

Журнал аналитической химии, 2021, T. 76, № 7, стр. 612-622
Экологически безопасное определение следов пищевых красителей в продуктах методом обращенно-фазовой ВЭЖХ с использованием перегретой воды в качестве элюента
Department of Chemistry, Faculty of Science, King Abdulaziz University
P.O. Box 80203, Jeddah, 21589 Saudi Arabia
* E-mail: lateefa_2003@hotmail.com
** E-mail: laalkhatib@kau.edu.sa
Поступила в редакцию 17.09.2019
После доработки 22.11.2019
Принята к публикации 28.01.2020
Аннотация
Разработана и валидирована простая и точная ВЭЖХ-методика разделения и определения следов пищевых красителей: тартразина (E102), желтого солнечного заката (E110) и красного очаровательного АС (E129) в образцах воды и напитков. Время разделения красителей на гибридной колонке ODS X-Вridge RP-C18 (150 × 3.5 мкм, внутренний диаметр 4.6 мм) составило менее 5 мин при использовании в качестве подвижной фазы перегретой дистиллированной воды, содержащей буферный раствор. Графики Вант-Гоффа линейны для всех изученных красителей в различных подвижных фазах, а факторы удерживания существенно не изменяются. Пределы обнаружения E129, E102 и E110 составляют 0.0004, 0.5 и 0.016 мг/мл соответственно. Оценены правильность и воспроизводимость методики. Методика применена для определения красителей в безалкогольных напитках и природных водах с удовлетворительными степенями извлечения – от 83.5 ± 1.9% до 114 ± 3%.
Безопасность пищевых продуктов вызывает озабоченность представителей органов здравоохранения во всем мире, поскольку она не только напрямую связана со здоровьем, но и серьезно влияет на развитие общества и экономики. В настоящее время искусственные красители, например тартразин (E102), желтый солнечный закат (ЖСЗ) (E110) и красный очаровательный АС (КО) (E129), широко применяют в пищевых продуктах из-за их высокой стабильности, низкой себестоимости, однородности цвета и малой токсичности [1–3], что используется для повышения потребительской привлекательности и увеличения продаж [3, 4]. Пищевые красители E110 и E129 используют для компенсации потери естественного цвета пищевых продуктов, поврежденных в ходе переработки [5–8]. Синтетические пищевые красители, получаемые из каменноугольной смолы, небезопасны для здоровья человека при длительном употреблении [9]. В зависимости от типа пищевого красителя и вида пищи допустимые уровни одобренных синтетических пищевых красителей составляют от 20 до 500 мг/кг [10].
Опубликовано много методик обнаружения следов пищевых красителей, включая капиллярный электрофорез [5], тонкослойную хроматографию [11, 12], ВЭЖХ [3–6, 13–15], ВЭЖХ с диодной матрицей [16–19] и масс-спектрометрическим детектированием [20, 21], ионную хроматографию [22], ВЭЖХ с ионным взаимодействием [23], автоматическую твердофазную экстракцию [24], экстракцию с помощью микроволнового излучения в сочетании с твердофазной экстракцией и обращенно-фазовой (ОФ) сверхвысокоэффективной жидкостной хроматографией [12, 25], твердофазную микроэкстракцию, жидкостно-жидкостную микроэкстракцию [26], вольтамперометрию [2, 8, 27–30], полярографию [31–34], потенциометрию [35], спектрофотометрию [36–39], мицеллярную микроэкстракцию [40], Рамановскую спектроскопию и спектроскопию гигантского комбинационного рассеяния [41]. Однако по-прежнему сложно напрямую использовать аналитические методы, такие как ВЭЖХ, для определения следов аналитов этого класса в объектах окружающей среды при очень низких концентрациях. В связи с этим для снижения пределов обнаружения пищевых красителей при определении методами ВЭЖХ со спектрофотометрическим (УФ−видимая области) или масс-спектрометрическим детектированием [3, 13–15] необходимо привлечение методов пробоподготовки, например жидкостно-жидкостной или твердофазной экстракции [24], твердофазной микроэкстракции, жидкостно-жидкостной микроэкстракции [26], микроволновой экстракции [12, 25] или сорбции на якоре мешалки. Методы пробоподготовки имеют ряд недостатков, например, необходимость использования больших количеств токсичных и легковоспламеняющихся органических растворителей и потери аналита при упаривании растворителей. Прямые спектрофотометрические методы страдают от наложения полос в спектрах [36, 37, 39], тогда как хроматографические [12, 26, 42] и полярографические методы [33, 34] требуют использования токсичных органических растворителей, вольтамперометрические [2, 8, 27–30] и потенциометрические методы [35] длительны и не позволяют определять следы пищевых красителей. С другой стороны, когда неподвижная фаза и аналит стабильны при высоких температурах, разделение можно проводить при высокой температуре, что дает ряд преимуществ, например, снижение противодавления, усиление массопереноса за счет снижения вязкости растворителя и коэффициента диффузии в подвижной фазе [43–45].
Сообщалось о создании ряда экологически чистых хроматографических методов с иcпользованием буферного раствора в подвижной фазе и без него, а также с небольшими объемами органических модификаторов в различных элюентах для разделения различных сложных частиц [45–47]. Вода является высокополярным элюентом при низкой температуре и имеет слабую элюирующую способность, что приводит к более длительному времени удерживания [45, 47, 48]. Сведения об использовании перегретой воды в качестве подвижной фазы в высокотемпературной ВЭЖХ для разделения E102, E110 и E129 отсутствуют.
Настоящее исследование посвящено разработке быстрых, экологичных методик разделения и одновременного определения ряда пищевых красителей с использованием гибридной стационарной фазы C18 и чистой воды в качестве элюента в ОФ-ВЭЖХ в реальных образцах при высоких температурах, валидацию предложенных методик определения выбранных пищевых красителей и, наконец, обоснование наиболее вероятного механизма удерживания при низком содержании органического растворителя и чистой воды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы. Растворители имели квалификацию ч. д. а. и использовались без дополнительной очистки. Пищевые красители тартразин (E102), желтый солнечный закат FCF (E110) и красный очаровательный АС (E129) приобретали у Sigma-Aldrich (Пул, Великобритания). Ацетонитрил (ACN) для ВЭЖХ получали от BDH (Пул, Великобритания) и использовали без дополнительной очистки. Стандартные исходные растворы (1 мг/мл) пищевых красителей готовили растворением навески в деионизированной воде. Более разбавленные стандартные растворы (0.1–500 мг/мл) пищевого красителя также готовили на деионизированной воде.
Аналитические приложения. Реальные коммерческие образцы апельсинов, безалкогольных напитков и фруктовых ароматизаторов, порошкового сока и конфет приобретали на Саудовском рынке в городе Джидда, Саудовская Аравия. Образец напитка объемом 10 мл переносили в мерную колбу емк. 50 мл и разбавляли деионизованной водой до метки. Образцы порошкового сока готовили растворением необходимой навески образца в деионизированной воде в мерной колбе емк. 50 мл. Конфеты растворяли в 10 мл деионизированной воды в небольшом химическом стакане и взбалтывали до полного перехода красителя в раствор. Образцы дегазировали перед разбавлением и фильтровали через мембрану 0.45 мкм перед измерениями.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние температуры. Взаимосвязь между удерживанием пищевых красителей и температурой подвижной фазы изучали в широком диапазоне температур колонки (30–150°C) с использованием урацила в качестве маркера для измерения мертвого времени при расчете коэффициента удерживания (k). Результаты измерения удерживания для E110 и E129 приведены в табл. 1 и 2. Типичные данные также показаны на рис. 1. Тартразин (E102) при 40°C разлагается, поэтому его не исследовали. Таким образом, исследование разделения было сосредоточено на двух других пищевых красителях. Удерживание изученных красителей снижалось с повышением температуры (рис. 1). Известно, что элюирование исследуемого красителя зависит от его гидрофобности [49]. С первой подвижной фазой, содержавшей 10% ACN, время удерживания E110 уменьшилось с 7.76 мин при 30°C до 1.45 мин при 150°C, тогда как для E129 время удерживания уменьшилось с 21.07 до 1.69 мин в том же диапазоне температур. Со второй подвижной фазой, а именно содержащей буферный раствор перегретой водой, время удерживания E110 уменьшилось с 5.64 до 1.42 мин и с 16.79 до 1.65 мин при увеличении температуры колонки от 120 до 170°C соответственно. Селективность (α) для пары E110/E129 при более высокой температуре была немного ниже для подвижной фазы, содержащей 10% ACN (табл. 1). Значение α возрастает при более высоких температурах при использовании перегретой воды в качестве подвижной фазы, как и ожидалось, из-за отсутствия ацетонитрила в подвижной фазе [49].
Таблица 1.
Влияние температуры на факторы удерживания пищевых красителей и селективность на колонке X-Bridge C18
| Краси-тель | Температура колонки, °C | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| комнат-ная | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | 110 | 120 | 130 | 140 | 150 | 120 | 130 | 140 | 150 | 160 | 170 | |
| Элюент – 10% ацетонитрил, pH 6.3 | Элюент – буферный раствор, pH 6.3 | |||||||||||||||||||
| E102 | 0.27 | 0.25 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| E110 | 5.74 | 3.44 | 2.55 | 1.90 | 1.37 | 0.92 | 0.72 | 0.59 | 0.47 | 0.39 | 0.42 | 0.25 | 0.30 | 0.27 | 3.09 | 1.54 | 0.60 | 0.46 | 0.26 | 0.12 |
| E129 | 18.10 | 11.03 | 7.69 | 5.55 | 3.49 | 2.20 | 1.77 | 1.32 | 0.99 | 0.74 | 0.66 | 0.44 | 0.34 | 0.32 | 11.10 | 5.11 | 2.27 | 1.35 | 0.76 | 0.29 |
| Фактор селективности α | 3.21 | 3.01 | 2.93 | 2.56 | 2.38 | 2.45 | 2.20 | 2.07 | 1.91 | 1.55 | 1.73 | 1.13 | 1.18 | 3.62 | 3.32 | 3.79 | 2.93 | 2.97 | 2.50 | |
Таблица 2.
Влияние температуры на термодинамические параметры пищевых красителей на колонке X-Bridge C18a
| Краситель | Элюент – 10%-ный ацетонитрил, pH 6.3 | Элюент – буферный раствор, pH 6.3 | ||||
|---|---|---|---|---|---|---|
| энтальпия (∆H°), кДж/моль | энтропия (∆S°), Дж/моль | коэффициент корреляции, R2 | энтальпия (∆H°), кДж/моль | энтропия (∆S°), Дж/моль | коэффициент корреляции, R2 | |
| E110 | −26.49 | −77.08 | 0.9976 | −35.99 | −89.47 | 0.9934 |
| E129 | −35.99 | −89.47 | 0.9977 | −38.43 | −90.53 | 0.9910 |
Рис. 1.
Разделение пищевых красителей при различных температурах с помощью смеси 10% (по объему) ацетонитрил−20 мМ фосфатный буферный раствор при pH 6.3 на колонке X-Bridge phenyl (4.6 × 150 мм). Соединения: урацил (1), желтый солнечный закат (2), красный очаровательный (3).
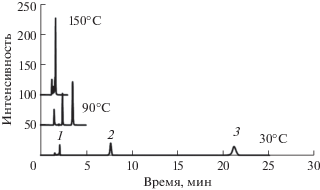
Для всех подвижных фаз ширина пика красителя уменьшалась с увеличением температуры колонки. Число теоретических тарелок (N) для красителя красный очаровательный увеличилось с 1.1 × 103 тарелок/м при 30°C до 9.9 × 103 тарелок/м при 150°C из-за коэффициента массопереноса (член C в уравнении Ван Деемтера [45, 50, 51]). Значения N для красителя желтый солнечный закат уменьшились с 1.3 × 104 до 8.6 × 103 тарелок/м при повышении температуры колонки с 30 до 80°C. Такие изменения можно объяснить уменьшением продольного коэффициента диффузии вещества в колонке. После повышения температуры колонки с 80 до 150°C аналиты проводили меньше времени в сорбированном состоянии, что уменьшало влияние фактора продольной диффузии вещества и увеличивало роль массопереноса [44, 48, 50]. Это может объяснить наблюдаемую тенденцию при разделении – аналиты проводят меньше времени на колонке, что снижает эффекты продольной диффузии, но увеличивает эффект массопереноса.
Время удерживания уменьшилось с 5 мин для ЖСЗ и 16 мин для КО при 120°C до менее чем 2 мин (1.43 и 1.70 мин) для обоих красителей при 170°C в подвижной фазе с перегретой водой. В чистой воде ширина пика также уменьшилась с 0.18 и 0.39 мм при 120°C до 0.09 и 0.10 мм при 170°C для ЖСЗ и КО соответственно. Разрешение двух красителей снизилось с 39.5 при 120°C до 0.3 при 170°C. В результате повышение температуры привело к снижению вязкости подвижной фазы, уменьшению продолжительности анализа и разрешающей способности, а также к улучшению формы пиков выбранных пищевых красителей (рис. 1).
В структуре красителя ЖСЗ имеется нафталиновое кольцо. Краситель ОК более гидрофобен, чем ЖСЗ и тартразин при 40°C. С другой стороны, тартразин разлагается при 40°C в 10% ACN (рис. 2). В последующей работе тартразин был исключен из смеси, а характеристики методики его определения рассматривали только при 30°C (табл. 3).
Рис. 2.
Хроматограммы первого (a) и второго пятен (б) тартразина (E102) с тонкослойной хроматограммы; подвижная фаза 10% ацетонитрил (по объему)−20 мМ фосфатный буферный раствор (pH 6.3) при 40°C.
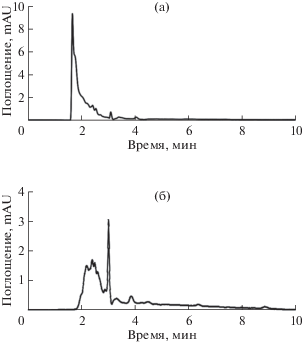
Таблица 3.
Аналитические характеристики методики определения пищевых красителей E102, E110 и E129a
| Параметр | E102 | E110 | E129 |
|---|---|---|---|
| Диапазон линейности, мг/л | 1.5–500 | 0.05–500 | 0.001–500 |
| Коэффициент детерминации R2 | 0.9951 | 0.9962 | 0.9950 |
| Наклон | 1.69 | 15.64 | 9.61 |
| Отсекаемый отрезок | 2.83 | −109.31 | −16.95 |
| Предел обнаружения, мг/л | 0.5 | 0.016 | 0.0004 |
| Нижняя граница определяемых содержаний, мг/л | 1.8 | 0.05 | 0.0013 |
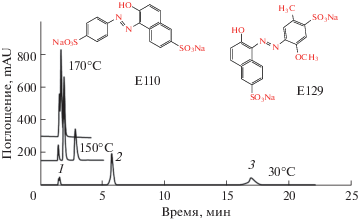
Разделение ЖСЗ (E110) и ОК (E129) проводили при разных температурах (120–170°C) с перегретой водой в качестве элюента на колонке X‑Bridge phenyl (4.6 × 150 мм) при скорости потока 1 мл/мин с УФ-детектированием при 244 нм (рис. 3). Коэффициенты удерживания E110 и E129 снизились с 3.09 до 0.12 мин в диапазоне температур колонки от 120 до 170°C, а также уменьшились для E129 с 11.17 до 0.29 мин при использовании чистой воды в качестве подвижной фазы при pH 6.3. В итоге для разделения и определения красителей E110 и E129 в реальных образцах приняли следующие оптимальные условия: использование содержащей буферный раствор перегретой воды в качестве подвижной фазы при 150°C.
Рис. 3.
Разделение E110 и E129 при различных температурах с помощью перегретой воды на колонке X-Bridge phenyl (4.6 × 150 мм) при скорости потока 1 мл/мин; фосфатный буферный раствор (pH 6.3), УФ-детектирование при 244 нм.
Исследование селективности. Селективность разработанного метода проверяли путем определения 1 ppm красителей в присутствии различных органических соединений, например лимонной кислоты, бензоата натрия, цитрата натрия, хлорида магния, аскорбиновой кислоты, глюкозы и сахарозы, поскольку они могут входить в состав продуктов совместно с пищевыми красителями в довольно высоких концентрациях. Относительная погрешность после добавления этих компонентов менее ±5% считалась допустимой. Результаты не выявили значительного мешающего влияния (<5%) даже в присутствии 100-кратного избытка мешающих веществ. С другой стороны, была достигнута удовлетворительная степень извлечения (90–97%) ± 2.9%, что подтверждает точность установленных процедур.
Метрологические характеристики методики. В оптимизированных условиях зависимости площадей пиков от концентраций тестируемых красителей (E102, E110, E129) линейны в диапазоне 1.5–500 мг/л для E102, 0.05–500 мг/л для E110 и 0.001–500 мг/л для E129 (табл. 3). Уравнения регрессии для E110 и E129 могут быть представлены уравнениями (1) и (2) [52]:
В табл. 4 дано сравнение эффективности разработанной методики с опубликованными в литературе, в основе которых лежат методы ВЭЖХ, капиллярного электрофореза [5], хроматографические [11, 53], вольтамперометрические [2, 8, 27–30], полярография [31, 33, 34], потенциометрия [35], спектрофотометрия [36–39], спектрофотометрия точки помутнения [40], цифровое изображение [54] и применение функционализированного наночастицами оксида индия пористого полимерного монолита в сочетании с методом ВЭЖХ−МС/МС [55]. Рассчитанные на основе данных работы [52] значения пределов обнаружения (смин) оказались равными 0.02 и 0.0004 мг/л, тогда как нижние границы определяемых содержаний (НГОС) составили 0.05 и 0.001 мг/л для E110 и E129 соответственно. Достигнуты удовлетворительные значения смин, НГОС и ширина линейного динамического диапазона. Предел обнаружения красителей по предлагаемой методике ниже максимально допустимого смин для воды (10 мкг/л), установленного Всемирной организацией здравоохранения и Агентством по охране окружающей среды США для водорастворимых пищевых красителей. Предлагаемая методика предлагает более широкий линейный диапазон, более низкие значения предела обнаружения и нижней границы определяемых содержаний по сравнению с описанными методами (R2 > 0.9997). Кроме того, предлагаемая методика проста и требует использования небольшого количества органического растворителя или вовсе его не требует, в отличие от многих других опубликованных хроматографических методов (табл. 4).
Таблица 4.
Сравнение аналитических характеристик предложенного и опубликованных методов определения E102, E110 и E129 в сложных матрицах
| Метод | Время удерживания | Диапазон линейности, мг/л | Предел обнаружения, мг/л | Примечания | Литера-тура |
|---|---|---|---|---|---|
| ОФ-ВЭЖХ, UV-DAD, Zorbax ODS, изократическое элюирование с использованием смеси метанол (45%)–водный раствор CH3COONH4 (55%) | 2 мин для E102 3 мин для E110 |
1–100 (E102) 1–100 (E110) |
0.04 (E102), 0.05 (E110) |
Дорогостоящий и токсичный элюент | [18] |
| ОФ-ВЭЖХ, DAD, синтетические пищевые красители в алкогольных напитках | 11 мин | 0.007–120 (E102) 0.016–160 (E110) |
2.1 × 10–3 0.0048 |
Подвижная фаза метанол−ацетат аммония (40 мМ) буферный раствор с использованием градиентного элюирования, используется большой объем органического растворителя для ВЭЖХ | [17] |
| ОФ ВЭЖХ с DAD, пищевые красители в продуктах питания | 29 мин | 0.006–21 (E102) 0.013–55 (E110) 0.023–51 (E129) |
1.87 × 10–3 (E102), 4.41 × 10–3 (E110), 7.46 × 10–3 (E129) |
Градиентное элюирование, подвижная фаза ацетонитрил−метанол (20 : 80, по объему)−ацетат аммония (1%, мас./об.), pH 7.5; трудоемкость, использование 100%-ных органических растворителей | [3] |
| ОФ ВЭЖХ с детектором DAD, программа градиента с использованием смеси 20 мМ ацетат аммония–90%-ный ацетонитрил для 14 синтетических пищевых красителей | Не более 7 мин | 0.0001–0.001 (E102) 0.0005–0.005 (E129) |
0.00007 (E102), 0.00007 (E129) |
Высокая чувствительность; использование 90%-ного ацетонитрила (токсичность) | [23] |
| ОФ ВЭЖХ с анионообменной разделительной колонкой, градиентное элюирование с использованием 2 М HCl−ацетонитрил | Время сокращено за счет увеличения содержания органических растворителей | 0.001–0.04 (E102) 0.002–0.04 (E110) 0.002–0.04 (E129) |
0.00005 (E102), 0.002 (E110), 0.002 (E129) |
Высокая чувствительность, высокая концентрация органического растворителя и сильной кислоты в подвижной фазе | [22] |
| ОФ ВЭЖХс детектором DAD, изократическое элюирование с использованием водного раствора Triton X-100 (0.25%) при pH 7 | 2.1, 3.6 и 7.0 мин для E102, E110, E129 | 0–50 | 0.125 (E102), 0.143 (E129) |
Низкая чувствительность, длительное достижение равновесия | [46] |
| ОФ ВЭЖХ с DAD и подвижной фазой, содержащей ACN (10%)–20 мМ буфер при pH 6.3 и 30°C | При низкой
температуре (30°C) 2.15 мин (E102), 7.76 мин (E110) и 21.07 мин (E129) |
1.5–500 (E102) 0.05–500 (E110) 0.001–500 (E129) |
0.5 (E102), 0.016 (E110), 0.0004 (E129) |
Меньшая продолжительность анализа, низкое содержание органического модификатора (низкая токсичность), низкая температура колонки с диоксидом кремния-С18 для увеличения срока ее службы | Данная работа |
Валидация методики. При валидации методики оценивали промежуточную (в пределах суток) и межсуточную прецизионность при концентрациях ЖСЗ и ОК 10 мг/л. Результаты представлены в табл. 5. Относительное стандартное отклонение (sr) менее 5.0% указывает на приемлемую сходимость. Кроме того, определяли межсуточную правильность и прецизионность в разные дни в течение 5 дней методом введено–найдено. Было найдено 99–108% и 105–109% от введенного количества для ЖСЗ и ОК соответственно. Сходимость результатов можно считать превосходной, поскольку относительное стандартное отклонение не превышает 5%.
Таблица 5.
Межсуточная и внутрисуточная прецизионность определения пищевых красителей Е110 и Е129 (10 мг/л) по разработанной методикеa
| № | Пищевой красительb | Промежуточная прецизионность (в пределах суток, n = 4) | Межсуточная прецизионность (n = 3) | ||
|---|---|---|---|---|---|
| найдено, мг/мл | sr, % | найдено, мг/мл | sr, % | ||
| 1 | E110 | 8.5 | 3.8 | 9.0 | 3.2 |
| E129 | 12.5 | 4.3 | 12.4 | 4.7 | |
| 2 | E110 | 11.5 | 2.9 | 11.4 | 3.6 |
| E129 | 11.0 | 3.3 | 11.9 | 4.0 | |
| 3 | E110 | 9.5 | 2.9 | 9.5 | 2.9 |
| E129 | 11.3 | 4.1 | 11.3 | 3.8 | |
Механизм разделения. Корреляция между коэффициентом удерживания (k) и температурой (график Вант-Гоффа) для данной подвижной фазы выражается уравнением (3) [51, 56, 57]:
где k – измеренный коэффициент удерживания, ΔH° и ΔS° − стандартные парциальные молярные энтальпия и энтропия переноса растворенного вещества из подвижной в неподвижную фазу соответственно, R – газовая постоянная, T – абсолютная температура (K), Φ – соотношение объемов неподвижной и подвижной фаз. На основании графиков Вант-Гоффа (рис. 4) сделали заключение об отсутствии значительных изменений в механизме удерживания аналита при повышении температуры колонки в чистой воде и растворе аналита. Отрицательные значения ΔH° (табл. 2) указывают на экзотермический процесс, т.е. в неподвижной фазе соединения удерживаются сильнее, чем в подвижной. Наблюдаемое уменьшение значений ΔH° при уменьшении концентрации ACN в подвижной фазе свидетельствует о сильном взаимодействии подвижной и неподвижной фаз (табл. 2). Эти данные согласуются с ранее опубликованными [44, 58]. Для каждого состава подвижной фазы (10% ACN и чистая вода) график Вант-Гоффа линеен с высоким коэффициентом корреляции (r2 > 0.99). Энтальпии (ΔH°) переноса растворенного вещества из подвижной фазы в неподвижную можно рассчитать по наклону и отсекаемому отрезку, представляющему собой энтропию плюс соотношение фаз колонки, которое является постоянным для обращенно-фазовой колонки [42, 57].Рис. 4.
Контролируемое разделение урацила (1), пищевых красителей E110 (2) и E129 (3) на колонке X‑Bridge phenyl C18 с использованием чистой воды при 130°C (a) и 10% ацетонитрила (б).
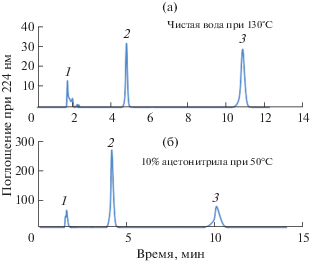
Энтальпии составляли –26.49 кДж/моль для E110 и –35.99 кДж/моль для E129 с использованием подвижной фазы 10% ACN на колонке X-Bridge C18. Найдены энтальпии для E110 и E129 в подвижной фазе, содержащей чистую воду с буферным раствором, составили –35.99 и –38.43 кДж/моль соответственно (табл. 2).
ΔH° и ΔS° отрицательны для всех растворенных веществ в изученных условиях. Как и ожидалось, значения ΔH° становятся более отрицательными с уменьшением содержания ацетонитрила в подвижной фазе. Это показывает, что сильное взаимодействие между подвижной и неподвижной фазами возникает, когда доля органического модификатора в элюенте уменьшается и в механизме разделения начинает преобладать гидрофобный эффект, что согласуется с наблюдениями других исследователей [50–53]. С другой стороны, энтропии составлют –77.08 и –89.47 Дж/моль⋅К в 10%-ном ACN; в чистой воде энтропии составляли –89.47 и –90.53 Дж/моль⋅К для E110 и E129 соответственно. Значения ΔS° являются мерой упорядоченности цепочек неподвижной фазы. Таким образом, в случае всех составов подвижной фазы значения ΔH° и ΔS° для всех растворенных веществ отрицательны в изученных условиях, а процесс удерживания контролируется энтальпией, которая играет более значительную роль в процессе удерживания, чем энтропия [45, 48].
Аналитическое применение. Разработанную методику опробовали при определении пищевых красителей в стандартных концентрациях (от 10 до 100 мг/л), добавленных к реальным образцам (безалкогольные напитки, леденцы и порошок сока). Красители ЖСЗ и ОК в образцах определяли до и после введения добавок. Тартразин (E102) не определяли из-за его разложения при 40°C [59, 60]. Данные приведены в табл. 6, а репрезентативные результаты показаны на рис. 5. Найденное количество красителей всегда было выше 82%, а стандартное отклонение находилось в диапазоне 2.0–3.5%, что свидетельствует о приемлемых характеристиках разработанной методики. Результаты определения красителя ЖСЗ предложенным и стандартными методами [6] удовлетворительно соответствуют друг другу. Значения экспериментального t-критерия Стьюдента (texp 1.72–2.28) и F-критерия (Fexp 1.20–2.33) с доверительной вероятностью 95% (n = 5) не превышало табличных значений t- (2.31) и F-критериев (6.38).
Таблица 6.
Определение красителей E110 и E129 в напитках, восстановленном соке и конфетах по разработанной методике (n = 3)а
| Образецb | Введено, мг/л | Найдено, мг/л | Средний результат |
|---|---|---|---|
| Краситель E110 | |||
| Апельсиновый напиток | 10 | 23 ± 4 | 84 |
| 50 | 63 ± 3 | 106 | |
| 100 | 113.2 ± 2.3 | 99 | |
| Апельсиновые конфеты | 10 | 12 ± 3 | 115 |
| 50 | 52 ± 4 | 97 | |
| 100 | 102.0 ± 2.6 | 101 | |
| Сок апельсина (восстановленный) | 10 | 18 ± 3 | 94 |
| 50 | 57.8 ± 2.5 | 84 | |
| 100 | 108 ± 4 | 104 | |
| Краситель E129 | |||
| Красный безалкогольный напиток | 10 | 14 ± 4 | 90 |
| 50 | 54.1 ± 1.9 | 81 | |
| 100 | 104.1 ± 1.4 | 105 | |
| Конфеты с добавкой красного красителя | 10 | 39.9 ± 1.4 | 85 |
| 50 | 79.9 ± 2.2 | 109 | |
| 100 | 129.9 ± 2.3 | 98 | |
| Красный восстановенный сок | 10 | 11.9 ± 1.4 | 110 |
| 50 | 52 ± 3 | 88 | |
| 100 | 101.9 ± 1.4 | 103 | |
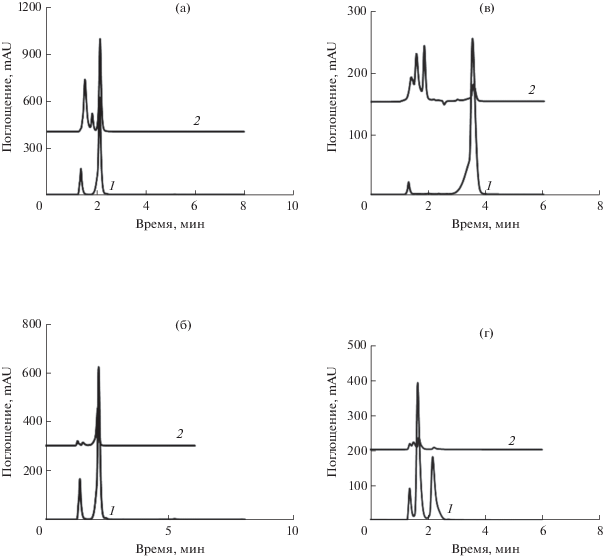
Рис. 5.
ВЭЖХ-хроматограммы стандартных пищевых красителей и реальных образцов безалкогольных напитков, порошкового сока и конфет. Условия: чистая вода при 150°C в качестве подвижной фазы при pH 6.3 на колонке X-Bridge C18 при скорости потока 1 мл/мин и УФ-детектирование при 244 нм. (а): Апельсиновый напиток: 1 – стандарт Е110, 2 – безалкогольный напиток, Е-110; (б): Апельсиновые конфеты: 1 – стандарт Е110, 2 – конфеты, Е110; (в) Фруктовый напиток: 1 – стандарт Е129, 2 – безалкогольный напиток, Е-129; (г): Фруктовый ароматизированный порошок сока: 1 – стандарты E110 и E129, 2 – соки, E110 и E129.
* * *
Разработана и валидирована гибридная ODS-хроматографическая методика для эффективного, простого, надежного и быстрого обнаружения красителей в реальных образцах − в конфетах и напитках − с использованием перегретой воды в качестве экосовместимого и экономичного элюента. Для ускорения массопереноса и диффузии, повышения эффективности разделения и сокращения продолжительности анализа использовали повышенную температуру и элюент, представляющий собой водный буферный раствор с малым содержанием ацетонитрила или без него. Предлагаемая методика представляет собой эффективное дополнение к методам определения водорастворимых красителей с точки зрения пригодности и экспрессности.
Работа поддержана деканатом научных исследований Университета короля Абдул-Азиза, Саудовская Аравия, при финансировании в рамках гранта № 130-073-33. Автор благодарит деканат научных исследований (DSR) Университета короля Абдул-Азиза, Саудовская Аравия, за предоставленную финансовую поддержку.
Список литературы
Patsovskii A., Rudometova N., Kamentsev Y.S. Electrophoretic determination of synthetic dyes in alcoholic beverages // J. Anal. Chem. 2004. V. 59. № 2. P. 150.
Florian M., Yamanaka H., Carneiro P., Zanoni M.V.B. Determination of brilliant blue FCF in the presence and absence of erythrosine and quinoline yellow food colours by cathodic stripping Vtammetry // Food Addit. Contam. 2002. V. 19. № 9. P. 803.
Minioti K.S., Sakellariou C.F., Thomaidis N.S. Determination of 13 synthetic food colorants in water-soluble foods by reversed-phase high-performance liquid chromatography coupled with diode-array detector // Anal. Chim. Acta. 2007. V. 583. № 1. P. 103.
Tripathi M., Khanna S.K., Das M. Surveillance on use of synthetic colours in eatables vis a vis Prevention of Food Adulteration Act of India // Food Control. 2007. V. 18. № 3. P. 211.
Huang H.-Y., Shih Y.-C., Chen Y.-C. Determining eight colorants in milk beverages by capillary electrophoresis // J. Chromatogr. A. 2002. V. 959. № 1. P. 317.
Ma M., Luo X., Chen B., Su S., Yao S. Simultaneous determination of water-soluble and fat-soluble synthetic colorants in foodstuff by high-performance liquid chromatography–diode array detection–electrospray mass spectrometry // J. Chromatogr. A. 2006. V. 1103. № 1. P. 170.
Fuh M.-R., Chia K.-J. Determination of sulphonated azo dyes in food by ion-pair liquid chromatography with photodiode array and electrospray mass spectrometry detection // Talanta. 2002. V. 56. № 4. P. 663.
Ghoreishi S.M., Behpour M., Golestaneh M. Simultaneous determination of Sunset yellow and Tartrazine in soft drinks using gold nanoparticles carbon paste electrode // Food Chem. 2012. V. 132. № 1. P. 637.
Nagaraja T.N., Desiraju T. Effects of chronic consumption of metanil yellow by developing and adult rats on brain regional levels of noradrenaline, dopamine and serotonin, on acetylcholine esterase activity and on operant conditioning // Food Chem. Toxicol. 1993. V. 31. № 1. P. 41.
Official Journal of the European Communities. Commission Directive 1994/36/EC and 2006/33/EC.
Oka H., Ikai Y., Ohno T., Kawamura N., Hayakawa J., Harada K., Suzuki M. Identification of unlawful food dyes by thin-layer chromatography-fast atom bombardment mass spectrometry // J. Chromatogr. A. 1994. V. 674. № 1–2. P. 301.
Kucharska M., Grabka J. A review of chromatographic methods for determination of synthetic food dyes // Talanta. 2010. V. 80. № 3. P. 1045.
Prado M., Godoy H. Validation of the methodology to determine synthetic dyes in foods and beverages by HPLC // J. Liq. Chromatogr. Relat. Technol. 2002. V. 25. № 16. P. 2455.
Kiseleva M., Pimenova V., Eller K. Optimization of conditions for the HPLC determination of synthetic dyes in food // J. Anal. Chem. 2003. V. 58. № 7. P. 685.
Esen B., Oymak T., Dural E. Determination of food colorings in pharmaceutical preparations and food additives by a validated HPLC method // IJSER. 2018. V. 9. № 8. P. 72.
Kirschbaum J., Krause C., Pfalzgraf S., Brückner H. Development and evaluation of an HPLC-DAD method for determination of synthetic food colorants // Chromatographia. 2003. V. 57. № 1. P. S115.
Zhang J., Gao N., Zhang Y. Method development and validation for the determination of five synthetic food colorants in alcoholic beverages by reversed-phase high performance liquid chromatography coupled with diode-array detector // Anal. Lett. 2007. V. 40. № 16. P. 3080.
Alves S.P., Brum D.M., de Andrade E.C.B., Netto A.D.P. Determination of synthetic dyes in selected foodstuffs by high performance liquid chromatography with UV-DAD detection // Food Chem. 2008. V. 107. № 1. P. 489.
Ma M., Luo X., Chen B., Su S., Yao S. Simultaneous determination of water-soluble and fat-soluble synthetic colorants in foodstuff by high-performance liquid chromatography–diode array detection–electrospray mass spectrometry // J. Chromatogr. A. 2006. V. 1103. № 1. P. 170.
Hull C.K., Martin P.D., Warwick M.J., Thomas E. Quantification of the N-desmethyl metabolite of rosuvastatin in human plasma by automated SPE followed by HPLC with tandem MS detection // J. Pharm. Biomed. Anal. 2004. V. 35. № 3. P. 609.
Zou T., He P., Yasen A., Li, Z. Determination of seven synthetic dyes in animal feeds and meat by high performance liquid chromatography with diode array and tandem mass detectors // Food Chem. 2013. V. 138. № 2. P. 1742.
Chen Q.-C., Mou S.-F., Hou X.-P., Riviello J.M., Ni Z.-M. Determination of eight synthetic food colorants in drinks by high-performance ion chromatography // J. Chromatogr. A. 1998. V. 827. № 1. P. 73.
Gennaro M.C., Gioannini E., Angelino S., Aigotti R., Giacosa D. Identification and determination of red dyes in confectionery by ion-interaction high-performance liquid chromatography // J. Chromatogr. A. 1997. V. 767. № 1. P. 87.
González M., Gallego M., Valcárcel M. Liquid chromatographic determination of natural and synthetic colorants in lyophilized foods using an automatic solid-phase extraction system // J. Agric. Food Chem. 2003. V. 51. № 8. P. 2121.
Sun H., Sun N., Li H., Zhang J., Yang Y. Development of multiresidue analysis for 21 synthetic colorants in meat by microwave-assisted extraction–solid-phase extraction–reversed-phase ultrahigh performance liquid chromatography // Food Anal. Methods. 2013. V. 6. № 5. P. 1291.
Sha O., Zhu X., Feng Y., Ma W. Aqueous two-phase based on ionic liquid liquid–liquid microextraction for simultaneous determination of five synthetic food colourants in different food samples by high-performance liquid chromatography // Food Chem. 2015. V. 174. P. 380.
Mo Z., Zhang Y., Zhao F., Xiao F., Guo G., Zeng B. Sensitive Vtammetric determination of Sudan I in food samples by using gemini surfactant–ionic liquid–multiwalled carbon nanotube composite film modified glassy carbon electrodes // Food Chem. 2010. V. 121. № 1. P. 233.
Sheikhshoaie M., Karimi-Maleh H., Sheikhshoaie I., Ranjbar M. Voltammetric amplified sensor employing RuO2-nano road and room temperature ionic liquid for amaranth analysis in food samples // J. Mol. Liq. 2017. V. 229. P. 489.
Ni Y., Bai J., Jin L. Multicomponent chemometric determination of colorant mixtures by Vtammetry // Anal. Lett. 1997. V. 30. № 9. P. 1761.
Karimi-Maleh H., Ahanjan K., Taghavi M., Ghaemy M. A novel Vtammetric sensor employing zinc oxide nanoparticles and a new ferrocene-derivative modified carbon paste electrode for determination of captopril in drug samples // Anal. Methods. 2016. V. 8. № 8. P. 1780.
Gan T., Sun J., Cao S., Gao F., Zhang Y., Yang Y. One-step electrochemical approach for the preparation of graphene wrapped-phosphotungstic acid hybrid and its application for simultaneous determination of Sunset Yellow and Tartrazine // Electrochim. Acta. 2012. V. 74. P. 151.
Songyang Y., Yang X., Xie S., Hao H., Song J. Highly-sensitive and rapid determination of Sunset Yellow using functionalized montmorillonite-modified electrode // Food Chem. 2015. V. 173. P. 640.
Combeau S., Chatelut M., Vittori O. Identification and simultaneous determination of Azorubin, Allura Red and Ponceau 4R by differential pulse polarography: Application to soft drinks // Talanta. 2002. V. 56. № 1. P. 115.
Chanlon S., Joly-Pottuz L., Chatelut M., Vittori O., Cretier J.L. Determination of Carmoisine, Allura Red and Ponceau 4R in sweets and soft drinks by differential pulse polarography // J. Food Compos. Anal. 2005. V. 18. № 6. P. 503.
Abu Shawish H.M., Ghalwa N.A., Saadeh S.M., Harazeen, H.E. Development of novel potentiometric sensors for determination of Tartrazine dye concentration in foodstuff products // Food Chem. 2013. V. 138. № 1. P. 126.
Nevado J.J., Cabanillas C.G., Salcedo A.M. Simultaneous spectrophotometric determination of three food dyes by using the first derivative of ratio spectra // Talanta. 1995. V. 42. № 12. P. 2043.
Berzas Nevado J.J, Rodriguez Flores J., Guiberteau Cabanillas C., Villasenor Llerena M.J., Salcedo A.C. Resolution of ternary mixtures of Tartrazine, Sunset Yellow and Ponceau 4R by derivative spectrophotometric ratio spectrum-zero crossing method in commercial foods // Talanta. 1998. V. 46. № 5. P. 933.
Ni Y., Wang Y., Kokot S. Simultaneous kinetic spectrophotometric analysis of five synthetic food colorants with the aid of chemometrics // Talanta. 2009. V. 78. № 2. P. 432.
Turak F., Ustun Ozgur M. Validated spectrophotometric methods for simultaneous determination of food colorants and sweeteners // J. Chem. 2013. V. 2013. P. 1.
El-Shahawi M.S., Hamza A., Al-Sibaai A.A., Bashammakh, A.S., Al-Saidi, H.M. A new method for analysis of Sunset Yellow in food samples based on cloud point extraction prior to spectrophotometric determination // J. Ind. Eng. Chem. 2013. V. 19. № 2. P. 529.
Ai Y.-J., Liang P., Wu Y.-X., Dong Q.-M., Li J.-B., Bai Y., Xu B.-J., Yu Z., Ni D. Rapid qualitative and quantitative determination of food colorants by both Raman spectra and surface-enhanced Raman scattering (SERS) // Food Chem. 2018. V. 241. P. 427.
Coym J.W., Dorsey J.G. Reversed-phase retention thermodynamics of pure-water mobile phases at ambient and elevated temperature // J. Chromatogr. A. 2004. V. 1035. № 1. P. 23.
McNeff C.V., Yan B., Stoll D.R., Henry R.A. Practice and theory of high temperature liquid chromatography // J. SeP. Sci. 2007. V. 30. № 11. P. 1672.
Al-Khateeb L.A., Smith R.M. High-temperature liquid chromatography of steroids on a bonded hybrid column // Anal. Bioanal. Chem. 2009. V. 394. P. 1255.
Al-Khateeb L.A., Smith R.M. Superheated water chromatography on phenyl bonded hybrid stationary phases // J. Chromatogr. A. 2008. V. 1201. № 1. P. 61.
Vidotti E.C., Costa W.F., Oliveira C.C. Development of a green chromatographic method for determination of colorants in food samples // Talanta. 2006. V. 68. № 3. P. 516.
Li J., Carr P.W. Effect of temperature on the thermodynamic properties,kinetic performance, and stability of polybutadiene-coated zirconia // Anal. Chem. 1997. V. 69. № 5. P. 837.
Guillarme D., Heinisch S., Rocca J.L. Effect of temperature in reversed phase liquid chromatography // J. Chromatogr. A. 2004. V. 1052. № 1. P. 39.
Li J., Carr P.W. Evaluation of temperature effects on selectivity in RPLC separations using polybutadiene-coated zirconia // Anal. Chem. 1997. V. 69. № 11. P. 2202.
Sanagi M.M., See H.H., Ibrahim W.A.W., Naim A.A. High temperature liquid chromatography of triazole fungicides on polybutadiene-coated zirconia stationary phase // J. Chromatogr. A. 2004. V. 1059. № 1. P. 95.
Liu Y., Grinberg N., Thompson K.C., Wenslow R.M., Neue U.D., Morrison D., Walter T.H., O’Gara J.E., Wyndham K.D. Evaluation of a C18 hybrid stationary phase using high-temperature chromatography // Anal. Chim. Acta. 2005. V. 554. № 1. P. 144.
Miller J.N., Miller J.C. Statistics for Analytical Chemistry, Essex, UK: Pearson Education Limited, 2000.
Yanuka Y., Shalon Y., Weissenberg E., Nir-Grosfeld I. The isolation and separation of dyes from foodstuffs by column chromatography // Analyst. 1963. V. 88. № 1052. P. 872.
Sorouraddin M.-H., Saadati M., Mirabi F. Simultaneous determination of some common food dyes in commercial products by digital image analysis // J. Food Drug Anal. 2015. V. 23. № 3. P. 447.
Qi R., Zhou X., Li X., Ma J., Lu C., Mu J., Zhang X., Jia Q. Rapid identification of synthetic colorants in food samples by using indium oxide nanoparticle-functionalized porous polymer monolith coupled with HPLC-MS/MS // Analyst. 2014. V. 139. № 23. P. 6168.
Cole L.A., G. Dorsey J., Dill K. Temperature dependence of retention in reversed phase liquid chromatography // Anal. Chem. 1992. V. 64. P. 1324.
Heinisch S., Rocca J.-L. Sense and nonsense of high-temperature liquid chromatography // J. Chromatogr. A. 2009. V. 1216. № 4. P. 642.
McNeff C.V., Yan B., Stoll D.R., Henry R.A. Practice and theory of high temperature liquid chromatography // J. SeP. Sci. 2007. V. 30. № 11. P. 1672.
Cheng Y., Xu, Q., Liu J., Zhao C., Xue F., Zhao Y. Decomposition of five phenolic compounds in high temperature water // J. Braz. Chem. Soc. 2014. V. 25. P. 1.
Rao M.P., Wu J.J., Asiri A.M., Anandan S. Photocatalytic degradation of tartrazine dye using CuO straw-sheaf-like nanostructures // Water Sci. Technol. 2017. V. 75. № 5–6. P. 1421.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии