраВЭЖХ, которые на данный момент доступны не всем лабораториям.
Цель данной работы – разработка методики количественного определения 20 протеиногенных аминокислот, иона аммония, цистина, а также дипептида аланил-глутамин, являющегося источником глутамина для клеток яичников китайского хомячка (Chinese hamster ovary cell, CHO), в культуральной жидкости. Для дериватизации аминокислот использовали флуоресцентную метку 6‑аминохинолил-N-гидроксисукцимидил карбамат (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, AQC) [22].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объект анализа, реагенты и оборудование. Объектом исследования являлась КЖ, полученная при культивировании клона-продуцента экулизумаба (ООО “Фармапарк”, Россия). Последний получали методом введения генетической конструкции, содержащей синтетический ген тяжелой и легкой цепей экулизумаба в клетки суспензионной линии CHO (Invitrogen, США).
Для дериватизации аминокислот применяли набор AccQ-Fluor Reagent Kit (Waters, США). Использовали библиотеку аминокислот (17 аминокислот + ион аммония с концентрацией 2.5 мМ) также производства Waters; индивидуальные аминокислоты, триэтиламин, ацетат натрия, сульфат аммония, фосфорную, хлористоводородную и 6‑аминокапроновую кислоты (Sigma-Aldrich, США); ацетонитрил (Panreac AppliChem, Испания). Применяли хроматографическую систему ВЭЖХ Dionex Ultimate 3000 (Thermo Fisher Scientific, Германия), оснащенную флуориметрическим FLD-3100 и диодно-матричным DAD-3000 детекторами.
Приготовление стандартного образца. Виалу с библиотекой аминокислот размораживали и инкубировали до достижения комнатной температуры в течение 30 мин, перемешивали, 40 мкл раствора помещали в пластиковую пробирку типа Эппендорф емк. 1.5 мл, содержащую 730 мкл 20 мМ HCl. Прибавляли 30 мкл внутреннего стандарта (internal standard, IS) (2.5 мМ 6-аминокапроновая кислота), а также по 40 мкл свежеприготовленных 2.5 мМ растворов аспарагина, глутамина, триптофана, цистеина и аланил-глутамина в 20 мМ HCl, перемешивали. Использовали свежеприготовленный раствор.
Приготовление испытуемого образца. КЖ размораживали и инкубировали до достижения комнатной температуры в течение 30 мин, перемешивали и центрифугировали для осаждения клеточного дебриса. 20 мкл помещали в пластиковую пробирку типа Эппендорф емк. 1.5 мл, содержащую 950 мкл 20 мМ HCl, прибавляли 30 мкл раствора внутреннего стандарта и перемешивали. Использовали свежеприготовленный раствор.
По 10 мкл стандартного и испытуемого растворов помещали в стеклянные виалы для ВЭЖХ емк. 300 мкл, прибавляли 70 мкл боратного буферного раствора (поставлялся с набором для дериватизации) и 20 мкл раствора AQC (содержимое виалы, растворенное в 1 мл ацетонитрила), образцы инкубировали при 50°С в течение 10 мин и далее помещали в автоматический дозатор хроматографа с температурой 2–8°С.
Анализ проводили на обращенно-фазовой колонке AccQ-Tag 3.9 × 150 мм с зерном сорбента 4 мкм, расход подвижной фазы (ПФ) составлял 1 мл/мин, объем пробы – 4 мкл, температура колонки 37°С. Для элюирования использовали двухкомпонентную систему. ПФ А состояла из 20 мМ ацетата натрия, 4 мМ триэтиламина с pH 5.89 (создавали добавлением H3PO4). В качестве ПФ Б использовали ацетонитрил для градиентной ВЭЖХ. Элюирование осуществляли по программе, приведенной в табл. 1.
Таблица 1.
Программа градиентного элюирования
| Интервал времени, мин | ПФ А, % | ПФ Б, % | Форма градиента* |
|---|---|---|---|
| 0 → 0.5 | 100.0 → 99.0 | 0 → 1.0 | 5 |
| 0.5 → 22.0 | 99.0 → 95.0 | 1.0 → 5.0 | 9 |
| 22.0 → 28.0 | 95.0 → 91.0 | 5.0 → 9.0 | 6 |
| 28.0 → 28.1 | 91.0 → 88.0 | 9.0 → 12.0 | 6 |
| 28.1 → 35.0 | 88.0 → 85.0 | 12.0 → 15.0 | 8 |
| 35.0 → 45.0 | 85.0 → 75.0 | 15.0 → 25.0 | 8 |
| 45.0 → 45.1 | 75.0 → 15.0 | 25.0 → 85.0 | 9 |
| 45.1 → 51.0 | 15.0 | 85.0 | – |
| 51.0 → 59.0 | 100.0 | 0.0 | – |
Детектировали флуориметрически при длинах волн возбуждения λex = 250 нм и эмиссии λem = = 395 нм. Хроматограммы обрабатывали в ПО Chromeleon 7.0 (Thermo Scientific), а также Microsoft Office Exel.
Валидацию методики осуществляли в соответствии с рекомендациями ICH Q2(R1) [23], а также Государственной Фармакопеи РФ 14 издания [24].
Специфичность оценивали сравнением хроматограмм от ввода пробы стандартного раствора аминокислот, испытуемого раствора КЖ, 20 мМ HCl, раствора очищенного белка с концентрацией 0.4 мг/мл, индивидуальных растворов 20 аминокислот, внутреннего стандарта, аланил-глутамина и сульфата аммония, дериватизированных согласно методике. На хроматограмме 20 мМ HCl и раствора очищенного белка должны отсутствовать пики, накладывающиеся на пики аминокислот; разрешение между соседними пиками всех аминокислот должно быть не менее 1.5. На хроматограмме испытуемого образца должны присутствовать пики аминокислот с аналогичными стандартному образцу временами удерживания и разрешением.
Линейный диапазон методики оценивали для концентрации каждой аминокислоты от 10 до 800 мкМ (20–800 мкМ для триптофана и цистина). Для этого растворы с концентрациями каждой аминокислоты 10, 20, 50, 100, 200, 300, 500, 800 мкМ анализировали трижды, во всех случаях концентрация внутреннего стандарта была одинаковой и составляла 75 мкМ. Для каждой аминокислоты строили график зависимости отношения площади пика аминокислоты к площади пика внутреннего стандарта от концентрации. Коэффициент линейной регрессии (r) должен составлять не менее 0.99 для всех аналитов.
Правильность методики контролировали методом добавок. Для этого к образцу КЖ с установленным содержанием аминокислот прибавляли известное количество стандартного раствора и оценивали отклик. Значение отклика не должно выходить за пределы диапазона 85–115%.
Повторяемость результатов оценивали путем дериватизации образца КЖ в шести повторностях и анализа полученных растворов. По результатам количественного определения оценивали относительное стандартное отклонение (sr), которое не должно превышать 10% для каждого аналита.
Внутрилабораторную прецизионность оценивали путем дериватизации одного образца КЖ в шести повторностях в двух сессиях, выполненных в разные дни разными сотрудниками, и анализа полученных растворов. По результатам количественного определения оценивали относительное стандартное отклонение между сессиями, которое не должно превышать 12%. Аналогичную процедуру осуществляли в другой лаборатории (ГосНИИ Генетики НИЦ “Курчатовский институт”) для оценки межлабораторной воспроизводимости, максимально допустимое значение sr при этом установили равным 15%.
Предел количественного определения (ПКО) оценивали расчетным путем, учитывая отношение сигнал/шум для пиков аминокислот с концентрацией 10 мкМ. Отношение сигнал/шум для ПКО должно составлять около 10, а также ПКО для всех аминокислот (за исключением цистина и триптофана) не должен превышать 5 мкМ (нижняя точка градуировки).
Надежность методики оценивали по следующим параметрам:
– Стабильность раствора пробы в течение 24 ч. Для этого раствор стандартного образца анализировали сразу после дериватизации и через 24 ч после дериватизации, при этом образец хранили в стеклянной виале в автоматическом дозаторе хроматографа при 2–8°С. Отклонение относительной площади пиков всех аналитов не должно превышать 5% от начальной.
– Вариация объема образца при дериватизации. Для этого анализировали растворы стандартного образца, объемы которых для дериватизации составляли 8 и 12 мкл (80 и 120% от номинального). Для таких образцов отклонение относительной площади пиков всех аналитов не должно превышать 5% от номинального.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выбор элюента. Применение нами коммерческого элюента Waters (AccQ-Tag Eluent A) для определения аминокислот привело к описанной выше проблеме со-элюирования Сер/Асн, Глн/Гис. Изменение pH элюента в пределах 5.00–5.80 не привело к разделению данных пар аминокислот (рис. 1). В связи с этим коммерческий элюент заменили на приготовленный из ацетата натрия и триэтиламина, который широко использовали для анализа смеси дериватизированных аминокислот методом обращенно-фазовой ВЭЖХ в работах [25, 26]. Варьирование концентраций триэтиламина и ацетата натрия в диапазонах 1–8 и 5–100 мМ соответственно, а также значений pH (создавали добавлением H3PO4) привело к следующим результатам (рис. 2). При концентрации модификаторов ниже 4 мМ триэтиламина и 20 мМ ацетата натрия происходит неполное разрешение и уширение пиков полярных аминокислот, а при концентрациях выше 5 мМ триэтиламина 50 мМ ацетата натрия – слияние пиков цистина, тирозина и внутреннего стандарта. В результате выбрали минимальную (для предотвращения возможного образования осадка в системе) концентрацию модификаторов, при которой происходит разделение всех аналитов – 4 мМ триэтиламина и 20 мМ ацетата натрия c pH 5.89. Небольшое изменение pH элюента в диапазоне от 5.75 до 6.00 не приводит к значительному изменению разрешения аминокислот, но вызывает изменение времен миграции пика 6-аминохинолина, таким образом способствуя его частичному со-элюированию с аспарагином (pH 5.75) либо с глицином (pH 6.00).
Рис. 1.
Влияние pH коммерческого элюента Waters на разрешение пар аминокислот Сер/Асн (a), Глн/Гис (б): 1 – коммерческий элюент без изменения pH (5.00), 2 – pH 5.83, 3 – pH 5.40, 4 – pH 5.20.
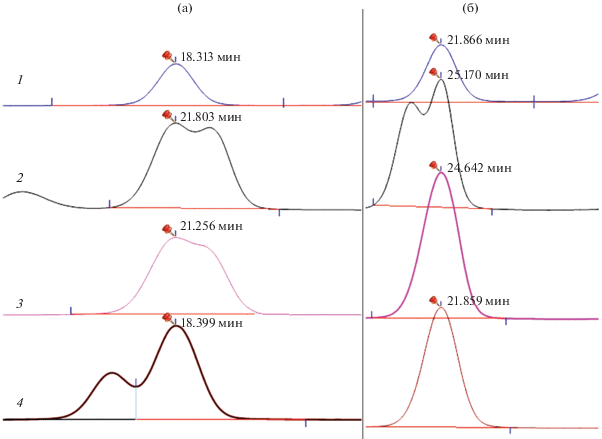
Рис. 2.
Хроматограммы стандарта аминокислот, полученные с элюентами: 20 мМ ацетат натрия, 4 мМ триэтиламин (стандартный) (а); 10 мМ ацетат натрия, 2 мМ триэтиламин (б); 5 мМ ацетат натрия, 1 мМ триэтиламин (в); 100 мМ ацетат натрия, 8 мМ триэтиламин (г). Идентификация пиков: 1 – Глу, 2 – Асп, 3 – Сер, 4 – Асн, 5 – пик от дериватизирующего реагента AQC (6-аминохинолин), 6 – Гли, 7 – Глн, 8 – Гис, 9 – аммоний, 10 – Тре, 11 – Арг, 12 – Ала, 13 – Ала-Глн, 14 – Про, 15 – Цис, 16 – Цистин, 17 – Тир, 18 – 6-аминокапроновая кислота (IS), 19– Вал, 20 – Мет, 21 – Иле, 22 – Лей, 23 – Лиз, 24 – Фен, 25 – Три.
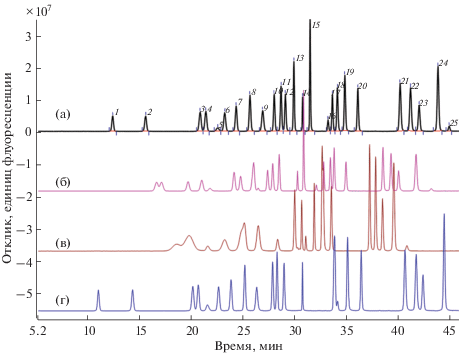
Валидация методики. На рис. 3 приведена хроматограмма стандартного раствора, а также результаты оценки специфичности методики. Видно, что происходит полное разделение AQC-производных всех 20 аминокислот, внутреннего стандарта, цистина, иона аммония, аланил-глутамина и пика дериватизирующего реагента, разрешение между соседними пиками аналитов составляет не менее 1.5 (табл. 2). На хроматограмме 20 мМ HCl, как и на хроматограмме раствора очищенного белка, отсутствуют пики, накладывающиеся на пики пики аминокислот. Следует отметить, что на всех хроматограммах присутствовала незначительная примесь AQC-производного иона аммония, что связано, по-видимому, с примесью в самом дериватизирующем реагенте.
Рис. 3.
Оценка специфичности методики. (а) – Хроматограмма стандартного образца, (б) – хроматограмма 20 мМ HCl, (в) – хроматограмма раствора аланил-глутамина с внутренним стандартом, (г) – хроматограмма раствора сульфата аммония с внутренним стандартом, (д) – хроматограмма раствора очищенного белка с концентрацией 0.4 мг/мл. Идентификацию пиков см. в подписи к рис. 2.
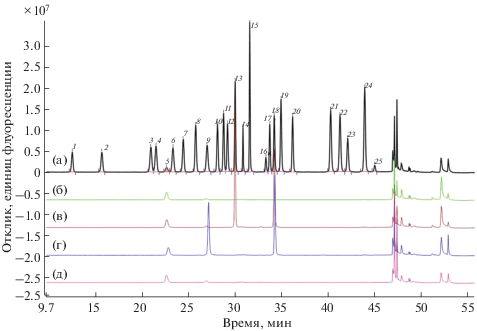
Таблица 2.
Оценка валидационных параметров методики
| Аминокислота | Специфичность | Линей-ность (r2) |
Правильность (отклик, %) | Повторя-емость (sr, %) | Промежуточная прецизионность (sr, %) |
Предел количественного определения, мкМ |
||
|---|---|---|---|---|---|---|---|---|
| время удержи- вания, мин |
разре-шение* | добавка 1 | добавка 2 | |||||
| Асп | 12.36 | 9.84 | 0.999 | 101.5 | 98.0 | 2.5 | 2.4 | 4.0 |
| Глу | 15.55 | 14.78 | 0.998 | 100.9 | 98.9 | 2.3 | 2.3 | 4.4 |
| Сер | 20.77 | 1.51 | 0.998 | 98.4 | 96.8 | 3.5 | 3.7 | 3.5 |
| Асн | 21.32 | 2.69 | 0.997 | 92.3 | 94.8 | 8.3 | 8.6 | 3.5 |
| 6-Аминохинолин | 22.59 | 1.63 | – | – | – | – | – | – |
| Гли | 23.13 | 3.32 | 0.994 | 99.4 | 99.6 | 6.0 | 6.2 | 3.5 |
| Глн | 24.23 | 4.43 | 0.992 | 100.0 | 94.9 | 9.8 | 11.2 | 2.9 |
| Гис | 25.64 | 3.74 | 0.996 | 101.2 | 99.0 | 5.2 | 5.9 | 2.1 |
| Аммоний | 26.86 | 3.6 | 0.990 | 100.2 | 98.8 | 2.0 | 2.1 | 2.5 |
| Тре | 27.99 | 2.7 | 0.994 | 100.7 | 98.9 | 3.7 | 4.2 | 1.9 |
| Арг | 28.68 | 1.78 | 0.992 | 95.8 | 93.3 | 4.2 | 4.5 | 1.7 |
| Ала | 29.07 | 3.79 | 0.993 | 90.2 | 93.3 | 5.3 | 5.1 | 1.9 |
| Ала-Глн | 29.91 | 5.44 | 0.995 | 93.2 | 95.3 | 4.8 | 4.8 | 1.1 |
| Про | 30.78 | 5.66 | 0.993 | 100.3 | 99.7 | 2.8 | 3.1 | 1.9 |
| Цис | 31.48 | 9.68 | 0.994 | 98.4 | 99.7 | 8.8 | 10.2 | 0.7 |
| Цистин | 33.21 | 2.03 | 0.99 | 87.4 | 92.6- | 9.2 | 11.7 | 7.8 |
| Тир | 33.62 | 2.23 | 0.998 | 97.9 | 98.7 | 3.7 | 3.9 | 1.5 |
| IS | 34.13 | 3.13 | – | – | – | – | – | – |
| Вал | 34.84 | 5.27 | 0.998 | 101.5 | 99.2 | 2.3 | 2.5 | 1.1 |
| Мет | 36.06 | 15.18 | 0.996 | 101.7 | 100.0 | 2.7 | 2.8 | 1.5 |
| Иле | 40.14 | 3.26 | 0.997 | 100.9 | 100.9 | 4.0 | 4.0 | 1.0 |
| Лей | 41.14 | 2.69 | 0.996 | 102.6 | 101.7 | 4.3 | 4.4 | 1.2 |
| Лиз | 41.98 | 6.11 | 0.999 | 100.8 | 99.7 | 4.4 | 4.6 | 2.1 |
| Фен | 43.77 | 3.93 | 0.995 | 98.2 | 98.4 | 3.9 | 3.9 | 0.8 |
| Три | 44.79 | – | 0.991 | 96.2 | 98.2 | 9.8 | 10.5 | 15.1 |
* Разрешение между двумя соседними пиками рассчитывали по формуле № 2, приведенной в Государственной фармакопее РФ [27].
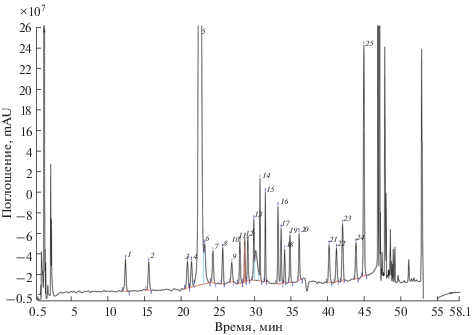
На рис. 4 приведена хроматограмма испытуемого образца КЖ, полученной при культивировании продуцента экулизумаба. Как видно, хроматограмма испытуемого образца на качественном уровне аналогична хроматограмме стандартного раствора. Времена удерживания и разрешение пиков всех аналитов близки к стандарту. Таким образом, специфичность методики подтверждена.
Рис. 4.
Хроматограмма испытуемого образца культуральной жидкости культуры клеток СНО. Идентификацию пиков см. в подписи к рис. 2.
Линейный диапазон оценивали путем анализа образцов стандартного раствора аминокислот с концентрациями, варьирующими в диапазоне от 10 до 800 мкМ (20–800 мкМ для триптофана и цистина). Коэффициенты линейной регрессии градуировочных графиков для всех аминокислот составляли более 0.99, что говорит о линейности градуировочных функций (табл. 2). При оценке правильности добавляли концентрированный раствор стандарта аминокислот (1000 мкМ) к образцу КЖ с предварительно установленным содержанием аминокислот (табл. 2). Для всех аналитов отклонение отклика лежало в пределах 85–115%, что подтверждает правильность методики.
Значения sr повторяемости и внутрилабораторной прецизионности (табл. 2) удовлетворяли установленным критериям для всех аналитов. Значения sr межлабораторной воспроизводимости не превышали 15% для всех аналитов, при этом наибольшие значения sr составляли 13.8 и 14.7% для глутамина и цистеина соответственно.
Предел количественного определения для большинства аналитов (табл. 2) составляет от 1 до 4 мкМ, за исключением триптофана и цистина.
При оценке надежности методики по параметру “стабильность раствора пробы в течение 24 ч” отклонение относительной площади пиков всех аналитов не превышало 5% от начальной. При вариации объема пробы в диапазоне 80–120% отклонение относительной площади пиков всех аналитов также не превышало 5% от номинального.
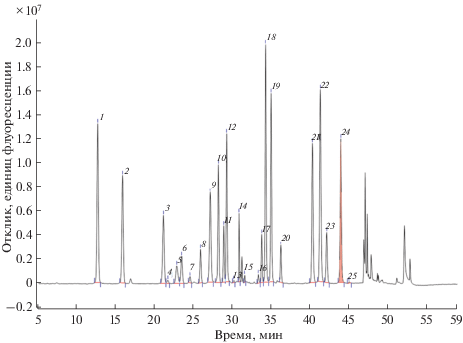
Помимо флуориметрического детектирования, AQC-дериватизированные аминокислоты можно определять спектрофотометрически, при этом чувствительность к большинству аминокислот уменьшается в 3–10 раз, за исключением триптофана, для которого чувствительность увеличивается в 6 раз, и цистина – остается неизменной. Детектирование глицина затруднено присутствием значительно большего (по сравнению с данными флуориметрического детектора) пика 6-аминохинолина (рис. 5). Аналогичный, но меньшего размера пик наблюдается вблизи пика аланил-глутамина, что также затрудняет точное определение глицина.
Рис. 5.
Хроматограмма стандартного образца аминокислот со спектрофотометрическим детектированием при 250 нм. Идентификацию пиков см. в подписи к рис. 2.
* * *
Таким образом, разработана методика количественного определения 20 протеиногенных аминокислот, аммония, цистина, а также источника глутамина – дипептида аланил-глутамина в культуральной жидкости культуры клеток СНО, продуцирующих рекомбинантное моноклональное антитело экулизумаб. Проведена валидация методики по параметрам специфичность, линейность, правильность, прецизионность, диапазон определяемых содержаний и предел количественного определения. Методика позволяет определять концентрации большинства аминокислот в диапазоне 10–800 мкМ с относительным стандартным отклонением от 2 до 5%.
Список литературы
Xing Z., Kenty B., Koyrakh I., Borys M., Pan S-H., Li Z.J. Optimizing amino acid composition of CHO cell culture media for a fusion protein production // Process Biochem. 2011. V. 46. № 7. P. 1423.
Wurm F.M. Production of recombinant protein therapeutics in cultivated mammalian cells // Nat. Biotechnol. 2004. V. 22. № 11. P. 1393.
Xie L., Wang D.I.C. Stoichiometric analysis of animal cell growth and its application of medium design // Biotechnol. Bioeng. 1994. V. 43. № 11. P. 1164.
Altamirano C., Illanes A., Casablancas A. Gámez X., Cairó J.J., Gòdia C. Analysis of CHO cells metabolic redistribution in a glutamate-based defined medium in continuous culture // Biotechnol. Prog. 2001. V. 17. № 6. P. 1032.
Nyberg G.B., Balcarcel R.R., Follstad B.D. Stephanopoulos G., Wang D.I.C. Metabolism of peptide amino acids by Chinese hamster ovary cells grown in a complex medium // Biotechnol. Bioeng. 1999. V. 62. № 3. P. 324.
Fan Y., Del ValI J., Muller C. Sen J.W., Rasmussen S.K., Kontoravdi C., Weilguny D., Andersen M.R. Amino acid and glucose metabolism in fed-batch CHO cell culture affects antibody production and glycosylation // Biotechnol. Bioeng. 2015. V. 112. № 3. P. 521.
Zunic G.D., Spasic S., Jelic-Ivanovic Z. Capillary electrophoresis of free amino acids in physiological fluids without derivatization employing direct or indirect absorbance detection // Methods Mol. Biol. 2012. V. 828. P. 243.
Шпак A.В., Пирогов А.В., Шпигун О.А. Определение аминокислот методом капиллярного электрофореза без предварительной дериватизации // Журн. аналит. химии. 2003. Т. 58. № 7. С. 729. (Shpak A.V., Pirogov A.V., Shpigun О.A. Determination of amino acids by capillary electrophoresis without preliminary derivatization // J. Analyt. Chem. 2003. V. 58. № 7. P. 729.)
Behjousiar A., Kontoravdi C., Polizzi K.M. In situ monitoring of intracellular glucose and glutamine in CHO cell culture // PLoS One. 2012. V. 7. № 4. Article e34512.
Frahm B., Brod H., Langer U. Improving bioreactor cultivation conditions for sensitive cell lines by dynamic membrane aeration // Cytotechnology. 2009. V. 59. № 1. P. 17.
Genzela Y., Koniga S., Reichla U. Amino acid analysis in mammalian cell culture media containing serum and high glucose concentrations by anion exchange chromatography and integrated pulsed amperometric detection // Anal. Biochem. 2004. V. 335. № 1. P. 119.
Macchi F.D., Shen F.J., Keck R.G., Harris R.J. Amino acid analysis, using postcolumn ninhydrin detection, in a biotechnology laboratory / Amino Acid Analysis Protocols. Methods in Molecular Biology / Eds. Alterman M.A., Hunziker P. London: Springer, 2012. 455 p.
Bartolomeo M.P., Maisano F. Validation of a reversed-phase HPLC method for quantitative amino acid analysis // J. Biomol. Tech. 2006. V. 17. № 2. P. 131.
Schwarza E.L., Robertsa W.L., Pasqualia M. Analysis of plasma amino acids by HPLC with photodiode array and fluorescence detection // Clin. Chim. Acta. 2005. V. 354. № 1–2. P. 83.
Mengerink Y., Kutlan D., Toth F., Csámpai A., Molnár-Perl I. Advances in the evaluation of the stability and characteristics of the amino acid and amine derivatives obtained with the o-phthaldialdehyde/3-mercaptopropionic acid and o-phthaldialdehyde/N-acetyl-L-cysteine reagents. High-performance liquid chromatography-mass spectrometry study // J. Chromatogr. A. 2002. V. 949. № 1–2. P. 99.
Kutlan D., Molnar-Perl I. Characteristics and stability of the OPA/3-mercaptopropionic acid and OPA/N-acetyl-L-cysteine derivatives of amino acids // Chromatographia. 2001. V. 53. Suppl. 1. P. S188.
Cohen S.A., Michaud D.P. Synthesis of a fluorescent derivatizing reagent, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and its application for the analysis of hydrolysate amino acids via high-performance liquid chromatography // Anal. Biochem. 1993. V. 211. № 2. P. 279.
Bosch L., Alegrla A., Farre R. Application of the (AQC) reagent to the RP-HPLC determination of amino acids in infant foods // J. Chromatogr. B. 2006. V. 831. № 1–2. P. 176.
Callejon R.M., Tesfaye W., Torija M.J., Mas A., Troncoso A.M., Morales M.L. HPLC determination of amino acids with AQC derivatization in vinegars along submerged and surface acetications and its relation to the microbiota // Eur. Food Res. Technol. 2008. V. 227. № 1. P. 93.
Hernandez-Orte P., Ibarz M.J., Cacho J., Ferreira V. Amino acid determination in grape juices and wines by HPLC using a modification of the 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) method // Chromatographia. 2003. V. 58. № 1–2. P. 29.
Dahl-Lassen R., van Hecke J., Jørgensen H., Bukh C., Andersen B., Schjoerring J.K. High-throughput analysis of amino acids in plant materials by single quadrupole mass spectrometry // Plant Methods. 2018. V. 14. № 8.
Krull I.S., Strong R. Derivatization / Encyclopedia of Separation Science. 1st Ed. / Ed. Wilson I. London: Academic Press, 2005. P. 583.
ICH Harmonised tripartite guideline. Validation of Analytical Procedures: Text and Methodology Q2(R1), 2005. C. 17.
Государственная фармакопея Российской Федерации. XIV издание. Ч. 1. ОФС 1.1.0012.15 “Валидация аналитических методик”. М.: Научный центр экспертизы средств медицинского применения, 2018. С. 276.
Zeng F., Ou J., Huang Y., Li Q., Xu G., Liu Z., Yang S. Determination of 21 free amino acids in fruit juices by HPLC using a modification of the 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) method // Food Anal. Methods. 2015. V. 8. № 2. P. 428.
Sánchez-Machado D., López-Cervantes J., López-Hernández J., Paseiro-Losada P., Simal-Lozano J. High-performance liquid chromatographic analysis of amino acids in edible seaweeds after derivatization with phenyl isothiocyanate // Chromatographia. 2003. V. 58. № 3–4. P. 159.
Государственная фармакопея Российской Федерации. XIV издание. Ч. 1. ОФС 1.2.1.2.0001.15 “Хроматография”. М.: Научный центр экспертизы средств медицинского применения, 2018. С. 850.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии