

Журнал аналитической химии, 2021, T. 76, № 5, стр. 421-432
Хромато-масс-спектрометрическая идентификация катионных и анионных поверхностно-активных веществ при микроэкстракционно-флуориметрическом скрининге проб воды и пищевых продуктов
В. Г. Амелин a, b, *, З. А. Ч. Шаока a, Д. С. Большаков b
a Владимирский государственный университет имени Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
b Федеральный центр охраны здоровья животных
600901 Владимир, мкр. Юрьевец, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 22.11.2020
После доработки 20.12.2020
Принята к публикации 25.12.2020
Аннотация
Предложен способ идентификации поверхностно-активных веществ (ПАВ) методом ультравысокоэффективной жидкостной хроматографии (УBЭЖХ) с масс-спектрометрическим детектированием высокого разрешения после скрининга проб воды и пищевых продуктов на суммарное содержание катионных и анионных ПАВ микроэкстракционно-флуориметрическим методом. Последний основан на использовании дисперсионной жидкостно-жидкостной микроэкстракции хлороформом ассоциатов ПАВ с органическими реагентами (эозин и акридиновый желтый), измерении флуоресценции полученных аддуктов с помощью смартфона, получении цветометрических характеристик RGB и определении суммарного содержания ПАВ. Установлены основные аналитические характеристики хромато-масс-спектрометрической идентификации катионных ПАВ (хлоридов алкилпириния, алкилтриметиламмония, алкилдиметилбензиламмония (бензалкония), алкилметилэтилбензиламмония, дидецилдиметиламмония, бензилдиметил[3-(миристоиламино)пропил]аммония, N,N-бис(3-аминопропил)додециламина) и анионных ПАВ (алкилбензолсульфонатов (сульфонола), алкилсульфатов, лауретсульфатов, алкилсульфонатов и алкилкарбоксилатов натрия) при выбранных условиях хроматографического разделения и масс-спектрометрического детектирования. Рассмотрены особенности хроматографического поведения полимергомологов ПАВ в условиях УBЭЖХ и градиентного элюирования.
Значительное число выявленных случаев загрязнения пищевой продукции и воды приходится на поверхностно-активные вещества. ПАВ синтезируют из природного сырья (фракции нефти, природные масла и жиры), поэтому они представляют собой сложные многокомпонентные смеси, состоящие из гомологов ПАВ и примесей исходных веществ. ПАВ активно используют на предприятиях пищевой промышленности не только с целью обеззараживания рабочих поверхностей, очистки технологического оборудования, но и в качестве неотъемлемого компонента рецептуры готовой (молочной и мясной) продукции. За счет бактерицидных, эмульгирующих, стабилизирующих и влагоудерживающих свойств применение различных ПАВ обеспечивает должную консистенцию, необходимые сроки годности и товарный вид реализуемых продуктов питания [1].
Определение конкретных ПАВ и оценку их суммарного содержания в водных объектах и пищевых продуктах проводят, главным образом, методом хромато-масс-спектрометрии [2–12].
В работе [2] описано определение хлоридов дидецилдиметиламмония и додецилбензилдиметиламмония в морской воде. Для извлечения аналитов использовали твердофазную экстракцию (ТФЭ) с последующим разделением полученной смеси методом жидкостной хроматографии–масс-спектрометрии. Экстракт очищали при помощи полимерных (Strata-X) картриджей. Степень извлечения определяемых компонентов составила 80–105%.
Предложена методика определения бензалкония в растворенных и взвешенных частицах проб городских ливневых стоков [3]. Анализ проводили методом жидкостной хроматографии в сочетании с тандемным масс-спектрометрическим детектированием (ЖХ–МС/МС). Установлено, что загрязнение стоков максимально (27 мг/л) во время первого дождя. Концентрация катионов бензалкония (сумма С12 и С14) остается высокой (до 1 мг/л) даже спустя 5 мес. после обработки. Содержание ПАВ в ливневых водах составляло 0.2 мкг/л.
Методом ЖХ–МС/МС исследовано распределение гомологов алкилтриметиламмония, бензилакилдиметиламмония и диалкилдиметиламмония в 52 пробах осадков городских сточных вод. Общие концентрации указанных соединений находились в диапазоне 0.38–293, 0.09–191 и 0.64–344 мкг/г сухого остатка [4].
Разработан простой и быстрый способ определения хлоридов бензалкония, дидецилдиметиламмония и бензэтония в образцах огурцов и апельсинов [5]. Для извлечения аналитов в работе использована дисперсионная твердофазная экстракция QuEChERS. Последующую идентификацию и определение проводили методом ВЭЖХ–МС/МС. Диапазоны определяемых содержаний составили 0.01–0.15 мг/кг, пределы обнаружения 0.4–1.0 мкг/кг, степень извлечения варьировалась в диапазоне 81–115%. Методика применена для анализа 30 образцов огурцов и апельсинов, в которых обнаружено 0.015–0.081 мг/кг хлорида бензалкония.
Сочетание пробоподготовки на основе методологии QuEChERS и метода УBЭЖХ–МС/МС использовано для определения катионных ПАВ (КПАВ) в сухом молоке [6]. Анализируемые образцы растворяли в воде, осаждали белки и проводили экстракцию целевых компонентов ацетонитрилом. Пределы обнаружения аналитов составили 0.4–14.5 мкг/г, степень извлечения – 91.2–115% при относительном стандартном отклонении 2.8–7.5%.
Разработана чувствительная и надежная методика определения хлоридов бензалкония и диалкилдиметиламмония в молочных продуктах [7], которая включает экстракцию ПАВ смесью ацетонитрила и этилацетата без дополнительной очистки. Для анализа полученного экстракта использовали метод ВЭЖХ–МС/МС. Предел обнаружения составил менее 1.9 мкг/кг, что существенно ниже максимально допустимого уровня ПАВ (0.1 мг/кг).
Часто определяемыми анионными ПАВ являются линейные алкилбензолсульфонаты (ЛАБС) и додецилсульфат. В работе [8] предложено определение ЛАБС и алкилбензолкарбоксилатов в сточных водах. Анионные ПАВ (АПАВ) определяли методом ВЭЖХ с флуориметрическим детектированием после ТФЭ-извлечения аналитов. Пределы обнаружения составили 0.2–0.4 мкг/л при объеме пробы 250 мл.
Разработана методика ВЭЖХ-разделения и определения индивидуальных (C10–C13) линейных алкилбензолсульфонатов [9]. Пределы обнаружения лежат в диапазоне от 1.5 нг/л (для C10 ЛАБС) до 11.5 нг/л (для C13 ЛАБС). Методику использовали для одновременного определения ЛАБС в различных пробах воды.
Сочетание ТФЭ с ЖХ легло в основу простой и экспрессной методики определения ЛАБС в водных образцах [10]. Подготовленные пробы анализировали методом ВЭЖХ с флуориметрическим детектированием. Пределы обнаружения ЛАБС составили 0.02–0.10 мкг/л.
Предложена методика определения индивидуальных ЛАБС и их суммы в пробах воды [13]. Особенностью методики является сочетание метода ТФЭ в автоматическом режиме, “lab-on-valve” модуля и системы капиллярного электрофореза (КЭ). Общее содержание ЛАБС определяли путем измерения собственного поглощения аналитов и окрашенных аддуктов после реакции со смесью метилового оранжевого и хлорида цетилпиридиния с пределами обнаружения 21 нг/л и 15 мкг/л соответственно. Метод определения индивидуальных ЛАБС при низких концентрациях основан на автоматическом предварительном концентрировании проб методом ТФЭ в “lab-on-valve” модуле, соединенном с системой КЭ. Полученные таким образом пределы обнаружения и определения находятся в диапазонах 1–21 и 4–70 нг/л соответственно. Методика апробирована на образцах очищенных сточных, сточных, поверхностных и морских вод.
Для идентификации 20 соединений ряда линейных алкилбензолсульфонатов в пробах воды, отобранных с очистных сооружений, предложено использовать метод газожидкостной хроматографии с масс-спектрометрическим детектированием в режиме электронной ионизации [12]. В данной работе представлены масс-спектры 20 соединений, предложен механизм образования диагностических ионов, полученных при электронной ионизации, и распределение индивидуальных изомеров в пробах воды.
На основе комбинации дисперсионной жидкостно-жидкостной микроэкстракции (ДЖЖМЭ) и ВЭЖХ в сочетании с тройной квадрупольной масс-спектрометрией разработана методика одновременного определения линейных алкилбензолсульфонатов (C10, C11, C12 и C13), нонилфенола, моно- и диэтоксилатов нонилфенола и ди-(2-этилгексил)фталата в водопроводной и сточной воде [13]. Для ЛАБС пределы определения составили 0.009–0.019 мкг/л.
Традиционные методы определения суммарного содержания ПАВ длительны [14–20]. Подобные методики основаны на жидкостной экстракции из пробы воды хлороформом ионных пар ПАВ с соответствующим красителем и определении суммарной концентрации аналитов по интенсивности флуоресценции полученного экстракта.
В условиях постоянно растущей потребности в оценке содержания остаточных количеств анионных и катионных ПАВ в пищевых продуктах необходима разработка современных экспресс-методов, обладающих высокими точностью и чувствительностью. Среди наиболее перспективных направлений можно выделить методы цветометрии при детектировании аналитического сигнала с применением современной цифровой и офисной техники [21, 22].
В работе [1] нами предложен способ определения катионных и анионных ПАВ в пищевых продуктах и воде, основанный на применении ДЖ-ЖМЭ ассоциатов ПАВ с органическими реагентами (эозин и акридиновый желтый), измерении флуоресценции полученных аддуктов с помощью смартфона и обработки данных с использованием специализированного программного обеспечения. В качестве аналитического сигнала (Ar) использовали значения цветометрических параметров в системе RGB: ${{A}_{r}}{\text{ }} = {\text{ }}\sqrt {{{{({{R}_{0}}{\text{ }} - {{R}_{x}})}}^{2}}{\text{ }} + {\text{ }}{{{({{G}_{0}}{\text{ }} - {\text{ }}{{G}_{x}})}}^{2}}{\text{ }} + {\text{ }}{{{({{B}_{0}}{\text{ }} - {{B}_{x}})}}^{2}}} .$ Пределы обнаружения и определения составляли 0.005–0.05 и 0.01–0.1 мг/л соответственно. Градуировочные зависимости линейны с коэффициентами достоверности аппроксимации ≥0.99.
Несмотря на высокую чувствительность и экспрессность данный подход не является избирательным по отношению к конкретным катионным или анионным ПАВ и предназначен для определения их суммарного содержания с использованием метода градуировочной зависимости (для одного из группы аналитов). Идентифицировать конкретные ПАВ можно методом УB-ЭЖХ с масс-спектрометрическим детектированием (УBЭЖХ–МС). В этом случае обе методики будут дополнять друг друга и составлять единый методологический подход: в случае положительного результата анализа при проведении микроэкстракционно-флуориметрического скрининга проб воды и пищевых продуктов идентификацию аналита проводят методом УBЭЖХ–МС (рис. 1).
Рис. 1.
Схема идентификации и определения катионных и анионных ПАВ в воде и пищевых продуктах микроэкстракционно-флуориметрическим и хромато-масс-спектрометрическим методами.

Цель настоящей работы состояла в идентификации индивидуальных катионных и анионных ПАВ в воде и пищевых продуктах методом УBЭЖХ–масс-спектрометрии высокого разрешения после проведения группового микроэкстракционно-флуориметрического определения катионных и анионных ПАВ с использованием дисперсионной жидкостно-жидкостной микроэкстракции и смартфона в качестве цветорегистрирующего устройства.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Для идентификации и определения ПАВ использовали ультравысокоэффективный жидкостной хроматограф UltiMate 3000 (Thermo Scientific, США), оснащенный квадруполь-времяпролетным масс-спектрометрическим детектором сверхвысокого разрешения maXis 4G. Электрораспылительную ионизацию при атмосферном давлении осуществляли с использованием устройства ionBooster (Bruker Daltonics, Германия). Хроматографическое разделение проводили на колонке ACQUITY UPLC BEH C18 (30 × 2.1 мм, 1.7 мкм) (Waters, США) в режиме градиентного элюирования.
В работе применяли аналитические весы Pioneer PA 214С специального класса точности с пределом взвешивания 0.1 мг (Ohaus Corporation, USA), дозаторы Proline Biohit одноканальные механические переменного объема 10–100 мкл, 100–1000 мкл, 1000–5000 мкл (Biohit, Финляндия), микрошприцы объемом 500 мкл (Hamilton Company, Япония), пробирки полипропиленовые емк. 15 мл (SPL Life Sciences Co., Корея).
Реактивы. Использовали стандартные образцы хлоридов алкилпиридиния, алкилтриметиламмония, алкилдиметилбензиламмония (бензалкония), алкилметилэтилбензиламмония, дидецилдиметиламмония, бензилдиметил[3-(миристоиламино)пропил]аммония, N,N-бис(3-аминопропил)додециламина, алкилбензолсульфонатов (сульфонола), алкилсульфатов, лауретсульфатов, алкилсульфонатов и алкилкарбоксилатов натрия (98–100%, Sigma-Aldrich, США). Основные стандартные растворы концентрацией 1 мг/мл готовили растворением точной навески препарата в деионизированной воде (не менее 18 МОм см, ОСТ 11 029.003-80). Рабочие стандартные растворы готовили последовательным разбавлением основных стандартных растворов деионизированной водой.
Использовали ацетонитрил (99.9%, Scharlab S.L., Испания), ЭДТА (этилендиаминтетраацетат натрия) (99%, ХИММЕД, Россия), трихлорметан (99.85%, Компонент-Реактив, Россия), хлорид натрия (х. ч., ХИММЕД, Россия), акридиновый желтый (ч. д. а., Союзхимпром, Россия), эозин (ч. д. а., Ленреактив, Россия), соляную кислоту (стандарт-титр, НПИП Уралхиминвест), тетраборат натрия (99.5%, Sigma-Aldrich, США).
Идентификация и определение. Идентификацию ПАВ по полученным хроматограммам проводили с использованием программного продукта DataAnalysis-4.1, TargetAnalysis (Bruker Daltonics, Германия), составление картины изотопного распределения аналита – с использованием IsotopePattern (Bruker Daltonics, Германия).
Условия хроматографического разделения. Подвижная фаза состояла из 0.1%-ной муравьиной кислоты в воде (элюент А) и 0.1%-ной муравьиной кислоты в ацетонитриле (элюент В). Осуществляли градиентное элюирование: 0 мин – 5% В, 0.5 мин – 5% В, 2 мин – 50% В, 5 мин – 100% В, 6 мин – 5% В, 8 мин – 5% В. Скорость потока подвижной фазы 0.4 мл/мин. Оптимальная температура хроматографической колонки 50°С, объем вводимой пробы 50 мкл. Температура термостата автоматического дозатора 10°С.
Условия ионизации и детектирования. Использовали электрораспылительную ионизацию в устройстве ionBooster (Bruker Daltonics, Германия). Установлены следующие оптимальные значения параметров ионизации: напряжение на щите капилляра 400 В, напряжение на капилляре 1000 В, давление газа-распылителя азота 4.76 атм, поток газа-осушителя азота 6 л/мин, температура газа-осушителя азота 200°С, поток газа-испарителя азота 250 л/ч, температура газа-испарителя азота 250°C.
Проводили регистрацию ионов в диапазоне значений m/z 100 до 1200. Для калибровки масс использовали раствор формиата натрия 10 мМ в смеси вода–изопропиловый спирт (1 : 1). Масс-спектр калибранта снимали в интервале времен хроматографирования от 9.5 до 10 мин. Катионные ПАВ регистрировали в режиме положительных ионов, анионные – в режиме отрицательных ионов.
Пробоподготовка [1]. Твердые продукты (мясо, фрукты), молоко. Навеску пробы массой 1.00 г помещали в пробирку емк. 15 мл, добавляли 1 мл этилового спирта, 9 мл деионизированной воды, встряхивали в течение 5 мин и центрифугировали 5 мин при 5000 об/мин. Отбирали 1 мл полученного экстракта в пробирку емк. 15 мл, добавляли 100 мкл 0.05%-ного раствора красителя, 100 мкл 2%-ного раствора тетрабората натрия (для системы КПАВ–эозин) или 1 М HCl (для системы АПАВ–акридиновый желтый), 100 мкл 10%-ного раствора NaCl (для системы КПАВ–эозин), 100 мкл раствора ЭДТА (10 мг/л). Смесь перемешивали, объем доводили до 10 мл деионизированной водой. С помощью шприца в подготовленную пробу впрыскивали 500 мкл экстрагирующей смеси хлороформ–ацетонитрил (1 : 4, по объему). Смесь тщательно встряхивали, затем центрифугировали в течение 5 мин при 5000 об/мин. При положительном результате с помощью микрошприца отбирали 50 мкл экстракта в микрофлакон для хроматографирования и упаривали досуха в токе азота. К сухому остатку добавляли 100 мкл ацетонитрила, 900 мкл деонизированной воды и тщательно перемешивали до растворения. Полученный раствор хроматографировали.
Вода. Пробу воды объемом 10 мл помещали в пробирку емк. 15 мл, приливали 100 мкл 0.05%-ного раствора красителя, 100 мкл 2%-ного раствора тетрабората натрия (для системы КПАВ–эозин) или 1 М HCl (для системы АПАВ–акридиновый желтый), 100 мкл 10%-ного раствора NaCl (для системы КПАВ–эозин), 100 мкл ЭДТА (10 мг/л). Смесь перемешивали, затем с помощью шприца в подготовленную пробу впрыскивали 500 мкл экстрагирующей смеси смеси хлороформ–ацетонитрил (1 : 4, по объему). Смесь тщательно встряхивали, затем центрифугировали в течение 5 мин при 5000 об/мин. При положительном результате с помощью микрошприца отбирали 50 мкл экстракта в микрофлакон для хроматографирования и упаривали досуха в токе азота. К сухому остатку добавляли 100 мкл ацетонитрила, 900 мкл деонизированной воды и тщательно перемешивали до растворения. Полученный раствор хроматографировали.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Условия хроматографического разделения во многом обусловлены особенностями используемого метода детектирования. Выбор элюентов для проведения хроматографического анализа с последующим масс-спектрометрическим детектированием крайне невелик. Использование более высокомолекулярных в сравнении с муравьиной кислотой и формиатом аммония уксусной кислоты и ацетата аммония является причиной значительного уменьшения эффективности ионизации определяемых соединений в условиях электрораспыления в устройстве ionBooster.
Учитывая особенности физико-химических свойств разделяемых соединений в качестве подвижной фазы использовали растворы 0.1%-ной муравьиной кислоты в воде (элюент А) и 0.1%-ной муравьиной кислоты в ацетонитриле (элюент В). Характер программы градиентного элюирования обусловлен не только природой определяемых компонентов, но и параметрами хроматографической колонки. В ходе серии экспериментов установлено, что оптимальной является следующая программа градиентного элюирования: 0 мин – 5% В, 0.5 мин – 5% В, 2 мин – 50% В, 5 мин – 100% В, 6 мин – 5% В, 8 мин – 5% В при скорости потока подвижной фазы 0.4 мл/мин, температуре хроматографической колонки 50°С и объеме вводимой пробы 50 мкл.
Кроме того, данные условия являются универсальными для большого числа соединений [24–27], поскольку при использовании микроколонки (30 × 2.1 мм, диаметр зерна сорбента 1.7 мкм) скорость подачи подвижной фазы и программа элюирования должны обеспечить равномерное вымывание соединений в соответствии с их гидрофобностью, а также очистку и кондиционирование колонки. Данные условия с позиции хроматографического анализа способствуют унификации множества подходов, предложенных ранее в работах [24–27]. Используя разные способы экстракции и концентрирования для определения аналитов различной природы аналитическая (испытательная) лаборатория может применять одно и тоже оборудование без смены колонки, детальной настройки параметров ионизации и электрораспыления. Данная особенность, в свою очередь, обеспечивает необходимую экспрессность, универсальность и экологичность метода УBЭЖХ–МС.
Идентификация ПАВ методом хромато-масс-спектрометрии. Идентификацию ПАВ проводили по полученным хроматограммам с использованием программы TargetAnalysis-1.3 (Bruker Daltonics, Германия), с помощью созданной базы данных (табл. 1) и идентификационным параметрам. Обработку хроматограмм по общему ионному току и хроматограмм извлеченных масс ионов проводили с использованием программы DataAnalysis-4.1 (Bruker Daltonics, Германия). Составление картины изотопного распределения аналитов выполняли с использованием программы IsotopePattern (Bruker Daltonics, Германия).
Таблица 1.
Аналитические характеристики катионных ПАВ, определяемых методом ультравысокоэффективной жидкостной хроматографии–масс-спектрометрии
| КПАВ | Ион | n | tR, мин | m/z | Δ, млн–1 | смин, мкг/л | Уравнения времен удерживания и характеристических ионов | R2 |
|---|---|---|---|---|---|---|---|---|
| Алкилпиридиний | [СnH2n+1NC5H5]+ | 10 | 4.5 | 220.2059 | 2 | 0.1 | tR = 0.2167n + 2.3333 | 0.9996 |
| 11 | 4.7 | 234.2216 | 3 | 0.1 | ||||
| 12 | 4.9 | 248.2373 | 3 | 0.1 | m/z = 14.016n + 80.049 | 0.9998 | ||
| 13 | 5.2 | 262.2529 | 4 | 0.1 | ||||
| 14 | 5.4 | 276.2686 | 2 | 0.01 | ||||
| 15 | 5.6 | 290.2842 | 3 | 0.01 | ||||
| 16 | 5.8 | 304.2999 | 1 | 0.01 | ||||
| Алкилтриметилам-моний | [СnH2n+1(СН3)3N]+ | 10 | 4.5 | 200.2373 | 2 | 0.1 | tR = 0.2167n + 2.3333 | 0.9996 |
| 11 | 4.7 | 214.2529 | 3 | 0.1 | ||||
| 12 | 4.9 | 228.2686 | 4 | 0.01 | m/z = 14.015n + 60.083 | 1.000 | ||
| 13 | 5.2 | 242.2842 | 3 | 0.01 | ||||
| 14 | 5.4 | 256.2998 | 3 | 0.01 | ||||
| 15 | 5.6 | 270.3155 | 2 | 0.01 | ||||
| 16 | 5.8 | 284.3312 | 2 | 0.01 | ||||
| Алкилдиметил-бензиламмоний (бензалконий) | [С6H5СН2N (CH3)2CnH2n+1]+ | 6 | 3.9 | 220.2060 | 2 | 0.1 | tR = 0.19n + 3.02 | 0.9972 |
| 8 | 4.5 | 248.2373 | 3 | 0.1 | ||||
| 10 | 4.9 | 276.2686 | 1 | 0.1 | m/z = 14.016n + 136.11 | 1.000 | ||
| 11 | 5.1 | 290.2842 | 2 | 0.1 | ||||
| 12 | 5.3 | 304.2999 | 3 | 0.01 | ||||
| 14 | 5.7 | 332.3312 | 4 | 0.01 | ||||
| 15 | 5.9 | 346.3468 | 3 | 0.1 | ||||
| 16 | 6.1 | 360.3625 | 3 | 0.1 | ||||
| 18 | 6.4 | 388.3940 | 2 | 0.1 | ||||
| Aлкилметилэтил-бензиламмоний | [С6H5СН2N(CH3) (C2H5)CnH2n+1]+ | 10 | 5.2 | 290.2842 | 2 | 0.1 | tR = 0.1558n + 3.5968 | 0.9933 |
| 11 | 5.3 | 304.2999 | 3 | 0.1 | ||||
| 12 | 5.4 | 318.3155 | 4 | 0.01 | m/z = 14.016n + 150.13 | 1.000 | ||
| 14 | 5.8 | 346.3468 | 3 | 0.01 | ||||
| 16 | 6.1 | 374.3781 | 2 | 0.1 | ||||
| 18 | 6.4 | 402.4094 | 4 | 0.1 | ||||
| N,N-бис(3-аминопропил)-додециламин | [С12Н25NH (C3H6NH2)2]+ | – | 3.9 | 300.3373 | 2 | 0.1 | – | – |
| Дидецилдиметил-аммоний | [(С10Н21)2N(CH3)2]+ | – | 5.9 | 326.3781 | 3 | 0.01 | – | – |
| Бензилдиметил[3-(миристоиламино) пропил]аммоний (мирамистин) | [С6Н5СН2N(CH3)2 С3Н6NHCOC13H27]+ | – | 5.3 | 403.3689 | 4 | 0.1 | – | – |
Идентификационными параметрами служили времена удерживания (±0.1 мин), точность моноизотопной массы иона (m/z) (±5 млн–1) и mSigma (<20). Параметр mSigma характеризует соответствие теоретического изотопного распределения практическому (рис. 2). Погрешность в определении масс ионов не превышала ±4 млн–1 (n = 3).
Рис. 2.
Масс-спектр бензалкония хлорида (С14), сгенерированный программой IsotopePattern (а) и полученный экспериментально (б).
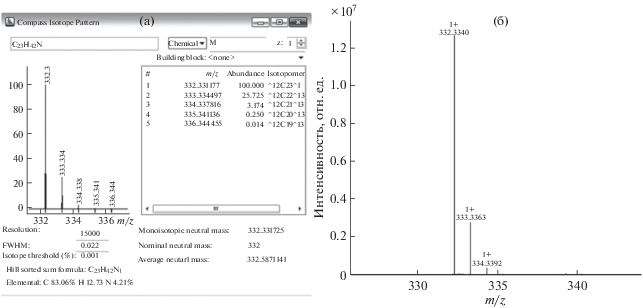
Пределы обнаружения (смин) рассчитывали по отношению аналитического сигнала (интенсивности пика) к шуму, равному 3. Пределы обнаружения составили 0.01–0.5 мкг/л (табл. 1, 2), что значительно ниже, чем в случае скринингового метода (10–50 мкг/л [1]).
Таблица 2.
Аналитические характеристики анионных ПАВ, определяемых методом ультравысокоэффективной жидкостной хроматографии–масс-спектрометрии
| АПАВ | Формула | n, m | tR, мин | m/z | Δ, млн–1 | смин, мкг/л | Уравнения времен удерживания и характеристических ионов | R2 |
|---|---|---|---|---|---|---|---|---|
| Алкилбензол-сульфонаты (сульфонол) | [СnH2n+1C6H4SO3]– | 10 | 6.4 | 297.1519 | 2 | 0.1 | tR = 0.341n + 2.990 | 0.9966 |
| 11 | 6.7 | 311.1675 | 3 | 0.01 | ||||
| 12 | 7.1 | 325.1832 | 1 | 0.01 | m/z = 14.016n + 157.010 | 0.9998 | ||
| 13 | 7.4 | 339.1988 | 2 | 0.01 | ||||
| 14 | 7.8 | 353.2144 | 3 | 0.5 | ||||
| Алкилсульфаты | [СnH2n+1SO4]– | 12 | 6.3 | 265.1470 | 4 | 0.01 | tR = 0.35n + 2.1 | 0.9989 |
| 14 | 7.0 | 293.1778 | 3 | 0.01 | m/z = 14.015n + 96.962 | 1.000 | ||
| Лауретсульфаты | [С12H25 (C2H4O)mSO4]– | 0 | 6.3 | 265.1470 | 2 | 0.01 | m/z = 44.026m + 265.147 | 0.9987 |
| 1 | 6.5 | 309.1730 | 3 | 0.1 | ||||
| 2 | 6.7 | 353.1992 | 1 | 0.1 | ||||
| 3 | 6.8 | 397.2255 | 2 | 0.1 | ||||
| 4 | 6.8 | 441.2517 | 3 | 0.1 | ||||
| 6 | 6.8 | 529.3041 | 4 | 0.5 | ||||
| 8 | 6.8 | 617.3565 | 3 | 0.5 | ||||
| 9 | 6.8 | 661.3827 | 3 | 0.5 | ||||
| [С14H29(C2H4O)mSO4]– | 0 | 7.0 | 293.1778 | 2 | 0.01 | tR = 0.17m + 7.02 | 0.9999 | |
| 1 | 7.2 | 337.2043 | 3 | 0.1 | m/z = 44.026m + 293.178 | 0.9978 | ||
| 2 | 7.4 | 381.2305 | 4 | 0.5 | ||||
| 3 | 7.5 | 425.2568 | 3 | 0.5 | ||||
| Алкилсульфонаты | [СnH2n+1SO3]– | 7 | 4.3 | 179.0736 | 2 | 0.01 | tR = 0.405n + 1.500 | 0.9933 |
| 12 | 6.5 | 249.1519 | 3 | 0.01 | ||||
| 14 | 6.9 | 277.1831 | 4 | 0.01 | m/z = 44.016n + 80.965 | 1.000 | ||
| Алкил-
карбоксилаты (мыла) |
[СnH2n+1СОО]– | 11 | – | 199.1693 | – | – | – | |
| 12 | – | 213.1849 | – | |||||
| 13 | – | 227.2006 | – | |||||
| 15 | 6.5 | 255.2319 | 3 | 0.1 | ||||
| 16 | – | 269.2475 | – | |||||
| 17 | – | 283.2631 | – |
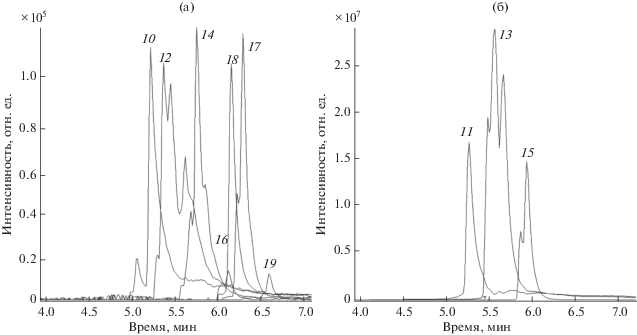
Катионные ПАВ. На рис. 3 и 4 представлены масс-хроматограмммы алкилдиметилбензиламмония (АДМБА, бензалкония) и алкилметилэтилбензиламмония (АМЭБА). Из рисунков следует, что данные препараты представляют собой смесь гомологов с длиной углеводородного радикала от С6 до С19. АДМБА содержит максимальное количество соединений с длиной углеводородного радикала С12 и С14, АМЭБА – С11, С13 и С15. Большинство гомологов представляют собой смесь изомерных форм, частично разделенных в данных условиях хроматографирования (рис. 3, 4). Особенно это характерно для АДМБА С11 и С13, для АМЭБА С10, С12, С13, С14, С15 и С17. Времена удерживания гомологов линейно увеличиваются с увеличением длины углеводородного радикала. Зависимость времени удерживания от числа атомов углерода в гомологе описывается уравнениями, представленными в табл. 1. Коэффициенты корреляции линейных зависимостей составляют не менее 0.99.
Рис. 3.
Масс-хроматограммы хлорида алкилдиметилбензиламмония (бензалкония) [С6H5СН2N(CH3)2CnH2n+ 1]+, где n = 6, 8, 10, 11, 13, 15, 16, 18 (a), n = 12, 14 (б) (с = 100 нг/мл).
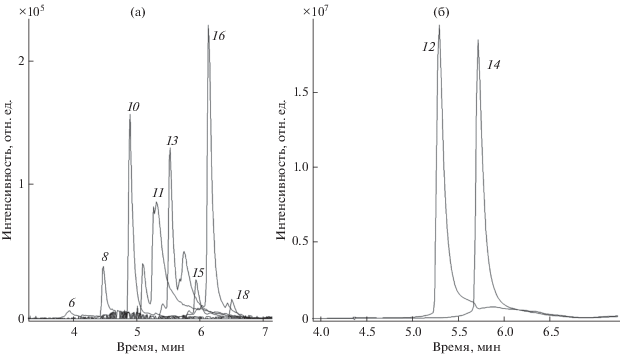
Рис. 4.
Масс-хроматограммы хлорида алкилметилэтилбензиламмония [С6H5СН2N(CH3C2H5)CnH2n+ 1]+, где n = 10, 12, 14, 16, 17, 18, 19 (a), n = 11, 13, 15 (б) (с = 100 нг/мл).
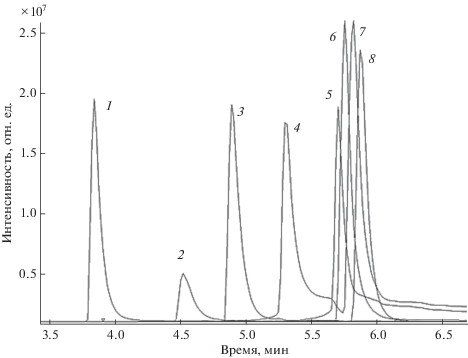
На рис. 5 показаны масс-хроматограммы индивидуальных КПАВ. Данные соединения не содержат изомерных форм, поэтому представлены симметричными хроматографическими пиками.
Рис. 5.
Масс-хроматограммы КПАВ: 1 – N,N-бис(3-аминопропил)додециламин, 2 – децилпиридиний, 3 – додецилтриметиламмоний, 4 – бензилдиметил[3-(миристоиламино)пропил]аммоний, 5 – бензалконий С14, 6 – цетилтриметиламмоний, 7 – цетилпиридиний, 8 – дидецилдиметиламмоний (с = 100 нг/мл).
Анализировали экстракты кожуры яблок, куриной грудки и томатов на наличие катионных ПАВ. По итогам предварительно проведенного скрининга микроэкстракционно-флуориметрическим методом в данных образцах установлено суммарное содержание КПАВ на уровне 2.5 ± 0.7, 2.20 ± 0.08 и 2.0 ± 0.5 мг/кг соответственно [1]. В результате хромато-масс-спектрометрического анализа в экстракте кожуры яблок идентифицированы катионы цетилтриметиламмония (m/z = = 284.3312), дидецилтриметиламмония (m/z = = 326.3781), цетилдиметилбензиламмония (m/z = = 360.3625) и октадецилдиметилбензиламмония (m/z = 388.3940) (рис. 6).
Рис. 6.
Масс-хроматограммы экстракта из кожуры яблок: 1 – цетилтриметиламмоний-катион (m/z = 284.3312), 2 – дидецилтриметиламмоний-катион (m/z = 326.3781), 3 – цетилдиметилбензиламмоний-катион (m/z = 360.3625) и 4 – октадецилдиметилбензиламмоний-катион (m/z = 388.3940).

В экстракте воды р. Клязьма обнаружены катионы дидецилтриметиламмония, цетилдиметилбензиламмония и октадецилдиметилбензиламмония.
Анионные ПАВ. Сульфонол представляет собой смесь гомологов с длиной углеводородного радикала от С10 до С14 (рис. 7). Продукт содержит С10 – 11%, С11 – 27%, С12 – 36%, С13 – 20%, С14 – 6%.

Лауретсульфат – амфифильное ПАВ, которое представляет собой смесь гомологов (преимущественно С12 и С14) с различной степенью этоксилирования (m = 0–9). Следует отметить несимметричность и раздвоение (расщепление) хроматографических пиков, что может свидетельствовать о существовании в данных препаратах изомерных форм (рис. 8).
Рис. 8.
Масс-хроматограммы лауретсульфата С12H25(C2H4O)m${\text{SO}}_{{\text{4}}}^{ - }$ (m = 0–9) (а) и С14H29(C2H4O)m${\text{SO}}_{{\text{4}}}^{ - }$ (m = 0–3) (б).

В табл. 2 приведены аналитические характеристики анионных ПАВ. Как следует из рис. 7 и 8, данные препараты представляют собой смесь гомологов с длиной углеводородного радикала от С10 до С14. Большинство гомологов представляют собой смесь изомерных форм, частично разделенных в данных условиях хроматографирования. Времена удерживания гомологов линейно увеличиваются с увеличением длины углеводородного радикала. Зависимость времени удерживания от числа атомов углерода в гомологе описывается уравнениями, представленными в табл. 2. Коэффициенты корреляции линейных зависимостей составляют не менее 0.99. Анион додецилсульфата на хроматограмме представлен одним пиком с временем удерживания 6.3 мин (табл. 2).
С учетом установленных в работе аналитических характеристик выполнили хроматографический анализ экстракта растительного продукта на основе овса “Nemoloko”. В ходе проведенного ранее исследования с использованием микроэкстракционно-цветометрического (флуориметрического) метода установлено суммарное содержание АПАВ на уровне (28 ± 2) мг/л [1]. На рис. 9 представлена масс-хроматограмма экстракта данного растительного продукта. В продукте выявлено присутствие сульфонола (С10–С13) и додецилсульфат-аниона.
Рис. 9.
Масс-хроматограмма экстракта растительного продукта на основе овса “Nemoloko”. Алкилбензолсульфонаты С10 (1), С11 (3), С12 (5), С13 (2), додецилсульфат (6).

В экстракте воды р. Клязьма обнаружены анионы алкилбензолсульфоната (С10–С13) и додецилсульфат.
Список литературы
Амелин В.Г., Шаока З.А.Ч., Большаков Д.С. Микроэкстракционно-цветометрическое (флуориметрическое) определение катионных и анионных ПАВ в пищевых продуктах // Журн. аналит. химии. 2021. Т. 76. № 3. С. 234.
Bassarab P., Williams D., Dean J.R., Ludkin E., Perry J.J. Determination of quaternary ammonium compounds in seawater samples by solid-phase extraction and liquid chromatography–mass spectrometry // J. Chromatogr. A. 2011. V. 1218. P. 673.
Van de Voorde A., Lorgeoux C., Gromaire M.C., Chebbo G. Analysis of quaternary ammonium compounds in urban stormwater samples // J. Environ Pollut. 2012. V. 164. P. 150.
Ruan T., Song S., Wang T., Liu R., Lin Y., Jiang G. Identification and composition of emerging quaternary ammonium compounds in municipal sewage sludge in China // J. Environ. Sci. Technol. 2014. V. 48. P. 4289.
Arrebola-Liebanas F.J., Abdo M.A., Moreno J.L., Martinez-Vidal J.L., Frenich A.G. Determination of quaternary ammonium compounds in oranges and cucumbers using QuEChERS extraction and ultra-performance liquid chromatography/tandem mass spectrometry // J. AOAC Int. 2014. V. 97. P. 1021.
Xian Y., Dong H., Wu Y., Guo X., Hou X., Wang B. QuEChERS-based purification method coupled to ultrahigh performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) to determine six quaternary ammonium compounds (QACs) in dairy products // J. Food Chem. 2016. V. 212. P. 96.
Slimani K., Feret A., Pirotais Y., Maris P., Abjean J.P., Hurtaud-Pessel D. Liquid chromatography-tandem mass spectrometry multiresidue method for the analysis of quaternary ammonium compounds in cheese and milk products: development and validation using the total error approach // J. Chromatogr. A. 2017. V. 1517. P. 86.
Leon V.M., Gonzalez-Mazo E., Gomez-Parra A. Handling of marine and estuarine samples for the determination of linear alkylbenzene sulfonates and sulfophenylcarboxylic acids // J. Chromatogr. A. 2000. V. 889. P. 211.
Wangkarn S., Soisungnoen P., Rayanakorn M., Grudpan K. Determination of linear alkylbenzene sulfonates in water samples by liquid chromatography–UV detection and confirmation by liquid chromatography–mass spectrometry // Talanta. 2005. V. 67. P. 686.
Hirayama Y., Ikegami H., Machida M., Tatsumoto H. Simple and rapid determination of linear alkylbenzene sulfonates by in-tube solid-phase microextraction coupled with liquid chromatography // J. Health Sci. 2006. V. 52. P. 228.
Moldovan Z., Avram V., Marincas O., Petrov P., Ternes T. The Determination of the linear alkylbenzene sulfonate isomers in water samples by gas-chromatography/mass spectrometry // J. Chromatogr. A. 2011. V. 1218. P. 343.
Martin J., Camacho-Munoz D., Santos J.L., Aparicio I., Alonso E. Determination of priority pollutants in aqueous samples by dispersive liquid–liquid microextraction // Anal. Chim. Acta. 2013. V. 773. P. 60.
Jimenez J.R., Luque de Castro M.D. Lab-on-valve for the automatic determination of the total content and individual profiles of linear alkylbenzene culfonates in water samples // Electrophoresis. 2008. V. 29. P. 590.
ГОСТ 31857-2012. Вода питьевая. Методы определения содержания поверхностно-активных веществ. М.: Стандартинформ, 2014. 21 с.
ПНД Ф 16.1:2:2.2:3.66-10. Количественный химический анализ почв. Методика измерений массовой доли анионных поверхностно-активных веществ в пробах почв, грунтов, донных отложений, илов, отходов производства и потребления экстракционно-фотометрическим методом. М.: ФГБУ “ФЦАО”, 2010, 18 с.
РД 52.24.439-2007. Массовая концентрация неионогенных синтетических поверхностно-активных веществ и полиэтиленгликолей в водах. Методика выполнения измерений экстракционно-фотометрическим методом. Ростов-на-Дону.: Росгидромет, 2007, 24 с.
ПНД Ф 14.1:2:4.15-95. Количественный химический анализ вод. Методика измерений массовой концентрации анионных поверхностно-активных веществ в питьевых, поверхностных и сточных водах экстракционно-фотометрическим методом. М.: ФГБУ “ФЦАО”, 2011, 13 с.
ПНД Ф 14.1:2.16-95. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации катионных поверхностно-активных веществ в пробах природных и очищенных сточных вод экстракционно-фотометрическим методом. М: ФГБУ “ФЦАО”, 2004, 13 с.
ПНД Ф 14.1:2.247-07. Количественный химический анализ вод. Методика выполнения измерений массовых концентраций неионогенных синтетических поверхностно-активных веществ (СПАВ) в пробах природных и сточных вод нефелометрическим методом. М.: ФГБУ “ФЦАО”, 2016, 12 с.
ПНД Ф 14.1:2:4.194-2003. Количественный химический анализ вод. Методика измерений массовой концентрации неионогенных поверхностно-активных веществ (НПАВ) в пробах питьевых, природных и сточных вод экстракционно-фотометрическим методом в присутствии анионоактивных ПАВ (АПАВ). М.: ФГБУ “ФЦАО”, 2012, 14 с.
Моногарова О.В., Осколок К.В., Апяри В.В. Цветометрия в химическом анализе // Журн. аналит. химии. 2018. Т. 73. № 11. С. 857.
Апяри В.В., Горбунова М.В., Исаченко А.И., Дмитриенко С.Г., Золотов Ю.А. Использование бытовых цветорегистрирующих устройств в количественном химическом анализе // Журн. аналит. химии. 2017. Т. 72. № 11. С. 963.
Иванов В.М., Кузнецова О.В. Химическая цветометрия: возможности метода, области применения и перспективы // Успехи химии. 2001. Т. 70. № 5. С. 411.
Амелин В.Г., Большаков Д.С. Экспресс-идентификация и определение N-нитрозаминов в пищевых продуктах методом ультравысокоэффективной жидкостной хроматографии/квадруполь-времяпролетной масс-спектрометрии высокого разрешения по точным массам протонированных ионов // Журн. аналит. химии. 2019. Т. 74. № 7S. С. S48.
Амелин В.Г., Большаков Д.С. Экспресс-определение аминогликозидов в молоке по точным массам ионов методом ультравысокоэффективной жидкостной хроматографии/квадруполь времяпролетной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2019. Т. 74. № 9S. С. S48.
Амелин В.Г., Большаков Д.С. Одновременное определение остаточных количеств хлорамфеникола и хлорамфеникола пальмитата в пищевых продуктах жидкостной хромато-масс-спектрометрией // Вестник Моск. ун-та. Серия 2. Химия. 2020. Т. 61. № 6. С. 420.
Амелин В.Г., Большаков Д.С., Подколзин И.В. Быстрый скрининг и определение остаточных количеств β-лактамных антибиотиков в пищевых продуктах методом ультравысокоэффективной жидкостной хроматографии – квадруполь-времяпролетной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2020. Т. 75. № 9. С. 806.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии