

Журнал аналитической химии, 2021, T. 76, № 3, стр. 234-243
Микроэкстракционно-цветометрическое (флуориметрическое) определение катионных и анионных поверхностно-активных веществ в пищевых продуктах
В. Г. Амелин a, b, *, З. А. Ч. Шаока a, Д. С. Большаков b
a Владимирский государственный университет имени Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
b Федеральный центр охраны здоровья животных
600901 Владимир, мкр. Юрьевец, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 30.07.2020
После доработки 21.08.2020
Принята к публикации 01.09.2020
Аннотация
Предложен способ определения катионных и анионных поверхностно-активных веществ (ПАВ) в пищевых продуктах, основанный на применении дисперсионной жидкостно-жидкостной микроэкстракции ассоциатов ПАВ с органическими реагентами (эозин и акридиновый желтый) и измерении цветометрических параметров флуоресценции экстрактов с помощью смартфона. При облучении ультрафиолетовым светом (365 нм) экстрактов ассоциатов анионное ПАВ–акридиновый желтый и катионное ПАВ–эозин наблюдается зеленая и желтая флуоресценция соответственно. В качестве аналитического сигнала (Ar) использовали значения цветометрических параметров в системе RGB: Ar = $\sqrt {{{{{\text{(}}{{R}_{{\text{0}}}} - {{R}_{x}}{\text{)}}}}^{3}}{\text{ + (}}{{G}_{{\text{0}}}} - {{G}_{x}}{{{\text{)}}}^{2}}{\text{ + (}}{{B}_{{\text{0}}}} - {{B}_{x}}{{{\text{)}}}^{2}}} .$ Разработаны методики определения суммарного содержания ПАВ в пищевых продуктах (молоко, овощи и фрукты, мясо, питьевая вода) на примере хлоридов цетилпиридиния, миристалкония, бензалкония, дидецилдиметиламмония, алкилдиметил(этилбензил)аммония, додецилсульфата и алкилбензолсульфоната натрия. Пределы обнаружения и определения находятся в диапазонах 0.005–0.05 и 0.01–0.1 мг/л соответственно. Градуировочные графики линейны в диапазоне концентраций 0.01–1 мг/л с коэффициентами достоверности аппроксимации ≥0.99. Продолжительность анализа 20–30 мин, относительное стандартное отклонение рузультатов анализа не превышает 0.24.
В аналитической химии пищевых продуктов значительное внимание уделяется хроматографическим методам анализа с различными вариантами детектирования. Это можно объяснить универсальностью данных методов, точностью, эффективностью разделения многокомпонентных смесей и стабильностью работы оборудования. С другой стороны, существует острая необходимость развития экспрессных и доступных методов, реализация которых не требует наличия дорогостоящего оборудования. Здесь следует выделить цветометрические методы анализа. Существенный интерес к цифровой цветометрии обусловлен возможностью использования в качестве цветорегистрирующей техники бытовых цифровых оптических устройств, которые не относятся к специализированным средствам измерения [1–4]. Техническая простота, экспрессность, достаточно высокие чувствительность и точность в значительной степени определили тенденции развития данного направления в целях оценки качества и безопасности продуктов питания.
Особое место в пищевом анализе занимает проблема определения поверхностно-активных веществ, которые могут попасть в продукты питания различными способами. Анионные поверхностно-активные вещества (АПАВ) могут оказаться в молоке в результате нетщательного отмывания доильных аппаратов и тары для перевозки молока от моющих средств. В качестве моющих средств применяют многокомпонентные растворы, большинство из которых содержит додецилсульфат и алкилбензолсульфонаты натрия. При производстве некоторых видов молочной продукции длительных сроков хранения, а также заменителей молочных продуктов АПАВ используют с целью фальсификации, в качестве стабилизаторов эмульсий для сохранения консистенции, обеспечения требуемой вязкости и полноты вкуса [5].
Способность ПАВ проявлять влагопоглощающие свойства объясняет их присутствие в мясной продукции и птице. В основном они попадают туда на стадии упаковки при поверхностной обработке продукта влагоудерживающими компонентами и эмульсиями антиоксидантов. Фрукты и овощи, особенно импортируемые, при хранении и перевозке многократно обрабатываются распыляемыми эмульсиями, стабилизированными ПАВ, с целью гарантированной защиты от увядания и усыхания, предотвращения поражений грибковыми заболеваниями и повреждения товарного вида насекомыми. Хлориды цетилпиридиния, миристалкония, бензалкония, дидецилдиметиламмония, алкилдиметил(этилбензил)аммония обладают бактерицидными свойствами и используются для обработки овощей, фруктов и мяса [6].
Для определения катионных и анионных ПАВ в пищевых продуктах используют в основном три группы методов: хромато-масс-спектрометрию, спектрофотометрию и цветометрию. Первый метод применяют для определения и идентификации конкретных ПАВ, второй и третий – для определения суммарного содержания ПАВ.
В работе [7] описано определение линейного додецилбензолсульфоната натрия в молоке методом ультра высокоэффективной жидкостной хроматографии (УВЭЖХ) с квадруполь-времяпролетным масс-спектрометрическим (МС) детектированием в диапазоне концентраций 24–240 мкг/л. Идентификация АПАВ проведена методом “отпечатков пальцев” по масс-хроматограммам экстракта молока с добавкой и без добавки АПАВ. Хлориды бензалкония (С10, С12, С14, С16), дидецилдиметиламмония, бензэтония в апельсинах и огурцах предложено определять с использованием экстракции QuEChERS и последующего анализа методом УВЭЖХ–МС/МС [8]. Диапазон определяемых содержаний составил 0.01–0.15 мг/кг. В работе [9] с использованием пробоподготовки QuEChERS определяли хлориды бензалкония (С12, С14, С16), дидецилдиметиламмония и додецилтриметиламмония в диапазоне концентраций 0.2–50 мкг/л методом УВЭЖХ–МС/МС. Хлориды бензалкония (С8,10,12,14,16) и диалкилдиметиламмония (С10,12,14,16,18) определяли в козьем молоке в диапазоне 5–150 мкг/кг методом ВЭЖХ–МС/МС [10]. Экстрагировали ПАВ из пробы во всех случаях ацетонитрилом, очистку экстракта проводили насыпными сорбентами PSA (смесь первичных и вторичных аминов) и сульфатом магния [7–10].
Для определения остаточных количеств хлорида цетилпиридиния в мясе кур и продуктах его переработки разработана методика [6], основанная на использовании метода ВЭЖХ с ультрафиолетовым детектированием при длине волны 260 нм после водно-спиртовой экстракции аналита из анализируемых проб и обезжиривания экстракта гексаном. Диапазон определяемых содержаний катинного ПАВ (КПАВ) составил 0.08–5 мг/кг.
Спектрофотометрический и цветометрический методы определения ПАВ основаны на образовании ионных ассоциатов ПАВ–анионный (катионный) краситель, экстракции нейтральной ионной пары и последующем определении спектрофотометрических (флуориметрических [11]) и цветометрических характеристик экстракта. В качестве красителей используют акридиновый желтый, эозин, азур A, патентованный голубой V, метиленовый синий [11–15]. Методы применены для определения ПАВ в питьевой воде и молоке.
Для количественной оценки содержания КПАВ и АПАВ в питьевой и природной водах установлен ряд стандартизованных методик [11]. Для извлечения соответствующих ионных ассоциатов ПАВ с органическими красителями используют жидкостно-жидкостную экстракцию хлороформом. В случае применения спектрофотометрического метода для определения АПАВ с метиленовым синим экстракты дополнительно обрабатывают кислотой для устранения мешающего влияния компонентов матрицы.
Среди недостатков хроматографических, спектрофотометрических и флуориметрических методов определения ПАВ в воде и пищевых продуктах следует выделить не только значительную стоимость аппаратурного обеспечения, но и необходимость использования больших количеств высокочистых и, как правило, токсичных веществ и растворителей.
Совершенствование экологичных подходов к определению ПАВ в практике испытательных и контрольных лабораторий возможно с внедрением методов жидкостной микроэкстракции и определения цветометрических характеристик экстракта с помощью смартфона [14, 15]. Для определения додецилсульфата в питьевой воде в диапазоне 10–70 мкг/л предложена дисперсионная жидкостно-жидкостная микроэкстракция (ДЖЖМЭ) 250 мкл смеси хлороформ–ацетонитрил (2 : 3, по объему) ионного ассоциата с метиленовым синим. Предел обнаружения составил 2.6 мкг/л [14]. При определении додецилсульфата натрия (ДДС) в синтетическом молоке для экстракции ассоциата ПАВ с метиленовым синим использовали 150 мкл хлороформа [15]. Диапазон линейности градуировочного графика 10–50 мг/л, предел обнаружения ДДС 2.2 мг/л.
Цель настоящей работы состояла в разработке способа микроэкстракционно-цветометрического (флуориметрического) определения катионных и анионных поверхностно-активных веществ в пищевых продуктах после извлечения и концентрирования аналитов в виде ассоциатов с органическими красителями путем дисперсионной жидкостно-жидкостной микроэкстракции и последующей регистрации цветометрических характеристик люминесценции с помощью смартфона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура и материалы. Для изучения оптических и цветометрических характеристик подготовленных экстрактов в качестве цветорегистрирующего устройства использовали смартфон iPhone X (Apple, США), оснащенный специализированным программным продуктом RGBer. Возбуждение флуоресценции проводили с помощью источника монохроматического излучения с длиной волны 365 нм Jialitte F114 5W 365NM UV (Shenzhen Jialite Technology Co., Китай).
В работе применяли аналитические весы Pioneer PA 214С специального класса точности с пределом взвешивания 0.1 мг (Ohaus Corporation, USA), лабораторную настольную центрифугу Jouan BBVV (Thermo Scientific, Франция), дозаторы Proline Biohit 1-канальные механические переменного объема 10–100, 100–1000, 1000–5000 мкл (Biohit, Финляндия), микрошприцы объемом 10, 100 и 500 мкл (Hamilton Company, Япония), политетрафторэтиленовые мембранные фильтры 25 мм диаметром пор 0.45 мкм (Pall Corporation, США), пробирки полипропиленовые емк. 15 мл (SPL Life Sciences Co., Корея), пробирки типа “Эппендорф” емк. 2.0 мл (GenFollower Biotech Co., Китай).
Реактивы. Использовали стандартные образцы хлоридов цетилпиридиния (ЦП), миристалкония, бензалкония, дидецилдиметиламмония, алкилдиметил(этилбензил)аммония, додецилсульфата натрия и сульфонола (98–100%, Sigma-Aldrich, США). Основные стандартные растворы с концентрацией 1 мг/мл готовили растворением точной навески препарата в деионизированной воде (не менее 18 МОм · см, ОСТ 11 029.003-80). Рабочие стандартные растворы готовили последовательным разбавлением основных стандартных растворов деионизированной водой.
Использовали ацетонитрил (99.9%, Scharlab S.L., Испания), метанол (PA-ACS-ISO, Panreac, ЕС), этилендиаминтетраацетат натрия (ЭДТА) (99%, ХИММЕД, Россия), трихлорметан (99.85%, Компонент-Реактив, Россия), этиловый спирт (х. ч., ХИММЕД, Россия), ацетон (99%, WarChem, Польша), хлорид натрия (х. ч., ХИММЕД, Россия), акридиновый желтый (ч. д. а., Союзхимпром, Россия), эозин (ч. д. а., Ленреактив, Россия), соляную кислоту (стандарт-титр, НПИИП Уралхиминвест), тетраборат натрия (99.5%, Sigma-Aldrich, США).
Схема установки. Для измерения цветометрических характеристик экстрактов ассоциатов использовали установку, представленную на рис. 1. Пробирку с экстрактом после микроэкстракционного концентрирования помещали в штатив, облучали ультрафиолетовым излучением (365 нм). С помощью смартфона iPhone X наводили фокус на экстракт, делали снимок и с применением программного продукта RGBer определяли цветометрические характеристики экстракта.
Рис. 1.
Установка для измерения цветометрических (флуориметрических) характеристик экстрактов: 1 – источник УФ-излучения, 2 – пробирка с экстрактом, 3 – смартфон, 4 – бокс из полипропилена (10 × 10 × 10 см).

Аналитический сигнал (Ar) в системе RGB рассчитывали по формуле:
Пробоподготовка. Твердые продукты (мясо, фрукты), молоко. Навеску пробы массой 1.00 г помещали в пробирку емк. 15 мл, добавляли 1 мл этилового спирта, 9 мл деионизированной воды, встряхивали в течение 5 мин и центрифугировали 5 мин при 5000 об/мин. Отбирали 1 мл полученного экстракта в пробирку емк. 15 мл, добавляли 100 мкл 0.05%-ного раствора красителя, 100 мкл 2%-ного раствора тетрабората натрия (для системы хлорид цетилпиридиния–эозин) или 1 М HCl (для системы додецилсульфат натрия–акридиновый желтый), 100 мкл 10%-ного раствора NaCl (для системы хлорид цетилпиридиния–эозин), 100 мкл раствора ЭДТА (10 мг/л). Смесь перемешивали, объем доводили до 10 мл деионизированной водой. С помощью шприца в подготовленную пробу впрыскивали 500 мкл экстрагирующей смеси. Смесь тщательно встряхивали, затем центрифугировали в течение 5 мин при 5000 об/мин.
Вода. Профильтрованную через мембранный фильтр пробу воды объемом 10 мл помещали в пробирку емк. 15 мл, приливали 100 мкл 0.05%-ного раствора красителя, 100 мкл 2%-ного раствора тетрабората натрия (для системы цетилпиридиний–эозин) или 1 М HCl (для системы додецилсульфат–акридиновый желтый), 100 мкл 10%-ного раствора NaCl (для системы цетилпиридиний–эозин), 100 мкл ЭДТА (10 мг/л). Смесь перемешивали, затем с помощью шприца в подготовленную пробу впрыскивали 500 мкл экстрагирующей смеси. Смесь тщательно встряхивали, затем центрифугировали в течение 5 мин при 5000 об/мин. С помощью смартфона измеряли цветометрические характеристики экстрактов. Концентрацию определяли методом градуировочного графика.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выбор условий дисперсионной жидкостно-жидкостной микроэкстракции. Выбор экстрагента. В качестве экстрагирующего растворителя в работе опробовали хлороформ, четыреххлористый углерод и хлористый метилен. Диспергирование проводили ацетонитрилом. Установлено, что проведение микроэкстракции различными растворителями обеспечивает близкие значения сигнала флуоресценции ассоциатов ДДС–акридиновый желтый и ЦП–эозин (рис. 2а). В дальнейших исследованиях использовали систему хлороформ–ацетонитрил как наиболее часто встречающуюся.
Рис. 2.
Выбор условий дисперсионной жидкостно-жидкостной микроэкстракции ассоциатов ДДС–акридиновый желтый (черный), ЦП–эозин (серый): экстрагента (а), объема экстрагента (б), диспергирующего растворителя (в), объема диспергирующего растворителя (г), ионной силы раствора (д), объемов 1 М HCl (черный) и 2%-ного раствора тетрабората натрия (серый) (е).
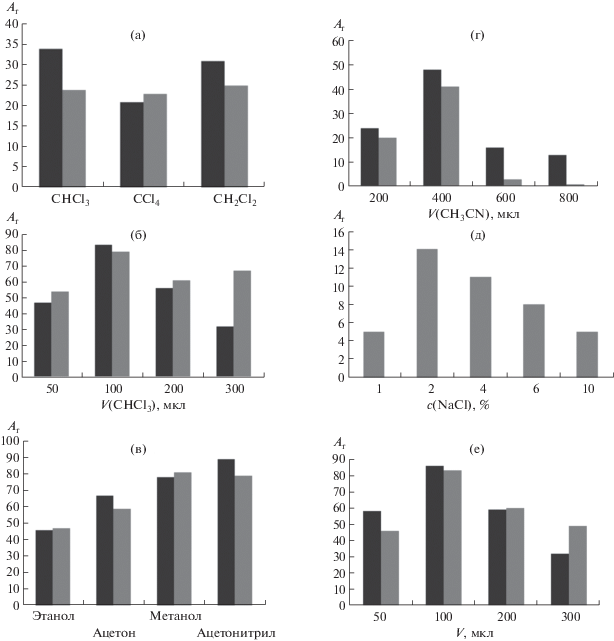
Выбор объема экстрагента. Для оптимизации объема экстрагирующего растворителя в качестве смеси для проведения дисперсионной микроэкстракции использовали комбинацию экстрагента CHCl3 и диспергирующего растворителя CH3CN. Для приготовления рабочих смесей в пробирку типа “Эппендорф” дозатором вносили 100, 200, 400 и 600 мкл хлороформа, объем до 1000 мкл доводили ацетонитрилом, затем смесь тщательно перемешивали. Полученные растворы объемом 500 мкл вводили с помощью шприца в рабочие пробы природной воды объемом 10 мл, содержащие 0 и 0.5 мг/л ПАВ. Затем измеряли цветометрические параметры экстрактов ассоциатов с помощью смартфона и рассчитывали аналитический сигнал Аr. Как показано на рис. 2б, максимальное значение аналитического сигнала флуоресценции можно наблюдать при проведении микроэкстракционного концентрирования в присутствии 100 мкл экстрагента CHCl3.
Выбор диспергирующего растворителя. В качестве диспергирующего растворителя испытали этанол, ацетонитрил, ацетон и метанол. Для этого в пробирки емк. 15 мл вносили 800 мкл этанола, ацетона, метанола и ацетонитрила, добавляли в каждую из них 200 мкл хлороформа, смесь тщательно перемешивали. Полученные растворы объемом 500 мкл вводили с помощью шприца в рабочие пробы природной воды объемом 10 мл, содержащие 0 и 0.5 мг/л ПАВ. Лучшие результаты (близкие значения аналитического сигнала флуоресценции) получены для метанола и ацетонитрила (рис. 2в). Учитывая меньшую токсичность ацетонитрила, дальнейшие исследования проводили с этим диспергирующим растворителем.
Выбор объема диспергирующего растворителя. В пробирки типа “Эппендорф” вносили 100 мкл CHCl3 и 200, 400, 600, 800 мкл CH3CN. Полученные растворы вводили с помощью шприца в рабочие пробы природной воды объемом 10 мл, содержащие 0 и 0.5 мг/л ПАВ. Установлено, что при объеме CH3CN 600 и 800 мкл не происходит разделение смеси и выполнить измерения при данном объеме диспергирующего растворителя невозможно (Аr < 5). Максимальное значение аналитического сигнала достигнуто в случае проведения ДЖЖМЭ при объеме диспергирующего растворителя CH3CN 400 мкл и экстрагирующего растворителя CHCl3 100 мкл (рис. 2г).
Влияние ионной силы исследовали добавлением 1–10% (мас./об.) NaCl к анализируемой пробе. Для системы ДДС–акридиновый желтый установлено, что внесение хлорида натрия в анализируемый раствор не изменяет значение аналитического сигнала. Для системы ЦП–эозин максимальное значение флуоресценции экстракта наблюдали при внесении в анализируемую систему 2% (мас./об.) раствора хлорида натрия (рис. 2д).
Влияние кислоты и тетрабората натрия. Образование ассоциатов ДДС–акридиновый желтый протекает в кислой среде, ЦП–эозин – в щелочной. Максимальное значение Ar наблюдали при добавлении в 10 мл раствора пробы 100 мкл 1 М HCl или 2%-ного раствора тетрабората натрия (рис. 2е).
Влияние концентрации красителя. При малой концентрации красителя ассоциаты с ПАВ практически не образуются, и аналитический сигнал равен нулю, а при высокой концентрации красителя наблюдается тушение люминесценции и иные отклонения от линейности. В данной работе оптимизировали концентрацию используемых красителей, которая при выбранных параметрах ДЖЖМЭ составила 100 мкл 0.05%-ного раствора.
Таким образом, оптимальные условия ДЖ-ЖМЭ: экстрагирующая смесь 500 мкл смеси хлороформ–ацетонитрил (1 : 4, по объему), 100 мкл 0.05%-ного раствора красителя и 100 мкл 1 М HCl (для системы ДДС–акридиновый желтый) или 100 мкл 2%-ного раствора тетрабората натрия, 100 мкл 10%-ного раствора хлорида натрия (для системы ЦП–эозин).
Определение анионных ПАВ. Сущность предлагаемого в работе подхода заключается в образовании ионных ассоциатов катионного красителя акридинового желтого с АПАВ, последующей микроэкстракции полученных ассоциатов хлороформом (при диспергировании ацетонитрилом), установлении флуориметрических характеристик полученного экстракта с помощью смартфона и определении концентрации АПАВ по градуировочному графику. При образовании ассоциата анионы додецилсульфата или другого АПАВ взаимодействуют с протонированными в кислой среде аминогруппами акридинового желтого (схема 1 ). Полученный таким образом нейтральный ассоциат экстрагируется в процессе ДДЖМЭ хлороформом. При облучении ультрафиолетовым светом (365 нм) экстракт ассоциата флуоресцирует зеленым цветом.

Схема 1 . Образование ионного ассоциата аниона додецилсульфата и протонированной формы акридиного желтого.
Для построения градуировочного графика готовили ряд стандартных растворов с концентрацией додецилсульфата натрия от 0.05 до 0.8 мг/л. Проводили микроэкстракционное концентрирование и измеряли цветометрические (флуориметрические) характеристики полученных экстрактов в системе RGB с УФ-облучением и без него. Канал G при использовании УФ-излучения не учитывали, так как при данных концентрациях наблюдали его максимальное значение (255). Градуировочные графики в выбранном диапазоне концентраций нелинейны и аппроксимируются логарифмической зависимостью (рис. 3). Установлено, что чувствительность определения повышается в несколько раз при измерении флуоресценции и градуировочный график линеен в диапазоне 0.01–0.20 мг/л (табл. 1).
Рис. 3.
Градуировочный график для микроэкстракционно-цветометрического (флуориметрического) определения додецилсульфата натрия с УФ-облучением (1) и без облучения (2).

Таблица 1.
Аналитические характеристики микроэкстракционно-флуориметрического определения ПАВ
| ПАВ | Реагент | смин, мг/л | сн, мг/л | ДОС, мг/л | Уравнение градуировочного графика | R2 |
|---|---|---|---|---|---|---|
| Додецисульфат натрия | Акридиновый желтый | 0.005 | 0.01 | 0.01–0.8 | Ar = 321.68c + 2.76 | 0.9907 |
| Сульфонол | Акридиновый желтый | 0.01 | 0.05 | 0.05–0.4 | Ar = 180.17c + 13.21 | 0.9937 |
| Цетилпиридиния хлорид | Эозин | 0.01 | 0.05 | 0.05–1 | Ar = 79.18с – 1.64 | 0.9900 |
| Миристалкония хлорид | Эозин | 0.02 | 0.06 | 0.06–1 | Ar = 1.59c + 14.86 | 0.9914 |
| Бензалкония хлорид | Эозин | 0.05 | 0.1 | 0.1–1 | Ar = 9.36с + 2.85 | 0.9953 |
| Дидецилдиметиламмония хлорид | Эозин | 0.02 | 0.05 | 0.05–1 | Ar = 121.52c – 1.04 | 0.9855 |
| Алкилдиметил (этилбензил)аммония хлорид | Эозин | 0.02 | 0.1 | 0.1–0.8 | Ar = 70.80с – 0.92 | 0.9913 |
Определение катионных ПАВ. Определение основано на образовании ионных ассоциатов анионного красителя эозина с КПАВ. При образовании ассоциата катион цетилпиридиния взаимодействует с окси- и карбоксигруппами эозина (схема 2 ). Нейтральный ассоциат экстрагируется хлороформом в результате ДЖЖМЭ. При УФ-облучении (365 нм) экстракт флуоресцирует желтым цветом.

Схема 2 . Образование ионного ассоциата катиона цетилпиридия и эозина.
Для построения градуировочного графика готовили ряд стандартных растворов хлорида цетилпиридиния в диапазонe концентраций от 0.05 до 1 мг/л. Проводили микроэкстракционное концентрирование и измеряли флуориметрические характеристики полученных экстрактов в системе RGB (табл. 1).
Аналитические характеристики определения ПАВ. В табл. 1 представлены аналитические характеристики методики определения катионных и анионных ПАВ. Пределы обнаружения (смин) и определения (сн) оценивали как концентрацию ПАВ, при которой значение аналитического сигнала превышает фоновый (для дистиллированной воды Аr = 1) в 2 и 5 раз соответственно. Предлагаемая методика характеризуется наибольшей чувствительностью по отношению к додецилсульфату натрия (смин = 0.005 мг/л), хлориду цетилпиридиния и сульфонолу (смин = 0.01 мг/л). Градуировочные графики линейны с коэффициентами достоверности аппроксимации ≥0.99.
Определение ПАВ в продуктах питания. Разработанную методику использовали для определения ПАВ в овощах, фруктах, мясе птице и молочной продукции. Ионы металлов, присутствующие в продуктах, могут мешать определению ПАВ. Для устранения мешающего влияния в анализируемый экстракт добавлялиу ЭДТА. Мешающее влияние белков устраняли их осаждением этанолом.
Следует отметить, что с использованием данного подхода в продукции определяли суммарное содержание ПАВ. В качестве стандарта при определении катионных ПАВ использовали хлорид цетилпиридиния, анионных ПАВ – додецилсульфат натрия. В табл. 2 и 3 представлены результаты анализа готовой продукции и водопроводной воды.
Таблица 2.
Результаты микроэкстракционно-флуориметрического определения анионных ПАВ в продуктах питания (n = 3, P = 0.95)
| Образец | Введено, мг/л | Цветометрические параметры | Ar | Найдено*, мг/л | sr | ||
|---|---|---|---|---|---|---|---|
| R (R0 – Rx) | G (G0 – Gx) | B (B0 – Bx) | |||||
| Напиток “Nemoloko” | H2O | 216 | 230 | 182 | – | ||
| – | 174(42) | 255(–25) | 98(84) | 97 | 28 ± 2 | 0.07 | |
| – | 172(44) | 255(–25) | 98(84) | 98 | |||
| – | 170(46) | 255(–25) | 95(87) | 102 | |||
| – | 220(–4) | 255(–25) | 100(82) | 86 | |||
| – | 228(–12) | 255(–25) | 95(87) | 92 | |||
| – | 223(–7) | 255(–25) | 101(81) | 85 | |||
| Молоко “Домик в деревне, 3.2%” | H2O | 197 | 228 | 192 | – | ||
| – | 197(0) | 237(–9) | 188(4) | 8 | <сн | – | |
| – | 200(–3) | 235(–7) | 189(3) | 8 | |||
| – | 193(4) | 233(–5) | 184(8) | 6 | |||
| Молоко “Добрая бурeнка, 4%” | H2O | 197 | 228 | 192 | – | ||
| – | 207(–10) | 240(–12) | 200(-8) | 8 | <сн | – | |
| – | 194(3) | 234(–6) | 186(6) | 9 | |||
| – | 202(–5) | 238(–10) | 185(7) | 7 | |||
| Вода водопроводная | H2O | 197 | 228 | 192 | – | ||
| – | 198(–1) | 219(9) | 186(6) | 11 | 0.03 ± 0.01 | 0.24 | |
| – | 200(–3) | 222(6) | 186(6) | 9 | |||
| – | 198(–1) | 224(4) | 182(10) | 11 | |||
| 0.2 | 189(8) | 255(–27) | 126(66) | 72 | 0.21 ± 0.04 | 0.07 | |
| 0.2 | 194(3) | 255(–27) | 128(64) | 70 | |||
| 0.2 | 207(–13) | 255(–27) | 134(58) | 65 | |||
Таблица 3.
Результаты экстрационно-флуориметрического определения катионных ПАВ в продуктах питания (n = 3, P = 0.95)
| Образец | Введено, мг/кг | Цветометрические параметры | Ar | Найдено*, мг/кг | sr | ||
|---|---|---|---|---|---|---|---|
| R (R0 – Rx) | G (G0 – Gx) | B (B0 – Bx) | |||||
| Куриная грудка | H2O | 229 | 210 | 205 | – | ||
| – | 214(15) | 204(6) | 202(3) | 16 | 2.20 ± 0.08 | 0.03 | |
| – | 214(15) | 204(6) | 202(3) | 16 | |||
| – | 221(8) | 199(11) | 199(6) | 15 | |||
| 10 | 243(–14) | 156(54) | 178(27) | 62 | 8.5 ± 0.2 | 0.10 | |
| 10 | 255(–26) | 147(63) | 177(28) | 74 | |||
| 10 | 252(–22) | 157(53) | 181(24) | 62 | |||
| Томат | H2O | 203 | 197 | 201 | – | ||
| – | 213(–10) | 205(–8) | 198(3) | 13 | 2.0 ± 0.5 | 0.11 | |
| – | 211(–8) | 198(–1) | 189(12) | 14 | |||
| – | 215(–12) | 198(–1) | 190(11) | 16 | |||
| 5 | 238(–35) | 204(–7) | 189(12) | 38 | 5.0 ± 0.1 | 0.02 | |
| 5 | 237(–34) | 195(2) | 186(15) | 37 | |||
| 5 | 238(–35) | 196(1) | 186(15) | 38 | |||
| Яблоко | H2O | 203 | 197 | 201 | – | ||
| – | 218(–15) | 208(–11) | 194(7) | 20 | 2.5 ± 0.7 | 0.10 | |
| – | 217(–14) | 207(–10) | 192(9) | 19 | |||
| – | 214(–11) | 202(–5) | 191(10) | 16 | |||
| 5 | 235(–32) | 211(–14) | 193(8) | 36 | 4.7 ± 0.3 | 0.04 | |
| 5 | 216(–13) | 196(1) | 171(30) | 33 | |||
| 5 | 217(–14) | 199(–2) | 170(31) | 34 | |||
| Питахайя | H2O | 203 | 197 | 201 | – | ||
| – | 209(–6) | 198(–1) | 192(9) | 11 | 1.6 ± 0.3 | 0.06 | |
| – | 209(–6) | 200(–3) | 193(8) | 10 | |||
| – | 215(–15) | 197(0) | 201(0) | 12 | |||
| 5 | 232(–29) | 206(–9) | 203(–2) | 35 | 4.6 ± 1.2 | 0.17 | |
| 5 | 228(–25) | 202(–5) | 197(4) | 26 | |||
| 5 | 237(–34) | 207(–10) | 203(–2) | 36 | |||
| Вода водопроводная | H2O | 210 | 205 | 202 | – | ||
| – | 213(–3) | 204(1) | 212(–10) | 11 | 0.16 ± 0.01 | 0.04 | |
| – | 218(–8) | 206(–1) | 207(–5) | 10 | |||
| – | 219(–9) | 203(2) | 208(–6) | 11 | |||
| 0.5 | 237(–27) | 153(52) | 182(20) | 62 | 0.52 ± 0.05 | 0.02 | |
| 0.5 | 234(–24) | 154(51) | 178(24) | 61 | |||
| 0.5 | 234(–24) | 157(48) | 182(20) | 57 | |||
Анионные ПАВ в количестве 28 ± 2 мг/л обнаружены только в напитке на основе овса “Nemoloko”. Молоко “Домик в деревне, 3.2%” и “Добрая буренка, 4%” содержат АПАВ в количествах, лежащих ниже предела обнаружения данной методики. Содержание АПАВ в водопроводной воде составило 0.03 ± 0.01 мг/л. Содержание катионных ПАВ в исследуемых продуктах находилось в диапазоне от 1.6 до 2.5 мг/кг. Наибольшее содержание обнаружено в яблоках, наименьшее – в питахайе. В водопроводной воде найдено 0.16 ± 0.01 мг/л КПАВ. Правильность анализа проверяли методом “введено–найдено”. Относительное стандартное отклонение результатов анализа не превышает 0.24. Продолжительность анализа 20–30 мин.
Список литературы
Иванов В.М., Кузнецова О.В. Химическая цветометрия: возможности метода, области применения и перспективы // Успехи химии. 2001. Т. 70. № 5. С. 411.
Моногарова О.В., Осколок К.В., Апяри В.В. Цветометрия в химическом анализе // Журн. аналит. химии. 2018. Т. 73. № 11. С. 857.
Апяри В.В., Горбунова М.В., Исаченко А.И., Дмитриенко С.Г., Золотов Ю.А. Использование бытовых цветорегистрирующих устройств в количественном химическом анализе // Журн. аналит. химии. 2017. Т. 72. № 11. С. 963.
McCracken K.E., Yoon J.-Y. Recent approaches for optical smartphone sensing in resource-limited setting: A brief review // Anal. Methods. 2016. V. 8. P. 6591.
Nascimento C.F., Santos P.M., Pereira-Filho E.R., Rocha F.R.P. Recent advances on determination of milk adulterants // Food Chem. 2017. V. 221. P. 1232.
МУК 4.1.2009-05. Определение остаточных количеств цетилпиридиний хлорида в мясе кур и продуктах его переработки. Определение остаточных количеств пестицидов в пищевых продуктах, сельскохозяйственном сырье и объектах окружающей среды: Сборник методических указаний. МУК 4.1.2009-4.1.2021-05. М.: Роспотребнадзор, 2009.
Tay M., Fang G., Chia P. L., Li S.F.Y. Rapid screening for detection and differentiation of detergent powder adulteration in infant milk formula by LC-MS // Forensic Sci. Int. 2013. V. 232. P. 32.
Arrebola-Liébanas F.J., Abdo M.A., Moreno J.L., Martínez-Vidal J.L., Frenich A.G. Determination of quaternary ammonium compounds in oranges and cucumbers using QuEChERS extraction and ultra-performance liquid chromatography/tandem mass spectrometry // J. AOAC Int. 2014. V. 97. P. 1021.
Xian Y., Dong H., Wu Y., Guo X., Hou X., Wang B. QuEChERS-based purification method coupled to ultrahigh performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) to determine six quaternary ammonium compounds (QACs) in dairy products // Food Chem. 2016. V.212 P. 96.
Slimani K., Féret A., Pirotais Y., Maris P., Abjean J.P., Hurtaud-Pessel D. Liquid chromatography-tandem mass spectrometry multiresidue method for the analysis of quaternary ammonium compounds in cheese and milk products: Development and validation using the total error approach. // J. Chromatogr. A. 2017. V. 1517. P. 86.
ГОСТ 31857-2012. Вода питьевая. Методы определения содержания поверхноcтно-активных веществ. М.: Стандартинформ, 2014. 21 с.
Barui A.K., Sharma R., Rajput Y.S. Detection of non-dairy fatin milk based on quantitative assay of azure A dye // Int. Dairy J. 2012. V. 24. P. 44.
Chennamsetty R., Kanchi S., Bisetty K., Nuthalapati V.N. Monitoring of cetylpyridinium chloride levels in surface waters: Patent blue-V as selective ligand for spectrophotometric determination // Asian J. Chem. 2016. V. 28. № 5. P.1039.
Lima M.J.A., Nascimento C.F., Rocha F.R.P. Feasible photometric measurements in liquid-liquid extraction by exploiting smartphone-based digital images // Anal. Methods. 2017. V. 9. № 14. P. 2220.
Acevedo M.S.F., Lima M.J.A., Nascimento C.F., Rocha F.R.P. A green and cost-effective procedure or determination of anionic surfactants in milk with liquid-liquid microextraction and smartphone-based photometric detection // Microchem. J. 2018. V. 143. P. 259.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии