

Журнал аналитической химии, 2021, T. 76, № 1, стр. 32-50
Хромато-масс-спектрометрические методы определения маркеров и биомаркеров отравляющих веществ
И. В. Рыбальченко a, b, *, Т. М. Байгильдиев a, И. А. Родин a
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b 27 Научный центр Министерства обороны Российской Федерации
105005 Москва, Бригадирский переулок, 13, Россия
* E-mail: rivrus@mail.ru
Поступила в редакцию 29.04.2020
После доработки 02.06.2020
Принята к публикации 09.07.2020
Аннотация
Рассмотрены публикации, посвященные разработке и совершенствованию методов определения маркеров и биомаркеров отравляющих веществ (ОВ) с помощью различных сочетаний и модификаций газовой и жидкостной хромато-масс-спектрометрии. Акцентировано внимание на значении выбора оптимальной стратегии анализа, оптимизации процедур пробоподготовки и режимов работы аналитических приборов. Обобщены литературные данные по выбору характерных маркеров фосфорорганических ОВ и сернистого иприта и методам их определения.
Вступление в силу в 1997 г. международной Конвенции о запрещении химического оружия (далее Конвенция) [1], к которой к настоящему времени присоединились 198 стран, вызвало появление в научной литературе большого количества публикаций, посвященных разработке методов аналитического контроля, предназначенных для выявления фактов несанкционированного хранения, использования или применения отравляющих веществ (ОВ). Эти разработки представляются особенно актуальными в связи с неоднократными случаями нарушения Конвенции в последние годы, сопровождающимися применением ОВ для достижения военных, политических или террористических целей [2–5].
До середины 80-х годов прошлого столетия наибольшее внимание исследователей привлекали экспресс-методы обнаружения боевых ОВ как таковых [6–8], что было продиктовано требованиями обеспечения мер безопасности и защиты как военнослужащих, так и населения при применении химического оружия в военных целях.
Вместе тем, поскольку химическое оружие (ХО) предназначено для нанесения быстрых поражений в зоне применения с последующим прекращением его действия, составляющие основу ХО отравляющие вещества в большинстве случаев относятся к нестойким органическим соединениям, которые подвергаются относительно быстрому разложению и/или преобразованию в окружающей среде и в организме. В связи с этим при формировании системы аналитического контроля за соблюдением Конвенции возникла необходимость создания лабораторных методов определения в различных объектах характерных долгоживущих продуктов деградации ОВ и их реакционных продуктов, получивших общее название “маркеров ОВ”; в случае биологических объектов их принято называть “биомаркерами”. Выявление маркеров ОВ в объектах окружающей среды (воздух, почва, вода, технологические материалы) и биомаркеров в биологических жидкостях (моча, кровь) позволяет подтвердить факт применения химического оружия, а их идентификация – определить тип исходного ОВ, которое предположительно было использовано. Результаты количественного анализа позволяют судить об уровне заражения среды, а также о токсической дозе, воздействующей на организм. Предпочтение отдаeтся методам, позволяющим идентифицировать маркеры и биомаркеры в возможно более отдалeнные сроки после экспозиции и/или отбора пробы, т.е. характеризующимся значительной “ретроспективностью”.
К расследованию предполагаемых фактов разработки, производства, хранения или применения ОВ международной Организацией по запрещению химического оружия (ОЗХО) привлекаются аккредитованные ею национальные лаборатории, владеющие арсеналом методов анализа проб окружающей среды (экологические пробы) [9], а также биологических сред (биомедицинские пробы) [10].
Подавляющее большинство исследований последних лет посвящено методам определения маркеров и биомаркеров фосфорорганических ОВ (ФОВ) и сернистого иприта (СИ), поскольку объявленные запасы этих ОВ в различных странах на момент вступления в силу Конвенции превосходили запасы других ОВ, а случаи их применения зафиксированы в ряде террористических актов и локальных конфликтов [11–13].
Маркеры и биомаркеры ОВ сильно различаются по химическим и физическим свойствам, среди них есть как полярные, так и менее полярные кислоты, основания, летучие и нелетучие вещества, содержащие в составе молекул гетероатомы фосфора, серы и азота, а также метаболиты и аддукты ОВ с аминокислотами и пептидами, входящими в состав белков и ферментов. Такое разнообразие состава и свойств объектов анализа обусловило формирование единого мнения подавляющего большинства исследователей о целесообразности применения для их определения наиболее универсальных физико-химических методов анализа, к которым отнесены сочетания газовой хроматографии (ГХ) и высокоэффективной жидкостной хроматографии (ВЭЖХ) с различными вариантами масс-спектрометрического (МС) детектирования.
В настоящем обзоре обсуждаются оригинальные и обзорные статьи, посвященные ГХ–МС- и ВЭЖХ–МС-методам определения маркеров и биомаркеров ФОВ и СИ.
ОПРЕДЕЛЕНИЕ МАРКЕРОВ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
Первоначально к маркерам ФОВ и СИ относили исключительно первичные и вторичные продукты их гидролиза и окисления (рис. 1–4). Однако приведенными на рис. 1–3 продуктами трансформации ОВ не ограничивается набор потенциальных маркеров, в число которых также входят продукты окисления и различного рода реакционные продукты взаимодействия с компонентами окружающей среды и с различными рецептурами, применяемыми для детоксикации ОВ. Так, в подробном обзоре [14] авторы приводят данные о 35 установленных продуктах деградации ФОВ (на примере вещеcтва VX) и 42 продуктах деградации СИ. Учитывая, что в запретительные списки Конвенции [1, Annex 1, P. 46] включены многочисленные аналоги и гомологи как ФОВ, так и СИ, можно говорить о сотнях тысяч потенциальных маркеров ОВ, подлежащих идентификации в различных средах [15]. В связи с этим в подавляющем большинстве исследований использованы наиболее универсальные методы – ГХ–МС и ВЭЖХ–МС – для определения в различных средах приоритетных маркеров, относящихся к продуктам трансформации табельных ОВ, подлежащих полному запрету и уничтожению.



Рис. 4.
Структурные формулы МФК и АМФК. МФК – метилфосфоновая кислота, ЭМФК – этилметилфосфоновая кислота; иПрМФК – изопропилметилфосфоновая кислота; ПинМФК – пинаколил метилфосфоновая кислота; иБуМФК – изобутилметилфосфоновая кислота.

В 1970–80-х годах вышли в свет первые публикации по ГХ-методам определения ОВ и продуктов их деструкции, искусственно внесенных в различные среды [16–20]. В 1990 г. был опубликован обзор хроматографических методов, нашедших применение для определения как интактных ОВ, так и некоторых их маркеров с использованием пламенно-фотометрического, азотно-фосфорного, атомно-эмиссионного, фотоионизационного и масс-спектрометрического детекторов [21]. Позднее появились публикации, посвященные применению для этих целей ИК-Фурье-спектрометрических [22–25], масс-спектрометрических [26–28] и других спектральных детекторов [29–34].
В связи с расследованием фактов применения в 1988 г. на севере Ирака зарина и иприта против курдского населения были опубликованы работы [35, 36], в которых впервые показана возможность эффективного ретроспективного (через четыре года после применения ОВ) применения газовой хроматографии с масс-спектрометрическим и тандемным масс-спектрометрическим (МС/МС) детектированием для определения следовых количеств зарина, иприта и продуктов их гидролиза в реальных пробах почвы, одежды и останков жертв химической атаки.
В этот период предпочтение отдавали методу газовой хроматографии по сравнению с ВЭЖХ из-за более высокого уровня аппаратурного оформления и доступности ГХ, а также универсальности, во многом благодаря наличию обширных библиотек масс-спектров [37], полученных в стандартных условиях с ионизацией электронами. Газовая хроматография с самого начала нашла широкое применение для определения интактных ОВ, подавляющее большинство которых относится к легколетучим малополярным соединениям – удобным объектам для анализа этим методом [38]. В то же время продукты трансформации ФОВ – алкиловые эфиры метилфосфоновой кислоты (АМФК), метилфосфоновая кислота (МФК) и диалкиламиноалкилсульфокислоты, а также продукты деструкции СИ – тиодигликоль (ТДГ) и его высшие производные, тиодигликолевая кислота (ТДГК) – полярные соединения, и их ГХ-определение возможно только после перевода в малополярные производные. Дериватизация представляет собой трудоемкую, время- и ресурсозатратную операцию, входящую в процедуру пробоподготовки. Разработке эффективных способов дериватизации полярных производных ФОВ и СИ посвящено множество работ. Описано получение метильных [39–42] триметилсилильных [43–45], трет-бутилдиметилсилильных [46], трифторацетильных [47, 48], гептафторбутирильных [49], пентафторбензоильных [50], пентафторбензильных [51–53] и некоторых других производных [54, 55]. Опыт многочисленных исследований в области дериватизации производных ФОВ и СИ для ГХ-анализа был обобщен в 2003 г. в обзорной статье [56] и позднее в книге [57].
В классическом варианте процедуре дериватизации в ходе пробоподготовки должны предшествовать процедуры извлечения аналита из матрицы, очистки пробы от мешающих примесей и концентрирования. Совмещение этих процедур позволяет существенно упростить и ускорить подготовку проб к газохроматографическому анализу. Этот подхода был реализован в ряде работ, посвященных созданию методик подготовки проб, содержащих маркеры ОВ, для ГХ–МС-анализа с использованием методов твeрдофазной экстракции [58–60], твердофазной микроэкстракции [61–63], жидкостной микроэкстракции [64–66], а также способов, позволяющих совмещать дериватизацию с термодесорбцией [67] или экстракцией [68].
Преимущество использования различных вариантов ВЭЖХ по сравнению с ГХ заключается в возможности прямого (без дериватизации) определения маркеров ОВ независимо от степени их полярности, а также применение более простой пробоподготовки [69, 70]. Хотя методами ГХ–МС можно надежно обнаруживать и определять многие продукты деструкции ОВ [71], они недостаточно универсальны, имеют существенные ограничения по летучести и термостабильности определяемых веществ [72]. При этом некоторые процедуры пробоподготовки весьма продолжительны (несколько часов), а ряд операций (очистка, экстракция, концентрирование, дериватизация) приводит к появлению неконтролируемых погрешностей при количественном анализе [73].
Недостатком ВЭЖХ–МС в сравнении с ГХ–МС является чрезвычайно высокая вариабельность параметров, которые необходимо учитывать при оптимизации условий анализа, что проявляется в недостаточном уровне стандартизации и в отсутствии унифицированных масс-спектральных электронных библиотек для этого метода. Тем не менее, существует ряд задач в рассматриваемой области, которые могут быть решены методом ВЭЖХ–МС и, особенно, ВЭЖХ–МС/МС с гораздо более высокой эффективностью (чувствительностью, селективностью) по сравнению с ГХ–МС (ГХ–МС/МС) [74, 75]. Все большее внимание привлекает такая модификация жидкостной хроматографии, как ультра-ВЭЖХ, т.е. работа при давлении в системе от 500 атм и выше [76]. В сочетании с тандемной масс-спектрометрией и масс-спектрометрией высокого разрешения [77] этот метод позволяет существенно сократить продолжительность анализа, добиться высочайшей эффективности разделения, а также определять следовые количества веществ с минимальными требованиями к процедуре очистки от мешающих примесей в процессе пробоподготовки.
В целом, учитывая критерии существующей системы качества [9, 10], согласно которым определение любого маркера ОВ должно быть подтверждено как минимум двумя независимыми методами, использование ВЭЖХ–МС (ВЭЖХ–МС/МС) наряду с ГХ–МС (ГХ–МС/МС) представляется разумной альтернативой.
Первые публикации, по определению маркеров ОВ с помощью ВЭЖХ с детекторами УФ, флуоресцентным (в виде производных), по светорассеянию и пламенно-фотометрическим [78–81] и ВЭЖХ–МС с ионизацией термораспылением [82] вышли в свет уже в конце 1980-х годов. С появлением методов мягкой ионизации, особенно электрораспылительной, в области ВЭЖХ–МС (ВЭЖХ–МС/МС) определения маркеров и биомаркеров ОВ было опубликовано множество статей экспериментального характера [83–98], а также ряд обзоров [99–103].
Жидкостная хроматография в применении к определению полярных соединений основана на гидрофильных взаимодействиях, когда стационарной фазой служит полярный силикагель или силикагель, модифицированный полярными аминопропильными или цианопропильными группами, а подвижная фаза состоит из воды со смешивающимися с ней растворителями: ацетонитрил, метанол и др. Вода образует иммобилизованный слой на поверхности стационарной фазы, в котором и происходит разделение заряженных молекул аналитов.
Для хроматографического разделения и надежного детектирования маркеров ОВ используют высокотехнологичное оборудование, при этом разделение основано либо на гидрофильно-связанной хроматографии (hydrophilic interaction liquid chromatography, HILIC), либо на обращенно-фазовой ВЭЖХ. В качестве оптимального способа фрагментации принято использовать электрораспылительную ионизацию в комбинации с тандемной масс-спектрометрией (ВЭЖХ–МС/МС) или масс-спектрометрией высокого разрешения (МСВР). Сравнение возможностей этих двух методов [77] показало, что их метрологические характеристики при определении АМФК близки, но метод МСВР имеет преимущества в отношении возможности идентификации новых соединений. В последние годы широко применяют хроматографы, в системе которых создается сверхвысокое давление [76], благодаря чему появляется возможность определять микроколичества компонентов в сложнейших матрицах. Для МС/МС-детектирования следовых количеств алкилфосфоновых кислот используют современные ВЭЖХ-тандемные (трехквадрупольные) масс-спектрометры, обеспечивающие скорость развертки не менее 1 скан/с с покрытием области регистрации ионов в диапазоне 20–600 m/z. Однако для надежного определения низких концентраций (менее 1 нг/мл) необходимо использовать источник электрораспылительной ионизации в комбинации с режимом сканирования типа “мониторинг множественных реакций” (SRM или MRM) и регистрацией отрицательных ионов, образующихся при фрагментации молекул аналитов. В случае использования спектрометров высокого разрешения (МСВР) приемлемым режимом детектирования является сканирование “по выделенным ионам” (SIM-режим) [77].
В зависимости от типа задач по разделению и идентификации маркеров ОВ в водных пробах в хроматографах применяют разнообразные ВЭЖХ-колонки с неподвижными фазами, включающими сорбенты с широкими спектрами состава и сорбционной емкости (табл. 1).
Таблица 1.
Параметры ВЭЖХ-определения алкиловых эфиров метилфосфоновой кислоты
| Метод анализа | Продукт деструкции | Предел обнаружения, нг/мл | Объект анализа | Характеристики колонки | Подвижная фаза | Лите-ратура |
|---|---|---|---|---|---|---|
| ВЭЖХ–МС | иПрМФК | 4 | Плазма | Kromasil 100 C18 column 250 × 1 мм, 5 мкм и Sinergi Hydro_RP column 150 × 2.1 мм, 5 мкм | Подвижная фаза А – 0.1 об. % HCOOH в воде, подвижная фаза Б – 0.1 об. % HCOOH в метаноле, градиент 0.2–0.3 мл/мин | [104] |
| ПинМФК | 0.6 | |||||
| иБуМФК | 1 | |||||
| ВЭЖХ–МС/МС | ЭМФК | 0.8 | Моча | Acclaim C18 150 × 2.1 мм, 2.2 мкм | Подвижная фаза А – 0.5 об. % HCOOH в воде, подвижная фаза Б – 0.5 об. % HCOOH в метаноле, градиент 0.45 мл/мин | [76] |
| иПрМФК | 0.5 | |||||
| ПинМФК | 0.1 | |||||
| иБуМФК | 0.4 | |||||
| ВЭЖХ–МС/МС | ЭМФК | 0.5 | Моча | Acclaim 120 C18 column 150 × 2.1 мм, 2.2 мкм | Подвижная фаза А – 0.5 об. % HCOOH в воде, подвижная фаза Б – 0.5 об. % HCOOH в метаноле, градиент 0.45 мл/мин | [98] |
| иПрМФК | 0.5 | |||||
| ПинМФК | 0.5 | |||||
| иБуМФК | 0.5 | |||||
| ВЭЖХ–МС/МС | МФК | 0.1 | Вода | Luna HILIC 200A 150 × 4.60 мм, 5 мкм | Подвижная фаза А – 10 мМ ацетат аммония в воде, подвижная фаза Б – ацетонитрил, градиент 0.4 мл/мин | [105] |
| ВЭЖХ–МС/МС | МФК | 0.2 | Грунто-пылевая смесь | Luna HILIC 200A 150 × 4.60 мм, 5 мкм | Подвижная фаза А – 10 мМ ацетат аммония в воде, подвижная фаза Б – ацетонитрил, градиент 1.5 мл/мин | [97] |
| ЭМФК | 0.07 | |||||
| иПрМФК | 0.008 | |||||
| ПинМФК | 0.005 | |||||
| иБуМФК | 0.008 |
Разработанные различными исследовательскими группами методы ГХ–МС (ГХ–МС/МС) и ВЭЖХ–МС (ВЭЖХ–МС/МС) определения маркеров ОВ были положены в основу исчерпывающих сборников рекомендованных рабочих процедур (проект “Голубая Книга”), выдержавших три издания [106–108]. Эти сборники методик пробоподготовки и анализа служат настольным руководством для национальных лабораторий, получивших аккредитацию ОЗХО на право анализа экологических проб (табл. 2).
Таблица 2.
Перечень национальных лабораторий, аккредитованных ОЗХО для анализа экологических проб (в редакции документа ОЗХО S/1866/2020 от 31 марта 2020 г.)
| № | Название лаборатории | Государство-участник |
|---|---|---|
| 1 | Belgium Defence Laboratories (DLD) | Бельгия |
| 2 | The Laboratory of Analytical Chemistry, Research Institute of Chemical Defence | Китай |
| 3 | Laboratory of Toxicant Analysis, Institute of Pharmacology and Toxicology, Academy of Military Medical Sciences | Китай |
| 4 | VERIFIN, Finnish Institute for Verification of the Chemical Weapons Convention | Финляндия |
| 5 | DGA Maоtrise NRBC, Analytical Chemistry Department, France | Франция |
| 6 | Bundeswehr Research Institute for Protective Technologies and NBC Protection | Германия |
| 7 | VERTOX Laboratory, Defence Research & Development Establishment | Индия |
| 8 | Defense Chemical Research Laboratory | Иран |
| 9 | TNO Defence, Secunty and Safety | Нидерланды |
| 10 | Analytical Laboratory, Defence Science Technology Organisation | Пакистан |
| 11 | Chemical Analysis Laboratory, CB Department, Agency for Defence Development | Республика Корея |
| 12 | Chemical, Biological and Radiological Defense Research Institute | Республика Корея |
| 13 | Scientific Research Center for CBRN Defense and Ecology. Chemical Analysis and Special Synthesis Laboratory | Румыния |
| 14 | Laboratory for the Chemical and Analytical Control of Military Research Centre | Российская Федерация |
| 15 | Central Chemical Weapons Destruction Analytical Laboratory of the Federal State Unitary Enterprise, “State Scientific Research Institute of Organic Chemistry and Technology” | Российская Федерация |
| 16 | Verification Laboratory, Defence Medical and Environmental Research Institute, DSO National Laboratories | Сингапур |
| 17 | LAVEMA (Laboratorio de Verificaciуn de Armas Quimicas), INTA Campus La Maraсosa | Испания |
| 18 | FOI, CBRN Defence and Security, Swedish Defence Research Agency | Швеция |
| 19 | Spiez Laboratory, Swiss NBC Defence Establishment | Швейцария |
| 20 | Defence Science and Technology Laboratory, Porton Down | Великобритания |
| 21 | Combat Capabilities Development Command, Chemical Biological Center (CBC) Forensic Analytical Laboratory | США |
| 22 | Lawrence Livermore National Laboratory | США |
ОПРЕДЕЛЕНИЕ БИОМАРКЕРОВ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
Цель анализа биомедицинских проб – установить, действительно ли люди или животные подверглись воздействию боевых ОВ, и по возможности идентифицировать тип ОВ и оценить его содержание. В случаях, когда доступ к месту предполагаемого применения ХО задерживается или невозможен, анализ биомедицинских проб, взятых у предположительно подвергшихся воздействию ОВ людей или животных, может оказаться единственным источником информации. С другой стороны, биомедицинские пробы представляют собой весьма сложный объект анализа в связи с чрезвычайно низкими концентрациями аналитов, а также сложностью состава биологических матриц, поэтому важнейшее значение имеет выбор наиболее информативных биомаркеров, эффективных методов пробоподготовки и анализа с учетом обстоятельств предполагаемого инцидента и типа полученной пробы.
Впервые биомаркеры СИ в реальных пробах мочи, взятых у пострадавших в зоне применения ХО на севере Ирака в 1984 г., удалось определить в 1990-x годах [109, 110].
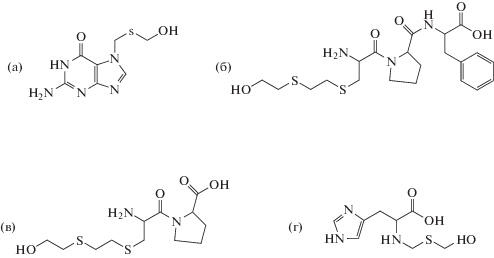
В 2002 г. Научно-консультативный комитет ОЗХО отметил особую актуальность разработок методов определения биомаркеров ОВ в биомедицинских пробах, что вызвало появление множества публикаций в этой области [111]. Первоначально внимание исследователей привлекли формирующиеся в организме низкомолекулярные метаболиты АМФК и МФК при воздействии ФОВ (рис. 1, 2, 4), а также ТДГ, его оксиды и β‑лиазные метаболиты 1,1'-сульфонилбис [2-S (N‑ацетилцистеинил)этан] (СБСНАЭ), 1,1'-сульфонилбис[2-(метилсульфинил)этан] (СБМСЭ) и 1-метилсульфинил-2-[2-(метилтиоэтилсульфонил]этан (МСМТЭСЭ) – при воздействии СИ (рис. 3, 5) [112]. Впоследствии больше внимания уделяли более долгоживущим аддуктам ОВ с белками и ДНК. Методам определения биомаркеров ОВ посвящен ряд исчерпывающих обзоров [113–115]. Отличительными особенностями анализа биомедицинских проб являются более жeсткие требования к пределам обнаружения биомаркеров на уровне 0.1–10 нг/мл, т.е. на три–четыре порядка ниже, чем для экологических проб, а также необходимость определения пептидных аддуктов ОВ, что обусловило применение термина “аддуктомика” для области определения биомаркеров ОВ [116].
Рис. 5.
Продукты взаимодействия иприта с белками в присутствии β-лиазы. СБСНАЭ – 1,1'-сульфонилбис[2-S (N‑ацетилцистеинил)этан], СБМСЭ – 1,1'-сульфонилбис[2-(метилсульфинил)этан] и МСМТЭСЭ – 1-метилсульфинил-2-[2-(метилтиоэтилсульфонил)этан].

Биомаркеры фосфорорганических отравляющих веществ. В качестве биомаркеров, подтверждающих факт воздействия ФОВ на людей или животных, рекомендуется рассматривать как свободные метаболиты этих ОВ, так и их аддукты с протеинами и аминокислотами [117–132]; перечень разработанных методов их определения приведен в табл. 3. Для определения свободных метаболитов (АМФК, МФК) пригодны более простые и доступные методы, но они характеризуются меньшей ретроспективностью. Ковалентно связанные остатки с протеинами (рис. 6) являются более долгоживущими, однако их определение требует применения более сложной аппаратуры и многостадийных процедур пробоподготовки. В отличие от экологических, для биомедицинских проб основным методом разделения является ВЭЖХ, результаты которой существенно зависят от выбранных параметров. Их влияние иллюстрируют примеры определения метаболитов ФОВ, представленные в табл. 1.
Таблица 3.
Биомаркеры фосфорорганических отравляющих веществ и методы их определения
| Проба | Биомаркер | Метод | Литература |
|---|---|---|---|
| ФОВ – зарин | |||
| Моча, кровь | иПрМФК | ВЭЖХ–МС/МС ГХ–МС/МС |
[120–124] |
| Кровь-бутирилхолинэстераза (F-регенерация) |
Фторангидрид иПрМФК |
ГХ–МС/МС | [125, 126] |
| Кровь-бутирилхолинэстераза | Аддукт иПрМФК–серин (в составе нонапептида) |
ВЭЖХ–МС/МС | [127, 128, 131] |
| Кровь-альбумин | Аддукт иПрМФК–тирозин | ВЭЖХ–МС/МС | [129, 130] |
| Кровь-альбумин | Аддукт иПрМФК–тирозин– треонин–лизин |
ВЭЖХ–МСВР | [132] |
| ФОВ – зоман | |||
| Моча, кровь | ПинМФК | ВЭЖХ–МС/МС ГХ–МС/МС |
[118–122] |
| Кровь-бутирилхолинэстераза (F-регенерация) |
Фторангидрид иПинМФК | ГХ–МС/МС | [120–124] |
| Кров-бутирилхолинэстераза | Аддукт ПинМФК–серин (в составе нонапептида) |
ВЭЖХ–МС/МС | [127, 128, 131] |
| Кровь-альбумин | Аддукт ПинМФК–тирозин | ВЭЖХ–МС/МС | [129, 130] |
| ФОВ – VX | |||
| Моча, кровь | ЭМФК | ВЭЖХ–МС/МС ГХ–МС/МС |
[118–122] |
| Кровь-бутирилхолинэстераза (F-регенерация) |
Фторангидрид ЭМФК | ГХ–МС/МС | [120–124] |
| Кровь-бутирилхолинэстераза | Аддукт ЭМФК–серин (в составе нонапептида) |
ВЭЖХ–МС/МС ВЭЖХ–МСВР |
[125, 126] |
| Кровь-альбумин | Аддукт ЭМФК–тирозин | ВЭЖХ–МС/МС ВЭЖХ–МСВР |
[129, 130] |
| Кровь-альбумин | Аддукт 2-(диалкиламино)этантиол–цистеин (в составе дипептида) | ВЭЖХ–МС/МС ВЭЖХ–МСВР |
[118] |
Рис. 6.
Альбуминовые аддукты фосфорорганических отравляющих веществ: (а) нонапептид–АМФК; (б) АМФК–тирозин; (в) 2-(диэтиламино)этантиол–цистеин–пролин; (г) АМФК–тирозин–треонин–лизин.

Биомаркеры сернистого иприта. Первоначально основными биомаркерами СИ считали тиодигликоль и его окисленные производные (рис. 3), однако впоследствии установили, что их наличие в биомедицинских пробах мочи не может являться однозначным доказательством поражения организма данным ОВ. Оказалось, что ТДГ и его окисленные производные, а также тиодигликолевая кислота (TDGK) могут содержаться в моче у лиц, которые воздействию СИ не подвергались. Это связано с возможностью естественного присутствия в пробах мочи отдельных людей достаточно высоких концентраций тиодигликоля и тиодигликолевой кислоты, что указывает на целый ряд профзаболеваний и почечную патологию [133, 134].
Активность ß-лиазы на бисцистеиновых конъюгатах СИ, получаемых из аддуктов глютатиона, ведет к выделению двух сульфоксидных/сульфоновых метаболитов, которые могут быть восстановлены до дериватов тиоэфира для последующего ГХ–МС/МС-анализа (рис. 5). Эти продукты (в совокупности) отсутствуют в моче у лиц, которые не подвергались воздействию СИ. Однозначные выводы об интоксикации ипритом можно сделать по результатам комплексного исследования [135–138], включающего выявление наличия в пробах мочи β-лиазных метаболитов [139–143], ДНК-аддуктов [144, 145] и протеиновых аддуктов [146, 147] (табл. 4).
Таблица 4.
Биомаркеры сернистого иприта и методы их определения
| Проба | Биомаркер | Метод | Литература |
|---|---|---|---|
| Моча, кровь | ТДГ | ГХ–МС/МС ВЭЖХ–МС/МС | [131, 140, 143] |
| Моча | 1,1'-Сульфонилбис [2-(метилсульфинил) этан] | ВЭЖХ–МС/МС ГХ–МС/МС |
[139, 140, 143, 148] |
| Моча | 1,1'-Сульфонилбис [2-S (N-ацетилцистеинил)этан] | ГХ–МС/МС ВЭЖХ–МС/МС | [139, 140, 145, 148] |
| Моча | 1-Метилсульфинил-2-[2-(метилтиоэтилсульфонил)этан] | ВЭЖХ–МС/МС | [140, 141, 143, 148] |
| Моча | Аддукт ГЭТЭ*–гуанин N7 | ВЭЖХ–МС/МС | [144] |
| Кровь | Аддукт СИ–валин | ВЭЖХ–МС/МС | [146] |
| Кровь | Аддукт ГЭТЭ–цистеин (в составе дипептида) |
ВЭЖХ–МС/МС | [147] |
| Кровь | Аддукт ГЭТЭ–цистеин (в составе трипептида) |
ВЭЖХ–МС/МС | [147] |
Для подготовки проб к хроматографическому разделению разработаны различные подходы [98, 113, 149]. Для ретроспективного и чувствительного определения неполярных и малополярных малых органических биомаркеров наиболее часто применяют методы ГХ–МС/МС и ВЭЖХ–МС/МС. Для сильнополярных соединений в основном используют метод ГХ–МС/МС в сочетании с процедурами дериватизации, применяемыми и для анализа экологических проб; для альбуминовых (рис. 6) и ДНК-аддуктов (рис. 7) – ВЭЖХ–МС/МС. Широкое использование масс-спектрометрических детекторов позволяет минимизировать количество ложноположительных и ложноотрицательных результатов. Прослеживается четкая тенденция к повышению эффективности и упрощению аналитических процедур, применяемых для анализа на уровне следовых содержаний ОВ [119, 148, 150–154].
Рис. 7.
ДНК и альбуминовые аддукты сернистого иприта: (а) N7-ГЭТЭ–гуанин; (б) [S-ГЭТЭ]–цистеин–пролин–фенилаланин; (в) [S-ГЭТЭ]–цистеин–пролин; (г) N3-ГЭТЭ–гистидин.
Анализ биомедицинских проб, в отличие от экологических, затруднeн вследствие необходимости их предварительной очистки от белков, солей и прочих мешающих примесей, характерных для матриц биологического происхождения. Этим обусловлено применение в ходе пробоподготовки различных вариантов твeрдофазной экстракции (ТФЭ), фильтрования, центрифугирования и прочих способов очистки [116] (рис. 8). Например, для определения альбуминового аддукта иприта (S-гидроксиэтилтиоэтил–цистеин–пролин–фенилаланин) в плазме крови человека, процедура пробоподготовки включает следующие стадии: осаждение белков в форме твердого осадка ацетоном с последующим центрифугированием и фильтрованием; добавление 0.1%-ного раствора муравьиной кислоты в воде для гомогенизации осадка после его высушивания при комнатной температуре; ферментативное расщепление белков протеиназой К при 50°C в слабощелочной среде в присутствии гидрокарбоната аммония в течение нескольких часов при постоянном перемешивании; центрифугирование, фильтрование, очистку на картриджах, заполненных полимерным сорбентом, модифицированным гидрофобными функциональными группами, с последующим элюированием аналитов ацетонитрилом; упаривание аликваты при пониженном давлении при 50°C; повторное растворение в минимальном объеме смеси ацетонитрил–вода (1 : 4) для последующего анализа. В результате выполнения процедуры удается достичь выхода аналита, близкого к 100%. Данный способ успешно использован для определения аддукта СИ с трипетидом методом жидкостной хроматографии в сочетании с тандемным масс-спектрометрическим детектированием.

Об уровне достигнутых пределов обнаружения ряда метоболитов можно судить из данных, приведенных в табл. 5.
Таблица 5.
Достигнутые пределы обнаружения некоторых биомаркеров
| Соединение | Метод анализа | Предел обнаружения, нг/мл | Литература |
|---|---|---|---|
| МФК ЭМФК иПрМФК ПинМФК |
ВЭЖХ–МС/МС | 0.1 | [132] |
| Тот же | 0.07 | [97] | |
| » | 0.008 | [97] | |
| » | 0.005 | [97] | |
| » | 0.139 | [151] | |
| » | 0.07 | [70] | |
| » | 0.04 | [70] | |
| » | 0.01 | [70] | |
| СБСНАЭ* СБМСЭ МСМТЭСЭ |
ВЭЖХ–МС/МС | 0.05 | [153] |
| Тот же | 2 | [98] | |
| » | 1 | [98] | |
| » | 0.5 | [148] | |
| » | 4 | [148] | |
| » | 4 | [148] | |
| ТДГK | ВЭЖХ–МС/МС | 10 | [96] |
| ГХ–МС | 20 | [58] | |
| ГХ–МС | 50 | [154] |
* Обозначения – см. рис. 5.
Методические указания по проведению пробоподготовки и анализа биомедицинских проб вошли в виде отдельных разделов в последнее издание “Рекомендованных операционных процедур…” [108] и служат руководством для национальных лабораторий, получивших аккредитацию ОЗХО в области анализа биомедицинских проб (табл. 6)
Таблица 6.
Перечень национальных лабораторий, аккредитованных ОЗХО для анализа биомедицинских проб (в редакции документа ОЗХО S/1661/2018 от 14 августа 2018 г.)
| № | Название лаборатории | Государство-участник |
|---|---|---|
| 1 | Defence Science and Technology Group | Австралия |
| 2 | Laboratory of Toxicant Analysis, Academy of Military Medical Sciences | Китай |
| 3 | Laboratory of Analytical Chemistry, Research Institute of Chemical Defence | Китай |
| 4 | Finnish Institute for Verification of the Chemical Weapons Convention (VERIFIN) | Финляндия |
| 5 | DGA Maitrise NRBC, Département D’analyses Chimiques | Франция |
| 6 | Bundeswehr Institute of Pharmacology and Toxicology | Германия |
| 7 | Vertox-Biochemistry Division, Defence Research and Development Establishment | Индия |
| 8 | TNO Defence, Security and Safety | Нидерланды |
| 9 | Chemical Analysis Laboratory, CB Department, Agency for Defence Development | Республика Корея |
| 10 | Laboratory for the Chemical and Analytical Control of Military Research Centre | Российская Федерация |
| 11 | Laboratory of Chemical Analytical Control and Biotesting, Research Institute of Hygiene, Occupational Pathology and Human Ecology (RIHOPHE) | Российская Федерация |
| 12 | Verification Laboratory, Defence Medical and Environmental Research Institute, DSO National Laboratories | Сингапур |
| 13 | Swedish Defence Research Agency (FOI) | Швеция |
| 14 | Defence Science and Technology Laboratory, Chemical and Biological Systems, Porton Down | Великобритания |
| 15 | Centers for Disease Control and Prevention | США |
| 16 | Edgewood Chemical and Biological Forensic Analytical Center | США |
| 17 | Lawrence Livermore National Laboratory | США |
Рассмотренные подходы к определению маркеров и биомаркеров основных ОВ (ФОВ и СИ) в экологических и биомедицинских пробах методом хромато-масс-спектрометрии прошли неоднократную апробацию как в системе профессионального тестирования ОЗХО, так и в ряде инцидентов, сопровождавшихся воздействием ОВ на окружающую среду, животных и людей.
В целом следует отметить, что определение маркеров и биомаркеров ОВ можно рассматривать как одну из наиболее освоенных и быстро развивающихся областей аналитической химии и токсикологии. При выборе метода анализа проблема заключается не в отсутствии соответствующих методик в литературных источниках, а в их многочисленности и разнообразии, а также в наличии соответствующего оборудования.
Разнообразие имеющихся современных методических подходов позволяет выбрать оптимальную стратегию анализа с учетом специфики решаемых задач и доступного оборудования. В целях доказательной диагностики при подозрениях на возможность интоксикации человека ФОВ или СИ существует потребность в разработке аттестованных методик, характеризующихся высокими точностью чувствительностью и воспроизводимостью. Остаются актуальными работы по поиску новых высокохарактеристичных биомаркеров. Очевидное преимущество использования аппаратуры последнего поколения на основе тандемной масс-спектрометрии или масс-спектрометрии высокого разрешения, отличающихся высокой чувствительностью и практическим отсутствием зависимости от влияния матрицы, заключается в возможности получения информации, достаточной для достоверной идентификации определяемых биомаркеров, на основании которой можно сделать однозначный вывод – подвергался ли организм интоксикации конкретным ОВ. Решение этой задачи вносит существенный вклад в совершенствование международной системы верификации, обеспечивающей контроль за полным запрещением и нераспространением химического оружия.
Работа выполнена при поддержке гранта РФФИ № 18-33-20068 мол_а_вед.
Список литературы
Convention on the Prohibition of the Development, Stockpiling and Use of Chemical Weapons and on their Destruction. Technical Secretariat of the Organisation for Prohibition of Chemical Weapons, The Hague, 1997. http://www.opcw.org. (22.05.2020).
Yanagisawa N., Morita H., Nakajima T., Okudera H., Shimizu M., Hirabayashi H., Nohara M., Midorikawa Y., Mimura S. Sarin poisoning in Matsumoto, Japan // Lancet. 1995. V. 346. P. 290.
Nagao M., Takatori T., Matsuda Y., Nakajima M., Iwase H., Iwadate K. Definitive evidence for the acute sarin poisoning diagnosis in the Tokyo subway // Toxicol. Appl. Pharmacol. 1997. V. 144. P. 198.
Sellström A., Cairns S., Barbeschi M. United Nations Mission to Investigate Allegations of the Use of Chemical Weapons in the Syrian Arab Republic, Final Report. https://unoda-web.s3.amazonaws.com/wp-content/uploads/2013/12/report.pdf. (27.01.2020).
Chai P.R., Boyer E.W., Al-Nahhas H., Erickson T.B. Toxic chemical weapons of assassination and warfare: nerve agents VX and sarin // Toxicol. Commun. 2017. V. 1. P. 21.
Brletich N.R., Waters M.J., Bowen G.W., Tracy M.F. US and foreign military CWA detectors / Worldwide Chemical Detection Equipment Handbook. Aberdeen, MD: Chemical and Biological Defense Information Analysis Center, 1995. 470 p.
Kanu A.B., Haigh P.E., Hill H.H. Surface detection of chemical warfare agent simulants and degradation products // Anal. Chim. Acta. 2005. V. 553. P. 148.
Рыбальченко И.В. Идентификация токсичных химикатов // Рос. хим. журн. (Журн. Рос. хим. о-ва им. Д.И. Менделеева). 2002. Т. 46. № 4. С. 113.
OPCW Quality Management System Document No.: QDOC/LAB/WI/PT04. 11 March 2019. Available from the OPCW Laboratory: proficiency@opcw.org (22.04.2020).
OPCW Quality Management System Document No.: QDOC/LAB/WI/BioPT04. 6 January 2020. Available from the OPCW Laboratory: proficiency@opcw.org (22.04.2020).
Tu A.T. Toxicological and chemical aspects of sarin terrorism in Japan in 1994 and 1995 // Toxin Reviews. 2007. V. 26. P. 231.
United Nations Document A/67/997–S/2013/553. Distr.: General 16 September 2013. 41 p.
John H., van der Schans M.J., Koller M., Spruit H.E.T., Worek F., Thiermann H., Noort D. Fatal sarin poisoning in Syria 2013: forensic verification within an international laboratory network // Forensic Toxicol. 2018. V. 36. P. 61.
Munro N.B., Talmage S.S., Griffin G.D., Waters, L.C., Watson A.P., King J.F., Hauschild V. The sources, fate, and toxicity of chemical warfare agent degradation products // Environ. Health Perspect. 1999. V. 107. P. 933.
Рыбальченко И.В., Киреев А.Ф., Цехмистер В.И. Химико-аналитический контроль в рамках Конвенции о запрещении химического оружия // Рос. хим. журн. (Журн. хим. об-ва им. Д.И. Менделеева). 1994. Т. 45. № 5. С. 11.
Harvey D.J., Horning M.G. Derivatives for the characterization of alkyl- and aminoalkylphosphonates by gas chromatography and gas chromatography-mass spectrometry // J. Chromatogr. 1973. V. 79. P. 65.
Sass S., Fisher T.L. Chemical ionization and electron impact mass spectrometry of some organophosphonate compounds // Org. Mass Spectrom. 1979. V. 14. P. 257.
Tingfa D. Gas chromatographic determination of O‑ethyl S-(N, N-diisopropylamino) ethyl methylphosphonothiolate and O,O-diisopropyl S-benzyl phosphorothiolate as corresponding phosphonofluoridate and phosphorofluoridate // Int. Environ. Anal. Chem. 1986. V. 27. P. 151.
Wils E.R.J., Hulst A.G. Mass spectra of some pinacolyl containing organophoshorus compounds // Org. Mass Spectrom. 1986. V. 21. P. 763.
Reynolds M.L., Little P.J., Thomas B.F., Bagley R.B., Martin B.R. Relationship between the disposition of [3H] soman and its pharmacological effects in mice // Toxicol. Appl. Pharmacol. 1985. V. 80. P. 409.
Witkiewicz Z., Mazurek M., Szulc J. Chromatographic analysis of chemical warfare agents // J. Chromatogr. 1990. V. 503. P. 293.
Söderström M.T., Ketola R.A. Identification of nerve agents and their homologues and dialkyl methylphosphonates by gas chromatography/fourier transform infrared spectrometry (GC-FTIR) // Fresenius J. Anal. Chem. 1994. V. 350. P. 162.
Söderström M.T., Ketola R.A., Kostiainen O. Identification of some nerve agent homologues and dialkyl methylphosphonates by gas chromatography/Fourier transform infrared spectrometry // Fresenius J. Anal. Chem. 1995. V. 352. P. 550.
Durst H.D., Mays J.R., Ruth J.L., Williams B.R., Duevel R.V. Micro-scale synthesis and in-situ spectroscopic characterization of some weapons related organophosphate compounds // Anal. Lett. 1998. V. 31. P. 1429.
Киреев А.Ф., Рыбальченко И.В., Савчук В.И., Суворкин В.Н., Холстов В.И. Идентификация производных алкилфосфоновых кислот методами ИК и масс-спектрометрии // Журн. аналит. химии. 2000. Т. 55. С. 933. (Kireev A.F., Rybal’chenko I.V., Savchuk V.I., Suvorkin V.N., Kholstov V.I. Identification of alkylphosphonic acid derivatives by IR and mass spectrometry // J. Analyt. Chem. 2000. V. 55. P. 837.)
Kientz C.E. Chromatography and Mass Spectrometry of Chemical Warfare Agents, Toxins and Related Compounds: State of the Art and Future Prospects // J. Chromatogr. A. 1998. V. 814. P. 1.
Hooijschuur E.W.J., Kientz C.E., Udo A.Th., Brinkman U.A.T. Analytical Separation Techniques for the Determination of Chemical Warfare Agents // J. Chromatogr. A. 2002. V. 982. P. 177.
D’Agostino P.A. Chemical warfare agents / Handbook of Analytical Separations. V. 6. Forensic Science / Ed. Bogusz M.J. Amsterdam: Elsevier B, 2008. P. 839.
Lakso H.A., Ng W.F. Determination of Chemical Warfare Agents in Natural Water Samples by Solid-Phase Microextraction // Anal. Chem. 1997. V. 69. P. 1866.
Kuitunen M.L., Hartonen K., Riekkola M.L. Analysis of chemical warfare agents in soil samples by off-line supercritical fluid extraction and capillary gas chromatography // J. Microcol. Sep. 1991. V. 3. P. 505.
Mazurek M., Witkiewicz Z., Popiel S., Sliwakowski M. Capillary gas chromatography–atomic emission spectroscopy–mass spectrometry analysis of sulphur mustard and transformation products in a block recovered from the Baltic Sea // J. Chromatogr. A. 2001. V. 919. P. 133.
Schoene K., Strihanses J., Bruch H.L., Konig A. Speciation of arsenic-containing chemical warfare agents by gas chromatographic analysis after derivatization with thioglycolic acid methyl ester // J. Chromatogr. 1992. V. 605. P. 257.
Eckert-Tilotta S.E., Hawthorne S.B., Miller D.J. Comparison of commercially available atomic emission and chemiluminescence detectors for sulfur-selective gas chromatographic detection // J. Chromatogr. A. 1992. V. 591. P. 313.
Wylie P.L., Sullivan J.J., Quimby B.D. An investigation of gas chromatography with atomic emission detection for the determination of empirical formulas // J. High Resolut. Chromatogr. 1990. V. 13. P. 499.
Black R.M., Clarke R.J., Cooper D.B., Read R.W., Utley D. Application of headspace analysis, solvent extraction, thermal desorption and gas chromatography–mass spectrometry to the analysis of chemical warfare samples containing sulphur mustard and related compounds // J. Chromatogr. A. 1993. V. 637. P. 71.
Black R.M., Clarke R.J., Read R.W. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to the analysis of chemical warfare samples, found to contain residues of the nerve agent sarin, sulphur mustard and their degradation products // J. Chromatogr. A. 1994. V. 662. P. 301.
OPCW Central Analytical Database, e-OCAD V. 2019, Technical Secretariat of the Organization for the OPCW, Heulweg, The Netherlands. 2019. Available from the OPCW Laboratory: proficiency@opcw.org. (22.04.2020).
Borrett V.T., Mathews R.J., Mattsson E.R. Verification of the Chemical Weapons Convention: mass spectrometry of alkyl methylphosphonofluoridates // Aust. J. Chem. 1994. V. 47. P. 2065.
Verweij A., Degenhardt C.E.A.M., Boter H.L. The occurrence and determination of PCH3-containing compounds in surface water // Chemosphere. 1979. V. 8. P. 115.
Tornes J.A., Johnsen B.A. Gas chromatographic determination of methylphosphonic acids by methylation with trimethylphenylammonium hydroxide // J. Chromatogr. A. 1989. V. 467. P. 129.
Amphaisri K., Palit M., Mallard G. Thermally assisted methylation and subsequent silylation of scheduled acids of chemical weapon convention for on-site analysis and its comparison with the other methods of methylation // J. Chromatogr. A. 2011. V. 1218. P. 972.
Valdez C.A., Leif R.N., Alcaraz A. Effective methylation of phosphonic acids related to chemical warfare agents mediated by trimethyloxonium tetrafluoroborate for their qualitative detection and identification by gas chromatography-mass spectrometry // Anal. Chim. Acta. 2016. V. 933. P. 134.
Bauer G., Vogt W. Gas chromatographic determination of acids derived from phosphorus by trimethylsilylation with N,O-bis(trimethylsilyl)trifluoroacetamide // Anal. Chem. 1981.V. 53. P. 917.
Rohrbaugh D.K., Sarver E.W. Detection of alkyl methylphosphonic acids in complex matrices by gas chromatography–tandem mass spectrometry // J. Chromatogr. A. 1998. V. 809. P. 141.
Creasy W.R., Rodrigues A.A., Stuff J.R., Warren R.W. Atomic emission detection for the quantitation of trimethylsilyl derivatives of chemical-warfare-agent related compounds in environmental samples // J. Chromatogr. A. 1995. V. 709. P. 333.
Purdon J.G., Pagotto J.G., Miller R.K. Preparation, stability and quantitative analysis by gas chromatography and gas chromatography–electron impact mass spectrometry of tert.-butyldimethylsilyl derivatives of some alkylphosphonic and alkyl methylphoshonic acids // J. Chromatogr. A. 1989. V. 475. P. 261.
Pardasani D., Palit M., Gupta A.K., Kanaujia P.K., Dubey D.K. Gas chromatography–mass spectrometry analysis of trifluoroacetyl derivatives of precursors of nitrogen and sulfur mustards for verification of chemical weapons convention // J. Chromatogr. A. 2004. V. 1059. P. 157.
Popiel S., Nawała J., Dziedzic D., Söderström M., Vanninen P. Determination of mustard gas hydrolysis products thiodiglycol and thiodiglycol sulfoxide by gas Chromatography-tandem mass spectrometry after trifluoroacetylation // Anal. Chem. 2014. V. 86. P. 5865.
Riches J., Read R.W., Black R.M. Analysis of the sulphur mustard metabolites thiodiglycol and thiodiglycol sulphoxide in urine using isotope-dilution gas chromatography-ion trap tandem mass spectrometry // J. Chromatogr. B. 2007. V. 845. P. 114.
Black R.M., Read R.W. Methods for the analysis of thiodiglycol sulphoxide, a metabolite of sulphur mustard, in urine using gas chromatography – mass spectrometry // J. Chromatogr. A. 1991. V. 558. P. 393.
Shih M.L., Smith J.R., McMonagle J.D., Dolzine T.W., Gresham V.C. Detection of metabolites of toxic alkylmethylphosphonates in biological samples // Biol. Mass Spectrom. 1991. V. 20. P. 717.
Fredriksson S., Hammarstrom L., Henriksson L., Lakso H. Trace determination of alkyl methylphosphonic acids in environmental and biological samples using gas chromatography/negative-ion chemical ionization mass spectrometry and tandem mass spectrometry // J. Mass Spectrom. 1995. V. 30. P. 1133.
Miki A., Katagi M., Tsuchihashi H., Yamashita M. Determination of alkylmethylphosphonic acids, the main metabolites of organophosphorus nerve agents, in biofluids by gas chromatography-mass spectrometry and liquid-liquid-solid-phase-transfer-catalyzed pentafluorobenzylation // J. Anal. Toxicol. 2009. V. 23. P. 86.
Nyholm J.R., Gustafsson T., Östin A. Structural determination of nerve agent markers using gas chromatography mass spectrometry after derivatization with 3-pyridyldiazomethane // J. Mass Spectrom. 2013. V. 48. P. 813.
Байгильдиев Т.М., Вокуев М.Ф., Орешкин Д.В., Браун А.В., Годовиков И.А., Рыбальченко И.В., Родин И.А. п-Метоксифенацилбромид – универсальный реагент для определения алкилфосфоновых и алкилметилфосфоновых кислот методами высокоэффективной жидкостной и газовой хроматографии с масс-спектрометрическим детектированием // Масс-спектрометрия. 2019. T. 16. C. 180.
Black R.M., Muir B. Derivatisation reactions in the chromatographic analysis of chemical warfare agents and their degradation products // J. Chromatogr. A. 2003. V. 1000. P. 253.
Kroening K.K., Easter R.N., Richardson D.D., Willison S.A., Caruso J.A. Analysis of Chemical Warfare Degradation Products. Chichester, UK: John Wiley & Sons Ltd., 2011. 146 p.
Saradhi U.V.R.V., Prabhakar S., Reddy T.J., Vairamani M. Ion-pair solid-phase extraction and gas chromatography-mass spectrometric determination of acidic hydrolysis products of chemical warfare agents from aqueous samples // J. Chromatogr. A. 2006. V. 1129. P. 9.
Saradhi U.V.R.V., Prabhakar S., Reddy T.J., Murty M.R.V.S. Gas chromatographic-mass spectrometric determination of alkylphosphonic acids from aqueous samples by ion-pair solid-phase extraction on activated charcoal and methylation // J. Chromatogr. A. 2007. V. 1157. P. 391.
Берзин И.А., Романов В.С., Савельева Е.И., Рыбальченко И.В., Новиков С.В., Василевский С.В., Гончаров В.М. Определение метаболитов фосфорорганических отравляющих веществ в биомедицинских пробах с использованием твердофазной экстракции // Судебная медицина. Т. 10. 2009. С. 44.
Lakso H.-Å, Ng W.F. Determination of chemical warfare agents in natural water samples by solid-phase microextraction // Anal. Chem. 1997. V. 69. P. 1866.
Sng M.T., Ng W.F. In-situ derivatisation of degradation products of chemical warfare agents in water by solid-phase microextraction and gas chromatographic-mass spectrometric analysis // J. Chromatogr. A. 1999. V. 832. P. 173.
Zygmunt B., Zaborowska A., Swiatlowska J., Namiesnik J. Solid phase microextraction combined with gas chromatography – A powerful tool for the determination of chemical warfare agents and related compounds // Curr. Org. Chem. 2007. V. 11. P. 241.
Lee H.S.N., Sng M.T., Basheer C., Lee H.K. Determination of degradation products of chemical warfare agents in water using hollow fibre-protected liquid-phase microextraction with in-situ derivatisation followed by gas chromatography–mass spectrometry // J. Chromatogr. A. 2007. V. 1148. P. 8.
Dubey D.K., Pardasani D., Gupta A.K., Palit M., Kanaujia P.K., Tak V. Hollow fiber-mediated liquid-phase microextraction of chemical warfare agents from water // J. Chromatogr. A. 2006. V. 1107. P. 29.
Palit M., Pardasani D., Gupta A.K., Dubey D.K. Application of single drop microextraction for analysis of chemical warfare agents and related compounds in water by gas chromatography/mass spectrometry // Anal. Chem. 2005. V. 77. P. 711.
Terzic O. Screening of degradation products, impurities and precursors of chemical warfare agents in water and wet or dry organic liquid samples by in-sorbent tube silylation followed by thermal desorption–gas chromatography–mass spectrometry // J. Chromatogr. A. 2010. V. 1217. P. 4987.
Pardasani D., Palit M., Gupta A.K., Kanaujia P.K., Sekhar K., Dubey D.K. Microemulsion mediated in situ derivatization-extraction and gas chromatography-mass spectrometric analysis of alkylphosphonic acids // J. Chromatogr. A. 2006. V. 1108. P. 166.
Black R.M., Read R.W. Analysis of Beta-Lyase metabolites of sulfur mustard in urine by electrospray liquid chromatography-tandem mass spectrometry // J. Chromatogr. A. 1998. V. 794. P. 233.
Roen B.T., Sellevag S.R., Lundanes E. On-line solid phase extraction-liquid chromatography-mass spectrometry for trace determination of nerve agent degradation products in water samples // Anal. Chim. Acta. 2013. V. 761. P. 109.
Савельева Е.И., Радилов А.С., Кузнецова Т.А., Волынец Н.Ф. Определение метилфосфоновой кислоты и ее эфиров как химических маркеров фосфорорганических отравляющих веществ // Журн. прикл. химии. 2001. Т. 74. № 10. С. 1671.
Kostiainen O. Gas chromatography in screening of chemicals related to the chemical weapon convention / Encyclopedia of Analytical Chemistry. John Wiley & Sons Ltd., 2016. https://doi.org/10.1002/9780470027318.a0412.pub3 (22.02.2020).
Василевский С.В., Киреев А.Ф., Рыбальченко И.В., Суворкин В.Н. Масс-спектрометрическая идентификация силилированных производных алкилфосфоновых, алкилтиофосфоновых и диалкиламидофосфоновых кислот // Журн. аналит. xимии. 2002. Т. 57. С. 597. (Vasilevskii S.V., Kireev A.F., Rybal’chenko I.V., Suvorkin V.N. Identification of sililated derivatives of alkilphosphonic, alkiltiophosphonic, and dialkilamidophosphonic acids by mass spectrometry // J. Analyt. Chem. 2002. V. 57. P. 491.)
Read R.W., Black R.M. Application of liquid chromatography-atmospheric pressure chemical ionization mass spectrometry, and tandem mass spectrometry, to the analysis and identification of degradation products of chemical warfare agents // J. Chromatogr. A. 1997. V. 759. P. 79.
Read R.W., Black R.M. Analysis of beta-lyase metabolites of sulfur mustard in urine by electrospray liquid chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 346.
Родин И.А., Браун А.В., Ставрианиди А.Н., Шпигун О.А., Рыбальченко И.В. Обнаружение маркеров нервно-паралитических отравляющих веществ методом ультра-высокоэффективной жидкостной хроматографии – тандемной масс-спектрометрии // Аналитика и контроль. 2012. Т. 16. С. 254.
Hamelin E.I., Bragg W., Shaner R L., Swaim L.L., Johnson R C. Comparison of high-resolution and tandem mass spectrometry for the analysis of nerve agent metabolites in urine // Rapid Commun. Mass Spectrom. 2013. V. 27. P. 1697.
Bossle P.C., Martin J.J., Sarver E.W., Sommer H.Z. Highperformance liquid chromatography of alkyl methylphosphonic acids by derivatization // J. Chromatogr. 1983. V. 267. P. 209.
Roach M.C., Ungar L.W., Zare R.N., Reiner L.M., Pompliano D.L., Frost J.W. Fluorescence detection of alkylphosphonic acids using p-(9-anthroyloxy)phenacyl bromide // Anal. Chem. 1987. V. 59. P. 1056.
Bossle P.C., Reutter D.J., Sarver E.W. Analysis of alkyl methylphosphonic acids in aqueous matrices by ion-pair reverse-phase ion chromatography // J. Chromatogr. 1988. V. 407. P. 399.
Kientz Ch.E., Verweij A., Boter H.L., Poppema A., Frei R.W., de Jong G.J., Brinkman U.A.Th. On-line flame photometric detection in micro-column liquid chromatography // J. Chromatogr. 1989. V. 467. P. 385.
Wils E.R.J., Hulst A.G. Determination of organophosphorus acids by thermospray-liquid chromatography-mass spectrometry // J. Chromatogr. 1988. V. 454. P. 261.
Read R.W., Black R.M. Analysis of the sulfur mustard metabolite 1,1'-sulfonylbis[2-S-(N-acetylcysteinyl)ethane] in urine by negative ion electrospray liquid chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 352.
D’Agostino P.A., Hancock J.R., Provost L.R. Analysis of o-ethyl s-[2-(diisopropylamino)ethyl] methylphosphonothiolate (VX) and its degradation products by packed capillary liquid chromatography-electrospray mass spectrometry // J. Chromatogr. A. 1999. V. 837. P. 93.
Brickhouse M.D., Creasy W.R., Williams B.R., Morrissey K.M., O’Connor R.J., Durst H.D. Multiple-technique analytical characterization of a mixture containing chemical weapons simulant from a munition // J. Chromatogr. A. 2000. V. 883. P. 185.
D’Agostino P.A., Hancock J.R., Provost L.R. Determination of sain, soman and their hydrolysis products in soil by packed capillary liquid chromatographyelectroapray mass spectrometry // J. Chromatogr. A. 2001. V. 912. P. 291.
D’Agostino P.A., Chenier C.L., Hancock J.R. Packed capillary liquid chromatography-electrospray mass spectrometry of snow contaminated with sarin // J. Chromatogr. A. 2002. V. 950. P. 149.
D’Agostino P.A., Hancock J.R., Chenier C.L. Mass spectrometric analysis of chemical warfare agents and their degradation products in soil and synthetic samples // Eur. J. Mass Spectrom. 2003. V. 9. P. 609.
D’Agostino P.A., Hancock J.R., Chenier C.L. Packed capillary liquid chromatography-electrospray ionization (tandem) mass spectrometry of mustard hydrolysis products in soil // J. Chromatogr. A. 2004. V. 1058. P. 97.
Ciner F.L., McCord C.E., Plunkett R.W., Jr., Martin M.F., Croley T.R. Isotope dilution LC/MS/MS for the detection of nerve agent exposure in urine // J. Chromatogr. B. 2007. V. 846. P. 42.
John H., Worek F., Thiermann H. LC-MS-based procedures for monitoring of toxic organophosphorus compounds and verification of pesticide and nerve agent poisoning // Anal. Bioanal. Chem. 2008. V. 391. P. 97.
Tak V., Pardasani D., Kanaujia P.K., Dubey D.K. Liquid-liquid-liquid microextraction of degradation products of nerve agents followed by liquid chromatography-tandem mass spectrometry // J. Chromatogr. A. 2009. V. 1216. P. 4319.
Koller M., Becker Ch., Thiermann H., Worek F. GC-MS and LC-MS analysis of nerve agents in body fluids: intra-laboratory verification test using spiked plasma and urine samples // J. Chromatogr. B. 2010. V. 878. P. 1226.
Schwarzenberg A., Ichou F., Cole R.B., Machuron-Mandard X., Junot C., Lesage D., Tabet J.-C. Identification tree based on fragmentation rules for structure elucidation of organophosphorus esters by electrospray mass spectrometry // J. Mass Spectrom. 2013. V. 48. P. 576.
Rodin I., Stavrianidi A., Smirnov R., Braun A., Shpigun O., Rybalchenko I. New techniques for nerve agent oxidation products determination in environmental water by high-performance liquid chromatography-mass spectrometry (HPLC-MS) and capillary electrophoresis (CE) with direct ultraviolet (UV) detection// Environ. Forensics. 2013. V. 14. P. 87.
Rodin I., Braun A., Stavrianidi A., Baygildiev T., Rybalchenko I., Shpigun O. A validated LC-MS/MS method for fast detection of thiodiglycolic acid in aqueous samples // Int. J. Environ. Anal. Chem. 2016. V. 96. P. 436.
Baygildiev T., Rodin I., Stavrianidi A., Braun A., Lebedev. A., Rybalchenko I., Shpigun O. Hydrophilic interaction liquid chromatography-tandem mass spectrometry methylphosponic and alkyl methylphosphonic acids determination in environmental samples after pre-column derivatization with p-bromophenacyl bromide // J. Chromatogr. A. 2016. V. 1442. P. 19.
Rybalchenko I., Rodin I., Baygildiev T., Stavrianidi A., Braun A., Morozik Y., Shpigun O. Novel analytical approaches to determination of chemical warfare agents and related compounds for verification of nonproliferation of chemical weapons // Pure Appl. Chem. 2017. V. 89. P. 1491.
Black R.M., Read R.W. Liquid chromatography/mass spectrometry in analysis of chemicals related to the chemical weapons convention / Chemical Weapons Convention Chemical Analysis / Ed. Mesilaakso M. Chichester, UK: Jon Wiley&Sons Ltd, 2005. P. 476.
Wood M., Laloup M., Samyn N., Fernandez M.M.R., Bruijn E.A., Maes R.A.A., Boeck G.D. Recent applications of liquid chromatography–mass spectrometry in forensic science // J. Chromatogr. A. 2006. V. 1130. P. 3.
Рыбальченко И.В. Роль аналитической химии в обеспечении международного контроля исполнения Конвенции о запрещении химического оружия // Рос. хим. журнал (Журн. хим. об-ва им. Д.И. Менделеева). 2007. Т. 51. № 2. С. 101.
Алексенко С.С. Жидкостная хроматография с масс‑ спектрометрическим детектированием для определения отравляющих веществ и продуктов их деструкции // Журн. аналит. химии. 2012. Т. 67. С. 116. (Aleksenko S.S. Liquid chromatography with mass-spectrometric detection for the determination of chemical warfare agents and their degradation products // J. Analyt. Chem. 2012. V. 67. P. 82.)
Oyler J., Maistros K., Oyler B., Kilgour D. Liquid chromatography–mass spectrometry in analysis of chemicals related to the chemical weapons convention // Wiley Online Library. 13 March 2020. 10.1002/9780470027318.a0408.pub3 (22.04.2020)
Родин И.А., Браун А.В., Ананьева И.А., Шпигун О.А., Савельева Е.И., Рыбальченко И.В., Болотов С.Л., Родченков Г.М. Обнаружение маркеров нервно-паралитических отравляющих веществ методом жидкостной хроматомасс-спектрометрии // Масс-спектрометрия. 2011. Т. 8. С. 45. (Rodin I.A., Braun A.V., Anan’eva I.A., Shpigun O.A., Savel’eva E.I., Rybal’chenko I.V., Bolotov S.L., Rodchenkov G.M. Detection of nerve agent markers by liquid chromatography-mass spectrometry // J. Analyt. Chem. 2011. V. 66. P. 1417.)
Rodin I., Baygildiev T., Stavrianidi A., Braun A., Rybalchenko I., Shpigun O. Hydrophilic interaction liquid chromatography tandem mass spectrometry methylphosphonic acid determination in water samples after derivatization with p-bromophenacyl bromide // Chromatographia. 2015. V. 78. P. 585.
Recommended Operating Procedures for Sampling and Analysis in the Verification of Chemical Disarmament / Ed. Rautio M. Helsinki: The Ministry for Foreign Affairs of Finland, 1994. 543 p. http://www.helsinki.fi/verifin/bluebook (12.12.2019).
Recommended Operating Procedures for Analysis in the Verification of Chemical Disarmament / Ed. Vanninen P. Helsinki: The Ministry for Foreign Affairs of Finland, 2011. 655 p. http://www.helsinki.fi/verifin/bluebook (12.12.2019).
Recommended Operating Procedures for Analysis in the Verification of Chemical Disarmament / Ed. Vanninen P. Helsinki: The Ministry for Foreign Affairs of Finland, 2017. 809 p. http://www.helsinki.fi/verifin/bluebook (12.12.2019).
Black R.M., Read R.W. Biological fate of sulphur mustard, 1,1'-thiobis(2-chloroethane): identification of beta-lyase metabolites and hydrolysis products in human urine // Xenobiotica. 1995. V. 25. P. 167.
Benschop H.P., Schans G.P., Noort D., Fidder A., de Jong L.P.A. Verification of exposure to sulfur mustard in two casualties of the Iran-Iraq conflict // J. Anal. Toxicol. 1997. V. 21. P. 249.
Barr J.R. Analysis of biological samples for chemical warfare agents // J. Anal. Toxicol. 2008. V. 32. P. 1.
Рыбальченко И.В., Хлебникова Н.С., Савельева Е.И., Радилов А.С., Рембовский В.Р. Газохроматографический анализ биологических проб. Определение метаболитов токсичных химикатов // Рос. хим. журн. (Журн. хим. об-ва им. Д.И. Менделеева). 2005. Т. 49. № 2. С. 26.
Black R.M. Historical and perspectives of bioanalytical methods for chemical warfare. Modifications to the organophosphorus nerve agent-protein adduct refluoridation. Method for retrospective analysis of nerve agent exposures // J. Anal. Toxicol. 2008. V. 32. P. 2.
Black R.M. Historical and perspectives of bioanalytical methods for chemical warfare // J. Chromatogr. B. 2010. V. 878. P. 1207.
Black R.M., Read R.W. Biological markers of exposure to organophosphorus nerve agents // Arch. Toxicol. 2013. V. 87. P. 421.
Golime R., Chandra B., Palit M., Dubey D.K. Adductomics: a promising tool for the verification of chemical warfare agents' exposures in biological samples // Arch. Toxicol. 2019. V. 93. P. 1473.
Fidder A., Hulst A.G., Noort D., de Ruiter R., Van der Schans M.J., Benschop H.P., Langenberg P. Retrospective detection of exposure to organophosphorus anti-cholinesterases: Mass spectrometric analysis of phosphylated human butyrylcholinesterase // Chem. Res. Toxicol. 2002. V. 5. P. 582.
Kranawetvogl A., Kuppers J., Siegert M., Gutschow M., Worek F., Thiermann H., Elsinghorst P.W., John H. Bioanalytical verification of V-type nerve agent exposure: simultaneous detection of phosphonylated tyrosines and cysteine-containing disulfide-adducts derived from human albumin // Anal. Bioanal. Chem. 2018. V. 410. P. 1463.
Rodin I., Braun A., Stavrianidi A., Baygildiev T., Shpigun O., Oreshkin D., Rybalchenko I. “Dilute-and-Shoot” RSLC-MS/MS method for fast detection of nerve and vesicant chemical warfare agent metabolites in urine // J. Anal. Toxicol. 2014. V. 39. № 1. P. 69.
Mawhinney D.B., Hamelin E.I., Sathya R.F., Antonis S.S., Pavlopoulos J., Kobelski R.J. The determination of organophosphonate nerve agent metabolites in human urine by hydrophilic interaction liquid chromatography tandem mass spectrometry // J. Chromatogr. B. 2007. V. 852. P. 223.
Lemire S.W., Barr J.R., Ashley D.L., Olson C.Y., Huyes T.L. Quantitation of biomarkers of exposure to nitrogen mustards in urine from rats dosed with nitrogen mustards and from an unexposed human population // J. Anal. Toxicol. 2004. V. 28. P. 320.
Swaim L.L., Johnson R.C., Zhou1 Y., Sandlin C., Barr J.R. Quantification of organophosphorus nerve agent metabolites using a reduced-volume, high-throughput sample processing format and liquid chromatography–tandem mass spectrometry // J. Anal. Toxicol. 2008. V. 32. P. 775.
Riches J., Morton I., Read R.W., Black R.M. The trace analysis of alkyl alkylphosphonic acids in urine using gas chromatography-ion trap negative ion tandem mass spectrometry // J. Chromatogr. B. 1998. V. 816. P. 251.
Subramaniam R., Östin A., Nilsson C., Åstot C. Direct derivatization and gas chromatography– tandem mass spectrometry identification of nerve agent biomarkers in urine samples // J. Chromatogr. B. 2013. V. 928. P. 98.
van der Schans M.J., Polhuijs M., Van Dijk C., Degenhardt C.E.A.M., Pleijsier K., Langenberg J.P., Benschop H.P. Retrospective detection of exposure to nerve agents: analysis of phosphofluoridates originating from fluoride-induced reactivation of phosphylated BuChE // Arch. Toxicol. 2004. V. 78. P. 508.
Holland K.E., Solano M.I., Johnson R.C., Maggio V.L., Barr J.R. Modifications to the organophosphorus nerve agent-protein adduct refluoridation method for retrospective analysis of nerve agent exposures // J. Anal. Toxicol. 2008. V. 32. P. 116.
van der Schans M.J., Fidder A., van Oeveren D., Hulst A.G., Noort D. Verification of exposure to cholinesterase inhibitors: Generic detection of OPCW schedule 1 nerve agent adducts to human butyrylcholinesterase // J. Anal. Toxicol. 2008. V. 32. P. 125.
Sporty J.L.S., Lemire S.W., Jakubowski E.M., Renner J.A., Evans R.A., Williams R.F., Schmidt J.G., van der Schans M.J., Noort D., Johnson R.C. Immunomagnetic separation and quantification of butyrylcholinesterase nerve agent adducts in human serum // Anal. Chem. 2010. V. 82. P. 6593.
Williams N.H., Harrison J.M., Read R.W., Black R.M. Phosphylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents // Arch. Toxicol. 2007. V. 81. P. 627.
Crow B.S., Pantazides B.G., González J.Q., Garton J.W., Carter M.D., Perez J.W., Watson C.M., Tomcik D.J., Crenshaw M.D., Brewer B.N., Riches J.R., Stubbs S.J., Read R.W., Evans Jerry D., Thomas R.A., Blake T.A., Johnson R.C. Simultaneous measurement of tabun, sarin, soman, cyclosarin, VR, VX, and VM adducts to tyrosine in blood products by isotope dilution UHPLC-MS/MS // Anal. Chem. 2014. V. 86. P. 10397.
Pantazides B.G., Watson C.M., Carter M.D., Crow B.S., Perez J.W., Blake T.A., Thomas J.D., Johnson R.C. An enhanced butyrylcholinesterase method to measure organophosphorus nerve agent exposure in humans // Anal. Bioanal. Chem. 2014. V. 406. P. 5187.
Ставрианиди А.Н., Браун А.В., Стекольщикова Е.А., Байгильдиев Т.М., Родин И.А., Рыбальченко И.В. Выбор условий регистрации и исследование фрагментации пептидного биомаркера зарина методом высокоэффективной жидкостной хроматомасс-спектрометрии высокого разрешения // Масс-спектрометрия. 2018. Т. 15. С. 44. (Stavrianidi A.N., Braun A.V., Stekolshchikova E.A., Baygildiev T.M., Rodin I.A., Rybalchenko I.V. Selection of recording conditions and study of fragmentation of a peptide biomarker of sarin by high-performance liquid chromatography–high-resolution mass spectrometry // J. Analyt. Chem. 2018. V. 73. P. 1357.)
Navratil T., Šenholdova-Dlaskova Z., Heyrovsky M., Pistoupilova K., Pistoupil T.I. Excretion of thiodiglycolic acid in urine influenced by some victuals and cetirizine // Anal. Lett. 2004. V. 37. P. 1093.
Muller G., Norpoth K., Kusters E., Herweg K., Versin E. Determination of thiodiglycolic acid in urine specimens of vinyl chloride exposed workers // Int. Arch. Occup. Environ. Health. 1978. V. 41. P. 199.
Gandor F., Gawlik M., Thiermann H., John H. Evidence of sulfur mustard exposure in human plasma by LC-ESI-MS-MS detection of the albumin-derived alkylated HETE-CP dipeptide and chromatographic investigation of its cis/trans isomerism // J. Anal. Toxicol. 2015. V. 39. P. 270.
Steinritz D., Striepling E., Rudolf K.D., Schroder-Kraft C., Puschel K., Hullard-Pulstinger A. Medical documentation, bio-analytical evidence of an accidental human exposure to sulfur mustard and general therapy recommendations. // Toxicol. Lett. 2016. V. 244. P. 112.
Mattes W.B., Hartley J.A., Kohn K.W. DNA sequence selectivity of guanine-N7 alkylation by nitrogen mustards // Nucleic Acids Res. 1986. V. 14. P. 2971.
Wei Y., Yue L., Liu Q., Chen J., Xie J. A sensitive high performance liquid chromatography-positive electrospray tandem mass spectrometry method for N7-[2-[(2-hydroxyethyl)thio]-ethyl]guanine determination // J. Chromatogr. B. 2011. V. 879. P. 1707.
Chang-Cai Liu, Shi-Lei Liu, Hai-Ling Xi, Hui-Lan Yu, Shi-Kun Zhou, Gui-Lan Huang, Long-Hui Liang, Jing-Quan Liua. Simultaneous quantification of four metabolites of sulfur mustard in urine samples by ultra-high performance liquid chromatography-tandem mass spectrometry after solid phase extraction // J. Chromatogr. A. 2017. V. 1492. P. 41.
Black R.M., Read R.W. Improved methodolgy for the detection and quantitation of urinary metabolites of sulphur mustard using gas chromatography tandem mass spectrometry // J. Chromatogr. B. 1995. V. 665. P. 97.
Hua Xu, Zhiyong Nie, Yajiao Zhang, Chunzheng Li, Lijun Yue, Wenfeng Yang, Ji Chen, Yuan Dong, Qin Liu, Ying Lin, Bidong Wu, Jianlin Feng, Hua Li, Lei Guo, Jianwei Xie. Four sulfur mustard exposure cases: Overall analysis of four types of biomarkers in clinical samples provides positive implication for early diagnosis and treatment monitoring // Toxicology Reports. 2014. V. 1. P. 533.
Daly J.D., O’Hehir M., Frame G.M. A sensitive method for quantitation of β-lyase metabolites of sulfur mustard as 1,1'-sulfonylbis[2-(methylthio)ethane] (SBMTE) in human urine by isotope dilution liquid chromatography-positive ion-electrospraytandem mass spectrometry // J. Chromatogr. B. 2007. V. 850. P. 120.
Boyer A.E., Ash D., Barr D.B., Young C.E., Driskell W.J., Whitehead R.D., Ospina J.M., Preston K.E., Woolfitt A.E., Martinez R.A. Quantitation of the sulfur mustard metabolites 1,1'-sulfonylbis[2-(methylthio)ethane] and thiodiglycol in urine using isotope-dilution Gas chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 327.
Орлова О.И., Савельева Е.И., Каракашев Г.В. Методы определения аддуктов сернистого иприта с ДНК // Журн. aналит. xимии. 2017. Т. 72. С. 209. (Orlova O.I., Savel’eva E.I., Karakashev G.V. Methods of determination of sulfur yperite–DNA adducts // J. Analyt. Chem. V. 72. P. 256.)
Zubel T., Hochgesand S., John H., Steinritz D., Schmidt A., Brkle A., Mangerich A. A mass spectrometric platform for the quantitation of sulfur mustard-induced nucleic acid adducts as mechanistically relevant biomarkers of exposure // Arch. Toxicol. 2019. V. 93. P. 61.
Nie Z., Liu Q., Xie J. Improvements in monitoring the N-terminal valine adduct in human globin after exposure to sulfur mustard // Talanta. 2011. V. 85. P. 1154.
John H., Siegert M., Gandor F., Gawlik M., Kranawetvogl A., Karaghiosoff K., Thiermann H. Optimized verification method for detection of an albumin-sulfur mustard adduct at cys(34) using a hybrid quadrupole time-of-flight tandem mass spectrometer after direct plasma proteolysis // Toxicol. Lett. 2016. V. 244. P. 103.
Kataoka M., Tsunoda N., Ohta H., Tsuge K., Takesako H., Seto Y. Effect of cation-exchange pretreatment of aqueous soil extracts on the gas chromatographic-mass spectrometric determination of nerve agent hydrolysis products after tert.-butyldimethylsilylation // J. Chromatogr. A. 1998. V. 824. P. 211.
Rodin I., Braun A., Savelieva E., Rybalchenko I., Ananieva I., Shpigun O. Rapid method for the detection of metabolite of sulfur mustard 1,1-sulfonylbis[2-S-(N-acetylcysteinyl)ethane] in plasma and urine by liquid chromatography–negative electrospray-tandem mass spectrometry // J. Liq. Chromatogr. Relat. Technol. 2011. V. 34. P. 1676.
Pantazides B.G., Crow B.S., Garton J.W., Quiñones-González J.A., Blake T.A., Thomas J.D., Johnson R.C. Simplified method for quantifying sulfur mustard adducts to blood proteins by ultrahigh pressure liquid chromatography–Isotope dilution tandem mass spectrometry // Chem. Res. Toxicol. 2015. V. 28. P. 256.
Noort D., Hulst A.G., Trap H.C., de Jong L.P.A., Benschop H.P. Synthesis and mass spectrometric identification of the major amino acid adducts formed between sulphur mustard and haemoglobin in human blood // Arch. Toxicol. 1997. V. 71. P. 171.
Niu Tian-qi, Matijasevic Z., Austin-Ritchie P., Stering A., Ludlum D.B. A 32P-postlabeling method for the detection of adducts in the DNA of human fibroblasts exposed to sulfur mustard // Chem. Biol. Interact. 1996. V. 100. P. 77.
Kubachka K.M., Richardson D.D., Heitkemper D.T., Caruso J.A. Detection of chemical warfare agent degradation products in foods using liquid chromatography coupled to inductively coupled plasma mass spectrometry and electrospray ionization mass spectrometry // J. Chromatogr. A. 2008. V. 1202. P. 124.
Halme M., Karjalainen M., Kiljunen H., Vanninen P. Development and validation of efficient stable isotope dilution LC-HESI-MS/MS method for the verification of β-lyase metabolites in human urine after sulfur mustard exposure // J. Chromatogr. B. 2011. V. 879. P. 908.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии