

Журнал аналитической химии, 2021, T. 76, № 8, стр. 701-707
Управление селективностью разделения антоцианов: замена ацетонитрила на метанол в подвижной фазе
В. И. Дейнека a, *, Е. Ю. Олейниц a, И. П. Блинова a, Л. А. Дейнека a
a Белгородский государственный национальный исследовательский университет
308015 Белгород, ул. Победы, 85, Россия
* E-mail: deineka@bsu.edu.ru
Поступила в редакцию 06.12.2020
После доработки 19.01.2021
Принята к публикации 19.01.2021
Аннотация
Исследована возможность замены ацетонитрила на метанол при разделении антоцианов в подвижных фазах, содержащих органический модификатор, 10 об. % муравьиной кислоты (регулятор рН) и воду в условиях обращенно-фазовой ВЭЖХ на С18-стационарной фазе (Symmetry®C18). Установлено, что при замене ацетонитрила на метанол необходимо использовать два компонента подвижной фазы – содержащие только метанол и только муравьиную кислоту, поскольку при совместном присутствии этих веществ наблюдается длительный дрейф удерживания, предположительно, из-за реакции ацилирования метанола. Установлено, что замена ацетонитрила на метанол приводит к небольшим изменениям в селективности разделения антоцианов с одинаковыми заместителями, но различными агликонами и с различными углеводными заместителями для одного и того же агликона. По эффективности разделения (по числу теоретических тарелок) антоцианов также нет существенных различий, что позволяет отказаться от дорогостоящего ацетонитрила в пользу метанола. Разработанная методика применена для определения антоцианов в плодах клюквы различной степени зрелости (по окраске). Определены особенности антоцианового состава плодов клюквы и показана необходимость контроля состава растворителя пробы для исключения появления артефактных пиков.
В обращенно-фазовой ВЭЖХ (ОФ ВЭЖХ) для элюирования аналитов необходимо использовать водно-органические смеси с регулирующими рН добавками, которые должны удовлетворять ряду требований по физическим свойствам, включая ограничения по вязкости, по летучести, по возможности применения спектрофотометрического детектирования в необходимом диапазоне длин волн. Для определения антоцианов в варианте ОФ ВЭЖХ элюирующую силу подвижной фазы наиболее часто регулируют изменением концентрации ацетонитрила. С недавних пор наблюдается тенденция отказа от этого растворителя – токсичного для человека и окружающей среды, стоимость которого существенно выросла в последние годы [1]. В этом отношении метанол, являясь токсичным для человека, менее опасен для окружающей среды [1, 2], и относится к дешевым растворителям. Ацетонитрил достаточно химически инертен, чтобы беспроблемно применяться в смеси с водой и разнообразными кислотами, включая муравьиную, добавка которой (до 10 об. %) необходима для перевода антоцианов во флавилиевую форму [3]. В отличие от ацетонитрила, метанол способен этерифицироваться карбоновыми кислотами с образованием существенно более гидрофобных сложных эфиров, поэтому времена удерживания аналитов могут уменьшаться в процессе использования смесей метанола с муравьиной кислотой. Впрочем, присутствие большого количества воды в приемлемых для определения антоцианов подвижных фазах смещает равновесие реакции этерификации в сторону исходных кислоты и спирта. Следует отметить, что в литературе описано использование для разделения антоцианов смесей метанола, муравьиной кислоты и воды в одном компоненте подвижной фазы, при этом отсутствуют указания на проблемы со стабильностью времен удерживания аналитов в изократическом режиме [4, 5], или градиентных режимах элюирования [6–10] в условиях обращенно-фазовой ВЭЖХ.
Цель настоящей работы – оценить возможность замены ацетонитрила на метанол в составе подвижных фаз, содержащих муравьиную кислоту, на разделение антоцианов и выявить изменение селективности разделения и эффективности (по числу теоретических тарелок) при такой замене в ОФ ВЭЖХ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали экстракты антоцианов из листьев церциса [11], плодов красной смородины [12] и вишни [13]. Экстракты готовили настаиванием растительного материала в 0.1 М водном растворе HCl и отделяли от растительного материала фильтрованием через бумажный фильтр. Частичную очистку проводили твердофазной экстракцией на концентрирующих насадках (патронах) ДИАПАК С18 (БиоХимМак СТ, Москва).
Разделение антоцианов осуществляли на хроматографе Agilent 1200 Infinity с диодно-матричным детектором. В работе использовали хроматографическую колонку 150 × 4.6 мм Symmetry®C18 с размером частиц 3.5 нм при температуре термостата колонок 40°С. Хроматограммы регистрировали и обрабатывали программой ChemStation.
Для приготовления подвижных фаз использовали метанол “analytical reagent grade”, ацетонитрил “HPLC Gradient grade” (Fisher Chemical, Германия); муравьиную кислоту, ч. (85%, Китай) и дистиллированную воду.
Для обозначения антоцианов использовали следующие сокрашения: антоцианидины: Dp (дельфинидин), Cy (цианидин), Pt (петунидин), Pn (пеонидин) и Mv (мальвидин); гликозиды: Glu (глюкозид), Sopho (2"-глюкозилглюкозид), Sam (2"-ксилозилглюкозид), Rut (6"-рамнозилглюкозид или рутинозид), XRut (2"-ксилозилрутинозид), GRut (2"-глюкозилрутинозид), с замещением в положении 3 агликона, см. схему 1 .
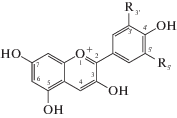
Схема 1 . Строение агликонов антоцианов (антоцианидинов).
Использовали следующую методику определения антоцианов плодов клюквы:
1. Антоцианы экстрагировали настаиванием плодов клюквы, разделенных на три группы по интенсивности окраски. Для этого к 7–8 г плодов добавляли 100 мл 0.1 М водного раствора HCl и смесь оставляли на ночь для настаивания. На следующий день экстракт отделяли от растительного материала и фильтровали через бумажный фильтр. Однократной экстракции было достаточно для практически полного извлечения антоцианов (более 98%).
2. Полученные экстракты использовали для спектрофотометрического определения антоцианов в пересчете на Cy3Glu [14], но без исключения абсорбции полимерных антоцианов при рН 4.5. Присутствие полимерных антоцианов оценивали хроматографическим методом [15]. По данному методу вклад полимерных антоциианов может быть найден по площади диффузного пика, начинающегося в диапазоне мертвого объема (времени); в результате получили не более 2% полимерных форм.
3. Следующим этапом была пробоподготовка к определению видового состава антоцианов методом обращенно-фазовой ВЭЖХ. Полученные в п. 1 экстракты сорбировали на активированном пропусканием 5 мл ацетона и кондиционированном пропусканием 10–15 мл 0.1 М водного раствора HCl концентрирующем патроне ДИАПАК С 18. Антоцианы реэкстрагировали с патрона 3 мл метанола, содержащего 10 об. % муравьиной кислоты. Реэкстракт разбавляли раствором муравьиной кислоты (10 об. %) в воде в соотношении 1 : 5 для последующего хроматографирования.
4. Далее проводили разделение антоцианов в условиях ОФ ВЭЖХ. Готовили раствор А, содержащий 30 об. % метанола в воде, и раствор Б, содержащий 20 об. % муравьиной кислоты в воде. Для элюирования использовали градиентный насос (двухканальный), но при постоянном соотношении растворов А и Б (1 : 1) при скорости подачи подвижной фазы 0.8 мл/мин. Хроматографическая колонка и температура разделения указаны выше. Хроматограммы записывали по три раза. Хроматограммы регистрировали, хранили и обрабатывали с помощью ПО “Agilent ChemStation”. Различие во временах удерживания основных компонентов не превышало 2 с, а расхождение между результатами определения доли антоцианов не превышало 0.5%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
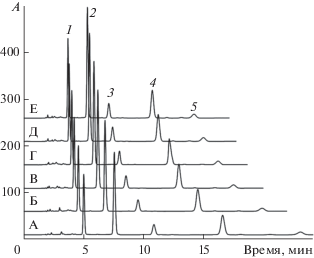
Возможность протекания реакции этерификации косвенно подтвердилась уже в первых опытах. При выводе хроматографической системы в режим в элюенте, содержащем из 12 об. % метанола и 10 об. % муравьиной кислоты в воде, дождаться прекращения дрейфа времен удерживания не удалось даже за 3 ч (рис. 1), хотя обычно для стабилизации удерживания антоцианов достаточно 20–30 мин. При этом наблюдается предсказанное выше уменьшение времен удерживания всех компонентов пробы.
Рис. 1.
Дрейф удерживания антоцианов в подвижной фазе CH3OH–HCOOH–H2O (12 : 10 : 78, по объему). Интервал между вводом проб А–Е 25–30 мин. Антоцианы: 1 – Dp3Glu; 2 – Cy3Glu; 3 – Pt3Glu; 4 – Pn3-Glu; 5 – Mv3Glu. Колонка 4.6 × 150 мм Symmetry C18, 3.5 мкм. Температура 40°С, скорость подачи подвижной фазы 0.8 мл/мин.
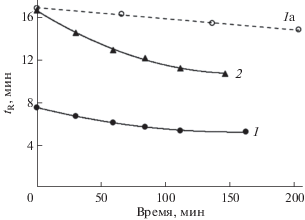
Используемая в ряде работ уменьшенная концентрация муравьиной кислоты (менее 10 об. %) также приводит к дрейфу времен удерживания, но более медленному (рис. 2). Отметим, что в литературе не обнаружены указания на дрейф вре-мен удерживания при работе с подобными смесями в составах подвижных фаз. Решение проблемы – смешивание двух растворов, один из которых содержит кроме воды только метанол, а второй представляет собой раствор муравьиной кислоты в воде. Такой подход использовали в некоторых работах по определению антоцианов в градиентных режимах [11, 12]. Нами экспериментально установлено, что указанный прием позволяет вывести хроматографическую систему в стационарный режим за обычное время (20–30 мин), но, к сожалению, одноканальные изократические насосы в этом случае становятся непригодными. Именно этот способ использовали в дальнейшем при исследовании возможности замены ацетонитрила метанолом.
Рис. 2.
Дрейф времен удерживания антоцианов в элюентах на основе метанола и муравьиной кислоты. Антоцианы: 1 и 1а – Cy3Glu, 2 – Pn3Glu. Подвижные фазы для 1 и 2 – 10 об. % HCOOH и 12 об. % CH3OH в воде, для 1a – 5 об. % HCOOH и 13 об. % CH3OH в воде.
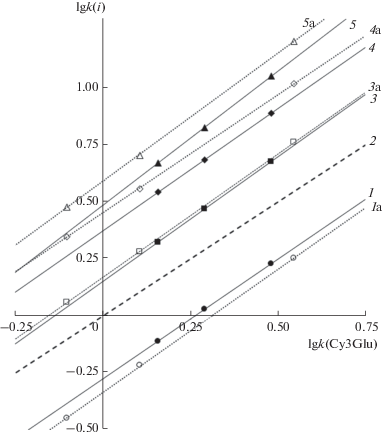
Сопоставление селективности разделения антоцианов. Для сопоставления селективности разделения 3-глюкозидов пяти из шести основных антоцианидинов при замене ацетонитрила на метанол наиболее эффективным к настоящему времени является метод сравнения карт разделения в варианте относительного удерживания, предложенный в работе [16] (рис. 3).
Рис. 3.
Карта разделения антоцианов для системы подвижных фаз I: метанол–10 об. % муравьиной кислоты–вода в сравнении с картой разделения антоцианов для системы II: ацетонитрил–10 об. % муравьиной кислоты–вода. Пунктирными линиями и цифрой с буквой а обозначено относительное удерживание в системе II. Антоцианы: 1 – Dp3Glu, 2 – Cy3Glu; 3 – Pt3Glu; 4 – Pn3Glu и 5 – Mv3Glu.
Оказалось, что замена ацетонитрила на метанол как на компонент с большей вязкостью приводит к небольшому росту давления (лишь на 10–15%) для использованных в работе составов подвижных фаз. При этом происходит более компактное элюирование в неизменном для обоих органических модификаторов подвижной фазы порядке:
Такая компактность может быть использована для сокращения времени, необходимого для разделения всех или некоторых компонентов природных смесей антоцианов, когда нет смысла в увеличении времени элюирования наиболее сильно удерживающегося компонента.
Добавка ОН-группы в кольцо В при переходе от Cy3Glu к Dp3Glu приводит при применении обоих типов органических модификаторов подвижной фазы к логичному уменьшению удерживания вследствие возрастания относительной гидрофильности сорбата. Поэтому линии трендов на карте разделения для Dp3Glu располагаются ниже линии тренда для Cy3Glu. Однако этот рост гидрофильности для подвижных фаз на основе метанола оказывается несколько меньшим, чем в случае подвижных фаз на основе ацетонитрила. При добавлении метоксигрупп линии тренда всегда располагаются выше, что можно интерпретировать как рост липофильности антоцианов. При этом линии трендов для Mv3Glu и Pn3Glu для подвижных фаз, содержащих метанол, располагаются ниже аналогичных линий трендов для подвижных фаз на основе ацетонитрила. Эти два фактора и приводят к уменьшению диапазона времен удерживания 3-глюкозидов (а, следовательно, и любых других однотипных гликозидов) пяти из шести основных природных антоцианидинов.
Строгое объяснение найденных изменений для одного состава подвижной фазы проблематично, поскольку в действительности каждый сорбат имеет свой баланс гидрофильных и гидрофобных свойств, т.е. его удерживание должно описываться минимум двухпараметрическим уравнением. Уравнения относительного удерживания [17] являются как раз двухпараметрическими:
где логарифм фактора удерживания сорбата i сопоставляется с логарифмом фактора удерживания сорбата, выбранного в качестве вещества сравнения (Cy3Glu). В этом уравнении параметр a1 может рассматриваться как относительная характеристика липофильности сорбата (по отношению к веществу сравнения). Параметр a0 не является однозначной характеристикой гидрофильности, для ее уточнения необходимо определить абсциссы точек конвергенции [18]. Однако в случае антоцианов гомологи, необходимые для нахождения координат таких точек, отсутствуют. Впрочем, сопоставляя наклоны линий трендов на рис. 3, можно установить, что на самом деле относительная липофильность антоцианов при добавлении метоксигрупп возрастает сильнее в случае подвижных фаз с метанолом.Для линий трендов на рис. 3 существует еще одна полезная для подтверждения их корректности особенность. Так, при аппроксимации вправо, т.е. на большие времена удерживания (или на малые содержания метанола или ацетонитрила) линии для одного и того же вещества должны пересекаться в точке, при которой концентрация органического модификатора (кроме муравьиной кислоты) обращается в ноль. И действительно, анализируя уравнения относительного удерживания, находим, что абсциссы точек пересечения для пар линий трендов для веществ Dp3G, Pn3G и Mv3G близки друг к другу: 1.99, 2.09 и 2.02, что удовлетворительно для линий, построенных только по трем экспериментальным точкам для каждой из зависимостей.
При исследовании влияния замены ацетонитрила на метанол на разделение различных типов гликозидов одного и того же антоцианидина (цианидина) линии трендов для одних и тех же соединений были близки (табл. 1). При этом наклон линии тренда для дигликозидов примерно на 0.1 больше, чем для 3-моногликозидa цианидина с заметным ростом для 3-рутинозида вследствие появления в углеводном заместителе метильной группы; для 3-тригликозидов наклон возрастает еще на 0.15, как и при использовании в качестве модификатора ацетонитрила. Таким образом, замена растворителя практически не приводит к изменению селективности разделения антоцианов при усложнении строения углеводного заместителя, а параметр a1, как и при использовании ацетонитрила [19], остается уникальным индикатором числа моноз в углеводном заместителе, что удобно при предварительной идентификации антоцианов без использования масс-спектрометрического детектирования.
Таблица 1.
Параметры уравнения относительного удерживания некоторых антоцианов для двух элюентных систем
| Антоциан | Система I | Система II | ||
|---|---|---|---|---|
| а1 | а0 | а1 | а0 | |
| Cy3Sam | 1.104 ± 0.008 | –0.062 ± 0.003 | 1.092 ± 0.007 | –0.053 ± 0.004 |
| Cy3Sopho | 1.083 ± 0.006 | –0.230 ± 0.002 | 1.089 ± 0.007 | –0.238 ± 0.004 |
| Cy3R | 1.146 ± 0.008 | 0.080 ± 0.004 | 1.127 ± 0.006 | 0.094 ± 0.003 |
| Cy3XR | 1.263 ± 0.007 | –0.029 ± 0.003 | 1.232 ± 0.005 | 0.003 ± 0.002 |
| Cy3GR | 1.267 ± 0.009 | –0.198 ± 0.004 | 1.254 ± 0.005 | –0.192 ± 0.004 |
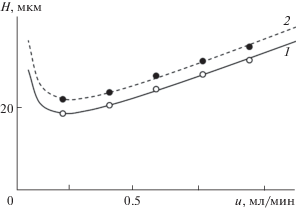
Сопоставление эффективности. Для исследования эффективности хроматографических систем обычно используют анализ экспериментальных данных по динамическому подходу по уравнению Ван Деемтера [20]. Такой анализ в нашем случае позволил установить, что минимальная высота теоретической тарелки достигается при нерационально малой скорости подачи подвижной фазы (менее 0.20 мл/мин) (рис. 4). В связи с этим для хроматографирования необходимо находить компромисс между степенью разделения (Rs) и временем хроматографирования, т.е. степень разделения может быть при необходимости увеличена за счет снижения скорости подачи подвижной фазы. Анализ данных рис. 4 показывает, что эффективность хроматографических систем даже немного увеличивается при замене ацетонитрила на метанол.
Рис. 4.
График Ван Деемтера для Cy3Glu в двух подвижных фазах. Подвижные фазы: 1 – 16 об. % CH3OH и 10 об. % HCOOH в воде; 2 – 8.8 об. % CH3CN и 10 об. % HCOOH в воде.
Определение антоцианов плодов клюквы. Относительную долю различных видов антоцианов в экстракте плодов клюквы можно определить при одной длине волны без введения поправок [15]. Это объясняется тем, что электронные спектры поглощения всех шести основных 3-гликозидов практически не различаются.
Разделение антоцианов плодов клюквы иллюстрирует рис. 5, а в табл. 2 приведены результаты определения антоцианов и их изменение по мере созревания клюквы. Кроме шести основных антоцианов, которые были обнаружены в целом ряде работ [21–25], на хроматограмме заметны также несколько минорных компонентов, доля которых в сумме антоцианов по площадям пиков не превышает 3–5%. Строение этих антоцианов в настоящей работе не определяли, но по анализу параметров линий трендов можно предположить, что они (кроме компонента а на рис. 5) являются моногликозидами цианидина, при этом их ацилирование некоторыми алифатическими кислотами не исключено. Ацилирование алифатическими кислотами (по нашему опыту) мало сказывается на наклоне линий трендов, но удерживание при этом сильно зависит от положения ацилирования углеводного заместителя. Учитывая, что при пробоподготовке и хранении пробы не исключено ацилирование антоцианов муравьиной кислотой, исследовали вариант, при котором пробу готовили, заменив муравьиную кислоту ортофосфорной. Оказалось, что такая замена никак не сказывается на виде хроматограммы, т.е. минорные пики являются не артефактами, а следствием особенностей биосинтеза антоцианов в плодах клюквы. Однако по мере созревания плодов (или в зависимости от интенсивности окраски) наблюдаются изменения в антоциановом составе. Особенно заметен рост относительной доли Cy3-Glu и Pn3Glu.
Рис. 5.
Разделение антоцианов плодов клюквы красной окраски в подвижной фазе 15 об. % CH3ОН и 10 об. % HCOOH в воде. Растворители проб – смеси метанола, муравьиной кислоты и воды в соотношениях: А – 20 : 10 : 70; Б – 50 : 10 : : 40 по объему. Детектирование при 215 нм. Соединения: 1 – Cy3Gala; 2 – Cy3Glu; 3 – Cy3Ara; 4 – Pn3Gala; 5 – Pn3Glu; 6 – Pn3Ara; a, b, c, d, e – не идентифицированы.
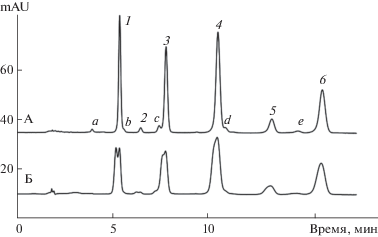
Таблица 2.
Антоцианы плодов клюквы окраски разной интенсивности
| Антоциан | Окраска плодов | ||
|---|---|---|---|
| темно-красная | красная | светло-красная | |
| Cy3Gala | 19.8* | 19.4 | 18.8 |
| Cy3Glu | 4.1 | 1.0 | 0.5 |
| Cy3Ara | 15.5 | 19.2 | 25.6 |
| Pn3Gala | 30.2 | 30.9 | 26.1 |
| Pn3Glu | 12.5 | 5.4 | 1.8 |
| Pn3Ara | 14.7 | 19.2 | 21.2 |
| Сумма неизвестных | 3.3 | 4.9 | 5.9 |
| Содержание суммы антоцианов** | 0.065 | 0.029 | 0.017 |
В одной из работ по ВЭЖХ антоцианов, выполненных в нашей лаборатории [26], показано, что при неверной пробоподготовке возможно появление на хроматограмме артефактных пиков. Они могут возникать при растворении пробы в растворителе, обладающем более высокой элюирующей способностью по сравнению с используемой подвижной фазой. При применении метанола оценка влияния состава растворителя на качество получаемой хроматограммы особенно важна, поскольку в литературе имеется немало публикаций, в которых рекомендуют в конце пробоподготовки растворять пробу в метаноле для последующего хроматографирования. Были приготовлены пробы, растворенные в растворителях на основе метанола (с 10 об. % муравьиной кислоты) с одинаковой концентрацией антоцианов, но различной концентрацией метанола (от 50 до 10 об. %). При последующем хроматографировании установлено, что даже при концентрации метанола в пробе на уровне 50% происходит раздвоение пиков антоцианов с меньшими временами удерживания и существенное уширение пиков антоцианов с большими временами удерживания (рис. 5б). Артефакты отсутствовали только при растворении пробы в смеси, приближенной по составу к используемой подвижной фазе; при этом условно допустимо изменение концентрации метанола лишь на ±50%. Поэтому не удивительно, что в работе [27], в которой хроматографировали пробу, растворенную в метаноле, на хроматограмме появляются раздвоенные пики веществ с наименьшими временами удерживания.
* * *
Таким образом, метанол является важной альтернативой экологически менее благоприятному ацетонитрилу при определении антоцианов в условиях обращенно-фазовой ВЭЖХ с 10 об. % муравьиной кислоты в качестве регулятора рН подвижной фазы.
Работа поддержана грантом Российского фонда фундаментальных исследований “Аспиранты”, номер 20-33-90031/20.
Список литературы
Bordonaba J.G., Crespo P., Terry L.A. A new acetonitrile-free mobile phase for HPLC−DAD determination of individual anthocyanins in blackcurrant and strawberry fruits: A comparison and validation study // Food Chem. 2011. V. 129. P. 1265.
Yabre M., Ferey L., Some I.T., Gaudin K. Greening reversed-phase liquid chromatography methods using alternative solvents for pharmaceutical analysis // Molecules. 2018. V. 23. P. 1065.
Pina F., Oliveira J., de Freitas V. Anthocyanins and derivatives are more than flavylium cations // Tetrahedron. 2015. V. 71. P. 3107.
Mozetič B., Trebše P., Hribar J. Determination and quantitation of anthocyanins and hydroxycinnamic acids in different cultivars of sweet cherries (Prunus avium L.) from Nova Gorica Region (Slovenia) // Food Technol. Biotechnol. 2002. V. 40. P. 207.
Koh K., Youn J.E., Kim H.-S. Identification of anthocyanins in black soybean (Glycine max (L.) Merr.) varieties // J. Food Sci. Technol. 2014. V. 51. P. 377.
Flamini R., Tomasi D. The anthocyanin content in berries of the hybrid grape cultivars Clinton and Isabella // Vitis. 2000. V. 39. P. 79.
Luczkiewicz M., Cisowski W. The RP−HPLC Analysis of anthocyanins // Chromatographia. 1998. V. 48. P. 360.
Valls J., Millán S., Martí M.P., Borràs E., Arola L. Advanced separation methods of food anthocyanins, isoflavones and flavanols // J. Chromatogr. A. 2009. V. 1216. P. 7143.
Revilla I., Pérez-Magariño S., González-Sanjosé M.L., Beltrán S. Identification of anthocyanin derivatives in grape skin extracts and red wines by liquid chromatography with diode array and mass spectrometric detection // J. Chromatogr. A. 1999. V. 847. P. 83.
Comandini P., Blanda G., Cardinali A., Cerretani L., Bendini A., Fiorenza M. Cabon CZE separation of strawberry anthocyanins with acidic buffer and comparison with HPLC // J. Sep. Sci. 2008. V. 31. P. 3257.
Doronin A.G., Deineka V.I., Deineka L.A., Tretiakov M.Y., Tokhtar V.K., Chulkov A.N. Peculiarities of use of bentonite clay at solid-phase purification of anthocyanins and flavonoids from leaves of plants // Adv. Biol. Sci. Res. 2019. V. 7. P. 90.
Дейнека В.И., Чулков А.Н., Дейнека Л.А., Сорокопудов В.Н. Определение антоцианового состава плодов красной смородины методом обращенно-фазовой ВЭЖХ // Сорбционные и хроматографические процессы. 2015. Т. 15. Вып. 4. С. 280.
Дейнека Л.А., Чулков А.Н., Дейнека В.И., Сорокопудов В.Н., Шевченко C.М. Антоцианы плодов вишни и родственных растений // Научные ведомости БелГУ. Сер. Естественные науки. 2011. № 9(104). Вып. 15/1. С. 364.
Monica Giusti M., Wrolstad R.E. Characterization and measurement of anthocyanins by UV-visible spectroscopy // Curr. Protoc. Food Anal. Chem. 2001. F1.2.1.
Дейнека В.И., Олейниц Е.Ю., Кульченко Я.Ю., Блинова И.П., Дейнека Л.А. Управление селективностью разделения и определение антоцианов плодов растений семейства Vaccinium с применением элюентов состава ацетонитрил–муравьиная кислота–вода // Журн. аналит. химии. 2020. Т. 75. С. 1021.
Дейнека В.И. Карта хроматографического разделения и инкрементные зависимости в методе относительного анализа удерживания в ВЭЖХ // Журн. физ. химии. 2006. Т. 80. С. 511.
Дейнека В.И. Экспериментальное обоснование метода относительного анализа удерживания в ВЭЖХ // Журн. физ. химии. 2006. Т. 80. С. 507.
Дейнека В.И., Дейнека Л.А., Туртыгин А.В. Метод относительного анализа удерживании: обращено-фазовая ВЭЖХ триглицеридов // Сорбционные и хроматографические процессы. 2008. Т. 8. Вып. 3. С. 465.
Дейнека В.И., Григорьев А.М. Определение антоцианов методом ВЭЖХ. Некоторые закономерности удерживания // Журн. аналит. химии. 2004. Т. 59. С. 305.
Katz E., Ogan K.L., Scott R.P.W. Peak dispersion and mobile phase velocity in liquid chromatography: the pertinent relationship for porous silica // J. Chromatogr. 1983. V. 270. P. 51.
Brown, P.N., Paul R. Shipley P.R. Determination of anthocyanins in cranberry fruit and cranberry fruit products by high-performance liquid chromatography with ultraviolet detection: Single-laboratory validation // J. AOAC Int. 2011. V. 94. P. 459.
Vorsa N., Polashock J.J. Alteration of anthocyanin glycosylation in Crabberrry through interspecific hybridization // J. Am. Soc. Hortic. Sci. 2005. V. 130. P. 711.
Jurikova T., Skrovankova S., Mlcek J., Balla S., Snopek L. Bioactive compounds, antioxidant activity, and biological effects of European cranberry (Vaccinium oxycoccos) // Molecules. 2019. V. 24. P. 24.
Česonienė L., Daubaras R., Jasutienė I., Miliauskienė I., Zych M. Investigations of anthocyanins, organic acids, and sugars show great variability in nutritional and medicinal value of European cranberry (Vaccinium oxycoccos) fruit // J. Appl. Bot. Food Qual. 2015. V. 88. P. 295.
Huopalahti R., Jrvenp E.P., Katina K. A novel solid-phase method for the analysis of anthocyanins and organic acid composition of Finnish cranberry // J. Liq. Chromatogr. Relat. Technol. 2000. V. 23. P. 2695.
Дейнека В.И., Сидоров А.Н., Дейнека Л.А., Тыняная И.И. Пробоподготовка при ВЭЖХ определении антоцианов и бетацианинов. Эффект растворителя образца // Сорбционные и хроматографические процессы. 2016. Т. 16. Вып. 3. С. 384.
Andrade P.B., Carvalho A.R.F., Seabra R.M., Ferreira M.A. A Previous study of phenolic profiles of quince, pear, and apple purees by HPLC diode array detection for the evaluation of quince puree genuineness // J. Agric. Food Chem. 1998. V. 46. P. 968.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии