

Журнал аналитической химии, 2021, T. 76, № 7, стр. 603-611
УФ-спектрометрическое определение суммарного содержания аренов в сточных водах
Т. В. Антонова a, В. И. Вершинин a, *, И. В. Власова a
a Омский государственный университет им. Ф.М. Достоевского
644077 Омск, просп. Мира, 55а, Россия
* E-mail: vyvershinin@yandex.ru
Поступила в редакцию 15.02.2021
После доработки 19.02.2021
Принята к публикации 19.02.2021
Аннотация
Предложены два способа оценки суммарного содержания (сAr) наиболее токсичных углеводородов (аренов) в сточных водах. Методики включают экстракцию нефтепродуктов н-гексаном, сорбционную очистку экстракта от фенолов и измерение светопоглощения аренов в УФ-области. Отделять алканы и циклоалканы не требуется. Первый способ позволяет быстро оценить сAr в пересчете на стандартное вещество Хст (о- или м-ксилол), например, с целью скрининга. Оптическую плотность измеряют при 250 нм, что уменьшает влияние внутригрупповой селективности и оставшихся в экстракте фенолов. Суммарные содержания аренов в н-гексановых растворах оцениваются этим способом довольно точно (погрешность δс < 7% при sr < 0.05). Однако анализ водных растворов (имитатов сточных вод) в интервале сAr от 0.1 до 50 мг/л приводит к δс > 50%. Результаты оценки сAr в имитатах и сточных водах оказываются заниженными, что в основном связано с потерями аренов в ходе пробоподготовки. Реализуя второй (более точный) способ, измеряют оптические плотности экстракта в области 240–280 нм при семи длинах волн. Величину сAr находят методом множественной линейной регрессии без пересчета на Хст. Многомерные градуировки строят, используя экстракты из 25 имитатов (обучающая выборка) и проводя их через все операции пробоподготовки, что снижает влияние потерь. При сAr > 1 мг/л погрешность анализа имитатов из тест-выборки не выше 20% при sr < 0.12. Продолжительность анализа – 1 ч. Методика проверена в анализе сточных вод предприятий разного профиля. Полученные результаты согласуются с результатами хроматографического анализа тех же проб.
Техногенное загрязнение водоемов нефтепродуктами контролируют, определяя суммарное содержание углеводородов (УВ) без учета их токсичности [1]. Специалисты рекомендуют дополнительно определять наиболее токсичные УВ – арены, преимущественно арены С6–С9 [2, 3]. Для этого после извлечения нефтепродуктов экстракт очищают от полярных соединений, фракционируют, упаривают и хроматографируют. Применяя пламенно-ионизационный детектор, арены С6–С9 определяют на уровне их ПДК (порядка 0.1 мг/л) [4]. Кроме обычной экстракции, применяют твердофазную микроэкстракцию [5] или анализируют равновесный пар [6]. К сожалению, из-за потерь в ходе пробоподготовки, а также из-за длительности и трудоемкости методик индивидуальные арены в ходе экологического мониторинга определяют очень редко. Так как разные арены имеют довольно близкие значения ПДК и сходные химико-аналитические свойства, можно контролировать их суммарное содержание (сAr), что упрощает и ускоряет анализ. Примером может быть методика экстракционно-хроматографического анализа сточных вод в диапазоне сAr от 0.1 до 100 мг/л [7]. Результаты хорошо воспроизводимы (sr < 0.10). Потери аренов в ходе пробоподготовки учитываются при построении градуировочной зависимости. Вследствие этого погрешность (δc, отн. %) не превышает 15%. Однако случайное совпадение времен удерживания аренов и других УВ (алканов, циклоалканов) может привести к намного большим значениям δc.
Мы предлагаем определять cAr экстракционно-спектрометрическим методом. В этом случае алканы и циклоалканы не помешают, так как они, в отличие от аренов, не поглощают свет в ближней УФ-области. Прямое измерение обобщенного сигнала аренов исключает идентификационные ошибки и вызванные ими погрешности количественного анализа. Кроме того, УФ-спектрометрия – более экспрессный, прецизионный и простой метод, хотя и уступающий газожидкостной хроматографии (ГЖХ) по чувствительности и селективности. УФ-спектрометрию с успехом используют в групповом анализе технических нефтепродуктов. Ее пытались применять и для определения содержания нефтепродуктов в водах [8], но результаты оказались очень неточными [9]. Для определения суммы аренов в водах УФ-спектрометрию ранее не применяли; публикации в этой области нам неизвестны, за исключением небольшой статьи [10].
Удельные коэффициенты светопоглощения разных аренов при одной и той же длине волны (К, л/(мг см)) различаются, в отличие от коэффициентов чувствительности при их пламенно-ионизационном детектировании. Это затрудняет прямой расчет сAr, но суммарное содержание аренов можно оценить в пересчете на стандартное вещество Хст. Соответствующий интегральный показатель СAr будет аналогичен углеводородному или фенольному индексам [11]. Естественно, неполнота экстракционного извлечения аренов и внутригрупповая селективность сигналов приведут к некоторой неопределенности нового показателя (“аренового индекса”), т.е. к непредсказуемым отличиям СAr от cAr. Однако способы снижения неопределенности интегральных показателей известны: в спектрометрии либо оптимизируют аналитическую длину волны (АДВ) и природу стандарта [12], либо переходят к многоволновым измерениям и многомерным градуировкам [13].
Мы считаем, что оценка cAr должна включать следующие этапы: а) экстракцию нефтепродуктов из исследуемой воды, б) сорбционную очистку экстракта от полярных компонентов, в) измерение светопоглощения очищенного экстракта в УФ-области; г) расчет CAr по предварительно построенной одномерной или многомерной градуировочной зависимости. На первых двух этапах анализ можно проводить так же, как и при определении показателя “нефтепродукты”.
Цель данной работы – проверка возможности точной оценки суммарного содержания аренов в водах экстракционно-спектрометрическим методом; разработка и проверка соответствующей методики анализа сточных вод.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Модельными веществами служили арены группы С6–С9: бензол (Б), толуол (ТЛ), о-ксилол (ОК), м-ксилол (МК), п-ксилол (ПК), этилбензол (ЭБ) и кумол (КМ). Многокомпонентные растворы аренов в н-гексане готовили по точным навескам реактивов х. ч., как описано в статье [7]. Суммарные концентрации аренов в модельных растворах (“смесях”) составляли от 40 до 1500 мг/л, массовые соотношения разных аренов в смеси не превышали 10 : 1. Всего было получено и проанализировано более 30 таких смесей. Их использовали для приготовления имитатов сточных вод, т.е. водных растворов с известными значениями сAr. Для этого к точно измеренному объему (обычно 10.0 мл) аренсодержащего модельного раствора добавляли дистиллированную воду, серную кислоту или щелочь до желаемого значения рН, тщательно перемешивали и доводили до 1000 мл дистиллированной водой. Суммарные концентрации аренов в имитатах находились в диапазоне 0.1–50 мг/л, что примерно соответствует составу очищенных сточных вод и не превышает равновесную растворимость аренов C6–C9 в воде. Примеры имитатов показаны в табл. 1. Некоторые имитаты готовили из водорастворимых государственных стандартных образцов (ГСО) с известным суммарным содержанием УВ и приблизительно известной долей аренов; примером может быть ГСО 7117-94. В другие имитаты к аренам добавляли известные количества фенолов (собственно фенол, п-крезол, о-крезол). Имитаты применяли: а) для оценки степени извлечения аренов и степени очистки экстракта от фенолов; б) в качестве градуировочных растворов при построении многомерных градуировок; в) для проверки правильности разработанных методик. Обучающая выборка включала 25 имитатов, тест-выборка – 10 других имитатов.
Таблица 1.
Состав некоторых имитатов (градуировочных водных растворов) для экстракционно-спектрометрического определения аренов
| № смеси | Концентрация арена, мг/л | сАr, мг/л | ||||
|---|---|---|---|---|---|---|
| Б | ТЛ | ОК | МК | ЭБ | ||
| 1 | – | 0.05 | 0.24 | – | 0.16 | 0.45 |
| 2 | 0.91 | 0.84 | 0.21 | – | – | 1.96 |
| 3 | 0.43 | 0.44 | 0.90 | 0.46 | 0.47 | 2.70 |
| 4 | – | – | 2.61 | 1.94 | – | 4.55 |
| 5 | 2.01 | 2.92 | 1.45 | 0.56 | 4.21 | 11.2 |
| 6 | – | 7.81 | 7.49 | 4.64 | 2.44 | 22.4 |
| 7 | 8.21 | 7.67 | 13.37 | 5.01 | 8.52 | 42.8 |
Пробы сточных вод (1.0 л) отбирали на предприятиях разного профиля сотрудники заводских контрольно-аналитических лабораторий, руководствуясь стандартными рекомендациями по определению показателя “нефтепродукты”. Сильно загрязненные пробы перед анализом разбавляли дистиллированной водой. Отобранные пробы анализировали трижды (по возможности в день пробоотбора). И пробы, и имитаты хранили на холоду не более трех суток. В ходе анализа сумму аренов, содержавшихся в 250 мл имитата или исследуемой воды, экстрагировали 25 мл н-гексана в присутствии высаливателя (NaCl). Время контакта фаз – 5 мин. Для очистки от полярных соединений обезвоженный экстракт пропускали через колонку с Al2O3. Объем очищенного экстракта доводили н-гексаном до 25.0 мл, далее экстракт не фракционировали и не упаривали. Методика экстракции детально описана в предыдущем сообщении [7], а методика сорбционной очистки экстрактов – в стандарте [14]. Спектры поглощения модельных растворов и экстрактов регистрировали с помощью спектрофотометра СФ-2000, применяя кварцевые кюветы (l = 10.0 мм). Спектры снимали против н-гексана. Чистоту растворителя контролировали, измеряя его оптическую плотность при 220 нм против пустой кюветы. Значимость отклонений от аддитивности светопоглощения (ΔА) проверяли по методике [15], используя 3s-критерий.
Применяя метод одноволновой УФ-спектрометрии, измеряли оптическую плотность экстрактов при выбранной АДВ (обычно при 250 нм). Суммарное содержание аренов в экстракте в пересчете на Хст находили с помощью заранее построенного по растворам Хст прямолинейного градуировочного графика. Конечный результат анализа (СAr) вычисляли с учетом объемов пробы и экстракта.
Анализируя имитаты и сточные воды методом многоволновой УФ-спектрометрии, использовали метод множественной линейной регрессии в варианте непрямой градуировки (МЛР-2). Обычно оптические измерения проводили при семи АДВ (например, при 243, 250, 260, 265, 270, 275 и 280 нм). В некоторых случаях автоматически измеряли оптическую плотность экстракта с шагом в 1 или 0.2 нм. Для построения многомерных градуировок и расчета результатов анализа применяли программы Excel [13] и Optic-MLR [16]. Градуировку А строили по данным о светопоглощении 10 гексановых растворов с известными значениями cAr. Градуировку Б строили по данным о светопоглощении 25 экстрактов из имитатов, прошедших все стадии пробоподготовки. В обоих случаях решали методом наименьших квадратов (МНК) переопределенную систему линейных уравнений вида
где сi – выраженное в мг/л суммарное содержание аренов в i-ом модельном растворе (градуировка А) или в i-ом имитате (градуировка Б); Аij – оптическая плотность i-го модельного раствора или экстракта из i-го имитата при j-ой АДВ; kj – j-й регрессионный коэффициент (в мг/л). Подставляя в формулу (1) найденные методом МНК оценки коэффициентов, получали искомые градуировки. Анализируя очередную пробу, в полученную градуировку подставляли оптические плотности фотометрируемого раствора при выбранных АДВ. По градуировке А находили содержание аренов в экстракте, а затем пересчитывали его на исходную пробу. По градуировке Б сразу находили содержание аренов в исследуемой воде, не вычисляя содержание аренов в экстракте. Статистическую обработку данных вели по традиционному алгоритму Стьюдента (n = 3, P = 0.95). Погрешности анализа имитатов (δс, отн. %) рассчитывали по формуле:(2)
$\delta c = 100 \times {{\left( {{{C}_{{{\text{Ar}}}}} - {{c}_{{{\text{Ar}}}}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{C}_{{{\text{Ar}}}}} - {{c}_{{{\text{Ar}}}}}} \right)} {{{c}_{{{\text{Ar}}}}}}}} \right. \kern-0em} {{{c}_{{{\text{Ar}}}}}}}.$Для проверки правильности результатов анализа сточных вод применяли метод стандартных добавок (введено–найдено). В качестве добавок использовали смеси аренов. Некоторые пробы дополнительно анализировали экстракционно-хроматографическим методом [7].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
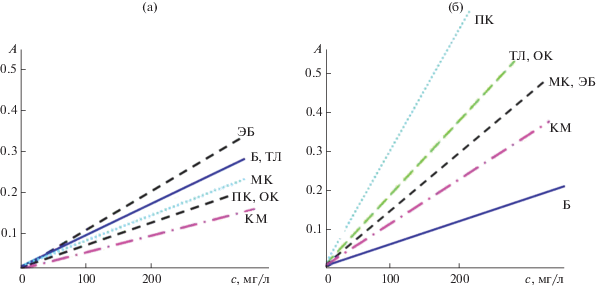
Одноволновая спектрометрия: выбор аналитической длины волны. Выбирая условия спектрометрической оценки сAr, следует учесть несколько факторов: чувствительность и прецизионность измерений при разных АДВ, выполнение закона Бугера−Ламберта−Бера, внутригрупповую и межгрупповую селективность сигналов, аддитивность светопоглощения смесей. Сильнее всего арены поглощают в области 180–220 нм, но результаты соответствующих измерений недостаточно воспроизводимы и плохо подчиняются закону Бугера−Ламберта−Бера. При λ > 280 нм некоторые моноциклические арены (например, бензол) не поглощают УФ-излучение, что исключает возможность точной оценки сAr. Для такой оценки следует использовать область 240–280 нм, где хорошо поглощают свет все арены, градуировочные графики прямолинейны (r > 0.99), результаты измерений оптической плотности характеризуются хорошей сходимостью (sr < 0.02) и приблизительно аддитивны (ΔA < 3s). К сожалению, чувствительность определения моноциклических аренов в области длин волн 240–280 нм невелика [17], причем при любой фиксированной АДВ удельные коэффициенты поглощения разных аренов различаются (рис. 1). Это приводит к образованию “веера градуировок” (рис. 2). Внутригрупповую селективность сигналов характеризовали безразмерным параметром Т, как принято при изучении интегральных показателей [18]:
(3)
$T = {{{{K}_{{\max }}}} \mathord{\left/ {\vphantom {{{{K}_{{\max }}}} {{{K}_{{\min }}}}}} \right. \kern-0em} {{{K}_{{\min }}}}}.$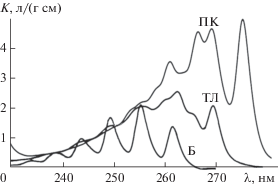
Рис. 2.
Градуировочные графики для определения индивидуальных аренов в гексановых растворах при 254 нм (а) и при 263 нм (б).
В формуле (3) символами Kmax и Kmin обозначены максимальное и минимальное значения удельных коэффициентов светопоглощения индивидуальных аренов при фиксированной АДВ. Параметр Т зависит от выбора АДВ. Так, для набора исследованных нами аренов при 263 нм Т ≈ 4, а при 254 нм Т ≈ 2. Внутригрупповая селективность оказалась минимальной при λ = 250 нм (Т ≈ 1.5). Пределы обнаружения разных аренов в модельных растворах при этой АДВ довольно близки и составляют 1–2 мг/л. Следовательно, при 10-кратном концентрировании аренов их пределы обнаружения в воде примерно соответствуют значениям ПДК, а нижние границы определяемых содержаний (НГОС) близки к 0.5 мг/л. Для сравнения укажем, что при определении аренов по методике [7] значения НГОС в пять раз ниже. Тем не менее чувствительность УФ-спектрометрии достаточна для определения аренов в очищенных сточных водах многих промышленных предприятий.
Дополнительным преимуществом выбранной АДВ является уменьшение влияния фенолов, часто сопутствующих аренам в сточных водах и не полностью отделяемых в ходе сорбционной очистки экстракта. Известно, что светопоглощение фенолов в неводных растворах максимально при 210–230 и 270–280 нм, тогда как в области 240–250 нм оно существенно ниже [19]. Проверка подтвердила, что при 250 нм смеси фенолов поглощают УФ-излучение слабее, чем смеси моноциклических аренов. Следует отметить, что при этой АДВ сильно поглощают тяжелые арены (например, нафталин и фенантрен) [17].
Выбор стандартного вещества. Для правильной спектрометрической оценки суммарного содержания однотипных аналитов в пересчете на Хст необходимо, чтобы удельный коэффициент поглощения стандартного вещества (Kст) отвечал условию:
Условию (4) отвечает множество веществ. Выбор лучшего стандарта облегчается, если концентрации всех аналитов искомой группы – величины одного порядка, а коэффициенты чувствительности для каждого аналита известны [12]. В этом случае для подбора оптимального стандарта пригодно эмпирическое соотношение:
(5)
${{K}_{{{\text{ст}}}}} \approx {{\left( {{{K}_{{\max }}} + {{K}_{{\min }}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{K}_{{\max }}} + {{K}_{{\min }}}} \right)} 2}} \right. \kern-0em} 2}.$Подстановка найденных при 250 нм значений Kmax и Kmin в формулу (5) приводит к выводу, что лучшие оценки сAr должны получаться в пересчете на о- или м-ксилол. При 250 нм эти арены определяются со средней и приблизительно одинаковой чувствительностью. Использование других стандартов должно приводить к систематически завышенным или систематически заниженным оценкам сAr, что и было подтверждено экспериментально. Результаты анализа одних и тех же модельных растворов, выраженные в пересчете на разные Хст, приведены в табл. 2. Все результаты характеризуются хорошей сходимостью (sr < 0.05). Если суммарное содержание аренов в гексановых растворах выражено в пересчете на о-ксилол или м-ксилол, то δс < 7 отн. % (по модулю). При неудачном выборе Хст погрешности намного выше. Так, при использовании толуола погрешности отрицательны и достигают 20 отн. %, при использовании кумола – погрешности положительны и достигают 40 отн. %.
Таблица 2.
Результаты и погрешности определения суммарного содержания аренов в модельных смесях (многокомпонентных гексановых растворах) в пересчете на разные Хст
| Номер смеси | сAr, мг/л | СAr в пересчете на Хст, мг/л | δс, отн. % | ||||
|---|---|---|---|---|---|---|---|
| ТЛ | ОК | МК | ТЛ | ОК | МК | ||
| 1 | 45 | 36 | 47 | 48 | –20.0 | 4.4 | 6.7 |
| 2 | 196 | 171 | 193 | 199 | –12.8 | –1.5 | 1.5 |
| 3 | 270 | 231 | 270 | 283 | –14.4 | 0.0 | 4.8 |
| 4 | 455 | 425 | 438 | 458 | –6.6 | –3.7 | –0.7 |
| 5 | 1120 | 1010 | 1090 | 1167 | –9.8 | –2.7 | 4.2 |
Одноволновая спектрометрия в анализе имитатов. Переход к анализу разбавленных водных растворов (в частности, имитатов сточных вод) существенно снижает точность оценки сAr. Поскольку анализ включает дополнительные операции, сходимость результатов ухудшается (sr < 0.12). Результаты анализа оказываются сильно заниженными (табл. 3); значения δс имеют преимущественно систематический характер и превышают 50% (по модулю). Мы считаем, что эти погрешности в основном вызваны потерями аренов в ходе экстракции. Известно, что моноциклические арены намного лучше растворяются в воде, чем другие УВ, что и снижает степень их извлечения. По нашим данным, потери аренов при однократной экстракции небольшим объемом н-гексана составляют от 40 до 70% в зависимости от природы извлекаемого арена. Это совпадает с данными [20]. Кроме того, до 10% массы извлеченных аренов теряется в ходе сорбционной очистки экстракта.
Таблица 3.
Результаты определения суммарного содержания аренов (в пересчете на м-ксилол) в имитатах сточных вод с применением одномерной градуировки (250 нм)
| № имитата | Арены | сAr, мг/л | сф, мг/л | СAr, мг/л | δс, отн. % |
|---|---|---|---|---|---|
| 13 | Б, ТЛ, ЭБ, ПК, КМ | 0.50 | – | 0.15 | –70 |
| 14 | ТЛ, ЭБ, КМ | 0.70 | – | 0.25 | –64 |
| 15 | Б, ОК, МК, ЭБ | 2.70 | – | 1.30 | –52 |
| 16 | Б, ОК, МК, ЭБ | 2.70 | 5.0 | 1.38 | –49 |
| 17 | Б, ТЛ, ОК, МК, ЭБ | 11.2 | – | 5.3 | –53 |
| 18 | Б, ТЛ, ОК, МК, ЭБ | 11.2 | 25.0 | 5.7 | –49 |
| 19 | Б, ТЛ, ОК, МК, ЭБ | 11.2 | 50.0 | 6.0 | –47 |
Добиться количественного извлечения аренов н-гексаном можно, увеличивая объем экстрагента при единичной экстракции и/или проводя повторные экстракции и объединяя экстракты. Однако указанные операции помешают сконцентрировать арены! Мы полагаем, что добиваться полного извлечения аренов нецелесообразно, так как соответствующие потери можно учесть при расчете результатов анализа. Речь идет не о введении эмпирических поправочных коэффициентов (хотя в некоторых случаях может быть полезен и этот прием), а о способе построения градуировочной зависимости. В качестве градуировочных растворов можно использовать водные растворы с известным содержанием Хст, проводя их через те же операции пробоподготовки, что и исследуемые пробы. По нашим данным, применение этого приема при построении одномерных градуировок снижает систематические погрешности анализа имитатов, но не устраняет их. Очевидно, потери аренов в ходе пробоподготовки не являются единственным источником систематических погрешностей. Другим источником может быть различие в экстрагируемости индивидуальных аренов (при выборе АДВ и Хст этот фактор не учитывали). Чтобы снизить его влияние, желательно проводить многоволновые измерения и строить многомерные градуировки (см. следующий раздел).
Менее серьезной проблемой оказалось влияние фенолов. Степень их извлечения из водных растворов меньше, чем степень извлечения аренов, особенно в щелочной среде [19]. После сорбционной очистки экстракта в нем остается лишь 5–10% от массы ранее извлеченных фенолов. Кроме того, при 250 нм оставшиеся фенолы поглощают УФ-излучение слабее, чем арены С6–С9. Если исходный имитат содержит примерно одинаковые количества фенолов и аренов, результат определения аренов в пересчете на Хст повышается всего на 3–5% по сравнению с имитатом, не содержащим фенолов. При этом результаты определения аренов остаются заниженными (см. табл. 3, нижние строки). При очень большом избытке фенолов могут быть получены и завышенные оценки сAr. В частности, высокие соотношения сф/cAr характерны для сточных вод предприятий, производящих фенолы или фенолформальдегидные смолы. Проблема влияния посторонних веществ на результаты экстракционно-спектрометрического определения аренов в водах разного типа требует специального изучения и будет рассмотрена в следующем сообщении.
Использование многоволновых измерений и многомерных градуировок. Как уже отмечалось, в экстракционно-спектрометрическом анализе сточных вод (или их имитатов) внутригрупповая селективность должна проявляться сильнее, чем в анализе модельных растворов. К различиям эталонных УФ-спектров индивидуальных аренов добавляются различия в экстрагируемости этих веществ. Как показано в предыдущих исследованиях нашей группы [13, 17], в подобных случаях для правильной оценки суммарных содержаний желательно измерять оптическую плотность при нескольких длинах волн, а результаты анализа рассчитывать с помощью многомерных градуировок без пересчета на Хст. На рис. 3 приведена схема такого анализа применительно к определению суммарного содержания аренов.
Рис. 3.
Схема экстракционно-спектрометрического анализа сточной воды с построением многомерной градуировки типа Б.
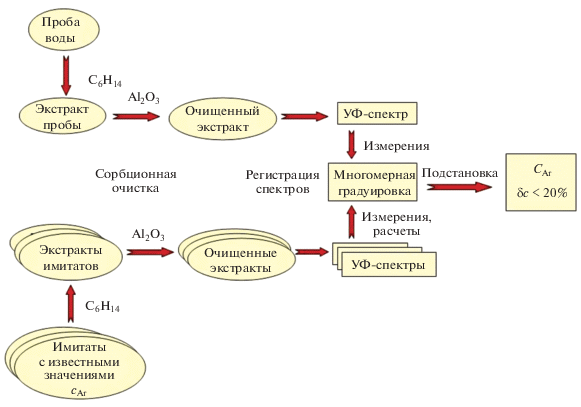
Сравнение табл. 3 и 4 показывает, что переход к многоволновым измерениям и многомерным градуировкам значительно улучшает результаты анализа имитатов. Отметим, что многомерные градуировки должны быть построены по результатам измерений, проведенных именно на том спектрофотометре, который будет в дальнейшем использован для анализа реальных проб; пользоваться литературными данными в таких случаях нельзя. В связи с этим в настоящей статье громоздкие формулы многомерных градуировок А и Б не приводятся.
Таблица 4.
Результаты определения суммарного содержания аренов в имитатах сточных вод с помощью многомерных градуировок A и Б (без пересчета на Хст)
| № имитата | сAr, мг/л | СAr, мг/л | δс, отн. % | ||
|---|---|---|---|---|---|
| А | Б | А | Б | ||
| 13 | 0.50 | 0.30 | 0.42 | –40 | –16 |
| 14 | 0.70 | 0.51 | 0.87 | –27 | 24 |
| 15 | 2.70 | 1.75 | 2.54 | –35 | –6 |
| 11 | 4.55 | 3.20 | 5.05 | –30 | 11 |
| 17 | 11.2 | 10.5 | 11.0 | –6 | –2 |
| 12 | 22.6 | 11.8 | 20.8 | –48 | –8 |
В табл. 4 сопоставлены результаты экстракционно-спектрометрического анализа имитатов из тест-выборки, полученные с помощью разных градуировок. Для расчетов использовали одни и те же исходные данные (значения оптической плотности гексанового экстракта при семи длинах волн). При использовании градуировки А погрешности не превышали 40 отн. %. Еще лучшие результаты были получены при использовании градуировки Б, построенной с помощью набора имитатов из обучающей выборки. В этом случае δс < 20% при sr < 0.12. Погрешности анализа разных имитатов различались и по абсолютной величине, и по знаку. При определении аренов на уровне сAr < 1 мг/л погрешности иногда повышались до 30%. Тем не менее точность полученных результатов отвечает установленным в РФ нормам погрешностей при определении микропримесей в водах [21].
Дополнительную проверку разработанной методики проводили, анализируя водные растворы ГСО 7117-94 с известным содержанием нефтепродуктов. Найденное с помощью программы Optic-MLR суммарное содержание аренов (от 1 до 10 мг/л) хорошо соответствовало данным по групповому составу этого ГСО [10].
Увеличение числа АДВ до нескольких десятков (или даже сотен) и последующее применение хемометрического алгоритма PLS дополнительно повышают точность определения сAr. Однако мы не считаем целесообразным усложнять новую методику, поскольку это может затруднить ее внедрение в практику работы контрольно-аналитических лабораторий.
Результаты анализа сточных вод. Пробы очищенных и неочищенных сточных вод четырех промышленных предприятий отбирали в омском регионе в 2017–2019 гг.; отбор и анализ некоторых проб повторяли с интервалами в несколько месяцев. Профили предприятий, составы их сточных вод и технологии водоочистки существенно различались, что влияло на общее содержание и соотношение концентраций разных аренов. Одноволновую УФ-спектрометрию мы использовали для скрининга: если найденные значения СAr были менее НГОС, пробы далее не исследовали. Остальные пробы анализировали путем многоволновых измерений с применением многомерной градуировки Б. Продолжительность единичного анализа – 1 ч. Для большинства проб относительное стандартное отклонение в условиях сходимости (sr) не превышало 0.12. Для слабо загрязненных вод (СAr < 1.0 мг/л) sr < 0.20. Результаты анализа 11 проб приведены в табл. 5. Для сравнения представлены результаты анализа тех же проб хроматографическим методом [7].
Таблица 5.
Результаты анализа сточных вод разных предприятий
| № пробы | Тип сточной воды | Профиль предприятия | Период пробоотбора | Результаты анализа, СAr, мг/л | |
|---|---|---|---|---|---|
| ГЖХ* | УФ** | ||||
| 1-1 | Неочищенная | Машиностроение | Весна 2017 | 2.4 ± 0.3 | 2.8 ± 0.6 |
| 1-2 | Неочищенная | Машиностроение | Осень 2017 | 2.7 ± 0.4 | 3.7 ± 0.6 |
| 1-3 | Очищенная | Машиностроение | Осень 2018 | 0.5 ± 0.2 | 0.6 ± 0.3 |
| 2-0 | Частично очищенная | Нефтепереработка | Весна 2018 | 26 ± 4 | 35 ± 6 |
| 2-1 | Очищенная | Нефтепереработка | Весна 2017 | 3.3 ± 0.3 | 3.6 ± 0.4 |
| 2-2 | Очищенная | Нефтепереработка | Осень 2017 | 2.4 ± 0.2 | 2.2 ± 0.1 |
| 2-3 | Очищенная | Нефтепереработка | Зима 2018 | 2.7 ± 0.2 | 3.9 ± 0.3 |
| 3-1 | Неочищенная | Энергетика | Лето 2018 | 0.8 ± 0.2 | 0.8 ± 0.3 |
| 3-2 | Очищенная | Энергетика | Лето 2018 | 0.12 ± 0.02 | Ниже НГОС |
| 4-1 | Неочищенная | Машиностроение | Весна 2017 | 2.0 ± 0.2 | 2.8 ± 0.3 |
| 4-2 | Неочищенная | Машиностроение | Осень 2017 | 1.2 ± 0.2 | 1.8 ± 0.5 |
* Определено по методике [7]; ** Рассчитано по градуировке Б.
Примечание: курсивом выделены практически совпадающие результаты.
Как видно из табл. 5, новая методика позволяет анализировать как неочищенные, так и очищенные сточные воды, что важно для правильной оценки эффективности очистки. Найденные спектрометрическим и хроматографическим методами значения СAr коррелируют между собой, линейная корреляция статистически значима (r > 0.99). Различия результатов для части проб статистически незначимы (эти результаты в табл. 5 выделены курсивом). Как правило, результаты определения сAr спектрометрическим методом были несколько выше, чем полученные для тех же проб методом ГЖХ. По-видимому, это связано с тем, что результаты экстракционно-спектрометрического анализа отражают суммарное содержание всех соединений, поглощающих УФ-излучение в области 240–280 нм, а не только содержание моноциклических аренов С6–С9, как в методике [7]. В частности, в указанной области УФ-излучение поглощают фенантрен, антрацен и другие полиарены, которые встречаются в сточных водах и тоже экстрагируются н-гексаном [17]. По методике [7] полиарены не определяются, так как при проведении хроматографического анализа экстракта опознают и учитывают лишь пики аренов С6–С9.
Спектры экстрактов из сточных вод очень похожи на спектры экстрактов из имитатов, содержащих смеси аренов С6–С9, и сильно отличаются от спектров поглощения полиаренов. По-видимому, вклад полиаренов в светопоглощение экстрактов из сточных вод невелик. Известно, что содержание полиаренов в природных и сточных водах намного меньше, чем содержание моноциклических аренов: из-за низкой растворимости в воде полиарены быстро переходят в донные отложения [22]. Тем не менее для некоторых сточных вод вклад полиаренов в светопоглощение экстрактов оказался статистически значимым.
Для проверки правильности результатов анализа сточных вод использовали способ добавок (табл. 6). В качестве добавок вводили водные растворы с известными значениями сAr. Погрешность при определении суммарного содержания аренов в добавках не превышала 25% (по модулю).
Таблица 6.
Проверка правильности результатов (мг/л) УФ-спектрометрического анализа двух проб сточной воды методом добавок (введено–найдено)
| № пробы | Найдено без добавки, СAr | Введено, сAr | Найдено с добавкой, СAr | Найдено в добавке, СAr | δс, отн. % |
|---|---|---|---|---|---|
| 4-1 | 2.82 ± 0.23 | 0.50 | 3.21 ± 0.20 | 0.39 ± 0.18 | –22 |
| 4-1 | 2.82 ± 0.23 | 1.00 | 3.95 ± 0.35 | 1.13 ± 0.21 | 13 |
| 4-2 | 1.80 ± 0.52 | 0.50 | 2.27 ± 0.16 | 0.47 ± 0.14 | –6 |
| 4-2 | 1.80 ± 0.52 | 1.00 | 2.89 ± 0.31 | 1.09 ± 0.13 | 9 |
* * *
Таким образом, УФ-спектрометрию можно использовать для приблизительной (δc > 50%) оценки суммарного содержания аренов в водах в пересчете на стандартное вещество Хст, например, с целью скрининга. Для точного (δc < 20%) и метрологически корректного (без пересчета на Хст) определения суммарного содержания аренов в водах следует проводить многоволновые измерения светопоглощения в области 240–280 нм и использовать соответствующие многомерные градуировки. При построении градуировок следует использовать экстракты из водных растворов с известными значениями сAr, проведенных через все операции пробоподготовки. Результаты УФ-спектрометрического анализа ряда сточных вод несколько превышают результаты анализа тех же проб методом ГЖХ, хотя полученные данные коррелируют друг с другом, а расхождения средних значений не всегда значимы. По-видимому, расхождения объясняются тем, что спектрометрическим методом определяется более широкая группа углеводородов, включающая и моноциклические, и полициклические арены, тогда как хроматографическим методом определяются лишь моноциклические арены С6–С9 (основные, но далеко не единственные ароматические соединения в соответствующих углеводородных смесях). Разработанная методика экстракционно-спектрометрического определения суммарного содержания аренов в диапазоне 0.5–50 мг/л пригодна для анализа сточных вод и может быть рекомендована контрольно-аналитическим лабораториям природоохранного профиля. Анализировать слабо загрязненные природные воды, а также контролировать состав питьевой воды по этой методике нельзя. Для повышения чувствительности и межгрупповой селективности предлагаемой методики необходимы дополнительные исследования.
Исследования проведены при финансовой поддержке РФФИ (грант 16-03-550479). Авторы благодарят к. ф.-м. н. С.М. Добровольского и к. х. н. С.В. Усову за ценные советы и замечания. В выполнении эксперимента принимали участие студентки Т.П. Доманина, О.А. Казакова, А.В. Мамонтова и $\boxed{П.А.\,\,Бурюкина}$.
Список литературы
Леоненко И.И., Антонович В.П., Андрианов А.М., Безлуцкая И.В., Цымбалюк К.К. Методы определения нефтепродуктов в водах и других объектах окружающей среды // Методы и объекты хим. анализа. 2010. Т. 5. № 2. С. 58.
Analysis of Petroleum Hydrocarbons in Environmental Media. Total Petroleum Hydrocarbon Criteria Working Group Series. V. 1 / Ed. Weisman W. Amherst, Mass., USA: Amherst Sci. Publ., 1998. 98 p.
Другов Ю.С., Родин А.А. Экологические анализы при разливах нефти и нефтепродуктов. СПб: Анатолия, 2000. 250 с.
Desideri P.G., Lepri L., Heimler D., Oianessi S., Checchini L. Concentration, separation and determination of hydrocarbons in sea water // J. Chromatogr. 1984. V. 284. № 10. P. 167.
Bianchin J.N., Nardini G., Merib J., Dias A.N., Martendal E., Carasek E. Simultaneous determination of polycyclic aromatic hydrocarbons and benzene, toluene, ethylbenzene and xylene in water samples using a new sampling strategy combining different extraction modes and temperatures in a single extraction solid phase microextraction – gas chromatography – mass spectrometry procedure // J. Chromatogr. A. 2012. V. 1233. P. 22.
РД.52.24.473-2012. Массовая концентрация летучих ароматических углеводородов в водах. Методика измерений газохроматографическим методом с использованием анализа равновесного пара. Ростов-на-Дону: ООО Вираж, 2012. 30 с.
Вершинин В.И., Усова С.В. Экстракционно-хроматографическое определение суммарного содержания моноциклических ароматических аренов в сточных водах // Журн. аналит. химии. 2021. Т. 76. № 3. С. 244. (Vershinin V.I., Usova S.V. Extraction–chromatographic determination of the total concentration of monocyclic arenes С6–С9 in wastewater // J. Anal. Chem. 2021. V. 76. № 3. P. 337.)
Гомеля Н.Д., Калабина Л.В., Хохотва А.П. Экстракционно-спектрофотометрический метод определения суммарного содержания тяжелых нефтепродуктов в воде // Химия и технология воды. 1999. Т. 21. № 6. С. 611.
Кленкин А.А., Павленко Л.Ф., Темердашев З.А. Некоторые методические особенности определения уровня нефтяного загрязнения водных экосистем // Заводск. лаборатория. Диагностика материалов. 2007. Т. 73. № 2. С. 31.
Бурюкина П.А., Власова И.В., Миргалеева Р.Р. Оценка суммарного содержания аренов в технических водах методом спектрофотометрии в сочетании с алгоритмом множествен-ной линейной регрессии // Вестник Омского университета. 2016. № 3. С. 54.
Baena J.R., Valcarcel M. Total indices in analytical sciences // Trends Anal. Chem. 2003. V. 22. № 10. P. 641.
Вершинин В.И. Формирование групп и выбор стандартных веществ при определении суммарных содержаний однотипных соединений в виде интегральных показателей // Журн. аналит. химии. 2017. Т. 72. № 9. С. 816. (Vershinin V.I. Group formation and choice of standard substances in the determination of total concentrations of similar compounds as total indices // J. Anal. Chem. 2017. V. 72. № 9. P. 947.)
Vershinin V.I., Petrov S.V. The estimation of total petroleum hydrocarbons in waste waters by multiwave IR spectrometry with multivariate calibrations // Talanta. 2016. V. 148. P. 163.
ГОСТ 51797-2001. Вода питьевая. Метод определения содержания нефтепродуктов / Контроль качества воды. Сб. ГОСТов. М.: Стандартинформ, 2010. 15 с.
Вершинин В.И., Цюпко Т.Г., Власова И.В. Выявление отклонений от аддитивности в спектрофотометрическом анализе неразделенных смесей // Методы и объекты химического анализа. 2010. Т. 5. № 4. С. 226.
Власова И.В., Цюпко Т.Г., Шелпакова А.С. Определение суммарного содержания антиоксидантов полифенольного типа по УФ спектру поглощения смеси // Методы и объекты химического анализа. 2012. Т. 7. № 1. С. 18.
Clar E. Polycyclic hydrocarbons. V. 1. London, New York: Academic Press, 1964. 486 p.
Vershinin V.I. Total indices as a tool to estimate sum content of similar analytes. Review // Talanta. 2015. V. 131. № 1. P. 293.
Алукер Н.Л., Лаврентьева А.Л., Суздальцева Я.М. Прямые оптические методы исследования в аналитике фенолов // Оптика и спектроскопия. 2020. Т. 128. № 3. С. 435. (Aluker N.L., Lavrentieva A.L., Suzdaltseva Ya.M. Direct optical research methods in the analytics of phenol // Opt. Spectrosс. 2020. V. 128. № 3. P. 422.)
Антонова Т.В., Усова С.В. Потери моноциклических ароматических углеводородов при экстракционном извлечении из водной фазы // Аналитика и контроль. 2017. № 4. С. 307.
ГОСТ 27384-2002. Вода. Нормы погрешности измерений показателей состава и свойств. М.: Стандартинформ, 2010. 10 с.
Adeniji A.O., Okoh O.O., Okoh A.I. Analytical methods for polycyclic aromatic hydro-carbons and global trend of distribution in water and sediment / Recent Insights in Petroleum Science and Engineering / Ed. El-Sayed Abdul-Raouf. 2018. DOI (дата обращения 18.02.2021).https://doi.org/10.5772/intechopen.71163
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии