

Журнал аналитической химии, 2021, T. 76, № 5, стр. 413-420
Прямой атомно-эмиссионный метод определения серы в углеродных материалах с дуговым источником излучения
Д. О. Антонов a, *, Э. Г. Силькис b, Б. К. Зуев a
a Институт геохимии и аналитической химии Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
b Институт спектроскопии Российской академии наук
108841 Троицк, Москва, ул. Физическая, 5, Россия
* E-mail: d.antonov3756@gmail.com
Поступила в редакцию 14.10.2020
После доработки 18.11.2020
Принята к публикации 05.12.2020
Аннотация
Для прямого экспрессного определения серы в чистых графитовых порошках использован эмиссионный метод с аргоновым дуговым источником, позволяющим существенно подавить помехи молекулярного спектра CN. Предложен метод временной селекции аналитического сигнала серы на линии 921.286 нм (регистрация сигнала в определенном интервале 9.8 с). Предел обнаружения серы – 5 × 10–4 мас. %. Диапазон определяемых содержаний 5 × 10–4–2 × 10–2 мас. %, который может быть расширен до 1 мас. %. Время единичного анализа (3 параллельных измерения) – 10 мин. Работа выполнена на компактном спектрометре МС-300 с эффективным в области 908–930 нм диодным линейным фотоприемником типа S13496.
В современной промышленности углеродные материалы широко применяются как антифрикционные, конструкционные и функциональные материалы для работы в агрессивных средах; для создания графитополимерных композитов; для получения карбидокремниевых материалов, обладающих исключительно высокими механическими характеристиками; как замедлители в атомной энергетике [1].
Одна из наиболее важных характеристик углеродных материалов на разных технологических стадиях является концентрация серы. Примесь серы содержится в прекурсорах искусственного графита – пеке и коксе, получаемых из природного углеводородного сырья, всегда содержащего серу. Сера существенно влияет на технологические характеристики углеродного материала: ее присутствие изменяет свойства прекурсоров углеродных матреиалов, она воздействует на процессы формования и карбонизации, а также катализирует графитацию и ингибирует силицирование [2–4].
Для определения серы в углеродных материалах и их прекурсорах стандартными являются гравиметрический метод Эшка [5] и метод с алкилметрическим титрованием [6]. В АО “НИИГрафит” разработаны кулонометрический и рентгенофлуоресцентный методы [7, 8]. Кроме того, описано определение серы в углеродных материалах методами иодометрического титрования [9], атомно-эмиссионной и масс-спектрометрии с индуктивно связанной плазмой [10], молекулярной флуоресцентной спектроскопии [11]. Все указанные методы, кроме рентгенофлуоресцентного, требуют сложной и длительной пробоподготовки вследствие высокой химической стойкости углеродных материалов и невозможности полного их перевода в раствор или газ. Пробоподготовка связана со сжиганием углеродного материала или химической экстракцией серы, что представляет собой длительный (порядка нескольких часов) процесс, не исключающий возможности потери серы.
В углеродной промышленности часто возникает необходимость быстрого контроля сырья, полуфабрикатов и продукции, поэтому разработка экспрессного метода определения серы и соответствующего прибора, обладающего высокими чувствительностью и точностью, весьма актуальна. Атомно-эмиссионный спектральный анализ с применением современных многоканальных систем регистрации спектров позволяет проводить быстрые измерения и обладает высокой чувствительностью. В промышленной и лабораторной практике распространено определение металлических примесей в углеродных материалах эмиссионным методом; с определенными модификациями этот метод может применяться и для определения неметаллических примесей.
Особенность эмиссионного спектра серы состоит в том, что самые интенсивные атомные линии лежат в вакуумной ультрафиолетовой и ближней инфракрасной областях [12]. Для определения примесей серы в сталях используют атомные линии серы 180.73 и 182.03 нм [13]. К сожалению, работа в этом диапазоне требует вакуумного или газонаполненного (Ar, N) дорогостоящего и сложного спектрографа.
Вне вакуумного ультрафиолетового диапазона, наиболее интенсивными являются атомные линии 921.286, 922.809 и 923.753 нм [14–16]. В работах [17, 18] по определению серы в геологических образцах с помощью высокотемпературного гелиевого плазмотрона эти линии также выбраны в качестве аналитических.
Цель данной работы – разработка методики определения содержания серы в графитовых порошках на дуговом аргоновом спектрометре. Основное внимание уделяли достижению высокой пороговой чувствительности и созданию недорогого целевого эмиссионного спектрометра, построению рабочей градуировки в диапазоне 5 × 10–4–2 × 10–2 мас. % для практической работы с чистыми графитовыми порошками. При создании установки использовали компактный спектрометр МС-300 [19] с системой регистрации на высокочувствительном диодном линейном фотоприемнике S13496.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Схема спектрометра изображена на рис. 1. Источником возбуждения спектра в образцах служила дуга постоянного тока между угольными электродами с испарением порошкообразного образца из кратера нижнего электрода (метод фракционной дистилляции). В качестве нижнего электрода применяли фассонные графитированные спектральные угли класса ос. ч. 7-4, тип IV диаметром 6 мм с кратером диаметром 4 и глубиной 4 мм, в качестве верхнего – тип II, заточенный на усеченный конус с острием диаметром 2 мм. Масса навески образца составила 50 мг. Разрядный промежуток (3 мм) выбирали таким образом, чтобы максимально снизить попадание излучения нагретых электродов на входную щель спектрометра. Для доочистки от остаточных примесей в электродах их подвергали обжигу в дуге постоянного тока силой 20 А на воздухе в течение 15 с. Такой метод подготовки электродов при анализе углеродных материалов описан в работе [20]. Для создания разряда в среде аргона применяли устройство типа Stallwood jet [21], поставляемое в комплекте со штативом УШТ-4. На него устанавливали защитный цилиндр из кварцевого стекла, отделяющий область разряда от окружающего пространства. Применение такого цилиндра позволило получать стабильный разряд при низком расходе аргона на уровне 1 л/мин, исключить попадание воздуха в разрядный промежуток и сдувание плазмы. Верхний электрод включался катодом, нижний – анодом. Для определения наилучшего отношения сигнал/шум регистрации аналитической линии измерения проводили в диапазоне значений тока 15–24 А; выбрано рабочее значение в 20 А. Работа при весьма высоких силах тока обусловлена необходимостью быстрого и стабильного прогрева кратера с порошкообразным образцом, что важно при получении спектров углеродных материалов в условиях фракционной дистилляции образца.
Рис. 1.
Схема спектрометра для измерения концентрации S в графитах: 1 – катод, 2 – анод, 3 – дуговой разряд, 4 – кварцевый цилиндр, 5 – устройство ввода аргона в разрядный промежуток, 6 – рефокусатор, 7 – светофильтр КС-19, 8 – оптическое волокно, 9 – спектрометр МС-300, 10 – система регистрации спектра.

Для получения и регистрации спектров применяли компактный, перестраиваемый по длинам волн спектрометр МС-300, выполненный по вертикально симметричной схеме Эберта с дифракционной решеткой 1200 штр/мм. Система регистрации спектров выполнена на линейном диодном детекторе типа S13496 фирмы “Hamamatsu” (Япония) [22], позволяющем одновременно регистрировать спектр на участке 75 нм со спектральным разрешением 0.1 нм в диапазоне от 190 до 1000 нм [19].
По сравнению с ранее применявшимся в МС300 ПЗС-детектором TCD1304DG фирмы “Toshiba” (Япония) [23] величина сигнала увеличилась в 5 раз при длине волны 921 нм. Динамический диапазон регистрации также увеличился более чем в 2.5 раза. В электронике управления и обработки сигнала применяли 16-разрядный АЦП [24].
Для освещения входной щели спектрографа использовали кварцевый рефокусатор с F = 25 мм, отображение разряда осуществлялось на оптическое волокно диаметром 400 мкм [25]. Для отсечения спектров второго порядка спектрографа, накладывающихся на исследуемую область, применяли фильтр из цветного стекла марки КС-19 с областью пропускания от 700 нм. Спектры регистрировали в режиме временной развертки (режим “серия” в ПО “МОРС”) [19]. Оптимальное время единичного кадра серии выбирали для получения максимальной чувствительности и предотвращения перегрузки сигнала аналитических линий при высоких концентрациях серы в образце; оно составило 240 мс. Предварительную калибровку шкалы осуществляли с помощью лампы с полым катодом Fe–Ne, а калибровку выбранного рабочего спектрального участка – по линиям серы, углерода и аргона.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 2 показан участок спектра 902–934 нм, полученный на созданном спектрометре, при этом дуговой источник работал в среде воздуха. По канту 914.203 нм определено, что в области от канта до 930 нм регистрируются линии молекулы CN [26, 27], являющиеся помехами при регистрации линии S 921.286. На рис. 3а показан участок спектра в диапазоне 912–927 нм во втором кадре рабочей серии, где есть примесь воздуха и соответственно присутствует молекулярный спектр CN, на рис. 3б показана сумма спектров третьего и четвертого кадров этой же серии, уже без СN.
Рис. 2.
Спектр углерода с примесью 1.76 × 10–2 мас. % серы в среде воздуха, регистрируемого в рабочем поле спектрометра. Диапазон 902–934 нм.
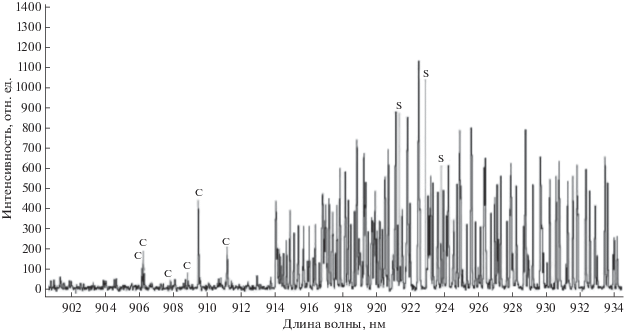
Рис. 3.
Участок эмиссионного спектра (912–927 нм) углеродного материала, содержащего серу, полученный на дуговом источнике в среде аргона: (а) – в присутствии примеси воздуха; (б) – в отсутствие примеси воздуха, тот же спектр (два суммированных кадра серии) без спектра CN.

Применение аргона позволило максимально ослабить молекулярный спектр CN. Другим источником помех является тепловое излучение электродов, представляющее собой непрерывный спектр, средний уровень интенсивности которого искажается интерференцией (предположительно на входном стекле диодного линейного детектора) (рис. 3б), флуктуации этого спектра ухудшают значение предельной чувствительности по сере.
Соотношение амплитуд сигналов двух линий S 921.286 и 923.749 составляет 2 : 1, в качестве аналитической выбрана линия 921.286 нм, линий помех по этой линии в данных спектрах нет. Третья линия S 922.811 в данном спектре находится на склоне линии Ar 922.450 нм. Измерения в воздушной среде показывает, что амплитуда этой линии составляет порядка 0.75 от амплитуды линии 921.286 нм.
Аттестовнные образцы сравнения типа “сера в чистом графите” для построения градуировочной кривой не разработаны и не производятся. Для изготовления таких образцов в определенном диапазоне концентраций тщательно проанализировали образец порошкообразного графита класса ос. ч. 8-4 (образец с наименьшей концентрацией) и образец с концентрацией, близкой к 2 × 10–2 мас. %. Для определения концентрации серы в образце ос. ч. 8-4 использовали спектрометр ЕА5000 (молекулярный УФ-флуоресцентный метод) с чувствительностью по сере на уровне 6 × 10–5 мас. %. Концентрация серы в этом образце составила 5 × 10–4 мас. %. Для анализа образцов с большой концентрацией серы применяли гравиметрический метод Эшка (ООО “ГрафитЭл-МЭЗ”), рентгенофлуоресцентный метод (АО “НИИГрафит”), атомно-эмиссионную спектроскопию с индуктивно связанной плазмой (ГЕОХИ РАН) и спектрометр ЕА5000. Диапазон полученных значений концентраций в образце с максимальной концентрацией серы составил 1.76 × 10–2–2 × 10–2 мас. %, за исходное приняли значение 1.76 × 10–2 мас. %, полученное на приборе ЕА5000, далее это образец 176.
Смешением образцов с концентрациями 1.76 × 10–2 и 5 × 10–4 мас. % получили еще четыре образца сравнения с концентрациями 1.5 × 10–3, 2.6 × 10–3, 4.7 × 10–3 и 9.0 × 10–3 мас. %. Таким образом, градуировочную кривую строили по шести рабочим точкам.
Оптимальную аналитическую экспозицию выбирали на основе анализа кривых испарения в зависимости от времени экспозиции. На рис. 4 представлены кривые испарения образцов сравнения (зависимость интегральной интенсивности линии S 921.286 нм). Как видно, кривая испарения S при сравнительно высоких концентрациях (1.76 × 10–2 мас. %), имеет максимум; максимальная интенсивность приходится на 1.2 с горения дуги (5 кадр). При более низкой концентрации 2.6 × × 10–3 мас. % процесс испарения серы происходит с той же закономерностью с максимумом также на 5 кадре. Время выгорания при разных концентрациях практически одинаково при данном способе получения эмиссионого спектра. В первые 0.72 с горения дуги (кадры 1–2) наблюдается слабый спектр СN, исчезающий к 3 кадру. Данное явление обусловлено десорбцией воздуха, попадающего в кратер электрода при набивке в него порошкообразного образца, при повышении температуры в кратере. Исчезновение спектра СN является сигналом к началу аналитической экспозиции. Оптимальным временем экспозиции для построения градуировочной кривой по образцам сравнения выбрали отрезок с 3 по 43 кадр, что соответствует 9.6 с горения дуги при длительности единичного кадра экспозиции 240 мс.
Рис. 4.
Изменение интенсивности спектральной линии S 921.286 в течение 24 с. Длительность одного кадра 240 мс; концентрации серы, мас. %: 1 – 1.76 × 10–2, 2 – 2.6 × 10–3. Выделена величина аналитической экспозиции, 3–43 кадры.

На рис. 5 приведены спектры для различных концентраций серы в графите. В спектре на рис. 5а при отсутствии пробы в графитовом электроде (холостое измерение) сигнал на линии S 921.286 не детектируется. При этом метка пика этой линии лежит в минимуме двух волн интерференции сигнала от спектроскопического фона. На рис. 5б, 5в приведены также участки спектров образцов cравнения с концентрациями 2.6 × 10–4 и 5 × 10–4 мас. % (ос. ч. 8-4) соответственно. Сигнал на линии S 921.286 детектируется и расчет интенсивности этой линии проводится по определенному алгоритму, а именно: необходимо вычесть интенсивность шума пикселей под линией S 921.286. Обычно вычитают сигналы пикселей, находящихся рядом с пикселями аналитической линии. Однако спектр холостого измерения позволяет найти группу пикселей на спектре, сигнал которых близок к сигналу от пикселей под линией S 921.286. Это делается по нескольким параллельным спектрам холостой пробы при достижении минимума между интегральной интенсивностью пикселей под линией и выбранной группой пикселей на спектре. Эта выбранная группа на рис. 5б, 5в обозначена серым цветом, пиксели линии окрашены в черный цвет.
Рис. 5.
Спектры холостой пробы и образцов сравнения в диапазоне 918–930 нм: (а) – спектр холостой пробы, (б) – участок спектра образца сравнения с концентрацией серы 2.6 × 10–3 мас. %, (в) – участок спектра ос. ч. 8-4 с концентрацией серы 5 × 10–4 мас. %.
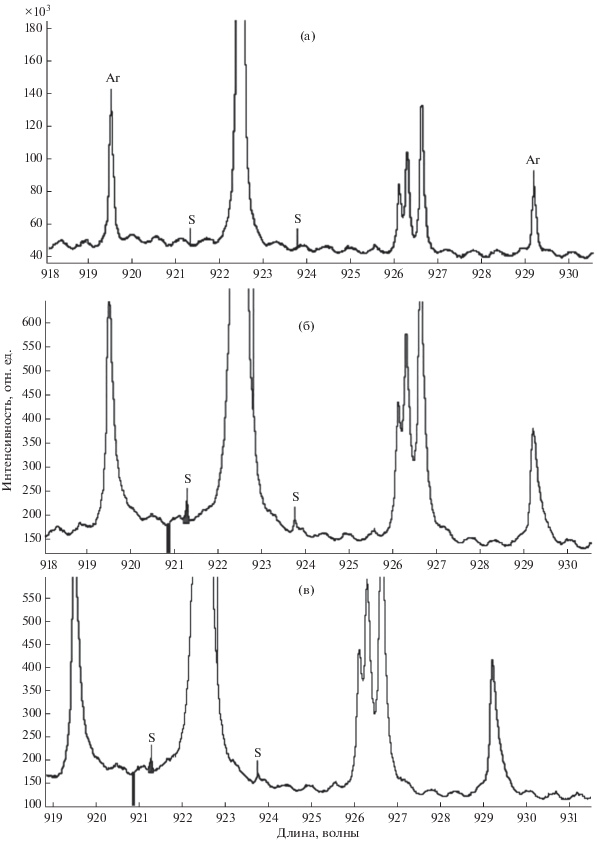
Три линии С (908.851, 911.156 и 940.573) и линия Ar 935.422 на рабочем участке спектра (рис. 2а) позволяют (в отсутствие линий серы при холостом измерении) откалибровать спектральную шкалу и определить центр тяжести линии S 921.286 с точностью до 0.2 пикселя. На откалиброванном спектре выбирается группа пикселей под линией S 921.286 нм и группа пикселей “учета фона”. Эти же линии позволяют корректировать шкалу при термосдвиге решетки. Две более близкие к линии S 921.286 линии Ar (рис. 5а) являются диффузными и их центр тяжести сдвинут относительно своей метки из базы линий (рис. 3б), поэтому они не могут применяться для коррекции шкалы при термосдвиге решетки МС-300. Результаты измерения таких сдвигов в спектрах графитовых порошков приведены в работе [28].
На рис. 6 приведена градуировочная кривая, построенная по трем параллельным измерениям каждого из шести образцов сравнения. Кривая построена с квадратичной аппроксимацией, наиболее точно передающей характер эмпирической зависимости, и может быть описана уравнением: I = –7971.676 + + 33 592 469.316c(S) – 686 275 939.602c(S)2, где I – интегральная интенсивность линии S 921.286 при экспозиции 40 кадров в отсчетах АЦП, c(S) – значение концентрации серы, мас. %.

Нижняя точка градировочной кривой определена по трем параллельным измерениям образца сравнения ос. ч. 8-4, среднее значение концентрации составило 4.99 × 10–4 мас. %, относительное стандартное отклонение – 10.91%. Среднее значение отношения сигнал/шум при измерении интенсивности линии S 921.286 нм для этой серии измерений составляет 3.3 : 1. Таким образом, значение концентрации серы в образце ос. ч. 8-4 5 × × 10–4 мас. % характеризует предел обнаружения серы для данного спектрометра [29].
Продолжительность анализа (время пробоподготовки, подготовки электродов и проведения трех параллельных измерений) составила 10 мин. Полученная градуировочная зависимость позволяет определять концентрации серы в чистых углеродных материалах в диапазоне 5 × 10–4–2 × 10–2 мас. %.
Диапазон определяемых концентраций может быть расширен до 1 мас. %. Прибор позволяет определять серу в полуфабрикатах и прекурсорах углеродных материалов. Он существенно проще и дешевле, чем дорогие аналитические приборы, применяемые для контроля содержания серы.
Список литературы
Колокольцев С.Н. Углеродные материалы. Свойства, технологии, применения. Долгопрудный, МО: ИД Интеллект, 2012. 296 с.
Claussea V., Bastienb T., Hoppeb S., Marêchéa J.F., Fierroa V., Celzard A. Investigation of pitch–sulphur mixtures used as binder in the preparation of black ceramics // Mater. Chem. Phys. 2009. V. 116. P. 619.
Brandtzæg R., Øye H.A. A possible mechanism of sulphur-induced graphitization // Carbon. 1988. V. 26. № 2. P. 163.
Tyumentsev V.A., Belenkov E.A., Shveikin G.P., Podkopaev S.A. The effects of sulfur and other impurities on carbon-graphite transitions // Carbon. 1998. V. 36. № 7–8. P. 845.
ISO 334:2013 Solid mineral fuels – Determination of total sulfur – Eschka method.
ISO 351:1996 Solid mineral fuels – Determination of total sulfur – High temperature combustion method.
ТУ 1915 – 019-54755093-2011 Графит кусковой.
Кутейников А.Ф., Машкович Л.А., Егорова В.А. Рентгенофлуоресцентное определение серы в коксах // Журн. аналит. химии. 1985. Т. 11. № 7. С. 1254.
Попов В.А., Пичугин В.В., Иванов Ю.А., Беневольский А.С. Способ определения общего содержания серы в углеродосодержащих материалах. Патент СССР № 874605. Заявка 2888241/23-26 от 27.02.1980, опубл. 28.10.1981.
Amais R.S., Donati G.L., Nobrega J.A. Sulfur determination in fuels by ICP-OES and ICP-MS to meet increasingly stricter legislation requirements // Spectroscopy. 2014. V. 29. № 3. P. 24.
https://www.analytik-jena.ru/ru/analiticheskoe-oborudovanie/produkcija/ehlementoorganicheskii-analiz-cnscl/ mikroehlementnyi-analiz/multi-ear-5000.html (09.10.2020).
https://www.nist.gov/pml/atomic-spectra-database (09.10.2020).
Gonzalez A., Oriz M., Campos J. Determination of Sulphur content in steel by laser-produced atomic emission spectroscopy // Applied Spectroscopy. 1995. V. 49. № 11. P. 1632.
Keane J.M., Fry R.C. Red and near infrared ICP emission spectra of F, Cl, Br, I and S with photodiode array detector // Anal. Chem. 1986. V. 58. P. 790.
Hughes S.K., Fry R.C. Near infrared atomic emissions of Sulfur and carbon in argon ICP // Appl. Spectrosc. 1981. V. 35. № 5. P. 494.
Schleisman A.J.J., Pivonka D.E., Fateley W.G., Fry R.C. Red/near-infrared atomic analysis for H, C, N, O, S, Cl and Br with a Fourier transform ICP emission spectrometer // Appl. Spectrosc. 1986. V. 40. № 4. P. 464.
Тюрин Д.А., Силькис Э.Г., Савинова Е.Н. Определение серы в геологических образцах и почвах с помощью высокотемпературного дугового плазмотрона // Журн. аналит. химии. 2018. Т. 73. № 5. С. 1.
Савинова Е.Н., Сукач Ю.С., Колесов Г.М., Тюрин Д.А. Новые возможности атомно-эмиссионной спектроскопии для определения треудновозбудимых элементов // Журн. аналит. химии. 2015. Т. 70. № 5. С. 502.
http://www.ooo-mors.ru/mc.html (09.10.2020).
Емелина О.И. Новые аналитические возможности атомно-эмиссионной спектроскопии с дуговым источником света. Дис. … канд. хим. наук. СПб: СПбГУ, 1996
Fagan A.W., Klein H.M. Rapid loading Stallwood Jet // Appl. Spectrosc. 1968. V. 22. № 4. P. 346.
https://www.hamamatsu.com/eu/en/product/type/ S13496/index.html (09.10.2020).
https://toshiba.semicon-storage.com/ap-en/semiconductor/product/linear-image-sensors/detail.TCD1304DG. html (09.10.2020).
http://trdc.com/ (09.10.2020).
http://www.optofiber.ru/ (09.10.2020).
Davis S., Wallace L., Brault J., Engleman R. The CN spectrum from the infrared to the ultraviolet / NSO Technical Report. 2005.
Pearse R.W.B., Gaydon A.G. Identification of molecular spectra. 3rd Ed. London: John Wiley & Sons, 1963. 347 p.
Силькис Э.Г. Сдвиг спектральных линий некоторых элементов в излучении плазмы источников возбуждения атомно-эмиссионных спектрометров // Аналитика и контроль. 2019. Т. 23. № 1. С. 43.
Oleneva E., Khayduhoja M., Ashina J., Yaroshenko I., Jahatspanian I., Legin A., Kirsanov D. A simple procedure to assess limit of detection for multisensor systems // Sensors. 2019. V. 19. № 6. P. 1359.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии