

Журнал аналитической химии, 2021, T. 76, № 10, стр. 927-936
ОДНОВРЕМЕННОЕ ОПРЕДЕЛЕНИЕ ГИДРАЗИНА, МЕТИЛГИДРАЗИНА И 1,1-ДИМЕТИЛГИДРАЗИНА В ВОДАХ МЕТОДОМ ВЭЖХ СО СПЕКТРОФОТОМЕТРИЧЕСКИМ ДЕТЕКТИРОВАНИЕМ С ПРИМЕНЕНИЕМ КАТАЛИЗА ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ
Ю. В. Тимченко a, *, А. В. Апенкина a, А. Д. Смоленков a, А. В. Пирогов a, О. А. Шпигун a
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, ГСП-1, Россия
* E-mail: yury_tim@mail.ru
Поступила в редакцию 11.04.2021
После доработки 23.04.2021
Принята к публикации 26.04.2021
Аннотация
Разработан простой, быстрый и чувствительный способ одновременного определения гидразина, метилгидразина и 1,1-диметилгидразина в водах, основанный на предколоночной дериватизации бензальдегидом и определении образующихся продуктов методом обращенно-фазовой ВЭЖХ со спектрофотометрическим детектированием при 300, 282 и 298 нм соответственно. Впервые применен эффект иминного и мицеллярного катализа для получения производных гидразинов при совместном присутствии. Выбраны условия дериватизации: тип, рН и концентрация буферной системы; концентрации реагента и поверхностно-активного вещества; температура и продолжительность реакции. Установлено, что реакция дериватизации протекает полностью при рН 9.4 в присутствии каталитической системы на основе 0.6 М аммония и 87 мМ додецилсульфата натрия, а также 3.5 мМ реагента при комнатной температуре за 5 мин. Количественный выход продуктов дериватизации подтвержден методом ионной хроматографии. Пределы обнаружения (S/N = 3) без дополнительного концентрирования составили 0.3, 2.3 и 1.3 мкг/л, а линейный диапазон 1–500, 7–1000 и 5–1000 мкг/л для гидразина, метилгидразина и 1,1-диметилгидразина соответственно.
Гидразин (Ги) и его метильные гомологи – метилгидразин (МГ) и несимметричный диметилгидразин (НДМГ) – являются продуктами многотоннажного производства и находят широкое применение в органическом синтезе, производстве лекарственных препаратов, регуляторов роста растений, а также в качестве компонентов ракетного топлива [1, 2]. При этом эти соединения обладают генотоксичной и мутагенной активностью по отношению к живым организмам [3, 4]. Американское агентство по защите окружающей среды и Европейское химическое агентство относят Ги, МГ и НДМГ к категории возможно канцерогенных для людей [5]. В России они относятся к веществам первого класса опасности и канцерогенам, вследствие чего их содержание в химической продукции и природных объектах строго контролируется. Так, для Ги в объектах хозяйственно-питьевого и культурно-бытового водопользования установлена ПДК 0.01 мг/л [6]. В связи с выявлением новых негативных эффектов долговременного воздействия гидразина и его производных на здоровье человека и живую природу, а также повышением требований к качеству жизни необходимо создание новых экспрессных и высокочувствительных подходов к определению Ги и его метилпроизводных.
Хроматографические методы, благодаря высокой селективности и чувствительности, хорошо зарекомендовали себя при определении следовых количеств гидразинов в объектах со сложной матрицей [7]. Прямое определение осложнено из-за их высокой полярности, термолабильности, склонности к окислению, отсутствия хромофорных групп и низкой молекулярной массы. Описано определение низких концентраций гидразинов в нативной форме [8, 9] с использованием вариантов ионной (ИХ) [10, 11], ион-парной [12, 13] и гидрофильной хроматографии (ГФ) [14] с амперометрическим детектированием (АД). Недостатками АД по сравнению с другими детекторами, применяемыми в жидкостной хроматографии, являются более низкая стабильность аналитического сигнала, а также перегрузка хроматограмм электроактивными примесями, присутствующими в сложных матрицах.
Альтернативным и наиболее популярным подходом к определению малых концентраций гидразинов является проведение предварительной дериватизации. Благодаря большому выбору реагентов дериватизацию широко используют в анализе для повышения устойчивости аналитов, а также улучшения характеристик разделения и детектирования [15].
Известны подходы к газохроматографическому определению гидразинов с использованием электронозахватного [16], азотно-фосфорного [17, 18], пламенно-ионизационного (ПИД) [19, 20] и масс-спектрометрического детекторов (МС) [21, 22]. Обязательная стадия замены растворителя в газовой хроматографии (ГХ) – очевидный и существенный недостаток данного метода, при этом продукты реакции выделяют и концентрируют из реакционной смеси. В случае обращенно-фазовой ВЭЖХ (ОФ ВЭЖХ) анализ возможен сразу после дериватизации без дополнительных трудоемких стадий. Предложены подходы к определению гидразина и его производных, основанные на применении метода ОФ ВЭЖХ со спектрофотометрическим (СФД) [23–28] и флуоресцентным детектированием [29, 30], МС [31] и тандемной МС [32, 33].
В качестве дериватизирующих реагентов используют галогенангидриды ароматических кислот, производные арилгалогенидов или карбонильные соединения: пентафторбензоилхлорид [16], бензоилхлорид [32], 4-хлор-5,7-динитробензофуразан [23] ацетон [17], глиоксаль [25], 2,3-нафталинальдегид [29], фурфурол [19, 22], 5-нитро-2-фуральдегид [24], бензальдегид [20, 22, 27, 28, 31] и его производные [18, 21, 22, 26, 30, 32]. Использование карбонильных соединений предпочтительно ввиду их более высокой селективности по отношению к гидразинам и устойчивости образующихся гидразонов.
Отметим, что пределы обнаружения (сmin) во всех указанных выше работах достигают значений порядка долей или нескольких мкг/л (мкг/кг) и выше, а подходов, позволяющих одновременно определять на этом уровне нескольких гидразинов с использованием группового реагента немного. Таких значений сmin удается достичь в основном за счет использования дорогого оборудования и детекторов, а также различных вариантов концентрирования. Для сокращения продолжительности реакции и увеличения выхода продуктов дериватизации нередко используют нагревание до 75°C в течение до 1.5 ч. Однако постоянный нагрев способствует разложению гидразонов [34], а в присутствии ионов металлов и кислорода интенсивно происходят окисление и трансформация гидразинов [35]. В целом этапу изучения и подбора условий реакций дериватизации уделяется недостаточное внимание, хотя от степени завершенности реакции напрямую зависят чувствительность и правильность результата определения. Количественный выход продуктов дериватизации особенно важен при определении следовых количеств аналитов.
Перспективный подход к сокращению продолжительности реакции – использование каталитического эффекта мицеллярных сред. Явление мицеллярного катализа (МК) широко используют в органическом синтезе для ускорения реакций конденсации, гидролиза, полимеризации и т.д. [36]. Так, каталитический эффект мицеллярных сред применили для определения гидразина в водных матрицах с предварительной дериватизацией 4-диметиламинобензальдегидом [37]. Авторы отмечают 100-кратное увеличении скорости реакции образования гидразона в присутствии додецилсульфата натрия (ДДСН). Другие подобные исследования реакций дериватизации в мицеллярных средах и их аналитического приложения для определения гидразинов на данный момент не проводились.
Цель данной работы – выбор условий реакций дериватизации Ги, МГ и НДМГ групповым реагентом бензальдегидом (БА), исследование возможности применения мицеллярного катализа для дериватизации гидразинов, а также разработка комбинированного похода к их одновременному определению в питьевой воде методом ОФ ВЭЖХ−СФД.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и материалы. Гидразина сульфат (>99%), МГ (>98%), НДМГ (>98%), безводный метиламин (>98%), три(гидроксиметил)аминометан (Трис) (>99.9%), бензальдегид (>99%), безводный цитрат натрия (>99%), формиат натрия (>99%), триэтаноламин (98%), декагидрат тетрабората натрия (>99.5%) были приобретены у “Sigma-Aldrich” (Германия). Ацетонитрил для хроматографии (99.9%), серная кислота (95%), ледяная уксусная кислота (99.7%), гидроксид натрия (98%), ацетат аммония (98%), дигидрат дигидрофосфата натрия (>99%), безводный гидрофосфат натрия (99%), ортофосфорная кислота (85%), муравьиная кислота (98%), соляная кислота (37%), хлорид аммония (99.5%) и додецилсульфат натрия (>85%) производства “Panreac” (Испания). Для приготовления всех растворов использовали высокочистую воду с удельным сопротивлением 18.2 МОм⋅см, полученную с применением системы очистки воды Milli-Q (Millipore, США).
Приготовление растворов. Исходные растворы Ги, МГ и НДМГ с концентрацией 10 мг/л готовили растворением точной навески в 10 мМ серной кислоте. Растворы хранили при +4°C не более месяца и использовали для приготовления всех необходимых рабочих растворов с меньшими концентрациями разбавлением 10 мМ серной кислотой непосредственно в день проведения соответствующего этапа эксперимента. Растворы БA с концентрациями 5 и 20 г/л готовили растворением точных навесок в ацетонитриле и хранили при +4°C не более недели.
Для обеспечения необходимого pH реакционной среды использовали следующие буферные растворы: формиатный (4.0 M; pH 3.0), цитратный (1.5 M; pH 4.0, 5.0), фосфатный (2.0 М; pH 6.0, 7.0), на основе триэтаноламина (4.0 М; pH 8.0), тетраборатный (0.1 М; pH 9.0, 10.0), а также раствор серной кислоты (10 мМ; pH 2.0). Применяли также каталитические буферные растворы с концентрацией 4 М на основе аммония, метиламина и Трис в диапазоне рН 5.0–11.5. Перечисленные выше растворы готовили растворением точного количества соответствующих твердых солей или чистых веществ в деионизированной воде, значение pH регулировали добавлением растворов кислоты или основания, контролируя его pH-метром PB-11 (Sartorius, Германия).
Хроматографический анализ. Использовали ВЭЖХ-систему Agilent 1100, состоящую из двухканального градиентного насоса со смешением по высокому давлению, термостата колонок, дегазатора подвижной фазы, спектрофотометрического детектора на диодной матрице (ДАД) и охлаждаемого автоматического инжектора с дозирующим устройством для ввода пробы (Agilent Technologies, США). Для управления хроматографом и обработки данных применяли программное обеспечение ChemStation (Agilent Technologies, США). Для разделения компонентов использовали хроматографическую колонку (50 × × 3.0 мм, диаметр зерна сорбента 1.8 мкм) ZORBAX Eclipse Plus C18 RRHD (Agilent Technologies, США), температура термостата колонки 30°C, скорость потока подвижной фазы – 0.4 мл/мин. Подвижной фазой являлась смесь 10 мМ аммонийно-ацетатного буферного раствора (рН 7) и ацетонитрила, содержание которого менялось по следующей градиентной программе, подобранной в предварительных экспериментах: 0–2 мин 25%, 2–7 мин линейный подъем до 80%, 7–9 мин 80%, 9–10 мин линейный спад до 25%, 10–12 мин 25%. Объем вводимой пробы составлял 100 мкл. Детектирование проводили при длинах волн, соответствующих максимумам поглощения БА-производных гидразинов: 300, 282 и 298 нм для Ги, МГ и НДМГ соответственно.
Использовали ВЭЖХ-систему с амперометрическим детектором “Цвет-Яуза” (НПО “Химавтоматика”, Россия). Объем петли крана ввода пробы составлял 100 мкл. Анализ проводили по измененной методике [10] с использованием колонки (250 × 4.6 мм, диаметр зерна сорбента 10 мкм) Luna SCX (Phenomenex, США). В качестве подвижной фазы применяли 100 мМ аммонийно-ацетатный буферный раствор (рН 5.4) с добавкой 25 об. % ацетонитрила. Скорость потока подвижной фазы составляла 1.0 мл/мин. Потенциал амперометрического детектора +1.3 В.
Выбор условий реакции дериватизации. В рамках однофакторной оптимизации последовательно подбирали условия проведения реакции дериватизации, варьируя требуемый параметр при постоянных значениях других. Дериватизацию проводили непосредственно в хроматографических виалах из темного стекла.
Значение рН. К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 200 мкл соответствующего буферного раствора, 20 мкл раствора БА с концентрацией 5 г/л. Полученные смеси оставляли без доступа света при комнатной температуре (20 ± 2°C) и анализировали методом ВЭЖХ−СФД через 30 мин после добавления реагента.
Концентрация буферного раствора. К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 2.5, 5, 10, 25, 50, 100, 150, 200, 300, 400, 500 мкл аммонийного буферного раствора с рН 9.4, 20 мкл раствора реагента с концентрацией 5 г/л. Полученные смеси оставляли без доступа света при комнатной температуре (20 ± 2°C) и анализировали методом ВЭЖХ−СФД через 5 мин после добавления реагента.
Концентрация реагента. К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 200 мкл аммонийного буферного раствора с рН 9.4 и 5, 10, 20, 50 мкл (5 г/л) или 25, 50 и 100 мкл (20 г/л ) раствора БА. Полученные смеси оставляли без доступа света при комнатной температуре (20 ± 2°C) и анализировали методом ВЭЖХ−СФД через 10 мин.
Температура и продолжительность реакции. К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 200 мкл аммонийного буферного раствора с рН 9.4 и 25 мкл (20 г/л) раствора БА. Пробы выдерживали при комнатной температуре (20 ± 2°C) и 40, 60, 80°C в твердотельном термостате T-1 (Biosan, Латвия) и через 2, 5, 10, 15, 30, 45, 60, 90 и 120 мин анализировали методом ВЭЖХ−СФД.
Концентрация поверхностно-активного вещества (ПАВ). К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 200 мкл аммонийного буферного раствора с рН 9.4 и навеску ДДСН (0.003, 0.0150, 0.030, 0.060 г). Смесь выдерживали в ультразвуковой (УЗ) ванне в течение 1 мин до полного растворения ДДСН, после чего добавляли 20 мкл (5 г/л) раствора БА. Реакцию проводили в течение 2 мин при комнатной температуре (20 ± 2°C), а затем анализировали методом ВЭЖХ−СФД.
Дериватизация в мицеллярной среде. К 1 мл растворов гидразинов с концентрациями по 1 мг/л добавляли 200 мкл аммонийного буферного раствора с рН 9.4 и 0.030 г ДДСН. Пробу выдерживали в УЗ-ванне в течение 1 мин, после чего вносили 25 мкл (20 г/л) раствора БА. Полученные смеси оставляли без доступа света при комнатной температуре (20 ± 2°C) и анализировали методом ВЭЖХ−СФД через 2, 5, 10, 15, 30, 60, 90 и 120 мин после добавления реагента.
Процедура определения гидразинов в образцах вод. К 1 мл образца или градуировочного раствора с заданными концентрациями гидразинов добавляли 200 мкл аммонийного буферного раствора с рН 9.4 и 0.030 г ДДСН. Каждую пробу выдерживали в УЗ-ванне в течение 1 мин, после чего вносили 25 мкл (20 г/л) раствора БА. Полученные смеси оставляли без доступа света при комнатной температуре (20 ± 2°C) в течение 5 мин, после чего проводили анализ методом ВЭЖХ−СФД.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выбор условий определения гидразинов методом обращено-фазовой ВЭЖХ со спектрофотометрическим детектированием. Благодаря наличию свободной аминогруппы, гидразины способны участвовать в реакции конденсации с ароматическими альдегидами с образованием соответствующих гидразонов (схема 1 ).

Схема 1 . Реакции гидразина, метилгидразина и 1,1-диметилгидразина с бензальдегидом.
Данная реакция полностью обратима и катализируется как кислотами, так и основаниями, а в основе механизма лежит стадия нуклеофильной атаки свободной аминогруппой гидразинов атома углерода карбонильной группы с последующим отщеплением молекулы воды [34, 38]. Гидразин реагирует сразу по двум аминогруппам с образованием бензалазина. Образующиеся производные более гидрофобны, чем исходный БА за счет наличия дополнительного бензольного кольца в структуре бензалазина или метильных групп в случае гидразонов МГ и НДМГ. В результате удерживание в условиях ОФ ВЭЖХ БА-производных гидразинов увеличивается в ряду МГ < < НДМГ < Ги. Образующиеся диметилгидразоны имеют в своей структуре атомы азота, которые могут участвовать в полярных взаимодействиях со свободными силанольными группами матриц гидрофобизированных силикагелей, что приводит к ухудшению формы пика и, как следствие, к снижению сmin. В связи с этим для разделения гидразонов выбрали колонку ZORBAX Eclipse Plus с привитыми октадецильными группами и двойным эндкэппингом, а использование подвижной фазы с pH 7 исключает кислотный гидролиз БА-производных во время анализа [39].
С использованием ДАД получили электронные спектры поглощения гидразонов в диапазоне длин волн от 190 до 700 нм. Максимумы поглощения Ги, МГ и НДМГ производных составили 300, 282 и 298 нм соответственно. Данные длины волн выбрали для детектирования.
Значение pH. Реакция образования гидразонов – реакция конденсации, в которой принимают участие ионы водорода [38]. В связи с этим изучали влияние кислотности реакционной среды в широком диапазоне pH 2–10 на выход дериватов. Выход продукта дериватизации (φ) рассчитывали как отношение площади пика гидразона к максимальной площади пика, полученной в оптимальных условиях для данной концентрации аналита. Концентрация БА в реакционной смеси составляла примерно 0.9 мМ.
Как видно из рис. 1а, при использовании буферных систем без катализатора наибольшие выходы дериватов достигнуты в диапазоне pH 4–7 (слабокислая среда). Максимум для Ги соответствует рН 5, а для МГ и НДМГ смещен к рН 7. Однако даже при продолжительности реакции 30 мин эти выходы малы и для МГ и НДМГ составляют не больше 7%, что делает невозможным практическое применение БА как группового реагента для определения гидразинов.
Рис. 1.
Зависимости выхода гидразонов (φ) от рН реакционной среды с использованием буферных систем без иминного катализатора (а) и на основе Трис (б), аммония (в) и метиламина (г). Продолжительность реакции 5 мин (б), (в), (г) и 30 мин (а) (n = 3, Р = 0.95).

Известно, что скорость образования гидразонов увеличивается в присутствии первичных аминов в результате иминного катализа [40]. Суть его заключается в образовании имина из амина и карбонильного соединения как промежуточного соединения, скорость реакции гидразинов с которым выше, чем с исходным карбонильным соединением. С целью уменьшения продолжительности реакции опробовали буферные системы на основе потенциальных иминных катализаторов, таких как аммиак (аммоний), метиламин и Трис. При проведении реакции в течение 5 мин при рН 5–7 без катализа выходы БА-производных для Ги, МГ и НДМГ составляют примерно 2.0, 0.6 и 0.4% соответственно. В то же время применение буферных систем на основе метиламина или аммония значительно ускоряет реакции образования БА-производных гидразинов (в 5–10 и 50–150 раз соответственно). Максимумы зависимостей выхода реакции соответствуют области рН 7–10 (слабощелочная среда). Присутствие Трис оказывает каталитический эффект только по отношению к МГ, что, вероятно, связано с меньшими стерическими затруднениями для нуклеофильной атаки МГ соответствующего имина. Для дальнейших исследований использовали аммонийный буферный раствор с pH 9.4, поскольку в этом случае реализуются наилучшие условия протекания реакции для всех гидразинов.
Концентрации буферного раствора и реагента. Поскольку скорость образования гидразонов зависит от концентраций всех участвующих в этом процессе веществ, изучали зависимости выхода гидразонов от концентрации аммонийного буферного раствора в конечной реакционной смеси (рис. 2). При концентрации меньше 0.03 М эффект иминного катализа не наблюдается. Из представленных зависимостей видно, что при концентрации аммонийного буферного раствора больше 0.6 М выход всех БА-производных значимо не меняется, поэтому концентрацию 0.67 М выбрали для дальнейших исследований.
Рис. 2.
Зависимости выхода гидразонов (φ) от концентрации буферного раствора (сбуф) в реакционной смеси (с учетом разбавления пробы). Продолжительность реакции 5 мин (n = 3, Р = 0.95).

Ввиду обратимости реакции [34] для достижения высоких выходов необходим значительный избыток дериватизирующего агента. Изучено влияние концентрации БА в диапазоне 0.2–14 мМ в реакционной смеси на выходы продуктов дериватизации гиздразинов (табл. 1).
Таблица 1.
Влияние концентрации бензальдегида в реакционной смеси на выход гидразонов (φ) (продолжительность реакции 10 мин, n = 3, Р = 0.95)
| с(БА)*, мM | φ(Ги), % | φ(МГ), % | φ(НДМГ), % |
|---|---|---|---|
| 0.2 | 13.2 ± 0.5 | 73.4 ± 1.9 | 8.8 ± 0.6 |
| 0.4 | 25.6 ± 0.7 | 87.6 ± 1.1 | 17.2 ± 1.0 |
| 0.9 | 55.1 ± 1.5 | 99.2 ± 1.0 | 34.9 ± 1.0 |
| 1.7 | 88.9 ± 0.9 | 99.0 ± 2.0 | 65.8 ± 2.6 |
| 3.5 | 92.4 ± 0.9 | 98.7 ± 3.0 | 92.3 ± 1.8 |
| 6.8 | 92.8 ± 0.9 | 94.9 ± 0.7 | 93.6 ± 0.8 |
| 14.0 | 83.7 ± 3.3 | 94.0 ± 1.1 | 92.0 ± 0.9 |
При концентрации БА в реакционной смеси 3.5 мМ достигаются почти количественные выходы производных, которые при дальнейшем увеличении содержания реагента в системе значимо не меняются. Концентрацию БА 3.5 мМ выбрали для дальнейших исследований.
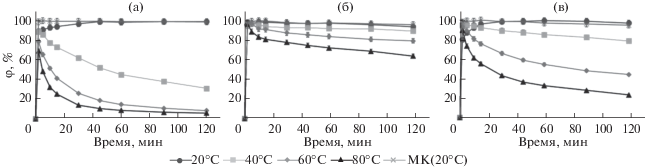
Температура. Равновесие реакции дериватизации достигается быстрее при более высоких температурах, однако образующиеся гидразоны могут разлагаться в этих условиях [34]. Зависимости выхода продуктов дериватизации от времени при различных температурах представлены на рис. 3. Как видно, реакция во всех случаях протекает быстрее при повышенных температурах, однако термическое воздействие приводит к уменьшению выхода диметилгидразонов, что связано с разрушением гидразонов и/или исходных веществ в результате ускорения побочных процессов в реакционной смеси, вероятно, окислительно-восстановительной природы. Авторы работ [24, 27, 33] использовали нагревание реакционных смесей с целью уменьшения продолжительности реакции, но это сильно сказывается как на воспроизводимости, так и на потенциальной чувствительности определения. В связи с этим выбрали комнатную температуру в качестве оптимальной для проведения дериватизации. Как видно из рис. 3, в этих условиях для всех гидразинов реакция протекает количественно (выход деривата >99%) примерно за 45 мин.
Рис. 3.
Зависимости выходов производных с бензальдегидом (φ) гидразина (а), метилгидразина (б) и 1,1-диметилгидразина (в) от температуры и продолжительности реакции (n = 3, Р = 0.95). МК – мицеллярный катализ.
Для подтверждения количественного протекания реакции в выбранных условиях проводили ионохроматографический анализ реакционной смеси. Пределы обнаружения Ги, МГ и НДМГ по методике [10] составили 1, 2.5 и 5 мкг/л соответственно. Концентрация свободных форм гидразинов оказалась ниже предела обнаружения, что говорит о том, что в выбранных условиях реакция протекает количественно для каждого аналита (>99%).
Концентрация поверхностно-активного вещества. Додецилсульфат натрия относится к типу сульфоанионных ПАВ и в водной среде образует прямые мицеллы. Существование мицелл в растворе возможно только при определенных условиях, а именно при концентрации ПАВ выше критической концентрации мицеллообразования (ККМ). Для выбора количества ДДСН изучили влияние его концентрации в реакционной смеси на выход БА-производных при концентрации БА 0.8 мМ и продолжительности реакции 2 мин (табл. 2).
Таблица 2.
Влияние концентрации додецилсульфата натрия в реакционной смеси на выход производных гидразина, метилгидразина и 1,1-диметилгидразина (φ) с бензальдегидом (продолжительность реакции 2 мин, n = 3, Р = 0.95)
| c(ДДСН)*, мМ | φ(Ги), % | φ(МГ), % | φ(НДМГ) , % |
|---|---|---|---|
| 0 | 14.3 ± 1.6 | 40.8 ± 1.6 | 8.1 ± 1.2 |
| 8.7 | 22.5 ± 1.5 | 56.0 ± 2.2 | 12.1 ± 1.4 |
| 43 | 35.8 ± 1.1 | 68.2 ± 1.4 | 17.5 ± 1.2 |
| 87 | 36.9 ± 1.3 | 69.4 ± 1.4 | 20.3 ± 1.7 |
| 173 | 36.4 ± 1.0 | 69.6 ± 2.8 | 20.1 ± 1.3 |
Известно, что значения первой и второй ККМ и для ДДСН составляют 8.3 и ~80 мМ соответственно [41]. При концентрации ДДСН 87 мМ достигается максимальная скорость образования соответствующих БА-производных, что соответствует второй ККМ. При дальнейшем увеличении концентрации ДДСН выход реакций дериватизации не меняется, поэтому концентрацию ДДСН 87 мМ выбрали для оценки эффективности мицелл в катализе реакций с гидразинами в ранее подобранных условиях (рис. 3). Обнаружили, что применение МК значительно сокращает время достижения количественного выхода дериватов, которое в этих условиях составляет всего 5 мин.
Анализ образцов воды. Для оценки метрологических характеристик и апробации предложенного комбинированного подхода проанализировали образцы водопроводной воды методом ВЭЖХ−СФД. Аналитическим сигналом для построения градуировочной зависимости служила площадь пика соответствующего гидразона. Градуировочные растворы в диапазоне концентраций 1–1000 мкг/л готовили добавлением стандартных растворов гидразинов к пробам водопроводной воды. Предел обнаружения оценивали по отношению сигнал/шум (S/N) = = 3. Нижнюю границу определяемых концентраций определяли как S/N = 10. Правильность предложенных подходов подтверждали методом введено–найдено (рис. 4). Сходимость рассчитывали по трем параллельным результатам анализа пробы в течение одного дня (табл. 3).
Рис. 4.
Наложение экспериментальных хроматограмм образцов водопроводной воды с добавкой по 5 мкг/л каждого компонента (1), по 1 мкг/л (2) и без добавки (3). Длина волны детектирования 300 нм.
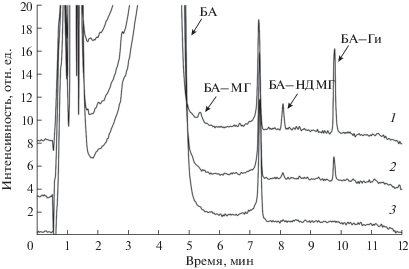
Таблица 3.
Характеристики определения гидразина, метилгидразина и 1,1-диметилгидразина в воде методом ВЭЖХ со спектрофотометрическим детектированием с предварительной дериватизацией бензальдегидом (n = 3, P = 0.95)
| Аналит | ЛДОК*, мкг/л | S = kc + а | R2 | сmin, мкг/л | Введено, мкг/л |
Найдено, мкг/л |
sr, % |
|---|---|---|---|---|---|---|---|
| Ги | 1–500 | S = (6.8 ± 0.1)c | 0.999 | 0.3 | 1.00 | 0.97 ± 0.09 | 3.8 |
| 125 | 127 ± 6 | 1.9 | |||||
| МГ | 7–1000 | S = (1.96 ± 0.07)c | 0.998 | 2.3 | 7.0 | 6.4 ± 0.9 | 5.9 |
| 250 | 252 ± 19 | 3.0 | |||||
| НДМГ | 5–1000 | S = (1.84 ± 0.04)c | 0.999 | 1.3 | 5.0 | 5.0 ± 0.5 | 3.7 |
| 250 | 247 ± 14 | 2.2 |
Известная методика определения гидразина [20] в питьевых водах методом ГХ-ПИД с предварительной дериватизацией БА предполагает трудоемкую стадию экстракции и концентрирования деривата с упариванием растворителя, при этом дериватизацию проводят в кислой среде при рН 2 в течение 20 мин. Выше отмечено, что в этих условиях выход бензалазина очень низкий, поэтому нижняя граница определяемых концентраций составляет 5 мкг/л с погрешностью определения 40% после стадии сложной пробоподготовки (табл. 4). Другие подходы с использованием БА как дериватизирующего реагента [22, 27, 28, 31] также не позволяют одновременно определять гидразин и его алкилпроизводные, поскольку предполагают использование классических буферных систем, в которых выходы БА-производных алкилгидразинов крайне низки. Разработанный нами подход сопоставим по чувствительности с предложенными ранее [10, 24], но характеризуется лучшей воспроизводимостью в области низких концентраций, приемлемой правильностью и широким линейными диапазоном. Методика определения алкилгидразинов с предварительной дериватизацией глиоксалем [25] превосходит предложенную нами по чувствительности, однако она не позволяет определять Ги, поскольку он активно образует с данным реагентом продукты поликонденсации.
Таблица 4.
Сравнение методов определения гидразинов (гидразин/метилгидразин/1,1-диметилгидразин) в водных образцах
| Метод | Дериватизация | сmin, мкг/л | сн*, мкг/л | sr, % | Источник |
|---|---|---|---|---|---|
| ИХ ВЭЖХ−АД | – | 0.2/0.5/1 | –/–/4 | –/–/– | [10] |
| ГФ ВЭЖХ−МС/МС | – | –/17.6/12.8 | –/40/60 | 14/14/14 | [11] |
| ИХ ВЭЖХ−МС | – | 70/30/12 | 200/80/40 | 8/5/5 | [13] |
| ГХ-ПИД | Бензальдегид, рН 2, 20 мин, 20°C | –/–/– | 5/–/– | 40/–/– | [20] |
| ОФ ВЭЖХ−СФД | 5-Нитро-2-фуральдегид, рН 5, 40 мин, 60°C | 0.9/0.4/0.2 | 3.2/1.4/0.7 | 20/20/20 | [24] |
| ОФ ВЭЖХ−СФД | Глиоксаль, рН 3.5, 20 мин, 25°C | –/0.5/0.25 | –/1/0.5 | –/15/10 | [25] |
| ОФ ВЭЖХ−СФД | Бензальдегид, рН 7, 30 мин, 70°C | 17/–/– | 40/–/– | 5/–/– | [27] |
| ОФ ВЭЖХ−МС/МС | 2-Нитробензальдегид, рН 5.5, 45 мин, 75°C | –/–/– | 1/10/10 | 11/8/7 | [33] |
| ОФ ВЭЖХ−СФД | Бензальдегид, рН 9.4 (ИК**, МК), 5 мин, 20°C | 0.3/2.3/1.3 | 1/7/5 | 4/6/4 | Данная работа |
Разработанный в данной работе подход не требует применения дополнительных стадий концентрирования, прост, надежен, а реакция протекает количественно всего за 5 мин, сам анализ занимает не более 13 мин. Такого результата удалось добиться благодаря применению иминного и мицеллярного катализа и проведения дериватизации в оптимальных условиях.
* * *
Таким образом, предложен простой, быстрый и чувствительный комбинированный способ определения Ги, МГ и НДМГ в широком диапазоне концентраций (1–1000 мкг/л) методом ОФ ВЭЖХ−СФД с предколоночной дериватизацией БА. Впервые применен эффект иминного катализа буферной системы на основе аммония для получения производных гидразинов с последующим аналитическим приложением. Доказан, продемонстрирован и успешно применен эффект ускорения реакций образования гидразонов в присутствии анионного ПАВ – ДДСН. Это позволило не только значительно уменьшить общую продолжительность анализа, что крайне важно в рутинном анализе, но и обеспечить образование производных при низких концентрациях, а также существенно понизить нижнюю границу определяемых концентраций. Показана нецелесообразность нагревания реакционных смесей с целью уменьшения продолжительности реакции из-за разложения образующихся гидразонов. Подтвержден независимым методом ИХ ВЭЖХ-АД количественный выход продуктов дериватизации в оптимальных условиях. Разработанная методика не требует проведения трудоемких стадий концентрирования и выделения веществ, труднодоступных реагентов и оборудования, характеризуется приемлемыми правильностью, воспроизводимостью и чувствительностью, а также широким линейным диапазоном определяемых концентраций. Применение мицеллярного катализа перспективно как для совершенствования уже известных, так и при разработке новых способов определения гидразинов. Поиск новых систем для иминного катализа позволит интенсифицировать реакции образования гидразонов с другими малоактивными групповыми реагентами. В перспективе разработанные методики могут быть распространены на анализ не только вод, но и любых водных матриц, в том числе кислотных вытяжек и отгонов из почв, а также смывов с поверхностей и отгонов из строительных материалов.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-33-90120.
Список литературы
Schmidt E.W. Hydrazine and its Derivatives. 2nd Ed. N.Y.: John Wiley & Sons, Inc., 2001. 2121 p.
Rothgery E.F. Hydrazine and its derivatives / Kirk-Othmer Encyclopedia of Chemical Technology. N.Y.: John Wiley & Sons, Inc., 2004. P. 562.
Адушкин В.В., Александров Э.Л., Бурчик В.Н. Экологические проблемы и риски воздействий ракетно-космической техники на окружающую природную среду. М: Анкил, 2000. 640 с.
Белов А.А. К вопросу о токсичности и опасности гидразина и его производных (обзор) // Современные проблемы токсикологии. 2000. № 1. С. 25.
Choudhary G., Hansen H. Human health perspective of environmental exposure to hydrazines: A review // Chemosphere. 1998. V. 37. № 5. P. 801.
СанПиН 1.2.3685-21. Гигиенические нормативы и требования к обеспечению безопасности и (или) безвредности для человека факторов среды обитания, 2021.
Смоленков А.Д. Хроматографические методы определения гидразина и его полярных производных // Обзорный журнал по химии. 2012. Т. 2. № 4. С. 334. (Smolenkov A.D. Chromatographic methods of determining hydrazine and its polar derivatives // Rev. J. Chem. 2012. V. 2. № 4. P. 329.)
Smolenkov A.D., Shpigun O.A. Direct liquid chromatographic determination of hydrazines: A review // Talanta. 2012. V. 102. P. 93.
Смоленков А.Д., Родин И.А., Шпигун О.А. Спектрофотометрические и флуориметрические методы определения гидразина и его метилированных аналогов // Журн. аналит. химии. 2012. Т. 67. № 2. С. 133. (Smolenkov A.D., Rodin I.A., Shpigun O.A. Spectrophotometric and fluorometric methods for the determination of hydrazine and its methylated analogues // J. Anal. Chem. 2012. V. 67. № 2. P. 98.)
Smolenkov A.D., Krechetov P.P., Pirogov A.V., Koroleva T.V., Bendryshev A.A., Shpigun O.A., Martynova M.M. Ion chromatography as a tool for the investigation of unsymmetrical hydrazine degradation in soils // Int. J. Environ. Anal. Chem. 2005. V. 85. № 14. P. 1089.
Kosyakov D.S., Ul’yanovskii N.V., Bogolitsyn K.G., Shpigun O.A. Simultaneous determination of 1,1-dimethylhydrazine and products of its oxidative transformations by liquid chromatography–tandem mass spectrometry // Int. J. Environ. Anal. Chem. 2014. V. 94. № 12. P. 1254.
Пономаренко С.А., Смоленков А.Д., Шпигун О.А. Определение 1,1-диметилгидразина и продуктов его разложения методом ион-парной хроматографии // Вестн. Моск. ун-та. Сер. 2: Химия. 2009. Т. 50. № 3. С. 185. (Ponomarenko S.A., Smolenkov A.D., Shpigun O.A. Determination of 1,1-dimethylhydrazine and its decomposition products using ion-pair chromatography // Moscow Univ. Chem. Bull. 2009. V. 64. № 3. P. 147.)
Смоленков А.Д., Родин И.А., Смирнов Р.С., Татаурова О.Г., Шпигун О.А. Применение ионной и ион-парной хроматографии с масс-спектрометрическим детектированием для определения нессиметричного диметилгидразина и продуктов его трансформации // Вестн. Моск. ун-та. Сер. 2: Химия. 2012. Т. 53. № 5. С. 312. (Smolenkov A.D., Rodin I.A., Smirnov R.S., Tataurova O.G., Shpigun O.A. Use of ion and ion-pair chromatography with mass spectrometric detection to determine unsymmetrical dimethylhydrazine and its transformation products // Moscow Univ. Chem. Bull. 2012. V. 67. № 5. P. 229.)
Kosyakov D.S., Pikovskoi I.I., Ul’yanovskii N.V., Kozhevnikov A.Y. Direct determination of hydrazine, methylhydrazine, and 1, 1-dimethylhydrazine by zwitterionic hydrophilic interaction liquid chromatography with amperometric detection // Int. J. Environ. Anal. Chem. 2017. V. 97. № 4. P. 313.
Sternson L.A. General aspects of precolumn derivatization with emphasis on pharmaceutical analysis / Chemical Derivatization in Analytical Chemistry: Chromatography. 1st Ed. / Eds. Frei R.W., Lawrence J.F. Boston: Springer, 1981. P. 127.
Newsome W.H., Collins P. An improved method for the determination of 1,1-dimethyl hydrazine in apple and cherry products // Int. J. Environ. Anal. Chem. 1988. V. 34. № 2. P. 155.
Holtzclaw J.R., Rose S.L., Wyatt J.R., Rounbehler D.P., Fine D.H. Simultaneous determination of hydrazine, methylhydrazine, and 1,1-dimethylhydrazine in air by derivatization/gas chromatography // Anal. Chem. 1984. V. 56. № 14. P. 2952.
Сотников Е.Е., Московкин А.С. Газохроматографическое определение несимметричного диметилгидразина в воде // Журн. аналит. химии. 2006. Т. 61. № 2. С. 139. (Sotnikov E.E., Moskovkin A.S. Gas-chromatographic determination of 1,1-dimethylhydrazine in water // J. Anal. Chem. 2006. V. 61. № 2. P. 129.)
Mazur J.F., Podolak G.E., Heitke B.T. Use of a GC concentrator to improve analysis of low levels of airborne hydrazine and unsymmetrical dimethylhydrazine // Am. Ind. Hyg. Assoc. J. 1980. V. 41. № 1. P. 66.
ПНД Ф 14.1:2.4.191-03. Методика выполнения измерений массовой концентрации гидразина в пробах питьевых, природных и сточных вод газохроматографическим методом, 2003.
Faughnan K.T., Woodruff M.A. Modified gas chromatographic/mass spectrometric method for determination of daminozide in high protein food products // J. Assoc. Off. Anal. Chem. 1991. V. 74. № 4. P. 682.
Cathum S., Atamaniouk V., Ananieva L., Ladanowski C., Whittaker H. Gas chromatography–mass spectrometric determination of unsymdimethylhydrazine in soil and water by derivatization with aromatic aldehydes // Can. J. Chem. Eng. 1998. V. 76. №. 3. P. 680.
Евгеньев М.И., Евгеньева И.И., Гармонов С.Ю., Исмаилова Р.Н., Белов П.Е. Сорбционно-хроматографическое определение гидразина и его замещенных в воздухе // Журн. аналит. химии. 2006. Т. 61. № 5. С. 492. (Evgen’ev M.I., Evgen’eva I.I., Garmonov S.Y., Ismailova R.N., Belov P.E. Sorption-chromatographic determination of hydrazine and its substituted derivatives in air // J. Anal. Chem. 2006. V. 61. № 5. P. 452.)
Амосов А.С., Ульяновский Н.В., Косяков Д.С., Шпигун О.А. Одновременное определение гидразина, метилгидразина и 1,1-диметилгидразина методом высокоэффективной жидкостной хроматографии с пред- и постколоночной дериватизацией 5-нитро-2-фуральдегидом // Журн. аналит. химии. 2018. Т. 73. № 5. С. 389. (Amosov A.S., Ul’yanovskii N.V., Kosyakov D.S., Shpigun O.A. Simultaneous determination of hydrazine, methylhydrazine, and 1,1-dimethylhydrazine by high-performance liquid chromatography with pre-and post-column derivatization by 5-nitro-2-furaldehyde // J. Anal. Chem. 2018. V. 73. № 5. P. 497.)
Смирнов Р.С., Смоленков А.Д., Болотник Т. А., Шпигун О.А. Применение глиоксаля и глиоксиловой кислоты для определения методом высокоэффективной жидкостной хроматографии // Вестн. Моск. ун-та. Сер. 2: Химия. 2013. Т. 54. № 1. С. 22. (Smirnov R.S., Smolenkov A.D., Bolotnik T.A., Shpigun O.A. The use of glyoxal and glyoxylic acid to determine N-and N,N-alkyl-substituted hydrazines by liquid chromatography // Moscow Univ. Chem. Bull. 2013. V. 68. № 1. P. 17.)
Филиппов О.А., Тихомирова Т.И., Смоленков А.Д., Цизин Г.И., Формановский А.А., Шпигун О.А. Выбор условий динамического сорбционного концентрирования производного гептила - N,N-диметилгидразона 4-нитробензальдегида, на гидрофобизированном кремнеземе // Журн. аналит. химии. 2001. Т. 56. №. 12. С. 1238. (Filippov O.A., Tikhomirova T.I., Smolenkov A.D., Tsizin G.I., Formanovskii A.A., Shpigun O.A. Selection of conditions for the dynamic sorption preconcentration of a 1,1-dimethylhydrazine derivative (4-nitrobenzaldehyde N,N-dimethylhydrazone) on hydrophobized silica // J. Anal. Chem. 2001. V. 56. № 12. P. 1070.)
Shustina R., Lesser J.H. Liquid chromatographic determination of hydrazine, carbohydrazide and thiocarbohydrazide in aqueous solutions // J. Chromatogr. A. 1991. V. 464. P. 208.
Tamás K., Wachter-Kiss E., Kormány R. Hydrazine determination in allopurinol using derivatization and SPE for sample preparation // J. Pharm. Biomed. Anal. 2018. V. 152. P. 25.
Smolenkov A.D., Chernobrovkina A.V., Smirnov R.S., Chernobrovkin M.G., Shpigun O.A. A sensitive chromatographic determination of hydrazines by naphthalene-2, 3-dialdehyde derivatization // Int. J. Environ. Anal. Chem. 2013. V. 93. № 12. P. 1286.
Christofi M., Markopoulou C.K., Tzanavaras P.D., Zacharis C.K. UHPLC-fluorescence method for the determination of trace levels of hydrazine in allopurinol and its formulations: Validation using total-error concept // J. Pharm. Biomed. Anal. 2020. V. 187. P. 113354.
Cui L., Jiang K., Liu D.Q., Facchine K.L. Simultaneous quantitation of trace level hydrazine and acetohydrazide in pharmaceuticals by benzaldehyde derivatization with sample ‘matrix matching’followed by liquid chromatography–mass spectrometry // J. Chromatogr. A. 2016. V. 1462. P. 73.
Susinskis I., Mekss P., Hmelnickis J. Method development for the determination of 1,1-dimethylhydrazine by the high-performance liquid chromatography–mass spectrometry technique // Eur. J. Mass Spectrom. 2018. V. 24. № 4. P. 352.
An Z., Li P., Zhang X., Liu L. Simultaneous determination of hydrazine, methylhydrazine, and 1,1-dimethylhydrazine in rat plasma by LC–MS/MS // J. Liq. Chromatogr. Relat. Technol. 2014. V. 37. № 9. P. 1212.
Gojon C., Dureault B. spectrophotometric study of the reaction between hydrazine and p. dimethylaminobenzaldehyde // J. Nucl. Sci. Technol. 1996. V. 33. № 9. P. 731.
Lunn G., Sansone E.B. Oxidation of 1,1-dimethylhydrazine (UDMH) in aqueous solution with air and hydrogen peroxide // Chemosphere. 1994. V. 29. № 7. P. 1577.
Dwars T., Paetzold E., Oehme G. Reactions in micellar systems // Angew. Chem. Int. Ed. 2005. V. 44. № 44. P. 7174.
Yatsimirsky A.K., Yatsimirskaya N.T., Kashina S.B. Micellar catalysis and product stabilization in hydrazone formation reactions and micellar-modified determination of hydrazine and phenylhydrazine // Anal. Chem. 1994. V. 66. № 14. P. 2232.
Sayer J.M., Peskin M., Jencks W.P. Imine-forming elimination reactions. I. General base acid catalysis and influence of the nitrogen substituent on rates and equilibria for carbinolamine dehydration // J. Am. Chem. Soc. 1973. V. 95. № 13. P. 4277.
Yildiz I. A DFT approach to the mechanistic study of hydrozone hydrolysis // J. Phys. Chem. A. 2016. V. 120. № 20. P. 3683.
Erkkilä A., Majander I., Pihko P.M. Iminium catalysis // Chem. Rev. 2007. V. 107. № 12. P. 5416.
Miura M., Kodama M. The second CMC of the aqueous solution of sodium dodecyl sulfate. I. Conductivity // Bull. Chem. Soc. Jpn. 1972. V. 45. № 2. P. 428.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии