

Журнал аналитической химии, 2021, T. 76, № 9, стр. 771-787
Методы разделения и концентрирования при определении высокотоксичных органических соединений (отравляющих веществ)
М. А. Ленинский a, *, М. Д. Шачнева a, Е. И. Савельева a, Н. Л. Корягина a
a Научно-исследовательский институт гигиены, профпатологии и экологии человека Федерального
медико-биологического агентства России
188663 п. Кузьмоловский, ст. Капитолово, Всеволожский район, Ленинградская обл., Россия
* E-mail: m.leninskii@yandex.ru
Поступила в редакцию 06.04.2021
После доработки 21.04.2021
Принята к публикации 21.04.2021
Аннотация
В обзоре рассмотрены и обобщены методы разделения и концентрирования при определении токсикантов 1-го класса опасности, а именно отравляющих фосфорорганических веществ нервно-паралитического (группа изомеров Vx, зарин, циклозарин, зоман, табун) и кожно-нарывного действия (сернистый иприт).
Отравляющие вещества (ОВ) или так называемые токсичные химикаты были предназначены для поражения живой силы противника во время военных действий [1]. В соответствии с международной Конвенцией о запрещении химического оружия (ХО) [2] к настоящему времени его запасы преимущественно уничтожены, но действие Конвенции продолжается в направлении контроля неиспользования и нераспространения ХО. Аналитические процедуры, используемые для определения ОВ и продуктов их трансформации, широко представлены в оригинальных статьях и обзорах.
Актуальность высокочувствительного и надежного определения ОВ в объектах окружающей среды и биопробах обусловлена требованиями химической безопасности при перепрофилировании бывших объектов уничтожения химического оружия, верификационной деятельностью в соответствии с Конвенцией о запрещении химического оружия и необходимостью противодействия химическому терроризму.
Процедуры определения ОВ и продуктов их трансформации в объектах окружающей среды и биопробах непрерывно совершенствуются. Основным методическим пособием для лабораторий, специализирующихся на определении ОВ, является сборник рекомендованных операционных процедур (Recommended Operating Procedures, ROP) [3]. Процедуры пробоподготовки, представленные в ROP, прошли апробацию в рамках решения задач международных тренировочных и квалификационных тестов, проводимых Организацией по запрещению химического оружия (ОЗХО), и были успешно применены при расследовании случаев применения ХО. Сборник содержит информацию о методах извлечения, концентрирования и определения ОВ и продуктов их трансформации в матрицах различной природы – воде, почве, полимерных материалах, биопробах и др. Основными методами извлечения аналитов и очистки образцов от матричных компонентов, представленными в ROP, являются: фильтрация, центрифугирование, замена растворителя, твердофазная экстракция (ТФЭ), твердофазная микроэкстракция (ТФМЭ), жидкофазная микроэкстракция. Концентрирование экстрактов, содержащих целевые вещества, предлагается осуществлять в токе азота, в роторных испарителях, а также в центрифужных концентраторах.
Ввиду беспрецедентной чувствительности и селективности для определения ОВ преимущественно применяют хромато-масс-спектрометрические (ХМС) методы. В 2021 г. Рыбальченко с соавт. опубликовал обзор [4], посвященный ХМС-методам определения маркеров и биомаркеров ОВ. В обзоре обсуждены достоинства и недостатки различных вариантов ХМС-анализа для определения ОВ и продуктов их трансформации, представлена система профессионального тестирования лабораторий ОЗХО. В то же время процедуры разделения и концентрирования, применяемые при определении ОВ в сложных матрицах, в современной литературе обсуждаются значительно реже, чем техника ХМС-анализа, хотя они имеют особое значение в аналитической химии ОВ. Ввиду чрезвычайно высокой токсичности ОВ даже летальные их дозы соответствуют крайне низким концентрациям, а высокая реакционная способность приводит к необратимой сорбции материалами или стремительному разложению в них. В биологических пробах ОВ могут быть обнаружены в виде низкомолекулярных метаболитов или конъюгатов с биомолекулами. В обоих случаях извлечение метаболитов из биоматриц и их концентрирование, как правило, представляют собой комбинации последовательных стадий, на каждой из которых существует опасность потери аналитов. В системе профессиональных тестов ОЗХО лаборатории-участники используют типовое ХМС-оборудование, в то время как применяемые процедуры разделения и концентрирования характеризуются значительным разнообразием. Что касается методов обнаружения ОВ в полевых (onsite) условиях, то при очевидной важности до настоящего времени они не охвачены системой профессионального тестирования ОЗХО.
КОНЦЕНТРИРОВАНИЕ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ В ПОЛЕВЫХ УСЛОВИЯХ ИЗ ВОЗДУШНОЙ СРЕДЫ
Известно, что ОВ не только чрезвычайно токсичны, но и способны к быстрому испарению и распространению в воздухе. Наиболее вероятный путь их воздействия как оружия массового поражения – ингаляционный. Технологии обнаружения и идентификации ОВ в воздухе наиболее востребованы в антитеррористических целях. В последние годы разработано большое количество мобильных приборов для обнаружения ОВ в полевых условиях [5]. При использовании большинства детекторов для onsite обнаружения ОВ в потоковом режиме их концентрирование не предусмотрено. При этом, однако, продолжаются поиски оптимальных решений для концентрирования ОВ из воздушной среды. Отбор и концентрирование ОВ из воздушных проб осуществляются либо в активном, либо в пассивном режиме. В первом случае отдающая среда принудительно прокачивается через принимающую. Примером могут служить портативные системы для анализа методом газовой хромато-масс-спектрометрии (ГХ-МС), снабженные приставкой для сорбции и термодесорбции [6]. Такие приборы серийно выпускаются и хорошо себя зарекомендовали, но сорбционное концентрирование в активном режиме требует специального оборудования и “захлебывается”, если в воздухе много пыли. Концентрирование органических соединений в пассивном режиме за счет диффузии осуществляется на полиуретановых пенах [7], сорбентах, импрегнированных полиуретановыми пенами [8], различных пористых полимерных сорбентах [9]. Наиболее часто используют ТФМЭ для пассивного отбора/концентрирования ОВ из воздушной среды [10]. В полевых условиях хорошо зарекомендовали себя микромеханические сенсорные системы на основе точного измерения массы связавшегося вещества [11]. Томас и Спицер [12] использовали этот метод для исследования механизма и кинетики адсорбции микроконцентраций ОВ на двусторонних консолях, покрытых нанотрубками с двуокисью титана. Оказалось, что геометрия нанотрубок в значительной степени обусловливает эффективность адсорбции имитаторов фосфорорганических ОВ (ФОВ). Диффузия ФОВ из воздуха в нанотрубки осуществляется последовательно через наружную поверхность, внутренние каналы и мезопоры. Далее происходит адсорбция ФОВ на активных центрах внутренней поверхности микропор нанотрубок. На этой стадии достигается равновесие, и именно она является лимитирующей по времени. Кинетические параметры очень важны, поскольку обнаружение высокотоксичных веществ в воздухе должно быть максимально приближено к режиму реального времени. До настоящего времени нет единого мнения об оптимальной кинетической модели адсорбции органических соединений из воздуха нанотрубками. Кинетическая модель псевдопервого порядка [13] основана на допущении, что скорость адсорбции пропорциональна количеству активных сайтов на сорбирующей поверхности. В рамках модели псевдовторого порядка [14] предполагается образование химической связи адсорбат–адсорбент, и этот процесс является лимитирующим. В обоих случаях скоростью диффузии молекул адсорбата к активным сайтам адсорбента пренебрегают [15]. Двухэкспоненциальная модель [16], описывающая диффузию и адсорбцию как два процесса, которые могут осуществляться параллельно либо последовательно, наиболее близка к практике.
При разработке новых подходов к анализу воздушной среды в полевых условиях в качестве аналитов рассматриваются заранее неизвестные ОВ, поэтому сорбирующие устройства должны быть универсальными, т.е. применимыми для улавливания и концентрирования в идеале любых летучих и среднелетучих ОВ. При этом в процессах хемосорбции учитывается, что все они являются сильными электрофилами. Анализ других объектов окружающей среды (вода, воздух, строительные материалы) чаще проводят в стационарных лабораториях, когда уже предположительно известно, какой тип ОВ мог послужить причиной инцидента. До настоящего времени чрезвычайные ситуации, обусловленные террористическими актами, локальными военными конфликтами или непреднамеренным контактом с захороненным или затопленным ХО, преимущественно были связаны с сернистым ипритом или ФОВ. Эти же вещества были накоплены в качестве ХО, затем подлежали уничтожению в соответствии с Конвенцией о его запрещении и, следовательно, должны были контролироваться в объектах природной и техногенной сред в целях обеспечения химической безопасности. В этой связи в настоящем обзоре рассмотрены методы разделения и концентрирования, применяемые в стационарных лабораториях при определении сернистого иприта и ФОВ, а также продуктов их трансформации.
СПОСОБЫ ИЗВЛЕЧЕНИЯ ИЗ МАТРИЦ И КОНЦЕНТРИРОВАНИЯ СЕРНИСТОГО ИПРИТА И ПРОДУКТОВ ЕГО ТРАНСФОРМАЦИИ ПРИ АНАЛИЗЕ ОБЪЕКТОВ ОКРУЖАЮЩЕЙ СРЕДЫ
Извлечение и концентрирование ОВ кожно-нарывного действия, в частности сернистого иприта (далее иприта), из неполярных матриц является сложной задачей в связи с липофильной природой данных ОВ, обусловливающей их сродство к неполярным матрицам. В доступной литературе имеется ограниченное количество публикаций на данную тему [3, 17]. Согласно рекомендациям [3] ТФЭ на силикатных картриджах и экстракция ацетонитрилом являются наиболее часто применяемыми процедурами для извлечения ОВ из образцов с высоким содержанием неполярных органических соединений. При этом отмечается, что сернистый иприт не удерживается эффективно на силикатной поверхности, что приводит к низкой степени его извлечения. В работе [18] исследован механизм сорбции иприта и показано, что на силикатной поверхности происходит его хемосорбция. В цитируемой работе также предложен новый способ эффективной экстракции иприта из неполярных матриц. Для этого был синтезирован сополимер, состоящий из метакриловой кислоты и этиленгликольдиметакрилата, который использовали в качестве сорбента для ТФЭ при извлечении иприта из дизельного топлива. В ряду экстрагентов опробовали дихлорметан, хлороформ, ацетон и этилацетат. Последний выбрали в качестве оптимального, поскольку он обеспечивал степень извлечения иприта с полимерного сорбента на уровне 75–87%, а также наилучший хроматографический профиль при разделении методом газовой хроматографии на капиллярной колонке с неполярной неподвижной фазой. Промывку картриджей после нанесения образцов осуществляли гексаном. Показано, что 1 мл гексана эффективно удаляет углеводородные матричные компоненты без потери аналита.
В работе [19] для извлечения десяти продуктов деструкции иприта из образцов воды был разработан сорбент на основе углеродного аэрогеля. Углеродный аэрогель является перспективным материалом для ТФЭ ввиду высокой пористости, очень низкой плотности и большой поверхности сорбирующего слоя. Элюирование десяти целевых продуктов деструкции иприта после нанесения образцов воды на картридж для ТФЭ с синтезированным сорбентом осуществляли 2 мл метанола. Степень извлечения аналитов составила не менее 83%, а для некоторых достигала 99% и более.
Извлечение ОВ из проб воды в работе [20] осуществляли методом магнитной дисперсионной ТФЭ (magnetic dispersive solid-phase extraction). Принцип метода заключается в диспергировании микро- и наноразмерных магнитных частиц, импрегнированных сорбентом, в воде. Эффективная адсорбция аналита на сорбенте происходит благодаря его большой удельной поверхности. После извлечения частиц сорбента из воды методом магнитной сепарации проводят элюирование аналита органическим растворителем. Магнитный гидрофильно-липофильный сорбент синтезировали из N-винилпирролидона и дивинилбензола в качестве мономеров и наночастиц оксида железа (Fe2O3) в качестве магнитного материала. Степень извлечения иприта при использовании синтезированного сорбента из образцов водопроводной, дистиллированной, дождевой и озерной воды с добавлением 5% NaCl во все образцы лежала в диапазоне 72–90%. Варьирование соотношения мономеров позволило получить сорбенты с желаемым гидрофильно-липофильным балансом для экстракции ОВ различной полярности. Широко используемыми коммерчески доступными гидрофильно-липофильными сорбентами являются Oasis HLB (Waters) и Supel-Select HLB (Supelco).
Магнитный нанокомпозитный сорбент Fe3O4–SiO2–G, синтезированный путем ковалентного связывания покрытого силикатами Fe3O4 с графеновыми подложками, применяли в работе [21] для концентрирования ОВ из образцов воды методом дисперсионной ТФЭ. Диспергирование 20 мг сорбента в 200 мл воды с добавлением 30% NaCl проводили в течение 20 мин. Элюирование сорбированного иприта осуществляли смесью метанол–хлороформ. Степень извлечения иприта из образцов дистиллированной, водопроводной и сточной воды составила 95, 90 и 69% соответственно. Предел обнаружения иприта был на уровне 0.11 нг/мл.
Коммерчески доступные микроволокна для ТФМЭ достаточно давно применяются для определения ОВ и продуктов их деструкции в различных матрицах [22–24], однако не всегда удается достичь удовлетворительной эффективности экстракции. С целью повышения селективности извлечения и концентрирования иприта и циклических продуктов его трансформации (схема 1 ) из образцов воды и донных отложений методом ТФМЭ в работе [25] разработано девять модифицированных типов микроволокон с сорбирующими покрытиями на основе полиакрилатов и полиметакрилатов: полиметилакрилата, полиметилметакрилата, полиэтилакрилата, полиэтилметакрилата, полибутилакрилата, полибутилметакрилата, а также сополимеров полиметилакрилата и полиметилметакрилата, полиэтилакрилата и полиэтилметакрилата, полибутилакрилата и полибутилметакрилата. Аналиты определяли методом тандемной газовой хромато-масс-спектрометрии (ГХ-МС/МС) в режиме ионизации электронами (ИЭ). Максимальную степень извлечения иприта и продуктов его трансформации обеспечивало микроволокно на основе полибутилакрилата. Пределы обнаружения аналитов составили 0.008–0.3 нг/г.
В последние годы ОЗХО инициировала исследования, направленные на определение ОВ и продуктов их трансформации в растениях. Байгильдиеву с соавт. [26] удалось продемонстрировать количественное извлечение некоторых продуктов трансформации иприта из гомогенизированных листьев кресс-салата с помощью жидкостной экстракции при ультразвуковом воздействии.
Схема 1 . Циклические продукты трансформации сернистого иприта в объектах окружающей среды.
РАЗДЕЛЕНИЕ И КОНЦЕНТРИРОВАНИЕ ПРИ ОПРЕДЕЛЕНИИ ФОСФОРОРГАНИЧЕСКИХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ В ОБЪЕКТАХ ОКРУЖАЮЩЕЙ СРЕДЫ
Как известно, ФОВ чрезвычайно токсичны, средняя летальная доза (ЛД50) для человека при трансдермальном пути поступления составляет: 4000 мг для табуна, 1700 мг для зарина, 300 мг для зомана, 10 мг для VX [27]. По этой причине процедуры определения ФОВ и продуктов их трансформации в объектах окружающей среды должны отвечать требованиям, предъявляемым к ультраследовому анализу. При проведении мониторинга ФОВ в объектах окружающей среды исследователи сталкиваются с чрезвычайным разнообразием анализируемых матриц. Данный аспект усложняет создание универсальной надежной процедуры пробоподготовки, в особенности это касается определения следовых количеств ФОВ. На сегодняшний день основными методами извлечения аналитов из образцов объектов окружающей среды, взаимодействовавших с ФОВ, являются фильтрация, дериватизация, центрифугирование, ультразвуковая обработка, жидкостная экстракция, изолирование на ионообменных картриджах, замена растворителей, концентрирование путем удаления растворителя и ТФЭ. Последняя практически повсеместно заменила жидкостную экстракцию ввиду возможности организации анализа в потоковом режиме.
Стоит отметить, что изначально в качестве маркеров воздействия на окружающую среду рассматривались сами интактные ФОВ, их первичные продукты гидролиза, соответствующие алкилметилфосфоновые кислоты (АМФК), и конечный продукт гидролиза – метилфосфоновая кислота (схема 2 ).
В настоящее время известно, что ФОВ G-типа практически не способны к депонированию в объектах окружающей среды, в то время как ФОВ V-типа могут сохраняться в материалах с пористой текстурой в течение длительного времени [28]. При этом количественное извлечение ФОВ V-типа из матриц, обладающих сорбционной активностью, как правило, не достигается. В работе [29] (2004 г.) достигнута рекордная степень извлечения ФОВ V‑типа из почвы, составившая 60% через три месяца после внесения VX в почву. В более поздних работах этот показатель так и не удалось улучшить. В качестве экстрагента использовали трис-буфер с рН 9, что близко к pKa VX. Далее из водного экстракта почвы VX извлекали смесью гексана и дихлорметана 85 : 15 (по объему). Наряду с VX в этих же условиях удалось извлечь основной продукт распада VX в почве – бис(2-диэтиламиноэтил)сульфид.
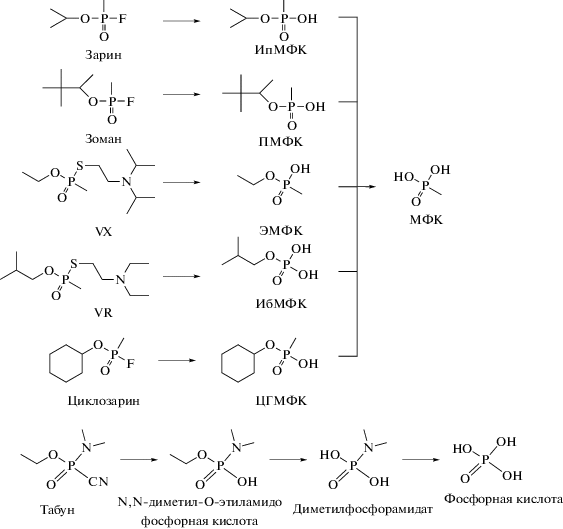
Схема 2 . Продукты гидролиза фосфорорганических отравляющих веществ (зарин – О-изопропилметилфторфосфонат; зоман – О-пинаколилметилфторфосфонат; VX – O-этил-S-(2-диизопропиламиноэтил)метилтиофосфонат; VR – O-изобутил-S-(2-диэтиламиноэтил)метилтиофосфонат; циклозарин – О-циклогексилметилфосфонат; табун – N,N-диметиламидо-О-этилцианфосфат; ИпМФК – О-изопропилметилфосфоновая кислота (продукт гидролиза зарина); ПМФК – О-пинаколилметилфосфоновая кислота (продукт гидролиза зомана); ЭМФК – О-этилметилфосфоновая кислота (продукт гидролиза VХ); ИбМФК – О-изобутилметилфосфоновая кислота (продукт гидролиза VR); ЦГМФ – О-циклогексилметилфосфоновая кислота (продукт гидролиза циклозарина); О-этил-N,N-диметиламид фосфат (продукт гидролиза табуна); МФК – метилфосфоновая кислота (универсальный маркер зарина).
Работы, относящиеся к 90-м годам прошлого века и к нулевым нынешнего, преимущественно выполнены с применением метода ГХ-МС на квадрупольных масс-анализаторах в режиме ИЭ [30]. Методами газовой хроматографии (ГХ) АМФК определяют в виде летучих производных, которые получают при действии различных дериватизирующих агентов [31]. Дериватизацию алкилфосфоновых и алкилметилфосфоновых кислот проводят не только в целях получения летучих производных для определения методами ГХ. В работе [32] дериватизацию этих соединений п-метоксифенацилбромидом проводили в целях повышения чувствительности анализа методом ВЭЖХ с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС).
Для концентрирования аналитов предпринимаются попытки создания высокоселективных сорбентов. Вызывает интерес работа [33], в которой в качестве метода концентрирования применили ТФЭ с молекулярным отпечатком. Данный метод заключается в комплексообразовании в растворе молекулы-шаблона с функциональными мономерами через нековалентные связи с последующей полимеризацией этих мономеров вокруг шаблона с помощью кросс-линкера. По окончании процесса полимеризации молекулы шаблона удаляют путем нарушения взаимодействия между шаблоном и мономерами, что приводит к образованию внутри полимера полостей с пространственным и функциональным распознаванием. В работе [34] с помощью ТФЭ с применением сорбента на основе диоксида циркония удалось достичь пределов определения 8.5–10 нг/мл для АМФК – продуктов гидролиза зарина, зомана, циклозарина и VX при ГХ-МС-детектировании.
На сегодняшний день ВЭЖХ-МС обеспечивает более высокочувствительное определение АМФК в воде [35, 36] и биожидкостях [37] по сравнению с ГХ-МС. В связи с этим в последние годы появилось много работ по определению маркеров ФОВ методом ВЭЖХ-МС. Так, в работе [38] авторы продемонстрировали в рамках одной процедуры возможность определения аналитов с существенно различающимися физико-химическими свойствами (зарин, зоман, VX, VR, циклозарин, табун и продукты их трансформации) в трех объектах: воде, желе (2 мас. % агара) и почве. Достигнуты пределы обнаружения аналитов от 1 до 10 нг/мл методом ВЭЖХ-МС высокого разрешения (ВР). Для концентрирования аналитов использовали улавливание на мембранных фильтрах из политетрафторэтилена (водные образцы), осаждение полисахаридов с последующим упариванием супернатанта (желе), 2-стадийную промывку с последующим концентрированием и перерастворением (почва). С помощью дериватизации 2-[(диметиламино)метил]фенолом [39] удалось достичь предела определения ФОВ G‑типа (зарин, зоман, циклозарин) 1 пг/мл для образцов воды при детектировании методом ВЭЖХ-МС/МС.
Всплеск интереса к методам микроэкстракции для концентрирования ФОВ и продуктов их трасформации пришелся на первое десятилетие XXI в. Из наиболее поздних представляет интерес работа [40], в которой представлен метод жидкостно-жидкостно-твердофазной микроэкстракции. Пористый полимерный сорбент пропитывали органическим растворителем и помещали в водную среду, содержащую имитаторы ФОВ. Аналиты детектировали методом ИК-Фурье спектроскопии непосредственно на сорбенте. Элюировать аналиты с сорбента для последующего ХМС-анализа, по-видимому, было затруднительно.
До настоящего времени применительно к экстракции продуктов гидролиза ФОВ метод микро-ТФЭ (MEPS), для осуществления которого используют специальные шприцы со сменными картриджами, не находил широкого применения. Недостатком метода является узкий диапазон типов сорбентов в таких картриджах. Преимущественно это разновидности полидиметилсилоксана, который неэффективен для извлечения высокополярных аналитов и АМФК в частности. В работе [41] для извлечения продуктов гидролиза зарина из водных сред использовали микрокартриджи, изготовленные самими авторами из графитизированного угля, подвергнутого специальной обработке.
Одним из основных направлений в разработке новых методов разделения и концентрирования при подготовке проб к определению ФОВ и продуктов их трансформации является изготовление композитных сорбентов с нанодисперсными включениями. Обобщенная информация о наиболее актуальных методах концентрирования и разделения ФОВ в объектах окружающей среды приведена в табл. 1.
Таблица 1.
Обобщенная информация о наиболее актуальных методах извлечения и разделения фосфорорганических отравляющих веществ в объектах окружающей среды
| Объект анализа | Аналит | Способ извлечения аналитов | Метод анализа | Достигнутые пределы обнаружения | Литература |
|---|---|---|---|---|---|
| Почва | VX | Жидкостно-жидкостная экстракция | ГХ-МС | 10 мкг/г | [28] |
| Вода | Продукты гидролиза зарина, зомана и VR | Дериватизация п-метоксифенацилбромидом | ВЭЖХ-МС/МС, ГХ-МС/МС |
От 0.02 до 0.2 нг/мл, от 10 до 50 нг/мл |
[31] |
| Вода | Продукты гидролиза зарина, зомана, циклозарина, табуна, VX | ТФЭ (сорбент на основе циркония) | ГХ-МС | От 8.5 до 10.0 нг/мл | [33] |
| Вода, гель (2 мас. % агара), почва | Зарин, зоман, VX, VR, циклозарин, табун и их продукты гидролиза | Водные образцы – мембранные фильтры из политетрафторэтилена. Агар-агар – осаждение полисахаридов с последующим упариванием супернатанта. Почва – 2-стадийная промывка с последующим извлечением и перерастворением |
ВЭЖХ-МС/МС-ВР | От 1 до 10 нг/мл | [37] |
| Вода/сыворотка плазмы крови | Продукты гидролиза нервно-паралитических агентов | ТФЭ и дериватизация п-бромфенацилом | ВЭЖХ-МС/МС | От 1 до 5 нг/мл | [38] |
МЕТОДЫ РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ ПРИ ОПРЕДЕЛЕНИИ МЕТАБОЛИТОВ СЕРНИСТОГО ИПРИТА В БИОПРОБАХ
Как уже отмечалось, процедура пробоподготовки, в особенности биомедицинских образцов, является критической и лимитирующей стадией химического анализа. Наиболее часто для извлечения и концентрирования всей линейки метаболитов ОВ из различных биологических матриц применяют ТФЭ. В литературе представлен большой объем публикаций по подбору условий ТФЭ и использованию данного способа экстракции для определения метаболитов сернистого иприта и других ОВ в моче и крови [3, 42–48] (табл. 2).
Таблица 2.
Обобщенные сведения о биомаркерах экспозиции сернистым ипритом и методах их извлечения и разделения
| Биомаркер | Объект анализа | Метод извлечения | Метод анализа | Литература |
|---|---|---|---|---|
| S-гидроксиэтилтиоэтилцистеин–пролин (HETE-CP) | Плазма крови | ТФЭ (Oasis HLB) | ВЭЖХ-МС/МС | [44] |
| S-гидроксиэтилтиоэтилцистеин–пролин–фенилаланин (HETE-CPF) | Плазма крови | ТФЭ (Oasis HLB) | ВЭЖХ-МС/МС | [43] |
| 1,1'-Сульфонилбис[2-(метилтио)этан] (СБМТЭ) | Моча | ТФЭ (Oasis HLB; Chromabond C8) | ГХ-МС/МС-ИЭ | [46, 47] |
| Тиодигликоль (ТДГ) | Моча, плазма крови | ТФЭ (EASY Chromabond; Oasis HLB; картриджи с сорбентом на основе активированных углеродных
волокон); ТФМЭ (100 мкм ПДМС) |
ВЭЖХ-МС/МС ГХ-МС-ИЭ ГХ-МС/МС-ХИ |
[46, 48, 51–53, 58] |
| 1,1'-Сульфонилбис-[2-(метилсульфинил)этан] (СБМСЭ) | Моча | ТФЭ (Oasis HLB; картриджи с сорбентом на основе активированных углеродных волокон, полимерный картридж Env+) | ВЭЖХ-МС/МС | [49, 51, 52] |
| 1-Метилсульфинил-2-[2-(метилтио)этилсульфонил]этан (МСМТЭСЭ) | Моча | ТФЭ (Oasis HLB; картриджи с сорбентом на основе активированных углеродных волокон, полимерный картридж Env+) | ВЭЖХ-МС/МС | [49, 51, 52] |
| 1,1'-Сульфонил-бис[2-S-(N-ацетилцистеинил)этан (СБСНАЭ) | Моча | ТФЭ (Oasis HLB; картриджи с сорбентом на основе активированных углеродных волокон; Supelclean ENVI-8) | ВЭЖХ-МС/МС | [50–52, 55] |
| N7-гидроксиэтилтиоэтилгуанин (N7-HETEG) | Моча, цельная кровь |
ТФЭ (Supelclean ENVI-8; С18) | ВЭЖХ-МС/МС | [43, 54, 55] |
| N-(2-гидроксиэтилтиоэтил)-DL-валин (HETE-Val) | Эритроциты | ТФЭ (Florisil) | ГХ-МС-ОХИ ГХ-МС/МС-ОХИ ГХ-МС/МС-ИЭ |
[56, 57] |
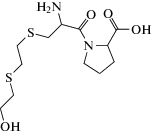
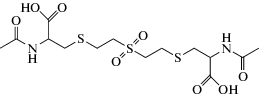
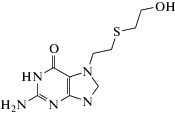
Сорбенты, способные обеспечивать гидрофильно-липофильный баланс (hidrophylic-lipophylic balance, HLB) могут удерживать воду, что важно для процесса сорбции, а также широкий спектр органических соединений различной природы. Картриджи для ТФЭ OASIS HLB (Waters) на основе сополимера дивинилбензола и N-винилпирролидона широко применяют для экстракции и концентрирования метаболитов иприта из образцов мочи и плазмы крови. Они подходят для индивидуального извлечения ТДГ из мочи [49], одновременного извлечения β-лиазных метаболитов [50] и СБСНАЭ [51] из подкисленной мочи. Подкисление мочи до pH ниже 3–4 необходимо для эффективного удерживания СБСНАЭ на картридже HLB, так как способствует возникновению ион-парного взаимодействия между группами –COOH и ацетилированными аминогруппами и приводит к повышению гидрофобности аналита [51]. Авторы работы [52] предложили способ одновременного определения ТДГ, МСМТЭСЭ, СБМСЭ и СБСНАЭ методом ультра ВЭЖХ-МС/МС после концентрирования аналитов из предварительно подкисленных аммонийно-ацетатным буферным раствором образцов мочи методом ТФЭ на картриджах OASIS HLB. Степень извлечения ТДГ, СБСНАЭ, СБМСЭ и МСМТЭСЭ составила 74, 86, 96 и 77% соответственно.
Работа [53] посвящена исследованию и оптимизации процессов извлечения из мочи и концентрирования четырех мочевых метаболитов иприта (ТДГ, МСМТЭСЭ, СБМСЭ и СБСНАЭ) с использованием сорбента на основе активированных углеродных волокон (АУВ). Благодаря микропористой структуре активированного угля и большой удельной площади поверхности АУВ достигается эффективное обратимое удерживание ОВ и их метаболитов. В качестве элюентов авторы исследовали дихлорметан, ацетон и метанол, а также их смеси в различных объемных соотношениях с добавками и без добавок кислот. Оптимальной для элюирования ТДГ и МСМТЭСЭ была смесь дихлорметан–ацетон (9 : 1), а для элюирования СБМСЭ и СБСНАЭ – 3%-ная HCl в метаноле. Анализ проводили методом ВЭЖХ-МС/МС в режиме электрораспытительной ионизации. Предел обнаружения ТДГ составил 5 нг/мл, остальных метаболитов – 1 нг/мл.
Авторы работы [54] определяли ТДГ в плазме крови, отобранной у пострадавших во время локального военного конфликта на Ближнем Востоке в 2015 г., после щелочного гидролиза и процедуры очистки и концентрирования аналита методом ТФЭ на картридже EASY Chromabond (Macherey-Nagel) на основе сополимера полистирола и дивинилбензола. Элюирование ТДГ осуществляли 500 мкл ацетонитрила. После дериватизации гептафтормасляным ангидридом (ГФМА) ТДГ анализировали методом ГХ-МС-ИЭ. В пробах двух пострадавших из семи обнаружили ТДГ.
Для оптимизации условий извлечения и концентрирования четырех аддуктов иприта с ДНК в моче – N7-гидроксиэтилтиоэтилгуанина (N7-HETEG), бис(2-этил-N7-гуанин)тиоэфира (Bis-G), N3-гидроксиэтилтиоэтиладенина (N3-HETEA), O6-гидроксиэтилтиоэтилгуанина (O6-HETEG) – авторы работы [55] приготовили серию картриджей для ТФЭ, наполненных 0.2, 0.5 и 1 г сорбента С18. Режим элюирования целевых соединений подбирали путем варьирования соотношения вода–метанол–муравьиная кислота. Показано, что степень извлечения аддуктов иприта с ДНК максимальна, а матричный эффект существенно снижен при двухстадийном элюировании аналитов 2 мл смеси 20%-ного метанола и 5%-ной муравьиной кислоты (1 : 1) и 1.5 мл смеси 50%-ного метанола и 5%-ной муравьиной кислоты (1 : 1) с картриджей, наполненных 1 г сорбента С18. Анализ осуществляли методом ультра ВЭЖХ-МС/МС-изотопного разбавления. Пределы обнаружения составили 2–5 пг/мл.
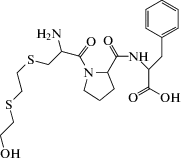
Возможность одновременного определения аддуктов иприта с ДНК (N7-HETEG) и с ацетилцистеином (СБСНАЭ) в моче, после экспозиции крыс ипритом в условиях терапии скавенджером исследована в работе [56]. Определяемые аналиты обладают разными физико-химические свойствами, поэтому процедуре подготовки образцов мочи для дальнейшего одновременного определения высокополярного СБСНАЭ и существенно менее полярного N7-HETEG уделили особое внимание. Были изучены пять типов коммерчески доступных ТФЭ-картриджей для извлечения и концентрирования аналитов (табл. 3). Для элюирования целевых соединений исследовали две системы растворителей (метанол–вода и ацетонитрил–вода) в различных объемных соотношениях. Максимальная эффективность одновременного концентрирования и извлечения N7-HETEG и СБСНАЭ из образцов мочи достигнута при элюировании аналитов с картриджа на основе силикагеля с привитыми группами С8 (Supelclean ENVI-8) подкисленной смесью ацетонитрил–вода (60 : 40). Анализ осуществляли методом ВЭЖХ-МС/МС-ВР. Авторы указывают на существенный матричный эффект при определении N7-HETEG. Его снижения не удалось добиться ни изменением условий удерживания аналита на сорбенте, ни подбором условий элюирования. От промывки картриджа после пропускания мочи пришлось отказаться, так как при этом не удавалось избежать потерь аналита. Незначительного снижения матричного эффекта достигли лишь при разбавлении готового к анализу образца равным количеством воды. Сильное матричное влияние было характерно для мочи крыс, поскольку даже в холостых образцах регистрировался сигнал иона с массовым числом m/z 256.08, совпадающим с m/z иона-прекурсора определяемого биомаркера N7-HETEG. По этой причине данный биомаркер может быть достоверно идентифицирован только по иону-продукту и с использованием внутреннего стандарта. Приведенный пример иллюстрирует необходимость разработки новых подходов к очистке проб от матричных компонентов без потери целевых аналитов, поскольку путем перебора известных вариантов ТФЭ эту проблему решить далеко не всегда удается.
Таблица 3.
Картриджи для твердофазной экстракции, используемые для извлечения и концентрирования N7-HETEG и СБСНАЭ [56]
| Торговое наименование картриджа (фирма) | Сорбент |
|---|---|
| OASIS HLB (Waters) | Сополимер дивинилбензола и N-винилпирролидона |
| Supel-Select HLB (Supelco) | Полистирол с гидрофильными группами |
| Supelclean ENVI-8 (Supelco) | Силикагель с привитыми октильными группами С8 |
| Supelclean LC18 (Supelco) | Силикагель с привитыми октадецильными группами С18 |
| Sep-Pak C18 (Waters) | Силикагель с привитыми октадецильными группами С18 |
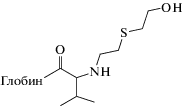
В работе [57] аддукт иприта с N-терминальным валином в глобине (HETE-Val) определяли в виде производного с пентафторфенилизотиоцианатом и ГФМА. Для оптимизации процедуры очистки и концентрирования методом ТФЭ исследовали два типа картриджей – на основе силикагеля и силанизированного силикагеля (Florisil). Несмотря на то, что картридж на основе силикагеля эффективно устраняет матричные компоненты, степень извлечения аддукта была неудовлетворительной, а при низких концентрациях происходила необратимая сорбция аналита. Подбор оптимального экстрагента осуществляли на картридже Florisil. Исследовали экстракционную способность этилацетата, диэтилового эфира, толуола, анизола, дихлорметана, ацетонитрила и их смесей в различных объемных отношениях. Наилучшая степень извлечения HETE-Val (56%) достигнута при элюировании аналита 2 мл смеси этилацетат–дихлорметан (1 : 9). Анализ проводили методом ГХ-МС в режиме отрицательной химической ионизации (ОХИ), а также методом ГХ-МС/МС в режимах ИЭ и ОХИ [58].
До настоящего времени для эффективного извлечения из биоматриц и концентрирования такого, казалось бы, простого вещества, как ТДГ, оптимального решения не найдено. В работе [59] для извлечения ТДГ из биоматриц применяли метод ТФМЭ после дериватизации аналита ГФМА непосредственно в биообразце. Анализ осуществляли методом ГХ-МС-ИЭ. Исследовали пять коммерчески доступных микроволокон с различной толщиной сорбирующего слоя и полярностью: 30 мкм полидиметилсилоксан (ПДМС), 100 мкм ПДМС, 85 мкм карбоксен/ПДМС, 70 мкм карбовакс/дивинилбензол и 85 мкм полиакрилат. Наиболее эффективным для концентрирования производного ТГД с ГФМА из образцов плазмы крови и мочи оказалось микроволокно 100 мкм ПДМС, с помощью которого был достигнут предел обнаружения аналита в исследуемых пробах, сравнимый с эндогенным уровнем ТДГ в биообразцах (1 нг/мл).
Определение метаболитов сернистого иприта в моче и плазме крови предлагалось в качестве задач первого (2016 г.) и шестого (2021 г.) квалификационных тестов ОЗХО по анализу биомедицинских образцов. Разбор результатов шестого теста позволит проследить эволюцию методов разделения и концентрирования, используемых ведущими лабораториями мира при решении этих сохраняющих актуальность, задач.
СТАНДАРТНЫЕ И ИННОВАЦИОННЫЕ ПОДХОДЫ К ОПРЕДЕЛЕНИЮ МЕТАБОЛИТОВ ФОСФОРОРГАНИЧЕСКИХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ В БИОПРОБАХ
При установлении факта применения ФОВ по результатам анализа биомедицинских проб следует исходить из того, что детектирование интактных ФОВ в неизменном виде практически не представляется возможным ввиду их стремительного метаболизма [60–62]. Гидролитические метаболиты ФОВ (АМФК) с применением процедур концентрирования могут быть обнаружены в организме в течение длительного периода времени, однако даже при воздействии высоких доз их прямое определение в биожидкостях возможно в течение нескольких суток, а чаще нескольких часов после отравления [63]. Опубликовано значительное количество методик определения этих метаболитов в плазме и моче. Выделение АМФК из плазмы или мочи возможно с применением жидкостной экстракции [63] или ТФЭ [64]. Эти соединения определяют с помощью жидкостной или газовой хроматографии. В последнем случае требуется проведение предварительной дериватизации [65]. В настоящее время, благодаря возможностям тандемной масс-спектрометрии, например МС/МС с тройным квадруполем и МС-ВР, возможно обнаружение продуктов гидролиза ФОВ в плазме крови и моче с пределом обнаружения вплоть до нескольких пг/мл [63, 66–68]. Такая высокая чувствительность определения позволит фиксировать продукты гидролиза ОВ в период до нескольких недель после воздействия [66].
В рамках ретроспективного анализа оптимальная стратегия, используемая для установления/подтверждения факта применения ФОВ, основана на механизме их связывания с белками крови, такими как ацетилхолинэстераза (АХЭ), бутирилхолинэстераза (БХЭ) и сывороточный альбумин (СА). Время жизни конъюгатов ФОВ с биомолекулами сопоставимо с временем жизни этих биомолекул в организме. Так, период полураспада БХЭ составляет примерно 12 дней [69], аддукты с АХЭ эритроцитов потенциально могут сохраняться на протяжении всего времени жизни эритроцитов – до 100 сут [70], сывороточный альбумин – 20–25 сут [71]. Это означает, что аддукты с белками могут служить в качестве ретроспективных биомаркеров воздействия ФОВ. Оптимальными биомаркерами являются аддукты ФОВ с БХЭ и СА, так как они присутствуют в плазме крови, которая является более стабильной матрицей для хранения, транспортировки и обработки образцов, чем цельная кровь. Важным соображением является и то, что концентрация БХЭ примерно в 10 раз выше концентрации АХЭ в крови [72]. Наиболее простым методом обнаружения аддукта ФОВ с БХЭ является реактивация фторид-ионом. Во время инкубации образца плазмы с ионами фтора ФОВ G-типа или фторангидрид ФОВ V-типа высвобождается и может быть экстрагирован ГХ-совместимым растворителем и впоследствии проанализирован с помощью ГХ. Минимальная обнаруживаемая степень ингибирования БХЭ, которая может быть установлена с помощью этого метода, зависит от типа ГХ-детектора. Типичные недорогие детекторы, такие как азотно-фосфорный, пламенно-фотометрический и масс-селективный, показывают абсолютные пределы обнаружения около 1 пг, что означает, что концентрация 1 нг/мл может быть обнаружена при введении в инжектор хроматографа пробы объемом 1 мкл. Основным недостатком метода является то, что не все аддукты ФОВ поддаются реактивации фторид-ионом; наиболее известным примером является состаренный аддукт зомана.
В 2002 г. в работе [73] описан метод, основанный на ВЭЖХ-МС/МС-определении фосфонилированного нонапептида, полученного после ферментативного гидролиза пепсином БХЭ, ингибированной ФОВ. Оптимальным методом определения фосфонилированного нонапептида является ВЭЖХ-МС/МС с использованием режима мониторинга заданных реакций (selected reaction monitoring, SRM). Масса исходного иона зависит от массы фрагмента ФОВ, конъюгированного с пептидом [74]. В процессе фрагментации сначала удаляется фосфильная часть ФОВ. Характеристичными ионами-продуктами являются ионы с массовыми числами m/z 778, 673 и 602, соответствующие фрагментам нативного пептида. Ионы-продукты фосфонилированного нонапептида могут быть успешно идентифицированы в пределах отклонения от теоретических массовых чисел 5 ppm. Фиддер с соавт. [73] описали процедуру извлечения БХЭ из плазмы с использованием самодельных прокаинамидных гелей. В последние несколько лет процедура пробоподготовки для выделения БХЭ из плазмы была еще более усовершенствована. В работе [75] продемонстрирована эффективность использования магнитных шариков, покрытых антителами, для выделения БХЭ из плазмы, что привело к лучшей очистке образца и повышению возможностей автоматизации метода. Метод иммуномагнитной сепарации оптимизирован [76] для количественного определения аддукта и повышения производительности анализа [77]. В 2019 г. опубликована работа [78], авторы которой использовали метод аффинной хроматографии для очистки БХЭ в качестве альтернативы выделению БХЭ с помощью антител. Анализ образующихся после фрагментирования ингибированной ФОВ БХЭ нонапептидных аддуктов в настоящее время также применяется для биомониторинга воздействия на организм других ингибиторов холинэстеразы. Например, в работах [79] и [80] использовали аналогичный пептид для диагностики воздействия три-о-крезилфосфата, нейротоксический эффект которого обусловлен его метаболизмом в печени с образованием продукта биотрансформации 2-(о-крезил)-4Н-1,3,2-бензодиоксафосфоран-2-он (салингенинфосфат), ингибирующего БХЭ.
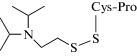
Другой подход представлен в работе [81]. Выделенный фосфонилированный сериновый остаток ингибированной ФОВ БХЭ может быть превращен в щелочных условиях в дегидроаланиновый остаток, который впоследствии может вступать в типовые реакции, образуя универсальный для всех ФОВ биомаркер. Сывороточный альбумин также является целевым белком для ФОВ, особенно при тирозине-411, но с более низким сродством к ФОВ, чем БХЭ. В этом случае проводят протеолиз с применением проназы, далее тирозиновый аддукт определяют с помощью ВЭЖХ-МС. Аддукты ФОВ с альбумином являются неподверженными старению биомаркерами воздействия ФОВ и пестицидов [82, 83]. В случае интоксикации VX уходящей группой при связывании с белками является диизопропиламиноэтантиол (ДИПАЭТ). Это соединение настолько специфично для VX, что может служить репрезентативным биомаркером воздействия VX. Свободная тиольная группа поддается реакции с белками, такими как альбумин, и связывается с различными цистеиновыми остатками с образованием дисульфидной связи. В работах [84, 85] сообщалось об определении аддукта ДИПАЭТ с альбумином в виде дипептида цистеин–пролин–ДИПАЭТ и трипептида метионин–пролин–цистеин–ДИПАЭТ, полученных в результате ферментативного гидролиза проназой. Эти биомаркеры являются наиболее специфичными для ФОВ V-типа.
При использовании технологий определения биомаркеров ОВ, представляющих собой фрагменты биомолекул, модифицированных остатками ОВ (табл. 4), наиболее сложной является стадия извлечения целевых аналитов и их очистки от коэлюируемых примесей.
Таблица 4.
Обобщенные сведения о биомаркерах экспозиции фосфорорганических отравляющих веществ и методах их извлечения и разделения (метод анализа – ВЭЖХ-МС/МС)
| Биомаркер | Метод извлечения | Литература |
|---|---|---|
| Объект анализа – плазма крови | ||
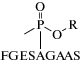
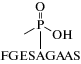
| Нонапептид, модифицированный по серину остатком ФОВ (FGES(AlkylMPA)AGAAS) Деалкилированный нонапептид, модифицированный по серину метил фосфоновой кислотой (МФК) (FGES(MPA)AGAAS) (F – фенилаланин, G – глицин, E – глутаминовая кислота, S – серин, A – аланин) | Ферментативный гидролиз пепсином БХЭ, ингибированной ФОВ | [72] |
| Иммуно-магнитная сепарация для выделения БХЭ из плазмы, ультрамикрофильтрация | [76] | |
| Выделение аддуктов АХЭ с использованием регенерируемого аффинного сорбента на основе хуприна (Hupresin) | [77] | |
| Тирозин, модифицированный ФОВ (AlkylMPA-Tyr) | Протеолиз с применением проназы, ТФЭ на картирджах Strata SDB-L (стирол дивинилбензол) | [82] |
| Дипептид, модифицированный по цистеину диизопропламиноэтантиолом (цистеин–пролин–ДИПАЭТ) Трипептид, модифицированный по цистеину диизопропламиноэтантиолом (метионин–пролин–цистеин–ДИПАЭТ) | Протеолиз с применением проназы, ультрамикрофильтрация | [84] |
| Объект анализа – моча | ||
| Алкилметилфосфоновые кислоты (АМФК) | Фильтрация на мембранном фильтре и разбавление деионизованной водой | [36] |
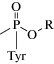
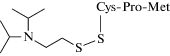
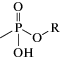
* * *
Для извлечения и концентрирования ОВ и продуктов их трансформации из водных сред, включая вытяжки из почвы и донных отложений, а также биопробы, наибольшие перспективы дальнейшего развития имеет метод ТФЭ с применением гидрофильно-липофильных полимерных сорбентов, которые могут быть использованы в широком диапазоне рН загружаемых растворов и элюентов и способны обратимо удерживать вещества с существенно различающимися физико-химическими свойствами. При общей тенденции к упрощению процедур подготовки маркеров ОВ к инструментальному анализу определение фрагментированных аддуктов ОВ с биомолекулами даже при использовании высокоселективных методов ГХ-МС/МС и ВЭЖХ-МС/МС-ВР требует выделения из биоматриц, концентрирования и очистки аналитов. Различные варианты хромато-масс-спектрометрического анализа для идентификации ОВ и продуктов их трансформации, определяются типом используемого оборудования, в то время как процедуры разделения и концентрирования, применяемые разными лабораториями, часто основаны на оригинальных разработках и представляют большой научный интерес.
Список литературы
Франке З. Химия отравляющих веществ. Пер. с нем. М.: Химия, 1973. 440 с.
Convention on the Prohibition of the Development, Stockpiling and Use of Chemical Weapons and on their Destruction. Technical Secretariat of the Organisation for Prohibition of Chemical Weapons, The Hague, 1997. http://www.opcw.org. (21.03.2021).
Recommended Operating Procedures for Analysis in the Verification of Chemical Disarmament / Ed. Vanninen P. Helsinki: The Ministry for Foreign Affairs of Finland, 2017. 809 p. http://www.helsinki.fi/veri-fin/bluebook. (13.04.2021).
Рыбальченко И.В., Байгильдиев Т.М., Родин И.А. Хромато-масс-спектрометрические методы определения маркеров и биомаркеров отравляющих веществ // Журн. аналит. химии. 2021. Т. 76. № 1. С. 32. (Rybal’chenko I.V., Baigil’diev T.M., Rodin I.A. Chromatography–mass spectrometry analysis for the determination of the markers and biomarkers of chemical warfare agents // J. Anal. Chem. 2021. V. 76. № 1. P. 26.)
Handbook of the Toxicology of Chemical Warfare Agents. 3rd Ed. / Ed. Gupta R.C. Amsterdam: Elsevier, 2020. 983 p.
Contreras J.A., Murray J.A., Tolley S.E., Oliphant J.L., Tolley H.D., Lammert S.A., Lee E.D., Later D.W., Lee M.L. Hand-portable gas chromatograph-toroidal ion trap mass spectrometer (GC-TMS) for detection of hazardous compounds // J. Am. Soc. Mass Spectrom. 2008. V. 19. P. 1425.
Shoeib M., Harner T. Characterization and comparison of three passive air samplers for persistent organic pollutants // Environ. Sci. Technol. 2002. V. 36. № 19. P. 4142.
Genualdi S., Lee S.C., Shoeib M., Gawor A., Ahrens L., Harner T. Global pilot study of legacy and emerging persistent organic pollutants using sorbent impregnated polyurethane foam disk passive air samplers // Environ. Sci. Technol. 2010. V. 44. № 14. P. 5534.
Zhang X., Barnes J., Lei Y.D., Wania F. Semivolatile organic contaminants in the Hawaiian atmosphere // Environ. Sci. Technol. 2017. V. 51. № 20. P. 11634.
Bryant C.K., LaPuma P.T., Hook G.L., Houser E.J. Chemical agent identification by field-based attenuated total reflectance infrared detection and solid-phase microextraction // Anal. Chem. 2007. V. 79. P. 2334.
Yaminsky I., Gorelkin P., Kiselev G. Concurrence of intermolecular foces in monolayers // Jpn. J. Appl. Phys. 2006. V. 45. № 3B. P. 2316.
Thomas G., Spitzer D. Double-side microcantilevers as a key to understand the adsorption mechanisms and kinetics of chemical warfare agents on vertically-aligned TiO2 nanotubes // J. Hazard. Mater. 2021. V. 406. P. 124672.
Viegas R.M., Campinas M., Costa H., Rosa M.J. How do the HSDM and Boyd’s model compare for estimating intraparticle diffusion coefficients in adsorption processes // Adsorption. 2014. V. 20. P. 737.
Soltani R., Marjani A., Shirazian S. Facile one-pot synthesis of thiolfunctionalized mesoporous silica submicrospheres for Tl(I) adsorption: Isotherm, kinetic and thermodynamic studies // J. Hazard. Mater. 2019. V. 371. P. 146.
Simonin J.-P. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics // Chem. Eng. J. 2016. V. 300. P. 254.
Tosun I. Ammonium removal from aqueous solutions by clinoptilolite: determination of isotherm and thermodynamic parameters and comparison of kinetics by the double exponential model and conventional kinetic models // Int. J. Environ. Res. Public Health. 2012. V. 9. P. 970–984.
Pardasani D., Palit M., Gupta A., Shakya P., Sekhar K., Dubey D. Sample preparation of organic liquid for off-site analysis of chemical weapons convention related compounds // Anal. Chem. 2005. V. 77. P. 1172.
Roy K., Goud D., Chandra B., Dubey D. Efficient extraction of sulfur and nitrogen mustards from nonpolar matrix and an investigation on their sorption behavior on silica // Anal. Chem. 2018. V. 90. № 14. P. 8295.
Joul P., Vaher M., Kuhtinskaja M. Evaluation of carbon aerogel-based solid-phase extraction sorbent for the analysis of sulfur mustard degradation products in environmental water samples // Chemosphere. 2018. V. 198. P. 460.
Singha V., Purohita A., Chinthakindia S., Taka R., Pardasania D., Shrivastavab A., Dubey D. Magnetic hydrophilic–lipophilic balance sorbent for efficient extraction of chemical warfare agents from water samples // J. Chromatogr. A. 2018. V. 1434. № 19. P. 39.
Chinthakindi S., Purohit A., Singh V., Tak V., Goud D., Dubey D., Pardasani D. Iron oxide functionalized graphene nano-composite for dispersivesolid phase extraction of chemical warfare agents from aqueoussamples // J. Chromatogr. A. 2015. V. 1394. P. 9.
Gregory L. Kimm, Gary L. Hook, Philip A. Smith. Application of headspace solid-phase microextraction and gas chromatography–mass spectrometry for detection of the chemical warfare agent bis(2-chloroethyl) sulfide in soil // J. Chromatogr. A. 2002. V. 971. P. 185.
Creek J., McAnoy A., Brinkworth C. Rapid monitoring of sulfur mustard degradation in solution by headspace solid-phase microextraction sampling and gas chromatography mass spectrometry // Rapid Commun. Mass Spectrom. 2010. V. 24. P. 3419.
Popiel S., Sankowska M. Determination of chemical warfare agents and related compounds in environmental samples by solid-phase microextraction with gas chromatography // J. Chromatogr. A. 2011. V. 1218. P. 8457.
Nawała J., Czuprynski K., Popiel S., Dziedzic D., Bełdowski J. Development of the HS-SPME-GC-MS/MS method for analysis of chemical warfare agent and their degradation products in environmental samples // Anal. Chim. Acta. 2016. V. 933. P. 103.
Baygildiev T., Vokuev M., Braun A., Rybalchenko I., Rodin I. Monitoring of hydrolysis products of mustard gas, some sesqui- and oxy-mustards and other chemical warfare agents in a plant material by HPLC-MS/MS // J. Chromatogr. B. 2021. V. 1162. P. 122452.
Vital de Oliveira O., Cuya Correa Ferreira E., da Silva Gonçalves A. Theoretical investigations of human acetylcholinesterase inhibition efficiency by neurotoxic organophosphorus compounds // Chem. Phys. Lett. 2018. V. 706. P. 82.
Савельева Е.И., Ленинский М.А., Васильева И.А., Каракашев Г.В., Самченко Н.А. Определение следовых количеств O-изобутил-S-[(2-диэтиламино)этил] метилфосфонотиоата и токсичного продукта его гидролиза методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием // Аналитика и контроль. 2021. Т. 25. № 1. С. 43.
Montauban C., Bégos A., Bellier B. Extraction of nerve agent VX from soils // Anal. Chem. 2004. V. 76. № 10. P. 2791.
Meyers R. Applications, theory, and instrumentation in encyclopedia of analytical chemistry. V. 2. West Sussex, UK: John Wiley & Sons Ltd., 2000. P. 1055.
Rohrbaugh D., Sarver E. Detection of alkyl methylphosphonic acids in complex matrices by gas chromatography-tandem mass spectrometry // J. Chromatogr. 1998. V. 809. № 1−2. P. 141.
Байгильдиев Т.М., Вокуев М.Ф., Орешкин Д.В., Браун А.В., Годовиков И.А., Рыбальченко И.В., Родин И.А. п-Метоксифенацилбромид – универсальный реагент для определения алкилфосфоновых и алкилметилфосфоновых кислот методами высокоэффективной жидкостной и газовой хроматографии с масс-спектрометрическим детектированием // Масс-спектрометрия. 2019. Т. 16. № 3. С. 180. (Baygildiev T.M., Vokuev M.F., Oreshkin D.V., Braun A.V., Godovikov I.A., Rybalchenko I.V., Rodin I.A. p-Methoxyphenacyl bromide as a versatile reagent for the determination of alkylphosphonic and alkylmethylphosphonic acids by high-performance liquid and gas chromatography with mass spectrometric detection // J. Anal. Chem. 2020. V. 75. № 13. P. 1708.)
Le Moullec S., Begos A., Pichon V., Bellier B. Selective extraction of organophosphorus nerve agent degradation products by molecularly imprinted solid-phase extraction // J. Chromatogr. A. 2006. V. 1108. P. 7.
Kanaujia P., Pardasani D., Tak V., Purohit A., Dubey D. Selective enrichment of the degradation products of organophosphorus nerve agents by zirconia based solid-phase extraction // J. Chromatogr. A. 2011. V. 1218. № 38. P. 6612.
Родин И.А., Браун А.В., Ставрианиди А.Н., Шпигун О.А., Рыбальченко И.В. Обнаружение маркеров нервно-паралитических отравляющих веществ методом ультра-высокоэффективной жидкостной хроматографии-тандемной масс-спектрометрии // Аналитика и контроль. 2012. Т. 16. № 3. С. 254.
Родин И.А., Браун А.В., Байгильдиев Т.М., Ананьева И.А., Шпигун О.А., Рыбальченко И.В. Определение продуктов гидролитической трансформации отравляющих веществ VX и VR в природных водах методом жидкостной хроматомасс-спектрометрии // Масс-спектрометрия. 2015. Т. 12. № 2. С. 100. (Rodin I.A., Braun A.V., Baygildiev T.M., Anan’eva I.A., Shpigun O.A., Rybalchenko I.V. Determination of the hydrolysis products of nerve agents in natural waters by liquid chromatography–mass spectrometry // J. Anal. Chem. 2015. V. 70. № 14. P. 1671.)
Корягина Н.Л., Савельева Е.И., Хлебникова Н.С., Уколов А.И., Уколова Е.С., Каракашев Г.В., Радилов А.С. Хроматомасс-спектрометрическое определение алкилметилфосфоновых кислот в моче // Масс-спектрометрия. 2015. Т. 12. № 4. С. 236. (Koryagina, N.L., Savel’eva, E.I., Khlebnikova, N.S., Ukolov A.I., Ukolova E.S., Karakashev G.V., Radilov A.S. Chromatography–mass spectrometry determination of alkyl methylphosphonic acids in urine // J. Anal. Chem. 2016. V. 71. № 14. P. 1309.)
Zhang M., Liu Y., Chen J., Liu H., Lu X., Wu J., Xie J. Sensitive untargeted screening of nerve agents and their degradation products using liquid chromatography–high resolution mass spectrometry // Anal. Chem. 2020. V. 92. № 15. P. 10578.
Weissberg A., Madmon M., Elgarisi M., Dagan S. Determination of trace amounts of G-type nerve agents in aqueous samples utilizing “in vial” instantaneous derivatization and liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2017. V. 1512. P. 71.
Garg P., Purohit A., Tak V., Kumar A., Dubey D. Liquid-liquid-solid microextraction and detection of nerve agent simulants by on-membrane Fourier transform infrared spectroscopy // Anal. Chim. Acta. 2012. V. 751. P. 71.
Li B., Wei J., Kong J., Qin M., Yang L., Li C. Rapid detection of Sarin hydrolysis products based on microextraction by packed sorbent combined with Nano-ESI mass spectrometry // Int. J. Mass Spectrom. 2021. V. 461. P. 116513.
Witkiewicz Z., Neffe S. Chromatographic analysis of chemical warfare agents and their metabolites in biological samples // Trends Anal. Chem. 2020. V. 130. P. 115960.
Chenga X., Liub C., Yang Y., Liang L., Chenb B., Yub H., Xiab J., Liub S., Li Y. Advances in sulfur mustard-induced DNA adducts: Characterization and detection // Tox. Lett. 2021. V. 344. P. 46.
Pantazides B., Crow B., Garton J., QuiñonesGonzález J., Blake T., Thomas J., Johnson R. A Simplified method for quantifying sulfur mustard adducts to blood proteins by ultra-high pressure liquid chromatography-isotope dilution tandem mass spectrometry // Chem. Res. Toxicol. 2015. V. 28. № 2. P. 256.
John H., Siegert M., Gandor F., Gawlik M., Kranawetvogl A., Karaghiosoff K., Thiermann H. Optimized verification method for detection of an albumin-sulfur mustard adduct at Cys34 using a hybrid quadrupole time-of-flight tandem mass spectrometer after direct plasma proteolysis // Tox. Lett. 2016. V. 244. P. 103.
Rodin I.A., Braun A.V., Savelieva E.I., Rybalchenko I.V., Ananieva I.A., Shpigun O.A. Rapid method for the detection of metabolite of sulfur mustard 1,1′-sulfonylbis[2-s-(n-acetylcysteinyl)ethane] in plasma and urine by liquid chromatography-negative electrospray-tandem mass spectrometry // J. Liq. Chromatogr. Relat. Technol. 2011. V. 34. № 16. P. 1676.
Boyer A., Ash D., Barr D., Young, C., Driskell W., Whitehead R., Ospina M., Preston K., Woolfitt A., Martinez R., Silks L., Barr J. Quantitation of the sulfur mustard metabolites 1,1'-sulfonylbis[2-(methylthio)ethane] and thiodiglycol in urine using isotope-dilution gas chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. № 5. P. 327.
Eyison R., Sezigen S., Ortatatli M., Kenar L. Optimized gas chromatography-tandem mass spectrometry for 1,1'-sulfonylbis[2-(methylthio) ethane] quantification in human urine // J. Chromatogr. Sci. 2019. V. 57. № 5. P. 397.
Riches J., Read R.W., Black R.M. Analysis of the sulphur mustard metabolites thiodiglycol and thiodiglycol sulphoxide in urine using isotope-dilution gas chromatography-ion trap tandem mass spectrometry // J. Chromatogr. B. 2007. V. 845. P. 114.
Read R.W., Black R.M. Analysis of beta-lyase metabolites of sulfur mustard in urine by electrospray liquid chromatography-tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 346.
Read R.W., Black R.M. Analysis of the sulfur mustard metabolite 1,1'-sulfonylbis[2-S-(N-acetylcysteinyl)ethane] in urine by negative ion electrospray liquid chromatography- tandem mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 352.
Liu C., Liu S., Xi H., Yu H., Zhou S., Huang G., Liang L., Liu J. Simultaneous quantification of four metabolites of sulfur mustard in urine samples by ultra-high performance liquid chromatography-tandem mass spectrometry after solid phase extraction // J. Chromatogr. A. 2017. V. 1492. P. 41.
Lee J., Lee Y. Solid-phase extraction of sulfur mustard metabolites using an activated carbon fiber sorbent // J. Anal. Toxicol. 2016. V. 40. P. 64.
John H., Koller M., Worek F., Thiermann H., Siegert M. Forensic evidence of sulfur mustard exposure in real cases of human poisoning by detection of diverse albumin–derived protein adducts // Arch. Toxicol. 2019. V. 93. P.1881.
Zhang Y., Yue L., Nie Z., Chen J., Guo L., Wu B., Feng J., Liu Q., Xie J. Simultaneous determination of four sulfur mustard–DNA adducts in rabbit urine after dermal exposure by isotope-dilution liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2014. V. 961. P. 29.
Орлова О.И., Каракашев Г.В., Савельева Е.И. Совместное определение аддуктов сернистого иприта с гуанином и ацетилцистеином в моче методом высокоэффективной жидкостной хроматографии–тандемной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2020. Т. 75. № 8. С. 714. (Orlova O.I., Karakashev G.V., Savelieva E.I. Simultaneous determination of sulfur mustard adducts with guanine and acetylcysteine in urine by high-resolution high-performance liquid chromatography–tandem mass spectrometry // J. Anal. Chem. 2020. V. 75. № 8. P. 1011.)
Nie Z., Liu Q., Xie J. Improvements in monitoring the N-terminal valine adduct in human globin after exposure to sulfur mustard and synthesis of reference chemicals // Talanta. 2011. V. 85. P. 1154.
Корягина Н.Л., Шачнева М.Д., Уколов А.И., Савельева Е.И., Хлебникова Н.С., Радилов А.С. Усовершенствованный способ обнаружения глобинового аддукта сернистого иприта методом тандемной газовой хроматомасс-спектрометрии // Масс-спектрометрия. 2017. Т. 14. № 4. С. 266. (Koryagina N.L., Shachneva M.D., Ukolov A.I., Savel’eva E.I., Khlebnikova N.S., Radilov A.S. An improved procedure for the gas chromatography–tandem mass spectrometry detection of the globin adduct of sulfur mustard // J. Anal. Chem. 2018. V. 73. № 13. P. 1269.)
Корягина Н.Л., Савельева Е.И., Хлебникова Н.С., Радилов А.С. Определение тиодигликоля и его оксида в биообразцах методом газовой хроматомасс-спектрометрии // Масс-спектрометрия. 2017. Т. 14. № 2. С. 124. (Koryagina N.L., Savelieva E.I., Khlebnikova N.S., Radilov A.S. Determination of thiodiglycol and its oxide in biomedical samples by gas chromatography–mass spectrometry // J. Anal. Chem. 2018. V. 73. № 13. P. 1209.)
Somani S., Lukey B.J., Romano J.A., Jr., Romano J.A., Salem H. Chemical Warfare Agents: Toxicity at Low Levels. Boca Raton, FL, USA: CRC Press, 2001. 464 p.
Romano J.A., Lukey B.J., Salem H. Chemical Warfare Agents, Chemistry, Pharmacology, and Therapeutics, 2nd Ed. Boca Raton, FL, USA: CRC Taylor & Francis, 2007. 752 p.
Lukey B.J., Romano J.A., Jr., Salem H. Chemical Warfare Agents: Biomedical and Psychological Effects, Medical Countermeasures, and Emergency Response. Boca Raton, FL: CRC Press, 2019. 848 p.
Barr J., Driskell W., Aston L. Quantitation of metabolites of the nerve agents sarin, soman, cyclohexylsarin, VX and russian VX in human urine using isotope-dilution gas chromatography-mass spectrometry // J. Anal. Toxicol. 2004. V. 28. P. 372.
Hamelin E., Schulze N., Shaner R. Quantitation of five organophosphorus nerve agent metabolites in serum using hydrophilic interaction liquid chromatography and tandem mass spectrometry // Anal. Bioanal. Chem. 2014. V. 406. № 21. P. 5195.
Black R., Muir J. Derivatisation reactions in the chromatographic analysis of chemical warfare agents and their degradation products // J. Chromatogr A. 2003. V. 1000. P. 253.
Riches J., Morton I., Read R.W. The trace analysis of alkyl alkylphosphonic acids in urine using gas chromatography-ion trap negative ion tandem mass spectrometry // J. Chromatogr. B. 2005. V. 816. № 1–2. P. 251.
Evans R., Jakubowski E., Muse W. Quantification of sarin and cyclosarin metabolites isopropyl methylphosphonic acid and cyclohexyl methylphosphonic acid in minipig plasma using isotope-dilution and liquid chromatography- time-of-flight mass spectrometry // J. Anal. Toxicol. 2008. V. 32. № 1. P. 78.
Hamelin E., Schulze N., Shaner R. Quantitation of five organophosphorus nerve agent metabolites in serum using hydrophilic interaction liquid chromatography and tandem mass spectrometry // Anal. Bioanal. Chem. 2014. V. 406. № 21. P. 5195.
Hall G., Wood G., Paterson J. Half-life of plasma cholinesterase // Br. J. Anaesth. 1984. V. 56. P. 903.
Мурашко Е.А., Дубровский Я.А., Бабаков В.Н. Аддукты фосфорорганических отравляющих веществ с белками крови как маркёры отравления (обзор) // Медицина экстремальных ситуаций. 2019. Т. 21. № 1. С. 124.
Schaller J., Gerber S., Kämpfer U., Lejon S., Trachsel Ch. Human Blood Plasma Proteins: Structure and Function. John Wiley & Sons, Ltd., 2008. 538 p.
Myers D. Studies on cholinesterase. Determination of the molar concentration of pseudo-cholinesterase in serum // Biochem. J. 1952. V. 51. P. 303.
Fidder A., Hulst A., Noort D. Retrospective detection of exposure to Organophosphorous anti-cholinesterases: Mass spectrometric analysis of phosphylated human butyrylcholinesterase // Chem. Res. Toxicol. 2002. V. 15. P. 582.
Корягина Н.Л., Савельева Е.И., Каракашев Г.В., Бабаков В.Н., Дубровский Я.А., Уколова Е.С., Хлебникова Н.С., Мурашко Е.А., Конева В.Ю., Уколов А.И., Копейкин В.А., Радилов А.С. Определение конъюгированных с белками метаболитов фосфорорганических отравляющих веществ в плазме крови // Журн. аналит. химии. 2016. Т. 71. № 8. С. 883. (Koryagina N.L., Savel’eva E.I., Karakashev G.V., Babakov V.N., Dubrovskii Y.A., Ukolova E.S., Khlebnikova N.S., Murashko E.A., Koneva V.Y., Ukolov A.I., Kopeikin V.A., Radilov A.S. Determination of protein adducts of organophosphorus nerve agents in blood plasma // J. Anal. Chem. 2016. V. 71. № 8. P. 849.)
Sporty J., Lemire S., Jakubowski E. Immunomagnetic separation and quantification of butyrylcholinesterase nerve agent adducts in human serum // Anal. Chem. 2010. V. 82. № 15. P. 6593.
Carter M., Crow B., Pantazides B. Direct quantitation of methyl phosphonate adducts to human serum butyrylcholinesterase by immunomagnetic-UHPLC-MS/MS // Anal. Chem. 2013. V. 85. № 22. P. 11106.
Knaack J., Zhou Y., Abney C., Jacob J., Prezioso S., Hardy K. A high-throughput diagnostic method for measuring human exposure to organophosphorus nerve agents // Anal. Chem. 2012. V. 84. № 21. P. 9470.
Schopfer L., Lockridge O., David E. Purification of human butyrylcholinesterase from frozen Cohn fraction IV-4 by ion exchange and Hupresin affinity chromatography // PLoS One. 2019. V. 14. № 1.
Schopfer L., Furlong C., Lockridge O. Development of diagnostics in the search for an explanation of aerotoxic syndrome // Anal. Biochem. 2010. V. 404. № 1. P. 64.
Johnson D., Carter M., Crow B. Quantitation of ortho-cresyl phosphate adducts to butyrylcholinesterase in human serum by immunomagnetic-UHPLC-MS/MS // J. Mass Spectrom. 2015. V. 50. № 4. P. 683.
Noort D., Fidder A., Van der Schans M. Verification of exposure to organophosphates: generic mass spectrometric method for detection of human butyrylcholinesterase adducts // Anal. Chem. 2006. V. 78. № 18. P. 6640.
Van der Schans M., Hulst A., Van der Riet-van Oeveren D. New tools in diagnosis and biomonitoring of intoxications with organophosphorothioates: Case studies with chlorpyrifos and diazinon // Chem. Biol. Interact. 2013. V. 203. № 1. P. 96.
Crow B., Pantazides B., Quinones-Gonzalez J. Simultaneous measurement of tabun, sarin, soman, cyclosarin, VR, VX, and VM adducts to tyrosine in blood products by isotope dilution UHPLC-MS/MS // Anal. Chem. 2014. V. 86. № 20. P. 10397.
ranawetvogl A., Kuppers J., Gutschow M. Identification of novel disulfide adducts between the thiol containing leaving group of the nerve agent VX and cysteine containing tripeptides derived from human serum albumin // Drug Test Anal. 2017. V. 9. № 8. P. 1192
Kranawetvogl A., Kuppers J., Siegert M. Bioanalytical verification of V-type nerve agent exposure: simultaneous detection of phosphonylated tyrosines and cysteine-containing disulfideadducts derived from human albumin // Anal. Bioanal. Chem. 2018. V. 410. № 5. P. 1463.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии