

Журнал аналитической химии, 2021, T. 76, № 8, стр. 708-722
Применение сверхсшитого полистирола для многокомпонентной твердофазной экстракции остатков 63 ветеринарных препаратов при их определении в курином мясе методом высокоэффективной жидкостной хроматографии–тандемной масс-спектрометрии
А. О. Мелехин a, b, В. В. Толмачева a, *, Е. Г. Шубина b, С. Г. Дмитриенко a, В. В. Апяри a, А. И. Грудев b
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, ГСП-1, Россия
b Центральная научно-методическая ветеринарная лаборатория
111622 Москва, ул. Оранжерейная, 23, Россия
* E-mail: nikatolm@mail.ru
Поступила в редакцию 24.12.2020
После доработки 18.01.2021
Принята к публикации 21.01.2021
Аннотация
Сверхсшитый полистирол предложен для многокомпонентной твердофазной экстракции остатков 63 ветеринарных препаратов различных классов (сульфаниламиды, тетрациклины, хинолоны, амфениколы, нитроимидазолы, β-лактамы, макролиды, линкозамиды и плевромутилины) при их определении в курином мясе методом ВЭЖХ–тандемной масс-спектрометрии. Пробоподготовка включает экстракцию аналитов буферным раствором Макилвейна, удаление жиров экстракцией гексаном и дальнейшую очистку экстрактов на картриджах, заполненных сверхсшитым полистиролом. Способ обеспечивает количественное выделение аналитов (степени извлечения составляют от 83 до 117%) и хорошую воспроизводимость (sr ≤ 0.12). Ветеринарные препараты идентифицировали по точным массам ионов аналитов, образующихся при электрораспылительной ионизации с переключением положительной и отрицательной полярности. Матричный эффект для всех ветеринарных препаратов был ниже 20%. Определение проводили методом матричной градуировки, пределы обнаружения и определения составили 0.01–0.3 и 0.02–1 нг/г соответственно. Анализ загрязненных образцов куриного мяса показал, что результаты удовлетворительно совпадают с данными, полученными методами, принятыми в Российской Федерации для определения различных групп антибиотиков.
Одной из основных проблем безопасности пищевых продуктов является присутствие в них остатков ветеринарных препаратов, которые применяют для профилактики и лечения заболеваний, а также в качестве стимуляторов роста [1]. Употребление в пищу таких продуктов связано с риском для здоровья потребителей, поскольку продукты могут вызывать аллергические реакции, индуцировать устойчивость патогенов к антибиотикам, оказывать токсическое микробиологическое действие, вызывать канцерогенные или тератогенные эффекты [2, 3]. По этой причине определению ветеринарных препаратов в пищевых продуктах животного происхождения уделяется большое внимание [4–7]. В Европейском и Таможенном союзах установлены максимально допустимые уровни (МДУ) остаточных количеств ветеринарных препаратов, которые варьируют от сотен мкг/кг до нескольких нг/кг [8, 9].
Перечень ветеринарных препаратов, которые необходимо контролировать в продуктах животного происхождения, включает сульфаниланамиды, тетрациклины, фторхинолоны, амфениколы, β-лактамы, аминогликозиды, макролиды и некоторые другие антибиотики [5]. Наиболее эффективные методы, используемые для одновременного определения остатков такого большого количества ветеринарных препаратов – это многокомпонентные методы анализа (Multiresidue methods). Анализ литературы указывает на то, что многокомпонентное определение чаще всего проводят методами ВЭЖХ в сочетании с тандемным масс-спектрометрическим детектированием (ВЭЖХ−МС/МС). Разработаны методики многокомпонентного ВЭЖХ−МС/МС-определения остатков ветеринарных препаратов разных классов в молоке [10–16], яйцах [13–21], мясе [15, 16, 20, 22–25].
Несмотря на высокую селективность масс-спектрометрического детектирования, в большинстве случаев такое определение проводят после сложной пробоподготовки, направленной на выделение целевых аналитов из пищевых продуктов с использованием различных растворителей, с последующим разбавлением или очисткой полученных экстрактов для минимизации матричных эффектов [26, 27]. Классическим методом очистки таких экстрактов является твердофазная экстракция (ТФЭ) [28]. Многокомпонентная ТФЭ большого числа ветеринарных препаратов различных классов, различающихся по физико-химическим свойствам, является сложной задачей. Проблемы связаны как с низкой удерживающей способностью обычно используемых обращенно-фазовых сорбентов по отношению к полярным соединениям, к которым относятся многие ветеринарные препараты, так и с неколичественной десорбцией гидрофобных аналитов [29]. В связи с этим круг используемых сорбентов невелик; в последнее время в этом варианте пробоподготовки все чаще используют гидрофильно-липофильный сбалансированный обращенно-фазовый сорбент Oasis HLB [10, 11, 15, 17, 18, 21, 24, 25]. Другие сорбенты, такие как С18 [16, 22, 23] или Strata X [20], применяют реже.
Как показали исследования последних лет, в том числе и проводимые в нашей научной группе, весьма перспективными сорбентами для твердофазной экстракции полярных органических соединений из водных растворов являются сверхсшитые полистиролы (ССПС) [30]. Эти сорбенты отличаются высокоразвитой удельной поверхностью в сочетании с аномально высоким значением площади поверхности, приходящейся на микропоры, а также существенно большим, чем у остальных сорбентов, сродством к полярным органическим соединениям и легкостью регенерации [31, 32]. Еще одно уникальное свойство сверхсшитого полистирола заключается в том, что он представляет собой так называемый “материал ограниченного доступа” (restricted access material) [33]. Его поры диаметром 1.5–3 нм доступны большинству низкомолекулярных веществ, но недоступны для таких крупных молекул, как белки, гликопротеины или полисахариды.
Цель данного исследования – показать возможность использования ССПС Диапак П-3 для многокомпонентного выделения ветеринарных препаратов различных классов (сульфаниламиды, тетрациклины, хинолоны, амфениколы, нитроимидазолы, β-лактамы, макролиды, линкозамиды и плевромутилины) из жидких экстрактов, получаемых в процессе пробоподготовки куриного мяса, перед их определением методом В-ЭЖХ–МС/МС. Ранее ССПС для этой цели не применяли.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Использовали высокоэффективный жидкостной хроматограф ExionLC (Shimadzu, Япония) в сочетании с тройным квадрупольным масс-спектрометрическим детектором SCIEX Triple QuadTM 5500 (AB Sciex, Сингапур), оснащенный бинарным насосом и автосамплером. Разделение проводили на колонке Acclaim™ 120 C18 (100 × 2.1 мм) c диаметром зерна сорбента 3.0 мкм (Thermo Scientific, США) в режиме градиентного элюирования. Применяли аналитические весы Sartorius AC 121S (Sartorius, Германия), систему подготовки деионизированной воды Milli-Q Synthesis (Millipore, США), центрифугу лабораторную Thermo Scientific SL40R (Thermo Scientific, США), систему упаривания закрытого типа TurboVapII.CaliperLifeSciences (Caliper Life Sciences, США), вакуумную установку для ТФЭ М6 (Манифолд, Россия), шейкер для пробирок MultiReax (Heidolph, Германия).
Реактивы. Использовали метанол, ацетонитрил, муравьиную кислоту, н-гексан (Fisher Scientific Inc., США), моногидрат лимонной кислоты (C6H8O7 · H2O), гидрофосфат натрия (Na2HPO4 · · 12H2O), ЭДТА (этилендиаминтетраацетат натрия, Na2EDTA) (Across organic, Бельгия), коммерчески доступный сверхсшитый полистирол (Диапак П-3, Био-ХимМак, Россия). ССПС перед использованием активировали ацетонитрилом. Очищенную воду получали с помощью системы Milli-Q Synthesis (Millipore, США).
Буферный раствор Макилвейна получали растворением 12.9 г C6H8O7 · H2O, 10.9 г Na2HPO4 · 12H2O и 37.2 г Na2EDTA в мерной колбе емк. 1000 мл примерно в 800 мл воды, далее добавляли 1 М NaOH до pH ∼ 5.0 и доводили до метки водой.
Использовали стандартные образцы следующих ветеринарных препаратов: сульфагуанидин, сульфаниламид, сульфадиазин, сульфатиазол, сульфапиридин, сульфамеразин, сульфамоксол, сульфаметазин, сульфаметоксипиридазин, сульфахлорпиридазин, сульфаметоксазол, сульфоксиэтоксипиридазин, сульфадиметоксин, сульфахиноксалин, триметоприм, окситетрациклин, тетрациклин, хлортетрациклин, доксициклин, флорфеникол амин, тиамфеникол, флорфеникол, хлорамфеникол, гидроксиметронидазол, метронидазол, гидроксиметилметронидазол, диметридазол, тернидазол, ронидазол, тинидазол, гидроксипронидазол, ипронидазол, амоксициллин, ампициллин, бензилпенициллин, феноксиметилпенициллин, оксациллин, клоксациллин, нафциллин, диклоксациллин, марбофлоксацин, офлоксацин, норфлоксацин, данофлоксацин, дифлоксацин, ципрофлоксацин, ломефлоксацин, энрофлоксацин, сарафлоксацин, пипемидовая кислота, оксолиновая кислота, налидиксовая кислота, флюмеквин, спирамицин, тилмикозин, эритромицин, тилозин, кларитромицин, тилвалозин, тиамулин, валнемулин, линкомицин, пирлимицин. В качестве внутренних стандартов использовали: сульфадиазин-D4, сульфатиазол-D4, сульфаметазин-D4, сульфаметоксазол-D4, триметоприм-D9, диметридазол-D3, ронидазол-D3, ипронидазол-D3, гидроксиметронидазол-D2, гидроксиметилметронидазол-D3, гидроксипронидазол-D3, бензилпенициллин-D7, хлорамфеникол-D5, дифлоксацин-D3, налидиксовая кислота-D5, норфлоксацин-D5, оксолиновая кислота-D5, сарафлоксацин-D8, ципрофлоксацин-D8, рокситромицин-D7, азитромицин-D3, клиндамицин-D3, валнемулин-D6, энрофлоксацин-D5, демеклоциклин. Все стандартные образцы с высокой степенью чистоты (≥95.0%) были получены от Sigma-Aldrich (США), Ehrenstorfer GmbH (Германия), European Pharmacopoeia (Франция), Toronto Research Chemicals (Канада) и Witega (Германия).
Исходные растворы ветеринарных препаратов с концентрацией 200 мкг/мл готовили растворением соответствующей навески в метаноле, смеси ацетонитрил–вода (1 : 1) (β-лактамы) и смеси метанол–10%-ный водный раствор уксусной кислоты (1 : 1) (хинолоны). Растворы хранили при –20°С в течение не более шести месяцев. Рабочий раствор смеси ветеринарных препаратов с концентрацией 1000 нг/мл готовили путем разбавления исходных растворов метанолом. Аналогичным образом готовили раствор смеси внутренних стандартов с концентрацией 1000 нг/мл. Срок хранения смесей составлял один месяц. Рабочие растворы готовили разбавлением исходных метанолом в день использования.
Анализируемые образцы. Использовали образцы куриного мяса, собранные Центральной научно-методической ветеринарной лабораторией (Москва, Россия) весной 2020 г. в процессе государственно мониторинга пищевой продукции. Образцы хранили при –20°С в холодильнике, перед анализом замороженные продукты гомогенизировали с использованием бытового миксера.
Условия хроматографического разделения и детектирования. Использовали подвижные фазы, состоящие из 0.5%-ной муравьиной кислоты в воде (А) и 0.5%-ной муравьиной кислоты в смеси ацетонитрила и метанола (50 : 50) (Б). Разделение проводили, применяя следующую программу градиентного элюирования: 0–0.2 мин, 10% Б; 0.2–9.4 мин, линейное увеличение до 60% Б; 9.4–9.5 мин 60% Б; 9.5–9.7 мин, линейное увеличение до 80% Б; 9.7–9.8 мин, линейное увеличение до 100% Б; 9.8–10.5 мин 100% Б; 10.5–11.0 мин, уменьшение до 10% Б; 11.0–12 мин, 10% Б. Скорость потока составляла 0.4 (0–9.8 мин) и 0.3 (9.8–12 мин) мл/мин. Температура колонки и автосамплера поддерживалась во время работы на уровне 40 и 15°C соответственно, объем вводимой пробы составлял 5 мкл.
Тройной квадрупольный масс-спектрометр (SCIEX Triple QuadTM 5500) был настроен на сбор данных в режиме мониторинга множественных реакций (ММР). Установлены следующие оптимальные значения параметров: напряжение на распыляющем капилляре – 4500 В; температура испарителя – 550°C; в качестве газа завесы и газа в ячейке использовали азот; давление газа соударений 10 фунтов на квадратный дюйм; давление газа завесы – 35 фунтов на квадратный дюйм; давление осушающего и распыляющего газов – 50 фунтов на квадратный дюйм; входной потенциал – 10 В.
Идентификация и определение. Ветеринарные препараты идентифицировали по полученным хроматограммам с использованием программного продукта Analyst 1.6.3 (AB Sciex, Сингапур). Неизвестную концентрацию аналита в пробе определяли методом градуировочного графика (матричная градуировка). Аналитическим сигналом служило отношение площади пика аналита к площади пика соответствующего внутреннего стандарта.
Пробоподготовка. В центрифужную пробирку емк. 15 мл вносили 1.00 ± 0.02 г тщательно измельченной пробы, добавляли 100 мкл раствора смеси внутренних стандартов, 400 мкл метанола и 8 мл буферного раствора Макилвейна. Содержимое пробирки перемешивали на шейкере в течение 15 мин. Затем для обезжиривания пробы к смеси добавляли 3 мл гексана, перемешивали на шейкере в течение 10 мин и центрифугировали при 4000 об/мин в течение 10 мин. Гексан удаляли, а водный слой очищали методом ТФЭ с помощью картриджа шприцевого типа, заполненного 30 мг ССПС (30 × 10 мм). Перед использованием картридж кондиционировали 2 мл ацетонитрила и 2 мл деионизированной воды. Твердофазную экстракцию проводили на вакуумной установке для ТФЭ (Манифолд М6, Россия). Перед элюированием картридж промывали 3 мл деионизированной воды, а затем элюировали аналиты 1.5 мл смеси ацетонитрил–метанол (1 : 1). Элюаты упаривали в атмосфере азота при 40°C досуха, перерастворяли в 1 мл смеси подвижных фаз A и Б (80 : 20) и использовали для дальнейшего ВЭЖХ–МС/МС-анализа.
Степени извлечения ветеринарных препаратов из куриного мяса определяли, вводя в образцы мяса, не содержащие исследуемых соединений, известные количества их добавок: 1, 2 и 200 нг/г. Степени извлечения (R, %) рассчитывали по формуле:
где с – найденное значение массовой концентрации аналита в образце, нг/г; с0 – значение массовой концентрации добавки, нг/г.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика сорбента. Морфология поверхности и структурные характеристики ССПС Диапак П-3 исследованы нами ранее [34]. По данным просвечивающей электронной микроскопии установлено, что частицы ССПС имеют правильную сферическую форму с диаметром ~60 мкм. Исследование пористой структуры указывает на то, что ССПС Диапак П-3 является микропористым сорбентом с большой удельной поверхностью (1132 м2/г) и бимодальным распределением пор, причем на долю микро- и мезопор приходится 53 и 42% площади поверхности соответственно. Наличие в структуре сорбента такого большого количества микропор обусловливает так называемую “структурную селективность” сорбента, влияющую на удерживание молекул сорбата, размеры которых сопоставимы с размерами пор. На этом сорбенте значительный вклад в механизм удерживания органических соединений вносят не только гидрофобные, но и π–π [33] и электростатические взаимодействия. Нами показано [34], что поверхность ССПС Диапак П-3 заряжена положительно при рН < 3.3 и отрицательно при более высоких значениях рН, причем наибольшего отрицательного значения ζ-потенциал достигает при рН ~ 8. Все сказанное позволяет рассматривать ССПС в качестве перспективного сорбента для многокомпонентной ТФЭ ветеринарных препаратов с разными физико-химическими свойствами.
Идентификация. Для идентификации 63 ветеринарных препаратов с помощью ВЭЖХ–МС/МС использовали метод MМР. Стандартные растворы аналитов (1.0 мкг/мл) вводили непосредственно в масс-спектрометр для получения ионов-предшественников и характерных дочерних ионов. В качестве ионов-предшественников выбрали характерные молекулярные ионы; для каждого соединения контролировали два иона-продукта. Для количественной оценки отслеживали наиболее интенсивный переход ММР, а также второй переход для подтверждения. Потенциал декластеризации (ПД) и энергия соударений (ЭС) двух наиболее распространенных переходов были оптимизированы в режимах положительных или отрицательных ионов.
Большинство ветеринарных препаратов рассматриваемых классов в условиях электрораспылительной ионизации образуют протонированные формы [M + H]+, к ним относятся сульфаниламиды, триметоприм, тетрациклины, нитроимидазолы, β-лактамы, хинолоны, макролиды, плевромутилины и линкозамиды. В режиме отрицательных ионов тиамфеникол, хлорамфеникол и флорфеникол образуют ион молекулы [M – H]–. Наилучшие условия ионизации получены при использовании муравьиной кислоты в качестве добавки. Основные характеристики 63 ветеринарных препаратов, определяемых методом ВЭЖХ–МС/МС с применением метода мониторинга множественных реакций, приведены в табл. 1. Параметры MМР для внутренних стандартов приведены в табл. 2.
Таблица 1.
Основные характеристики 63 ветеринарных препаратов, определяемых методом ВЭЖХ–МС/МС с применением метода мониторинга множественных реакций
| Аналит | Внутренний стандарт | tR, мин | Q1m/z | Q3m/z | ПД, B | ЭС, эВ |
|---|---|---|---|---|---|---|
| Сульфаниламиды и триметоприм | ||||||
| Сульфагуанидин | Сульфадиазин-D4 | 1.36 | 215.1(+) | 156.1/92.0 | 50/50 | 20/37 |
| Сульфаниламид | Сульфаметазин-D4 | 1.50 | 173.1(+) | 156.1/92.1 | 10/60 | 12/25 |
| Сульфадиазин | Сульфадиазин-D4 | 2.92 | 251.1(+) | 156.1/108.1 | 50/50 | 21/29 |
| Сульфатиазол | Сульфатиазол-D4 | 3.23 | 256.1(+) | 156.1/108.1 | 50/50 | 19/33 |
| Сульфапиридин | Сульфадиазин-D4 | 3.30 | 250.1(+) | 156.1/184.1 | 50/50 | 21/25 |
| Сульфамеразин | Сульфадиазин-D4 | 3.62 | 265.1(+) | 156.1/172.1 | 50/50 | 23/21 |
| Сульфамоксол | Сульфаметазин-D4 | 4.14 | 268.1(+) | 156.1/113.1 | 50/50 | 21/23 |
| Сульфаметазин | Сульфаметазин-D4 | 4.26 | 279.1(+) | 186.1/124.1 | 50/50 | 23/29 |
| Сульфаметоксипиридазин | Сульфаметаксазол-D4 | 4.57 | 295.1(+) | 156.1/108.1 | 50/50 | 25/35 |
| Сульфахлорпиридазин | Сульфаметазин-D4 | 5.28 | 284.9(+)/ 286.9 |
156.1 | 50/50 | 21/21 |
| Сульфаметаксазол | Сульфаметаксазол-D4 | 5.62 | 254.1(+) | 156.1/108.1 | 50/50 | 21/23 |
| Сульфаэтоксипоридазин | Сульфаметаксазол-D4 | 6.12 | 295.1(+) | 156.1/140.1 | 61/61 | 47/47 |
| Сульфадиметоксин | Сульфатиазол-D4 | 7.09 | 311.1(+) | 156.1/245.1 | 50/50 | 27/27 |
| Сульфахиноксалин | Сульфадиазин-D4 | 7.26 | 301.1(+) | 156.1/108.1 | 66/66 | 23/35 |
| Триметоприм | Триметоприм-D9 | 3.87 | 291.1(+) | 230.1/261.0 | 50/50 | 31/33 |
| Тетрациклины | ||||||
| Окситетрациклин | Демеклоциклин | 4.24 | 461.1(+) | 426.1/444.1 | 60/60 | 30/22 |
| Тетрациклин | Демеклоциклин | 4.49 | 445.1(+) | 428.1/410.0 | 60/60 | 30/27 |
| Хлортетрациклин | Демеклоциклин | 5.98 | 479.1(+) | 444.1/462.1 | 60/60 | 30/25 |
| Доксициклин | Демеклоциклин | 6.77 | 445.1(+) | 428.0/410.0 | 60/60 | 26/30 |
| Амфениколы | ||||||
| Флорфеникол амин | Хлорамфеникол-D5 | 1.31 | 248.1(+) | 230.1/130.1 | 140/140 | 20/46 |
| Тиамфеникол | Хлорамфеникол-D5 | 4.30 | 353.9(–) | 290.0/184.9 | –140/–140 | –19/–30 |
| Флорфеникол | Хлорамфеникол-D5 | 5.82 | 356.1(–) | 185.1/119.1 | –140/–140 | –27/–45 |
| Хлорамфеникол | Хлорамфеникол-D5 | 6.92 | 321.0(–) | 152.0/257.0 | –140/–140 | –23/–15 |
| Нитроимидазолы | ||||||
| Гидроксиметронидазол | Гидроксиметронидазол-D2 | 2.13 | 188.1(+) | 125.1/123.1 | 90/60 | 19/17 |
| Метронидазол | Гидроксиметронидазол-D2 | 2.36 | 172.1(+) | 128.1/82.1 | 100/100 | 20/30 |
| Гидроксиметил- метронидазол | Гидроксиметилмет- ронидазол-D3 | 2.51 | 158.1(+) | 140.1/55 | 100/80 | 20/29 |
| Диметридазол | Диметридазол-D3 | 2.60 | 142.1(+) | 81.1/96.1 | 50/90 | 30/22 |
| Тернидазол | Ронидазол-D3 | 2.98 | 186.1(+) | 128.1/82.1 | 90/90 | 27/35 |
| Ронидазол | Ронидазол- D3 | 3.02 | 201.1(+) | 140.1/110.1 | 40/50 | 15/21 |
| Тинидазол | Ронидазол-D3 | 3.90 | 248.1(+) | 121.1/93.0 | 90/100 | 22/25 |
| Гидроксиипронидазол | Гидроксиипронидазол-D3 | 4.83 | 186.1(+) | 168.1/122.1 | 100/70 | 17/28 |
| Ипронидазол | Ипронидазол-D3 | 5.75 | 170.1(+) | 124.1/109.1 | 103/66 | 25/33 |
| β-Лактамы | ||||||
| Амоксициллин | Бензилпенициллин-D7 | 2.48 | 366.1(+) | 114.1/379.0 | 56/56 | 29/13 |
| Ампициллин | Бензилпенициллин-D7 | 4.35 | 350.1(+) | 160.1/114.0 | 71/71 | 15/41 |
| Бензилпенициллин | Бензилпенициллин-D7 | 8.81 | 333.0(+) | 176.0/160.0 | 60/76 | 25/17 |
| Феноксиметилленициллин | Бензилпенициллин-D7 | 9.68 | 349.0(+) | 160.1/114.0 | 66/66 | 21/43 |
| Оксациллин | Бензилпенициллин-D7 | 9.98 | 400.1(+) | 160.1/243.1 | 71/71 | 17/17 |
| Клоксациллин | Бензилпенициллин-D7 | 10.53 | 390.1(+) | 160.0/277.0 | 81/81 | 19/19 |
| Нафциллин | Бензилпенициллин-D7 | 10.98 | 415.0(+) | 256.0/199.2 | 80/80 | 22/25 |
| Диклоксациллин | Бензилпенициллин-D7 | 11.07 | 468.0(+) | 160.0/310.9 | 81/81 | 25/23 |
| Хинолоны | ||||||
| Марбофлоксацин | Норфлоксацин-D5 | 4.11 | 363.1(+) | 345.1/320.1 | 21/21 | 29/23 |
| Офлоксацин | Энрофлоксацин-D5 | 4.41 | 362.1(+) | 318.1/261.0 | 86/86 | 27/37 |
| Норфлоксацин | Норфлоксацин-D5 | 4.44 | 320.1(+) | 302.1/276.0 | 66/66 | 27/23 |
| Данофлоксацин | Дифлоксацин-D3 | 4.87 | 358.1(+) | 340.2/82.1 | 42/42 | 35/67 |
| Дифлоксацин | Дифлоксацин-D3 | 5.41 | 400.2(+) | 356.2/299.1 | 121/121 | 29/41 |
| Ципрофлоксацин | Ципрофлоксацин-D8 | 4.61 | 332.0(+) | 231.0/324.2 | 81/40 | 49/35 |
| Ломефлоксацин | Энрофлоксацин-D5 | 4.86 | 352.2(+) | 265.1/308.1 | 86/86 | 33/25 |
| Энрофлоксацин | Энрофлоксацин-D5 | 4.96 | 360.2(+) | 342.2/316.1 | 45/45 | 31/27 |
| Сарафлоксацин | Сарафлоксацин-D8 | 5.44 | 386.2(+) | 342.1/299.1 | 96/96 | 27/37 |
| Пипемидовая кислота | Оксолиновая кислота-D5 | 6.50 | 304.2(+) | 286.1/217.0 | 61/61 | 25/29 |
| Оксолиновая кислота | Оксолиновая кислота-D5 | 7.24 | 262.1(+) | 216.0/160.0 | 51/51 | 39/51 |
| Налидиксовая кислота | Налидиксовая кислота-D5 | 8.69 | 233.1(+) | 215.0/187.0 | 40/40 | 19/33 |
| Флюмеквин | Норфлоксацин-D5 | 9.14 | 262.1(+) | 202.0/174.1 | 61/61 | 45/55 |
| Макролиды | ||||||
| Спирамицин | Азитромицин-D3 | 6.12 | 422.3(+) | 100.9/142.1 | 60/60 | 19/27 |
| Тилмикозин | Азитромицин-D3 | 7.38 | 869.6(+) | 174.3/696.5 | 37/37 | 61/55 |
| Эритромицин | Рокситромицин-D7 | 8.55 | 734.4(+) | 158.1/576.4 | 60/60 | 43/25 |
| Тилозин | Рокситромицин-D7 | 8.88 | 916.6(+) | 174.2/772.4 | 39/39 | 55/41 |
| Кларитромицин | Рокситромицин-D7 | 9.89 | 748.5(+) | 158.0/590.4 | 61/61 | 41/27 |
| Тилвазолин | Рокситромицин-D7 | 10.94 | 1042.6(+) | 229.1/174.3 | 86/86 | 55/47 |
| Плевромутилины | ||||||
| Тиамулин | Валнемулин-D6 | 8.85 | 494.3(+) | 192.2/119.0 | 43/43 | 31/53 |
| Валнемулин | Валнемулин-D6 | 10.15 | 565.4(+) | 263.1/147.1 | 37/37 | 21/53 |
| Линкозамиды | ||||||
| Линкомицин | Клиндамицин-D3 | 3.45 | 407.3(+) | 126.1/359.3 | 35/35 | 37/25 |
| Пирлимицин | Клиндамицин-D3 | 6.50 | 411.2(+) | 112.1/363.3 | 93/93 | 37/23 |
Таблица 2.
Основные характеристики внутренних стандартов, используемых для определения ветеринарных препаратов с применением метода мониторинга множественных реакций
| Внутренний стандарт | tR, мин | Q1m/z | Q3m/z | ПД, B | ЭС, эВ |
|---|---|---|---|---|---|
| Сульфадиазин-D4 | 2.89 | 255.1(+) | 160.1 | 50 | 23 |
| Сульфатиазол-D4 | 3.18 | 260.1(+) | 160.1 | 50 | 24 |
| Сульфаметазин-D4 | 4.21 | 283.1(+) | 186.1 | 50 | 26 |
| Сульфаметаксазол-D4 | 5.56 | 258.1(+) | 160.1 | 50 | 22 |
| Триметоприм-D9 | 3.80 | 300.1(+) | 250.1 | 50 | 33 |
| Диметридазол-D3 | 2.56 | 145.1(+) | 99.1 | 70 | 23 |
| Ронидазол-D3 | 3.02 | 204.1(+) | 143.1 | 45 | 15 |
| Ипронидазол-D3 | 5.70 | 173.1(+) | 127 | 110 | 25 |
| Гидроксиметронидазол-D2 | 2.13 | 190.1(+) | 125.1 | 90 | 19 |
| Гидроксиметилметронидазол-D3 | 2.50 | 161.1(+) | 143.1 | 70 | 20 |
| Гидроксиипронидазол-D3 | 4.79 | 189.1(+) | 171.1 | 70 | 18 |
| Бензилпенициллин-D7 | 8.73 | 340.0(+) | 183.1 | 76 | 17 |
| Хлорамфеникол-D5 | 6.88 | 326.1(-) | 157.0 | –140 | –23 |
| Демеклоциклин | 5.16 | 465.1(+) | 448.1 | 60 | 25 |
| Дифлоксацин-D3 | 5.41 | 403.2(+) | 359.0 | 86 | 27 |
| Налидиксовая кислота-D5 | 8.69 | 238.1(+) | 220.0 | 36 | 21 |
| Норфлоксацин-D5 | 4.44 | 325.1(+) | 281.1 | 76 | 23 |
| Оксалиновая кислота-D5 | 7.21 | 267.1(+) | 249.1 | 46 | 23 |
| Сарафлоксацин-D8 | 5.41 | 384.2(+) | 350.1 | 81 | 27 |
| Ципрофлоксацин-D8 | 4.59 | 340.0(+) | 296.1 | 91 | 25 |
| Энрофлоксацин-D5 | 4.93 | 365.2(+) | 245.1 | 30 | 37 |
| Рокситромицин-D7 | 9.98 | 428.3(+) | 686.5 | 46 | 29 |
| Азитромицин-D3 | 6.30 | 752.5(+) | 594.5 | 39 | 41 |
| Клиндамицин-D3 | 6.88 | 428.3(+) | 129.1 | 36 | 39 |
| Валнемулин-D6 | 10.14 | 571.4(+) | 269.2 | 37 | 25 |
Пробоподготовка. С учетом данных [25] и дальнейшего использования ТФЭ для очистки экстрактов, для выделения ветеринарных препаратов из куриного мяса использовали экстракцию буферным раствором Макилвейна. При оптимизации условий ТФЭ на картридже, заполненном ССПС, варьировали массу сорбента (30 и 50 мг), концентрацию аналитов (1, 2 и 200 нг/г), природу и объем элюента. Для минимизации объема элюента оказалось целесообразным использование картриджа, заполненного 0.030 г ССПС. С учетом проведенных нами ранее исследований [35, 36] в качестве элюирующего растворителя выбрали смесь ацетонитрила с метанолом (50 : 50).
Еще одним варьируемым параметром в рамках этого исследования был тип растворителя на стадии перерастворения. Модельные растворы ветеринарных препаратов в смеси ацетонитрила с метанолом (50 : 50) упаривали в атмосфере азота при 40°C досуха и вновь растворяли в 1 мл подвижной фазы A или Б и смеси подвижных фаз A и B в соотношении 80 : 20. Установлено, что наиболее полное повторное растворение всех аналитов наблюдается в смеси подвижных фаз A и B (80 : 20).
Степени извлечения ветеринарных препаратов оценивали с использованием образца куриного мяса, не содержащего остаточных количеств определяемых аналитов, с добавками определяемых соединений в количестве 1, 2 и 200 нг/г. Для определения внутридневной и междневной повторяемости готовили по 5 и 15 образцов для каждого уровня концентрации соответственно. Как видно из табл. 3, предлагаемый метод обеспечивает не только количественное выделение аналитов из анализируемых проб (степени извлечения аналитов составляют от 83 до 117%), но и отличается хорошей воспроизводимостью (sr ≤ 0.12).
Таблица 3.
Основные характеристики определения 63 ветеринарных препаратов в курином мясе после очистки экстрактов методом твердофазной экстракции с применением сверхсшитого полистирола
| Аналит | Содержа-ние, нг/г | Степень извлече- ния R, % | Внутридневная повторяемость (sr, n = 5) |
Междневная повторяемость (sr, n = 15) |
МЭ, % | сн,нг/г |
|---|---|---|---|---|---|---|
| Сульфаниламиды и триметоприм | ||||||
| Сульфагуанидин | 1/2/200 | 106/113/108 | 0.07/0.08/0.06 | 0.07/0.07/0.05 | –13.7 | 0.4 |
| Сульфаниламид | 1/2/200 | 112/113/107 | 0.09/0.11/0.06 | 0.08/0.06/0.05 | –18.2 | 1 |
| Сульфадиазин | 1/2/200 | 104/106/105 | 0.08/0.10/0.06 | 0.08/0.07/0.05 | 0.5 | 0.1 |
| Сульфатиазол | 1/2/200 | 101/105/102 | 0.08/0.07/0.08 | 0.07/0.06/0.06 | –0.7 | 0.2 |
| Сульфапиридин | 1/2/200 | 109/109/117 | 0.11/0.10/0.06 | 0.07/0.07/0.06 | 5.4 | 0.1 |
| Сульфамеразин | 1/2/200 | 100/93/98 | 0.11/0.12/0.08 | 0.06/0.06/0.05 | 21.2 | 0.2 |
| Сульфамоксол | 1/2/200 | 87/86/89 | 0.08/0.07/0.06 | 0.06/0.07/0.06 | –16.6 | 0.2 |
| Сульфаметазин | 1/2/200 | 103/104/103 | 0.10/0.11/0.07 | 0.08/0.06/0.05 | –1.3 | 0.2 |
| Сульфаметоксипиридазин | 1/2/200 | 88/87/95 | 0.08/0.09/0.06 | 0.07/0.06/0.06 | 13.9 | 0.2 |
| Сульфахлорпиридазин | 1/2/200 | 99/101/98 | 0.11/0.11/0.05 | 0.07/0.07/0.05 | –2.3 | 0.4 |
| Сульфаметоксазол | 1/2/200 | 97/95/97 | 0.11/0.11/0.05 | 0.07/0.07/0.06 | 1.1 | 0.4 |
| Сульфаэтоксипоридазин | 1/2/200 | 88/87/95 | 0.10/0.08/0.07 | 0.07/0.07/0.06 | 13.3 | 1 |
| Сульфадиметоксин | 1/2/200 | 86/84/92 | 0.11/0.09/0.05 | 0.07/0.07/0.06 | 14.4 | 0.1 |
| Сульфахиноксалин | 1/2/200 | 102/101/97 | 0.10/0.11/0.08 | 0.07/0.07/0.05 | 19.2 | 0.4 |
| Триметоприм | 1/2/200 | 103/108/105 | 0.10/0.12/0.06 | 0.07/0.07/0.06 | –0.8 | 0.2 |
| Тетрациклины | ||||||
| Окситетрациклин | 1/2/200 | 89/93/87 | 0.08/0.09/0.04 | 0.08/0.06/0.06 | –4.7 | 0.4 |
| Тетрациклин | 1/2/200 | 99/97/94 | 0.09/0.11/0.08 | 0.08/0.07/0.05 | –13.9 | 0.4 |
| Хлортетрациклин | 1/2/200 | 89/92/95 | 0.08/0.09/0.05 | 0.07/0.07/0.05 | –4 | 0.4 |
| Доксициклин | 1/2/200 | 84/87/94 | 0.11/0.09/0.07 | 0.08/0.07/0.06 | –13.9 | 0.2 |
| Амфениколы | ||||||
| Флорфеникол амин | 1/2/200 | 83/85/89 | 0.11/0.07/0.04 | 0.07/0.06/0.05 | –12.4 | 1 |
| Тиамфеникол | 1/2/200 | 93/90/98 | 0.09/0.09/0.08 | 0.07/0.07/0.05 | –12.2 | 1 |
| Флорфеникол | 1/2/200 | 90/91/96 | 0.10/0.12/0.06 | 0.07/0.07/0.06 | –6.2 | 0.8 |
| Хлорамфеникол | 1/2/200 | 102/105/104 | 0.11/0.11/0.05 | 0.07/0.06/0.05 | –1.1 | 0.8 |
| Нитроимидазолы | ||||||
| Гидроксиметронидазол | 1/2/200 | 103/105/103 | 0.09/0.08/0.06 | 0.07/0.07/0.06 | –0.6 | 0.3 |
| Метронидазол | 1/2/200 | 104/102/104 | 0.10/0.09/0.09 | 0.07/0.07/0.06 | –12.8 | 0.2 |
| Гидроксиметилметронидазол | 1/2/200 | 99/101/104 | 0.10/0.07/0.06 | 0.07/0.07/0.05 | 0.5 | 0.3 |
| Диметридазол | 1/2/200 | 95/97/97 | 0.10/0.10/0.06 | 0.06/0.07/0.05 | –0.3 | 0.2 |
| Тернидазол | 1/2/200 | 88/91/86 | 0.11/0.12/0.04 | 0.07/0.07/0.05 | 13.2 | 0.2 |
| Ронидазол | 1/2/200 | 99/95/96 | 0.11/0.08/0.04 | 0.07/0.07/0.05 | –1.2 | 0.4 |
| Тинидазол | 1/2/200 | 88/83/90 | 0.08/0.10/0.06 | 0.07/0.06/0.06 | 17.9 | 0.1 |
| Гидроксиипронидазол | 1/2/200 | 105/101/100 | 0.12/0.11/0.07 | 0.07/0.07/0.06 | 1 | 0.1 |
| Ипронидазол | 1/2/200 | 101/107/101 | 0.10/0.09/0.07 | 0.07/0.07/0.05 | 0 | 0.08 |
| β-Лактамы | ||||||
| Амоксициллин | 1/2/200 | 102/108/108 | 0.10/0.09/0.07 | 0.07/0.07/0.05 | –3.2 | 1 |
| Ампициллин | 1/2/200 | 101/110/107 | 0.09/0.10/0.08 | 0.08/0.06/0.06 | 9.3 | 1 |
| Бензилпенициллин | 1/2/200 | 98/101/103 | 0.07/0.10/0.06 | 0.07/0.07/0.06 | 0.3 | 1 |
| Феноксиметилпенициллин | 1/2/200 | 104/102/101 | 0.08/0.07/0.05 | 0.07/0.06/0.06 | –8.8 | 1 |
| Оксациллин | 1/2/200 | 103/108/109 | 0.12/0.11/0.05 | 0.07/0.07/0.06 | 0.6 | 1 |
| Клоксациллин | 1/2/200 | 105/106/108 | 0.10/0.09/0.07 | 0.08/0.06/0.05 | 4.5 | 1 |
| Нафциллин | 1/2/200 | 101/104/105 | 0.11/0.07/0.05 | 0.07/0.06/0.06 | –1.3 | 1 |
| Диклоксациллин | 1/2/200 | 100/107/103 | 0.10/0.11/0.08 | 0.08/0.07/0.05 | –9.1 | 1 |
| Хинолоны | ||||||
| Марбофлоксацин | 1/2/200 | 92/91/96 | 0.09/0.11/0.08 | 0.06/0.06/0.05 | –16.6 | 0.8 |
| Офлаксацин | 1/2/200 | 101/110/106 | 0.09/0.07/0.06 | 0.06/0.07/0.06 | –4.8 | 0.2 |
| Норфлоксацин | 1/2/200 | 99/101/104 | 0.07/0.11/0.04 | 0.07/0.06/0.05 | –1.3 | 1 |
| Данофлоксацин | 1/2/200 | 105/101/105 | 0.08/0.09/0.06 | 0.07/0.07/0.05 | 7.2 | 0.4 |
| Дифлоксацин | 1/2/200 | 106/104/107 | 0.10/0.12/0.06 | 0.07/0.07/0.06 | –0.5 | 0.2 |
| Ципрофлоксацин | 1/2/200 | 101/98/106 | 0.11/0.09/0.08 | 0.06/0.07/0.06 | 1.5 | 0.8 |
| Ломефлоксацин | 1/2/200 | 90/82/90 | 0.11/0.09/0.05 | 0.08/0.07/0.06 | –5.3 | 0.2 |
| Энрофлоксацин | 1/2/200 | 100/101/99 | 0.08/0.10/0.07 | 0.07/0.06/0.05 | 0.8 | 0.4 |
| Сарафлоксацин | 1/2/200 | 96/103/105 | 0.11/0.09/0.07 | 0.07/0.06/0.06 | 1.5 | 0.4 |
| Пипемидовая кислота | 1/2/200 | 102/103/106 | 0.08/0.08/0.06 | 0.07/0.06/0.06 | 6.4 | 1 |
| Оксолиновая кислота | 1/2/200 | 99/101/107 | 0.09/0.09/0.04 | 0.08/0.07/0.06 | 1 | 0.4 |
| Налидиксовая кислота | 1/2/200 | 104/99/104 | 0.08/0.11/0.05 | 0.07/0.07/0.05 | 0 | 0.08 |
| Флюмеквин | 1/2/200 | 94/94/98 | 0.10/0.12/0.08 | 0.06/0.07/0.06 | 2.5 | 0.1 |
| Макролиды | ||||||
| Спирамицин | 1/2/200 | 105/108/101 | 0.11/0.07/0.07 | 0.07/0.07/0.06 | 15 | 1 |
| Тилмикозин | 1/2/200 | 104/98/100 | 0.10/0.10/0.05 | 0.07/0.07/0.06 | 12.3 | 0.8 |
| Эритромицин | 1/2/200 | 96/103/101 | 0.09/0.07/0.05 | 0.07/0.06/0.05 | 18.8 | 0.8 |
| Тилозин | 1/2/200 | 86/87/91 | 0.10/0.08/0.05 | 0.07/0.07/0.06 | 28.7 | 0.8 |
| Кларитромицин | 1/2/200 | 97/101/101 | 0.11/0.08/0.04 | 0.07/0.07/0.05 | 0.6 | 0.04 |
| Тилвазолин | 1/2/200 | 90/86/88 | 0.08/0.06/0.06 | 0.08/0.06/0.06 | 10 | 0.8 |
| Плевромутилины | ||||||
| Тиамулин | 1/2/200 | 100/106/104 | 0.11/0.07/0.06 | 0.07/0.07/0.05 | 7.7 | 0.02 |
| Валнемулин | 1/2/200 | 101/104/98 | 0.11/0.07/0.06 | 0.07/0.07/0.06 | –0.9 | 0.08 |
| Линкозамиды | ||||||
| Линкомицин | 1/2/200 | 95/96/90 | 0.10/0.10/0.07 | 0.08/0.06/0.05 | 11.6 | 0.1 |
| Пирлимицин | 1/2/200 | 96/98/95 | 0.11/0.10/0.09 | 0.08/0.07/0.06 | –5.8 | 0.4 |
Матричный эффект (МЭ). Известно, что матричные эффекты, возникающие при анализе таких сложных матриц, как пищевые продукты, могут как увеличивать, так и понижать интенсивность сигнала аналита [37]. Для оценки МЭ использовали площади хроматографических пиков аналитов, полученные в условиях анализа экстрактов куриного мяса, не содержащего исследуемых соединений, с добавками ветеринарных препаратов (10 нг/г) и соответствующих водных растворов. Расчет проводили по формуле:
Как видно из табл. 3, в большинстве случаев МЭ ниже 20%, что может быть следствием не только использования метода внутреннего стандарта, но и эффективной очистки экстрактов.
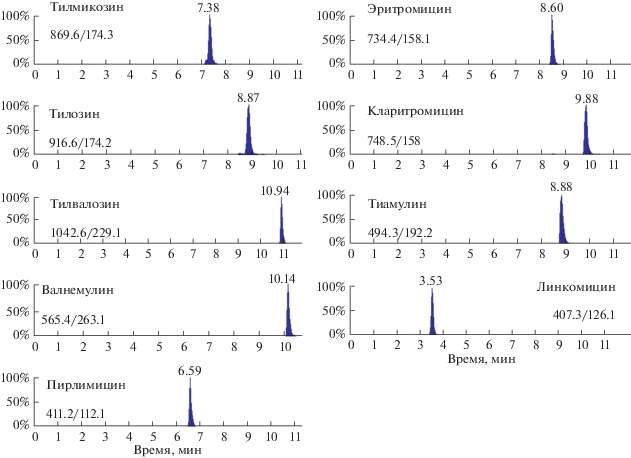
Определение. Количественный анализ проводили с использованием матричной градуировки. Линейность градуировочных графиков оценивали на модельном образце куриного мяса, не содержащего остаточных количеств определяемых аналитов, с добавками ветеринарных препаратов на уровнях концентраций 0.1, 1, 10, 50, 100 и 200 нг/г. Коэффициенты корреляции линейной зависимости площадей хроматографических пиков препаратов от их концентрации в анализируемом образце составили не менее 0.99. На рис. 1 представлены масс-хроматограммы экстракта куриного мяса с добавлением 50 нг/г 63 ветеринарных препаратов. Пределы обнаружения (смин) и определения (сн) рассчитывали по отношению аналитического сигнала (интенсивности пика) к шуму, равному 3 и 10 соответственно. Пределы обнаружения и определения составили 0.01–0.3 и 0.02–1 нг/г соответственно (табл. 3), что позволяет определять эти ветеринарные препараты на уровне, меньше чем МДУ (1–10 нг/г).
Рис. 1.
Масс-хроматограммы экстракта куриного мяса с добавлением 50 нг/г 63 ветеринарных препаратов после очистки методом твердофазной экстракции с применением сверхсшитого полистирола.
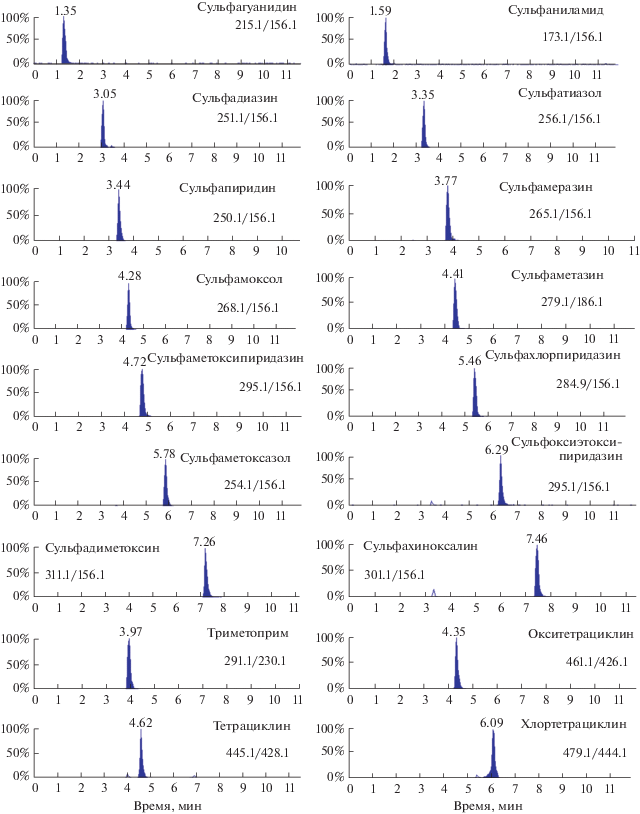
Рис. 1.
Продолжение
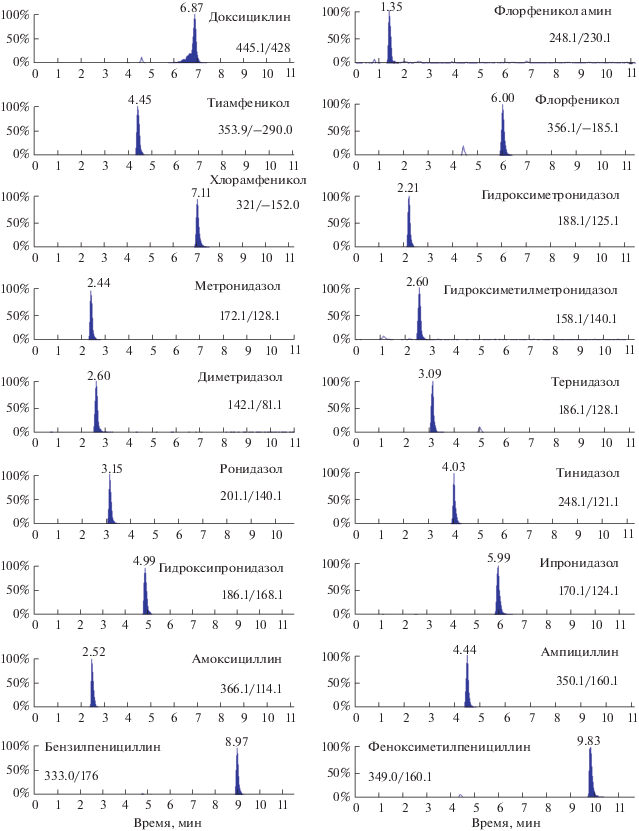
Рис. 1.
Продолжение
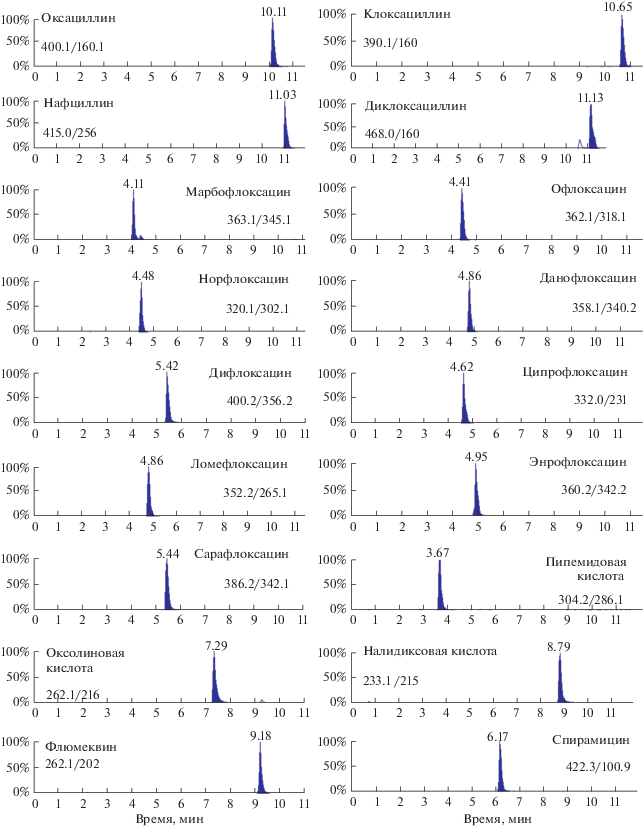
Рис. 1.
Окончание
Анализ реальных проб. Для анализа использовали 8 “положительных” образцов куриного мяса, в которых в ходе мониторинга пищевых продуктов Центральной научно-методической ветеринарной лабораторией установлено превышение МДУ по некоторым антибиотикам. Пробоподготовку образцов, очистку экстрактов и определение проводили по методике, описанной выше. Результаты определения идентифицированных в пробах ветеринарных препаратов приведены в табл. 4, из которой следует, что полученные результаты удовлетворительно совпадают с данными, полученными с использованием методов, рекомендованных в Российской Федерации для определения различных групп антибиотиков.
Таблица 4.
Сравнение результатов анализа образцов куриного мяса по разработанной методике и по ГОСТу (n = 3, P = 0.95)
| Образец куриного мяса | Аналит | Найдено по разработанной методике, нг/г (sr) | Найдено
по ГОСТу, нг/г (sr) |
Литература |
|---|---|---|---|---|
| 1 | Хлорамфеникол | 9 ± 1 (0.06) | 8 ± 2 (0.09) | [38] |
| 2 | Сульфаметазин | 2.2 ± 0.4 (0.07) | 2.3 ± 0.6 (0.10) | [38] |
| Офлаксацин | 5 ± 0.9 (0.07) | 5 ± 1 (0.11) | [39] | |
| Энрофлоксацин | 42 ± 6 (0.06) | 44 ± 10 (0.09) | [39] | |
| 3 | Сульфаметазин | 1.9 ± 0.3 (0.07) | 2.0 ± 0.6 (0.11) | [38] |
| 4 | Триметоприм | 4.6 ± 0.7(0.06) | 4.4 ± 0.9 (0.08) | [38] |
| 5 | Флюмеквин | 3.6 ± 0.7 (0.08) | 3.5 ± 0.9 (0.10) | [39] |
| 6 | Ципрофлоксацин | 2.5 ± 0.4 (0.07) | 2.7 ± 0.7 (0.10) | [39] |
| 7 | Офлаксацин | 29 ± 5 (0.07) | 32 ± 9 (0.11) | [39] |
| Линкомицин | 2.1 ± 0.5 (0.09) | 2.2 ± 0.7 (0.12) | [40] | |
| 8 | Доксициклин | 13 ± 3 (0.09) | 14 ± 4 (0.11) | [41] |
| Сульфадиазин | 5.2 ± 0.6 (0.05) | 5 ± 1 (0.09) | [38] |
* * *
Таким образом, отечественный, коммерчески доступный сорбент ССПС Диапак П-3 может быть использован для очистки жидких экстрактов, получаемых в процессе пробоподготовки куриного мяса при определении в этом объекте остатков ветеринарных препаратов методом В-ЭЖХ–МС/МС. Полученные данные указывают на то, что по эффективности ССПС Диапак П-3 не уступает такому сорбенту, как Oasis HLB; степени извлечения 63 ветеринарных препаратов, принадлежащих к 9 классам лекарственных соединений, составили 83–117% (sr ≤ 0.12). Применение этого сорбента в процессе очистки позволило уменьшить матричные эффекты: для всех ветеринарных препаратов они оказались ниже 20%. Полученные экспериментальные данные создают основу для дальнейшего исследования аналитических возможностей ССПС Диапак П-3 в качестве эффективного сорбента для многокомпонентной ТФЭ в процессе пробоподготовки других пищевых продуктов перед определением в них остатков ветеринарных препаратов методом ВЭЖХ–МС/МС.
Авторы выражают благодарность Министерству науки и высшего образования Российской Федерации и Совету по грантам Президента Российской Федерации для государственной поддержки молодых российских ученых и по государственной поддержке ведущих научных школ Российской Федерации за финансовую поддержку исследований (проект МД-1448.2021.1.3).
Список литературы
Ture M., Fentie T., Regassa B. Veterinary drug residue: the risk, public health significance and its management college of veterinary medicine and animal science // J. Vet. Sci. 2019. V. 13. № 2. P. 555856.
Baynes R.E., Dedonder K., Kissell L., Mzyk D., Marmulak T., Smith G., Riviere J.E. Health concerns and management of select veterinary drug residues // Food Chem. Toxicol. 2016. V. 88. P. 112.
Beyene T. Veterinary drug residues in food-animal products: its risk factors and potential effects on public health // J. Vet. Sci. 2016. V. 1. № 7. P. 285.
Stolker A.A.M., Zuidema T., Nielen M.W.F., Nielen M.W.F. Residue analysis of veterinary drugs and growth-promoting agents // Trends Anal. Chem. 2007. V. 26. P. 967.
Bogialli S., Di Corcia A. Recent applications of liquid chromatography−mass spectrometry to residue analysis of antimicrobials in food of animal origin // Anal. Bioanal. Chem. 2009. V. 395. № 4. P. 947.
Masia A., Suarez-Varela M.M., Llopis-Gonzalez A., Pico Y. Determination of pesticides and veterinary drug residues in food by liquid chromatography mass spectrometry: a review // Anal. Chim. Acta. 2016. V. 936. P. 40.
Rocca L.M., Gentili A., Pérez-Fernández V., Tomai P. Veterinary drugs residues: a review of the latest analytical research on sample preparation and LC−MS based methods // Food Addit. Contam. Part A: Chem. Anal. Control. Expo. Risk Assess. 2017. V. 34. P. 766.
Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin.
Технический регламент Таможенного союза (ТР ТС 034/2013) “О безопасности мяса и мясной продукции” (Принят Решением Совета Евразийской экономической комиссии от 9 октября 2013 г. N 68).
Han R.W., Zheng N., Yu Z.N., Wang J., Xu X.M., Qu X.Y., Li S.L., Zhang Y.D., Wang J.Q. Simultaneous determination of 38 veterinary antibiotic residues in raw milk by UPLC–MS/MS // Food Chem. 2015. V. 181. P. 119.
Wang J., Leung D., Chow W., Chang J., Wong J.W. Development and validation of a multiclass method for analysis of veterinary drug residues in milk using ultrahigh performance liquid chromatography electrospray ionization quadrupole orbitrap mass spectrometry // J. Agric. Food Chem. 2015. V. 63. P. 9175.
Амелин В.Г., Федина Н.М., Подколзин И.В., Коротков А.И. Быстрый скрининг и определение остаточных количеств ветеринарных препаратов в молоке методом ультравысокоэффективной жидкостной хроматографии-квадруполь-времяпролетной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2018. Т. 73. № 6. С. 461. (Amelin V.G., Fedina N.M., Podkolzin I.V., Korotkov A.I. Rapid screening and determination of residual veterinary drugs in milk by ultrahigh performance liquid chromatography–high-resolution quadrupole time-of-flight mass spectrometry // J. Anal. Chem. 2018. V. 73. № 6. P. 576.)
Амелин В.Г., Андоралов А.М., Волкова Н.М., Коротков А.И., Никешина Т.Б., Сидоров И.И., Тимофеев А.А. Идентификация и определение токсикантов с использованием стандартной добавки в пищевых продуктах, продовольственном сырье и кормах методом высокоэффективной жидкостной хроматографии /времяпролетной масс-спектрометрии высокого разрешения // Аналитика и контроль. 2015. Т. 19. № 2. С. 189.
Amelin V., Korotkov A., Andoralov A. Identification and determination of 492 contaminants of different classes in food and feed by high-resolution mass spectrometry using the standard addition method // J. AOAC Int. 2016. V. 99. № 6. P. 1600.
Chen D., Yu J., Tao Y., Pan Y., Xie S., Huang L., Peng D., Wang X., Wang Y., Liu Z., Yuan Z. Qualitative screening of veterinary anti-microbial agents in tissues, milk, and eggs of food-producing animals using liquid chromatography coupled with tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1017–1018. P. 82.
Kang J., Park S.J., Park H.C., Hossain M.A., Kim M.A., Son S.W., Cho B.H. Multiresidue screening of veterinary drugs in meat, milk, egg, and fish using liquid chromatography coupled with ion trap time-of-flight mass spectrometry // Biotechnol. Appl. Biochem. 2017. V. 182. № 2. P. 635.
Piatkowska M., Gbylik-Sikorska M., Gajda A., Jedziniak P., Bladek T., Zmudzki J., Posyniak A. Multiresidue determination of veterinary medicines in lyophilized egg albumen with subsequent consumer exposure evaluation // Food Chem. 2017. V. 229 P. 646.
Wang K., Lin K., Huang X., Chen M. A simple and fast extraction method for the determination of multiclass antibiotics in eggs using LC−MS/MS // J. Agric. Food Chem. 2018. V. 65 P. 5064.
Wang C., Li X., Yu F., Wang Y., Ye D., Hu X., Zhou L., Du J., Xia X. Multi-class analysis of veterinary drugs in eggs using dispersive-solid phase extraction and ultra-high performance liquid chromatography−tandem mass spectrometry // Food Chem. 2021. V. 334. 127598.
Peters R.J., Bolck Y.J., Rutgers P., Stolker A.A., Nielen M.W. Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry // J. Chromatogr. A. 2009. V. 1216. № 46. P. 8206.
Azzouz A, Ballesteros E. Multiresidue method for the determination of pharmacologically active substances in egg and honey using a continuous solid-phase extraction system and gas chromatography-mass spectrometry // Food Chem. 2015. V. 178. P. 63.
Geis-Asteggiante L., Lehotay S.J., Lightfield A.R., Dutko T., Chilton Ng., Bluhm L. Ruggedness testing and validation of a practical analytical method for >100 veterinary drug residues in bovine muscle by ultrahigh performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2012. V. 1258. P. 43.
Schneider M.J., Lehotay S.J., Lightfield A.R. Validation of a streamlined multiclass, multiresidue method for determination of veterinary drug residues in bovine muscle by liquid chromatography–tandem mass spectrometry // Anal. Bioanal. Chem. 2015. V. 407. P. 4423.
Tang Y.Y., Lu H.F., Lin H.Y., Shin Y.C., Hwang D.F. Development of a quantitative multi-class method for 18 antibiotics in chicken, pig, and fish muscle using UP-LC−MS/MS // Food Anal. Methods. 2012. V. 5. P. 1459.
Zhang Z., Li X., Ding S., Jiang H., Shen J., Xia X. Multiresidue analysis of sulfonamides, quinolones, and tetracyclines in animal tissues by ultra-high performance liquid chromatography–tandem mass spectrometry // Food Chem. 2016. V. 204. P. 252.
Kinsella B., O’Mahony J., Malone E., Moloney M., Cantwell H., Furey A., Danaher M. Current trends in sample preparation for growth promoter and veterinary drug residue analysis // J. Chromatogr. A. 2009. V. 1216. 7977.
Berendsen B.J.A., Stolker L.A.A.M., Nielen M.W.F., Nielen M.W.F. Selectivity in the sample preparation for the analysis of drug residues in products of animal origin using LC−MS // Trends Anal. Chem. 2013. V. 43. P. 229.
Faraji M., Yamini Y., Gholami M. Recent advances and trends in applications of solid‑phase extraction techniques in food and environmental analysis // Chromatographia. 2019. V. 82 P. 1207.
Rossi R., Saluti G., Moretti S., Diamanti I., Giusepponi D., Galarini R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: a review // Food Addit. Contam. Part A – Chem. 2018. V. 35. P. 241.
Дмитриенко С.Г., Тихомирова Т.И., Апяри В.В., Толмачева В.В., Кочук Е.В., Золотов Ю.А. Применение сверхсшитых полистиролов для концентрирования и разделения органических соединений и ионов элементов // Журн. аналит. химии. 2018. Т. 73. № 11. С. 830. (Dmitrienko S.G., Tikhomirova T.I., Apyari V.V., Tolmacheva V.V., Kochuk E.V., Zolotov. Yu.A. Application of hypercrosslinked polystyrenes to the preconcentration and separation of organic compounds and ions of elements: a review // J. Anal. Chem. 2018. V. 73. P. 1053.)
Даванков В.А., Цюрупа М.П. Сверхсшитые полистирольные сорбенты. Структура, свойства, применение. Саарбрукен, Германия: Palmarium Academic Publishing, 2012. 76 с.
Tsyurupa M.P., Davankov V.A. Porous structure of hypercrosslinked polystyrene: State-of-art mini-review // React. Funct. Polym. 2002. V. 53. P. 193.
Цюрупа М.П., Блинникова З.К., Проскурина Н.А., Пастухов А.В., Павлова Л.А., Даванков В.А. Сверхсшитый полистирол – первый нанопористый полимерный материал // Рос. нанотехнол. 2009. Т. 4. С. 109.
Tolmacheva V.V., Yarykin D.I., Serdiuk O.N., Apyari V.V., Dmitrienko S.G., Zolotov Yu. A. Adsorption of catecholamines from their aqueous solutions on hypercrosslinked polystyrene // React. Funct. Polym. 2018. V. 131. P. 56.
Дмитриенко С.Г., Кочук Е.В., Толмачева В.В., Апяри В.В., Золотов Ю.А. Сравнение сорбентов для концентрирования сульфаниламидов из водных растворов перед их определением методом ВЭЖХ // Журн. аналит. химии. 2013. Т. 68. № 10. С. 966. (Dmitrienko S.G., Kochuk E.V., Tolmacheva V.V., Apyari V.V., Zolotov Yu.A. Comparison of adsorbents for the preconcentration of sulfanilamides from aqueous solutions prior to HPLC determination // J. Anal. Chem. 2013. V. 68. № 10. P. 871.)
Удалова А.Ю., Дмитриенко С.Г., Натчук С.В., Апяри В.В., Золотов Ю.А. Концентрирование антибиотиков тетрациклиновой группы на сверхсшитом полистироле и их определение в водах методом высокоэффективной жидкостной хроматографии // Журн. аналит. химии. 2015. Т. 70. № 3. С. 273. (Udalova A.Yu., Dmitrienko S.G., Natchuk S.V., Apyari V.V., Zolotov Yu.A. Preconcentration of tetracycline antibiotics on a hyper-crosslinked polystyrene and their determination in waters by high-performance liquid chromatography // J. Anal. Chem. 2015. V. 70. № 3. P. 292.)
Ferrer C., Lozano A., Aguera A., Giron A.J., Fernandez A.R. Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables // J. Chromatogr. A. 2011. V. 1218. P. 7634.
ГОСТ 34533-2019. Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания сульфаниламидов, нитроимидазолов, пенициллинов, амфениколов с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором. М.: Стандартинформ, 2019. 20 с.
ГОСТ 32797-2014. Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания хинолонов с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором. М.: Стандартинформ, 2015. 23 с.
ГОСТ 34136-2017. Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания макролидов, линкозамидов и плевромутилинов с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием. М.: Стандартинформ, 2018. 18 с.
ГОСТ 31694-2012. Продукты пищевые, продовольственное сырье. Метод определения остаточного содержания антибиотиков тетрациклиновой группы с помощью высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектором. М.: Стандартинформ, 2013. 28 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии