

Журнал аналитической химии, 2022, T. 77, № 1, стр. 53-69
Новый способ нахождения пределов определения элементов, оценки динамического диапазона определяемых содержаний и выявления матричных и межэлементных влияний в спектральном анализе (атомно-абсорбционная спектрометрия и ИСП-методы анализа)
О. Н. Гребнева-Балюк *
Институт геохимии и аналитической химии им. В.И. Вернадского Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: grebneva@geokhi.ru
Поступила в редакцию 07.04.2021
После доработки 09.08.2021
Принята к публикации 09.08.2021
- EDN: IVRXXQ
- DOI: 10.31857/S0044450222010042
Аннотация
Рассмотрены общепринятые и инновационные варианты нахождения пределов обнаружения/определения элементов в спектральном анализе, в частности, для атомно-абсорбционной спектрометрии с пламенной атомизацией, атомно-эмиссионной спектрометрии с индуктивно связанной плазмой, масс-спектрометрии с индуктивно связанной плазмой. Предложен простой способ нахождения величины нижней границы определяемых содержаний – экспериментального предела определения (сопр. эксп). Применимость и правильность предложенного способа продемонстрированы на примере анализа водных растворов и растворов, содержащих неорганическую или органическую матрицу. Показана возможность использования предложенного способа для оценки динамического диапазона определяемых содержаний и выявления матричных и межэлементных влияний. Экспериментальные данные подтверждены теоретическими расчетами, выполненными с помощью уравнения Саха.
Минимально возможная определяемая концентрация элемента является одной из важных характеристик метода/методики/оборудования. Информация о достижимых пределах обнаружения/определения (собн/сопр) облегчает правильный выбор оборудования при комплектации аналитической лаборатории, особенно, если лаборатория ориентирована на определение следовых и ультраследовых концентраций аналитов [1].
Рассмотрим общепринятые подходы к нахождению собн/сопр и инновационные способы расчета этой характеристики. Одним из широко используемых терминов является “предел обнаружения, соответствующий минимальному значению концентрации, которое может быть обнаружено (с вероятностью Р) при использовании минимального статистически значимого сигнала как порогового значения. При этом пороговое значение понимается как минимальное статистически значимое значение “чистого” сигнала, который может быть зарегистрирован с вероятностью Р” [2]. собн рассчитывают по формуле [2]. Здесь и далее все формулы приводятся в авторском написании:
В России термины cобн/cопр регламентируются Национальным стандартом РФ ГОСТ Р 52361–2018 “Контроль объекта аналитический. Термины и определения” [3]. Так, согласно этому ГОСТу “предел обнаружения (аналита): наименьшее содержание аналита, при котором он может быть обнаружен по данной методике анализа вещества или материала объекта аналитического контроля с заданной доверительной вероятностью. Примечание: пределом обнаружения обычно считают содержание аналита, равное сумме результата холостого опыта и его стандартного отклонения, умноженного на коэффициент, соответствующий заданной доверительной вероятности”, т.е.:
где cобн – предел обнаружения, схол – результат холостого опыта, k – коэффициент, соответствующий заданной доверительной вероятности, σ – стандартное отклонение.Согласно ГОСТу [3] “предел определения (аналита): наименьшее содержание аналита, которое может быть количественно определено с помощью данной методики анализа вещества или материала объекта аналитического контроля с установленными значениями характеристик погрешности или неопределенности”:
Основные варианты расчета сопр в атомной спектрометрии рассмотрены в обзоре [4]. Автор провел детальный анализ пяти способов [4], а позже представил подробное описание своего метода расчета сопр и назвал его методом “профиля точности” [5]. Данный метод позволяет аналитику определять область достоверности, где самая низкая точка градуировочного графика есть предел количественного определения. Авторы работы [5] рассчитали пределы количественного определения палладия методом масс-спектрометрии с индуктивно связанной плазмой (МС-ИСП), а также урана и бора методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой (АЭС-ИСП); изучили параметры, которые могут влиять на значения сопр; сделали выводы о сопоставимости теоретически рассчитанных и полученных практически значений.
Инновационный способ расчета собн/сопр в МС-ИСП с использованием метода Монте-Карло и численного интегрирования рассмотрен в работах [6–8]. Авторы упростили формулы, ранее предложенные в работах [9–12], получив
Хорошо согласуются результаты теоретических расчетов пределов обнаружения и экспериментальные данные для АЭС-ИСП и МС-ИСП в работах [13–16], где рассчитаны стандартное отклонение и предел обнаружения с учетом флуктуации параметров спектрометра, линейности и нелинейности градуировочных графиков.
Таким образом, несмотря на большое количество описанных выше подходов к расчету собн/сопр элементов, сохраняется потребность в быстрой, простой, понятной, практической оценке предельных возможностей оборудования/методики.
В основе предложенной нами модели лежит описание предела определения “как самой низкой концентрации аналита, которая может быть определяется с приемлемым уровнем неопределенности и, следовательно, может быть установлена произвольно как требуемый нижний предел рабочего диапазона метода”, или другими словами, “предел определения – это наименьшая концентрация на градуировочном графике, измеренная в выбранных инструментальных условиях с учетом чистоты реактивов” [6, 17]. Наименьшая концентрация – это значение, соответствующее холостому опыту (бланку). Согласно определению ГОСТа [3] холостой опыт (бланк) – “проба вещества [материала] объекта аналитического контроля, аналогичная аналитической пробе, но не содержащая аналита”. Другими словами, это неизвестный образец, в котором нет аналита. Данное определение холостого опыта (бланка) не соответствует современным высокочувствительным спектральным методам анализа, в частности МС-ИСП с высоким разрешением, поскольку практически все элементы периодической системы присутствуют в любых анализируемых объектах на уровне нг/мл (ppb)–пг/мл (ppt). Принимая данную модель, формулу для расчета экспериментально полученного значения сопр. эксп можно представить следующим образом:
Модельные эксперименты проводили на системах, имитирующих состав геологических и технологических объектов, для элементного состава которых характерны межэлементные и матричные влияния.
Метод атомно-абсорбционной спектрометрии с пламенной атомизацией (ПААС) применяли при исследовании водных растворов, содержащих золото и железо в разных концентрациях. Пара золото–железо является хорошим примером межэлементных влияний при определении золота в железосодержащих объектах, в частности в геологических образцах. Методом АЭС-ИСП исследовали водные растворы и растворы с матричным содержанием циркония (1 мг/мл). Выбор циркония неслучаен. Этот элемент имеет богатый набор спектральных линий и при высоких концентрациях оказывает межэлементные и матричные влияния на спектральные линии большинства элементов периодической системы. Выявление и учет влияний актуален при определении примесей в технологических объектах, в частности в оксиде циркония и циркониевых сплавах. Исследования методом МС-ИСП проводили на примере водных растворов и растворов, содержащих органический углерод. Присутствие углерода в анализируемых растворах, даже после разложения и концентрирования, характерно для различных схем пробоподготовки как природных, так и технологических объектов. Ранее нами показано, что остаточный углерод, находящийся в растворах после разложения нефти разной вязкости, влияет на формирование аналитических сигналов элементов при МС-ИСП-определении [18]. В связи с этим важно выявить, оценить и учесть степень влияния углерода при определении элементов на уровне ультраследовых концентраций.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование и материалы. Определение элементов методом ПААС выполняли на спектрометре АА-7000 (Shimadzu, Япония). Рабочие условия определения золота: длина волны 242.8 нм; тип пламени (скорость подачи) ацетилен (1.8 л/мин)–воздух; коррекция фона дейтериевая; ток на лампе 10 мА; ширина щели 0.7 нм.
Для АЭС-ИСП-определений применяли спектрометры компаний “Thermo Scientific” (США) и “Teledyne Leeman Labs.” (США). Поскольку в статье приведены сравнительные данные, с целью исключения рекламы далее будем использовать обозначения “АЭС-ИСП спектрометр 1”, “АЭС-ИСП спектрометр 2” и “АЭС-ИСП спектрометр 3”.
Измерения методом МС-ИСП выполняли на спектрометре высокого разрешения с двойной фокусировкой ELEMENT XR (Finnigan MAT., Германия). Рабочие условия: мощность генератора – 1200 Вт; распылительная камера циклоновая; распылитель MicroMist (конический); конуса никелевые; скорости плазмообразующего вспомогательного и распылительного потоков 16, 0.9 и 1.010 соответственно, л/мин; разрешение 300, 4000, 10 000 (низкое, среднее, высокое).
С целью повышения качества определения следовых и ультраследовых концентраций веществ бидистиллированную воду и конц. HNO3 (ос. ч., Химмед, Россия) дополнительно очищали с помощью системы перегонки без кипения BSB-939-IR (Berghof, Германия).
Рабочие растворы для ИСП-определений готовили путем последовательного разбавления по массе с использованием стандартных многоэлементных растворов с содержанием 100 мкг/мл каждого элемента (High-Purity Standards, США). Для приготовления модельных растворов использовали одноэлементные растворы Au – 100 мкг/мл, Fe – 10 мг/мл, Сd – 10 мг/мл и Zr – 10 мг/мл (Inorganic Ventures, США). В качестве источника углерода для приготовления модельных растворов, содержащих органическую матрицу, использовали лимонную кислоту х. ч. (ЛенРеактив, Россия).
Все расчеты проводили с использованием программного обеспечения Excel.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Предложенный подход экспериментального нахождения предела определения включает анализ как минимум трех растворов с известными концентрациями аналита, а также анализ холостого раствора. Затем по формуле
Далее выполняют обработку полученных результатов. Если значения близки или имеют незначительное расхождение, то за предел определения принимают среднее арифметическое сопр. эксп. Если значения сопр. эксп имеют значительные расхождения, то можно сделать вывод о наличии матричных и межэлементных влияний и, следовательно, принять решение об использовании спектральной линии или изотопа. В случае, если из анализа невозможно исключить спектральную линию или изотоп, выявленные помехи можно учесть путем: 1) математической коррекции фона или межэлементного влияния, применяя выверенные коэффициенты, или 2) использования для калибровки спектрометра синтетических растворов сравнения, имитирующих состав анализируемого образца, или 3) отделения мешающего элемента, или 4) использования инструментальных возможностей спектрометра, в частности за счет применения реакционных/коллизионных ячеек в квадрупольных масс-спектрометрах.
Ниже приведены примеры нахождения предела определения различными спектральными методами анализа, а также продемонстрирована возможность использования предложенного подхода для оценки динамического диапазона определяемых содержаний и выявления матричных и межэлементных влияний.
Расчет предела определения методом атомно-абсорбционной спектрометрии с пламенной атомизацией. Для расчета сопр. эксп выбрали пару элементов с известным спектральным наложением – золото (242.795 нм) и железо (242.820 нм), которое необходимо учитывать, в частности, при анализе геологических проб. В табл. 1 приведены значения сопр. эксп для золота (242.8 нм), полученные при анализе модельных растворов – одноэлементных водных и растворов, содержащих кроме золота 100, 1000 и 5000 мкг/мл железа. Из данных табл. 1 видно, что с увеличением концентрации железа со 100 до 1000 мкг/мл сопр. эксп повышается как минимум в два раза.
Таблица 1.
Значения сопр. эксп, полученные для золота (242.8 нм) в водных растворах в отсутствие и в присутствии железа
| Раствор | Поглощение, A | сопр. эксп, мкг/мл |
|---|---|---|
| Холостой раствор (1%-ная HNO3) | 0.0011 | |
| 1 мкг/мл Au | 0.0492 | 0.022 |
| 2 мкг/мл Au | 0.0891 | 0.025 |
| 3 мкг/мл Au | 0.1312 | 0.025 |
| 4 мкг/мл Au | 0.1793 | 0.025 |
| Холостой раствор (100 мкг/мл Fe) | 0.0015 | |
| 1 мкг/мл Au + 100 мкг/мл Fe | 0.0466 | 0.032 |
| 2 мкг/мл Au + 100 мкг/мл Fe | 0.0898 | 0.033 |
| 3 мкг/мл Au + 100 мкг/мл Fe | 0.1355 | 0.033 |
| 4 мкг/мл Au + 100 мкг/мл Fe | 0.1831 | 0.033 |
| Холостой раствор (1000 мкг/мл Fe) | 0.0032 | |
| 1 мкг/мл Au + 1000 мкг/мл Fe | 0.0514 | 0.062 |
| 2 мкг/мл Au + 1000 мкг/мл Fe | 0.0972 | 0.066 |
| 3 мкг/мл Au + 1000 мкг/мл Fe | 0.1452 | 0.066 |
| 4 мкг/мл Au + 1000 мкг/мл Fe | 0.2030 | 0.067 |
| Холостой раствор (5000 мкг/мл Fe) | 0.0034 | |
| 1 мкг/мл Au + 5000 мкг/мл Fe | 0.0532 | 0.064 |
| 2 мкг/мл Au + 5000 мкг/мл Fe | 0.1083 | 0.063 |
| 3 мкг/мл Au + 5000 мкг/мл Fe | 0.1609 | 0.063 |
| 4 мкг/мл Au + 5000 мкг/мл Fe | 0.2129 | 0.064 |
Когда характеристическая концентрация (в нашем случае значения сопр. эксп) в реальной пробе с высокой концентрацией солевой матрицы отличается от подобного параметра в чистом водном растворе более чем на ±5%, можно говорить о влиянии матрицы на результат анализа [1]. Следовательно, для корректного определения золота в присутствии железа с концентрацией более 100 мкг/мл необходимо: 1) отделение железа или 2) переход на спектральную линию золота, свободную от влияния железа, либо 3) использование коэффициентов математической коррекции для учета влияний.
Расчет предела определения методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой. В табл. 2 приведены экспериментальные данные, полученные на трех АЭС-ИСП-спектрометрах разных производителей.
Таблица 2.
Найденные значения сопр. эксп элементов методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой
| Содержание элемента, мкг/мл | АЭС-ИСП-спектрометр 1 | АЭС-ИСП-спектрометр 2 | АЭС-ИСП-спектрометр 3 | |||||
|---|---|---|---|---|---|---|---|---|
| водные растворы | растворы, содержащие 1 мг/мл циркония (в том числе и холостой) | водные растворы | водные растворы | |||||
| I, имп/с | сопр. эксп, мкг/мл | I, имп/с | сопр. эксп. мкг/мл; влияние Zr |
I, имп/с | сопр. эксп, мкг/мл | I, имп/с | сопр. эксп, мкг/мл | |
| Al 308.2 нм | ||||||||
| Хол. р-р | 0.1245 | – | 25 112 | 160 | ||||
| 0.01 | – | – | – | 26 597 | 0.009 | – | ||
| 0.1 | 0.204 | 0.06 | – | – | 36 264 | 0.07 | 435 | 0.04 |
| 1 | 0.8722 | 0.14 | – | – | 126 877 | 0.2 | 3151 | 0.05 |
| 10 | 8.491 | 0.15 | – | – | 884 868 | 0.3 | 29 350 | 0.06 |
| Al 396.1 нм | ||||||||
| Хол. р-р | 0.0113 | 10.65 | 227 | 19 | ||||
| 0.01 | – | 0.01 | 10.86 | 0.01 | 2633 | 0.0009 | – | |
| 0.1 | 0.1027 | 0.01 | 14.88 | 0.07 | 33 776 | 0.0007 | 45.8 | 0.04 |
| 1 | 0.8801 | 0.01 | 57.14 | 0.19 | 333 927 | 0.0007 | 393 | 0.05 |
| 10 | 11.03 | 3057 485 | 0.0007 | 3695 | 0.05 | |||
| Zr 396.3 | ||||||||
| Ba 455.4 нм | ||||||||
| Хол. р-р | 0.0008 | 14.43 | 13 327 | 47 | ||||
| 0.01 | – | 26.83 | 0.005 | 301 027 | 0.0004 | – | ||
| 0.1 | 0.0931 | 0.0009 | 155.7 | 0.009 | 2 853 087 | 0.0005 | 1912 | 0.0025 |
| 1 | 0.9306 | 0.0009 | 1431 | 0.01 | 26 676 799 | 0.0005 | 18 638 | 0.0025 |
| 10 | 8.936 | 0.0009 | 214 947 297 | 0.0006 | 184 953 | 0.0025 | ||
| Zr 456.1 | ||||||||
| Ca 317.9 нм | ||||||||
| Хол. р-р | 0.2214 | – | 4797 | 78 | ||||
| 0.01 | – | – | – | 8344 | 0.006 | – | ||
| 0.1 | 0.2924 | 0.08 | – | – | 45 912 | 0.01 | 2169 | 0.004 |
| 1 | 0.9205 | 0.2 | 361 398 | 0.01 | 13 922 | 0.006 | ||
| 10 | 7.443 | 0.3 | – | – | 2 894 386 | 0.02 | 125 803 | 0.006 |
| Cd 214.4 нм | ||||||||
| Хол. р-р | 0.1339 | 3.539 | 180 | 2020 | ||||
| 0.01 | – | 4.528 | 0.008 | 3044 | 0.0006 | – | ||
| 0.1 | 5.541 | 0.002 | 13.39 | 0.03 | 28 866 | 0.0006 | 2300 | 0.09 |
| 1 | 61.1 | 0.002 | 104.9 | 0.03 | 272 389 | 0.0007 | 22 602 | 0.09 |
| 10 | 603.1 | 0.002 | 2 130 748 | 0.0008 | 208 711 | 0.09 | ||
| Zr 214.4 | ||||||||
| Cd 228.8 нм | ||||||||
| Хол. р-р | 0.0018 | 6.945 | 694 | – | ||||
| 0.01 | – | 8.401 | 0.008 | 2410 | 0.003 | – | ||
| 0.1 | 0.0888 | 0.002 | 21.95 | 0.08 | 18 021 | 0.004 | – | – |
| 1 | 0.984 | 0.002 | 162.9 | 0.04 | 168 128 | 0.004 | – | – |
| 10 | 8.878 | 0.002 | 1 368 411 | 0.005 | – | – | ||
| Zr 227.6 Zr 228.3 |
||||||||
| Ce 446.0 нм | ||||||||
| Хол. р-р | 0.2622 | 76.92 | – | – | ||||
| 0.01 | – | – | – | – | ||||
| 0.1 | 2.748 | 0.01 | 77.33 | 0.01 | – | – | – | – |
| 1 | 22.51 | 0.01 | 81.39 | 0.09 | – | – | – | – |
| 10 | 226.4 | 0.01 | 118.4 | 0.65 | – | – | – | – |
| Zr 445.7 Zr 446.6 |
||||||||
| Cr 267.7 нм | ||||||||
| Хол. р-р | 0.1218 | 11.41 | 623 | – | ||||
| 0.01 | – | 11.92 | 0.01 | 4297 | 0.001 | – | ||
| 0.1 | 3.522 | 0.004 | 16.81 | 0.07 | 29 759 | 0.002 | – | – |
| 1 | 29.61 | 0.004 | 64.11 | 5.5 | 270 483 | 0.002 | – | – |
| 10 | 280.1 | 0.004 | 2 201 210 | 0.003 | – | – | ||
| Zr 267.7 | ||||||||
| Cr 357.8 нм | ||||||||
| Хол. р-р | 0.0044 | 11.03 | – | 18 | ||||
| 0.01 | – | 11.54 | 0.01 | – | – | – | ||
| 0.1 | 0.1012 | 0.004 | 16.37 | 0.08 | – | – | 1442 | 0.001 |
| 1 | 0.8966 | 0.005 | 67.73 | 0.2 | – | – | 14 029 | 0.001 |
| 10 | 10.26 | 0.004 | – | – | 129 020 | 0.001 | ||
| Zr 358.5 | ||||||||
| Cu 224.7 нм | ||||||||
| Хол. р-р | 0.1521 | 2.757 | 550 | – | ||||
| 0.01 | – | 2.863 | 0.01 | 1173 | 0.005 | – | ||
| 0.1 | 0.7933 | 0.02 | 3.976 | 0.07 | 6143 | 0.009 | – | – |
| 1 | 7.405 | 0.02 | 15.99 | 0.2 | 50 137 | 0.01 | – | – |
| 10 | 81.21 | 0.02 | 400 434 | 0.01 | – | – | ||
| Zr 224.7 | ||||||||
| Cu 327.3 нм | ||||||||
| Хол. р-р | – | – | ||||||
| 0.01 | 0.055 | 31.44 | – | – | – | |||
| 0.1 | 6.126 | 0.0009 | 32.32 | 0.01 | – | – | – | – |
| 1 | 62.59 | 0.0009 | 41.45 | 0.08 | – | – | – | – |
| 10 | 661.5 | 0.0008 | 135.2 | 0.2 | – | – | – | – |
| Zr 327.9 | ||||||||
| Fe 240.4 нм | ||||||||
| Хол. р-р | 0.0522 | 7.125 | – | – | ||||
| 0.01 | – | 7.361 | 0.01 | – | – | – | ||
| 0.1 | 1.127 | 0.005 | 9.024 | 0.08 | – | – | – | – |
| 1 | 7.357 | 0.007 | 25.03 | 0.28 | – | – | – | – |
| 10 | 65.52 | 0.008 | – | – | – | – | ||
| Zr 240.5 | ||||||||
| Fe 259.9 нм | ||||||||
| Хол. р-р | 0.022 | 6.327 | 7294 | – | ||||
| 0.01 | – | 7.345 | 0.009 | 17 088 | 0.004 | – | ||
| 0.1 | 0.1582 | 0.01 | 14.46 | 0.04 | 69 577 | 0.01 | – | – |
| 1 | 0.999 | 0.02 | 83.33 | 0.08 | 546 404 | 0.01 | – | – |
| 10 | 8.802 | 0.03 | 4 347 905 | 0.02 | – | – | ||
| Zr 259.4 | ||||||||
| Ge 206.8 нм | ||||||||
| Хол. р-р | 0.0345 | 6.789 | – | – | ||||
| 0.01 | – | 6.925 | 0.01 | – | – | – | ||
| 0.1 | 0.1284 | 0.03 | 8.29 | 0.08 | – | – | – | – |
| 1 | 1.071 | 0.03 | 22.71 | 0.3 | – | – | – | – |
| 10 | 10.94 | 0.03 | – | – | – | – | ||
| Zr 206.9 | ||||||||
| Hf 232.2 нм | ||||||||
| Хол. р-р | 0.0065 | 4.254 | – | – | ||||
| 0.01 | – | 4.356 | 0.01 | – | – | – | ||
| 0.1 | 0.0859 | 0.008 | 5.369 | 0.08 | – | – | – | – |
| 1 | 0.796 | 0.008 | 15.41 | 0.27 | – | – | – | – |
| 10 | 7.775 | 0.008 | – | – | – | – | ||
| Zr 230.8 | ||||||||
| Hf 263.8 нм | ||||||||
| Хол. р-р | 0.0153 | 8.338 | – | – | ||||
| 0.01 | – | 8.653 | 0.009 | – | – | – | ||
| 0.1 | 0.1451 | 0.01 | 11.81 | 0.07 | – | – | – | – |
| 1 | 1.266 | 0.01 | 42.05 | 0.2 | – | – | – | – |
| 10 | 12.57 | 0.01 | – | – | – | – | ||
| Zr 262.8 Zr 264.4 |
||||||||
| Mg 285.2 нм | ||||||||
| Хол. р-р | 0.0618 | 15.54 | – | 0.3 | ||||
| 0.01 | – | 16.54 | 0.009 | – | – | – | ||
| 0.1 | 0.1419 | 0.04 | 30.59 | 0.05 | – | – | 72 | 0.0004 |
| 1 | 0.884 | 0.07 | 173 | 0.09 | – | – | 727 | 0.0004 |
| 10 | 8.533 | 0.07 | – | – | 7092 | 0.0004 | ||
| Mn 257.6 нм | ||||||||
| Хол. р-р | 0.0005 | 8.804 | 2589 | 0.7 | ||||
| 0.01 | – | 11.72 | 0.008 | 15 089 | 0.002 | – | ||
| 0.1 | 0.0842 | 0.0006 | 39.95 | 0.02 | 121 340 | 0.002 | 7068 | 0.00001 |
| 1 | 0.8037 | 0.0006 | 320.3 | 0.03 | 1 092 274 | 0.002 | 68 733 | 0.00001 |
| 10 | 7.752 | 0.0006 | 8 529 916 | 0.003 | 621 823 | 0.00001 | ||
| Mn 259.3 нм | ||||||||
| Хол. p-р | 0.1732 | 10.18 | – | 4 | ||||
| 0.01 | – | 12.66 | 0.008 | – | – | – | ||
| 0.1 | 15.17 | 0.001 | 36.86 | 0.03 | – | – | 5722 | 0.00007 |
| 1 | 143.8 | 0.001 | 277 | 0.04 | – | – | 55 958 | 0.00007 |
| 10 | 1388 | 0.001 | – | – | 506 824 | 0.00008 | ||
| Zr 259.4 | ||||||||
| Mo 202.0 нм | ||||||||
| Хол. р-р | 0.0273 | 6.445 | – | – | ||||
| 0.01 | – | 7.186 | 0.009 | – | – | – | ||
| 0.1 | 0.2293 | 0.01 | 14.18 | 0.05 | – | – | – | – |
| 1 | 2.671 | 0.01 | 85.91 | 0.08 | – | – | – | – |
| 10 | 18.04 | 0.015 | – | – | – | – | ||
| Zr 201.7 Zr 202.6 |
||||||||
| Pb 261.4 нм | ||||||||
| Хол. р-р | 0.0111 | 9.17 | – | – | ||||
| 0.01 | – | 11.27 | 0.008 | – | – | – | ||
| 0.1 | 1.497 | 0.0007 | 31.97 | 0.03 | – | – | – | – |
| 1 | 14.53 | 0.0008 | 237.6 | 0.04 | – | – | – | – |
| 10 | 163.3 | 0.0007 | – | – | – | – | ||
| Zr 262.0 | ||||||||
| Sc 361.3 нм | ||||||||
| Хол. р-р | 0.0003 | 15.93 | 1372 | – | ||||
| 0.01 | – | 26.31 | 0.006 | 80 830 | 0.0002 | – | ||
| 0.1 | 0.0995 | 0.0003 | 132.4 | 0.01 | 787 762 | 0.0002 | – | – |
| 1 | 0.9919 | 0.0003 | 1172 | 0.01 | 7 458 295 | 0.0002 | – | – |
| 10 | 9.471 | 0.0003 | 61 551 812 | 0.0002 | – | – | ||
| Ti 334.9 нм | ||||||||
| Хол. р-р | 0.0027 | 30.36 | – | 1368 | ||||
| 0.01 | – | 34.78 | 0.009 | – | – | – | ||
| 0.1 | 0.0991 | 0.003 | 79.54 | 0.04 | – | – | 4400 | 0.03 |
| 1 | 0.9588 | 0.003 | 476.6 | 0.06 | – | – | 45 333 | 0.03 |
| 10 | 8.414 | 0.003 | – | – | 433 112 | 0.03 | ||
| Ti 337.2 нм | ||||||||
| Хол. р-р | 0.8354 | 38.84 | 6126 | – | ||||
| 0.01 | – | 41.85 | 0.01 | 13 367 | 0.005 | – | ||
| 0.1 | 19.98 | 73.2 | 0.05 | 93 148 | 0.007 | – | – | |
| 1 | 185.2 | 0.004 | 315.9 | 0.1 | 858 975 | 0.007 | – | – |
| 10 | 1516 | 0.0045 | 7 221 122 | 0.008 | – | – | ||
| W 207.9 нм | ||||||||
| Хол. р-р | 1.044 | 6.351 | – | – | ||||
| 0.01 | – | 6.576 | 0.01 | – | – | – | ||
| 0.1 | 2.249 | 8.579 | 0.07 | – | – | – | – | |
| 1 | 14.05 | 0.05 | 28.51 | 0.2 | – | – | – | – |
| 10 | 131.4 | 0.07 | – | – | – | – | ||
| 0.08 | Zr 207.7 Zr 208.7 |
|||||||
Отметим, что если три полученных значения сопр. эксп близки, то можно считать, что, во-первых, спектральная линия свободна от помех и может быть успешно использована для определения элемента в анализируемой матрице, а во-вторых, все три полученные значения близки к истинному значению сопр. эксп в выбранных условиях. Если же полученные значения сопр. эксп расходятся, то это свидетельствует о наличии помех, вызванных матричными и/или межэлементными влияниями. При этом аналитик должен исключить ошибки, связанные с приготовлением растворов. Для обеспечения правильности измерения, т.е. исключения систематических погрешностей, растворы рекомендуется готовить с использованием разбавления с контролем их массы.
Анализ литературы показал, что существует несколько способов нахождения предела обнаружения/определения. Для сравнения рассчитали пределы определения некоторых элементов с использованием предложенного нами подхода и по формулам [19, 20]:
Рассчитанные значения сопр. эксп и LOQ приведены в табл. 3. Сравнение значений показывает, что сопр. эксп, полученные по предложенной нами формуле, ниже значений LOQ, рассчитанных по формулам [20]. Стоит обратить внимание на значения сопр. эксп и LOQ для разных концентраций аналита. Следуя предложенному нами подходу, различия в значениях сопр. эксп для одного и того же аналита при разных концентрациях свидетельствует о наличии спектральных помех, которые должны быть выявлены и учтены.
Таблица 3.
Пределы определения некоторых элементов в модельных водных растворах, найденные методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой
| Элемент; λ, нм | Содержание элемента, мкг/мл | I, имп/с | сопр. эксп, мкг/мл | LOQ*, мкг/мл |
|---|---|---|---|---|
| Ba 455.4 | Холостой р-р | 0.0008 | ||
| 0.1 | 0.0931 | 0.0009 | 0.001 | |
| 1 | 0.9306 | 0.0009 | 0.001 | |
| 10 | 8.936 | 0.0009 | 0.001 | |
| Cd 214.4 | Холостой р-р | 0.1339 | ||
| 0.1 | 5.541 | 0.002 | 0.007 | |
| 1 | 61.1 | 0.002 | 0.007 | |
| 10 | 603.1 | 0.002 | 0.007 | |
| Fe 238.2 | Холостой р-р | 0.1014 | ||
| 0.1 | 0.9828 | 0.01 | 0.1 | |
| 1 | 6.076 | 0.017 | 0.15 | |
| 10 | 53.27 | 0.019 | 0.17 |
* Расчет выполнен по формулам [20].
Расчет предела определения методом масс-спектрометрии с индуктивно связанной плазмой. Предложенный нами подход нахождения сопр. эксп использовали для обработки данных, опубликованных в книге [20] (табл. 4). Из полученных значений сопр. эксп можно сделать вывод о том, что выбранные условия обеспечивают правильное и воспроизводимое МС-ИСП-определение 208Pb+ в анализируемом растворе.
Таблица 4.
Результаты расчета сопр. эксп изотопа 208Pb+ при определении методом масс-спектрометрии с индуктивно связанной плазмой (квадруполь)*
| Содержание 208Pb+, мкг/л | I, имп/с | сопр. эксп, мкг/л |
|---|---|---|
| Холостой р-р | 565 | |
| 10 | 19 887 | 0.28 |
| 20 | 45 356 | 0.25 |
| 30 | 59 876 | 0.28 |
| 40 | 78 543 | 0.29 |
| 50 | 99 654 | 0.28 |
* Информация о концентрациях и интенсивностях взята из пособия [20].
Экспериментальные данные, полученные методом МС-ИСП c высоким разрешением, приведены в табл. 5. Поскольку масс-спектрометры высокого разрешения оснащены высокочувствительными детекторами при следовом и ультраследовом анализе важное значение имеют чистота используемой посуды, реактивов, помещения и т.д. [20], а также полное исключение эффекта памяти, который вносит значительную погрешность при расчете сопр [21].
Таблица 5.
Значения сопр. эксп элементов, найденные методом масс-спектрометрии с индуктивно связанной плазмой (с высоким разрешением)
| Содержание элемента, нг/мл | Водные растворы | Растворы, содержащие 520 мг/мл углерода (в том числе холостой) | ||
|---|---|---|---|---|
| I, имп/с | сопр. эксп, нг/мл | I, имп/с | сопр. эксп, нг/мл | |
| 89Y+ (LR) | ||||
| Холостой р-р | 572 | 1922 | ||
| 0.1 | 194 747 | 0.0003 | 244 698 | 0.0008 |
| 1.0 | 19 29 794 | 0.0003 | 2 184 714 | 0.0009 |
| 10.0 | 19 066 578 | 0.0003 | ||
| 139La+ (LR) | ||||
| Холостой р-р | 913 | 3464 | ||
| 0.1 | 203 958 | 0.0004 | 283 191 | 0.001 |
| 1.0 | 2 066 825 | 0.0004 | 2 513 923 | 0.001 |
| 10.0 | 22 825 000 | 0.0004 | ||
| 140Сe+ (LR) | ||||
| Холостой р-р | 748 | 2131 | ||
| 0.1 | 77 514 | 0.001 | 65 233 | 0.003 |
| 1.0 | 769 756 | 0.001 | 675 421 | 0.003 |
| 10.0 | 7 847 423 | 0.001 | ||
| 141Pr+ (LR) | ||||
| Холостой р-р | 264 | 1091 | ||
| 0.1 | 231 804 | 0.0001 | 328 552 | 0.0003 |
| 1.0 | 2 312 848 | 0.0001 | 2 903 756 | 0.0004 |
| 10.0 | 24 408 035 | 0.0001 | ||
| 143Nd+ (LR) | ||||
| Холостой р-р | 135 | 426 | ||
| 0.1 | 26 569 | 0.0005 | 38 783 | 0.001 |
| 1.0 | 260 487 | 0.0005 | 323 511 | 0.001 |
| 10.0 | 2 700 038 | 0.0005 | ||
| 147Sm+ (LR) | ||||
| Холостой р-р | 54 | 150 | ||
| 0.1 | 30 291 | 0.0002 | 44 073 | 0.0003 |
| 1.0 | 299 621 | 0.0002 | 378 159 | 0.0004 |
| 10.0 | 2 732 970 | 0.0002 | ||
| 151Eu+ (LR) | ||||
| Холостой р-р | 89 | 309 | ||
| 0.1 | 90 184 | 0.0001 | 138 396 | 0.0002 |
| 1.0 | 900 154 | 0.0001 | 1 180 331 | 0.0003 |
| 10.0 | 8 964 390 | 0.0001 | ||
| 157Gd+ (LR) | ||||
| Холостой р-р | 40 | 123 | ||
| 0.1 | 31 625 | 0.0001 | 48 006 | 0.0003 |
| 1.0 | 301 284 | 0.0001 | 403 879 | 0.0003 |
| 10.0 | 3 953 201 | 0.0001 | ||
| 159Tb+ (LR) | ||||
| Холостой р-р | 75 | 332 | ||
| 0.1 | 159 952 | 0.00005 | 260 497 | 0.0001 |
| 1.0 | 1 599 013 | 0.00005 | 2 257 719 | 0.0001 |
| 10.0 | 16 016 750 | 0.00005 | ||
| 161Dy+ (LR) | ||||
| Холостой р-р | 42 | 117 | ||
| 0.1 | 48 258 | 0.0001 | 72 872 | 0.0002 |
| 1.0 | 481 951 | 0.0001 | 626 324 | 0.0002 |
| 10.0 | 4 794 708 | 0.0001 | ||
| 165Ho+ (LR) | ||||
| Холостой р-р | 106 | 400 | ||
| 0.1 | 249624 | 0.00004 | 381 956 | 0.0001 |
| 1.0 | 2504921 | 0.00004 | 3 431 097 | 0.0001 |
| 10.0 | 26589003 | 0.00004 | ||
| 166Er+ (LR) | ||||
| Холостой р-р | 79 | 208 | ||
| 0.1 | 78 774 | 0.0001 | 122 904 | 0.0002 |
| 1.0 | 791 002 | 0.0001 | 1062 628 | 0.0002 |
| 10.0 | 7 905 443 | 0.0001 | ||
| 169Tm+ (LR) | ||||
| Холостой р-р | 69 | 338 | ||
| 0.1 | 230 991 | 0.00003 | 369 463 | 0.0001 |
| 1.0 | 2 401 725 | 0.00003 | 3 289 687 | 0.0001 |
| 10.0 | 2 300 7491 | 0.00003 | ||
| 172Yb+ (LR) | ||||
| Холостой р-р | 19 | 97 | ||
| 0.1 | 53 338 | 0.00004 | 80 660 | 0.0001 |
| 1.0 | 503 439 | 0.00004 | 684 741 | 0.0001 |
| 10.0 | 5 129 780 | 0.00004 | ||
| 175Lu+ (LR) | ||||
| Холостой р-р | 503 | 1085 | ||
| 0.1 | 202 753 | 0.0002 | 337 353 | 0.0003 |
| 1.0 | 2 115 302 | 0.0002 | 2 981 497 | 0.0003 |
| 10.0 | 25 157 030 | 0.0002 | ||
| 45Sc+ (MR) | ||||
| Холостой р-р | 29 | 44 | ||
| 0.1 | 7472 | 0.0004 | 7772 | 0.0006 |
| 1.0 | 70 650 | 0.0004 | 66 895 | 0.0007 |
| 51V+ (MR) | ||||
| Холостой р-р | 86 | 93 | ||
| 0.1 | 1358 | 0.006 | 1528 | 0.007 |
| 1.0 | 13 029 | 0.006 | 13 813 | 0.007 |
| 10.0 | 143 587 | 0.006 | ||
| 55Mn+ (MR) | ||||
| Холостой р-р | 989 | 2607 | ||
| 0.1 | 2486 | 0.04 | 3300 | 0.08 |
| 1.0 | 22 959 | 0.04 | 15 804 | 0.16 |
| 10.0 | 225 270 | 0.04 | ||
| 75As+ (HR) | ||||
| Холостой р-р | 23 | 182 | ||
| 0.1 | 71 | 0.03 | – | |
| 1.0 | 469 | 0.05 | 1993 | 0.2 |
| 10.0 | 4932 | 0.05 | 12 504 | 0.3 |
Рассмотрим значения, полученные для 55Mn+ и 75As+ (табл. 5), которые обычно присутствуют в качестве примесей в HNO3 ос. ч., используемой в качестве реагента. В настоящей работе рабочие растворы были приготовлены также с использованием этого реагента. Для выбранных изотопов характерны полиатомные влияния:
Как видно из табл. 5, значение сопр. эксп для 55Mn+ составляет 0.04 нг/мл (“среднее разрешение”) и для 75As+ – 0.05 нг/мл (“высокое разрешение”) в водных модельных растворах. Полученные значения сопр. эксп для 55Mn+ и 75As+ свидетельствуют об отсутствии влияния указанных выше полиатомных молекул на аналитические сигналы этих изотопов.
Одним из распространенных компонентов анализируемых растворов является углерод, который входит в состав исходных образцов, реактивов, воздуха и т.д. Нами показано [18], что углерод, содержащийся в анализируемых растворах, влияет на формирование аналитического сигнала как в АЭС-ИСП, так в МС-ИСП, причем влияния в МС-ИСП выражены сильнее. В табл. 5 приведены значения сопр. эксп, полученные для растворов, содержащих 520 мкг/мл углерода (лимонная кислота). Как видно, присутствие углерода повышает значения сопр. эксп большинства исследованных изотопов. Выявление причин подавления/усиления аналитических сигналов в присутствии углерода является отдельной темой исследований и выходит за рамки данной статьи.
Оценка динамического диапазона определяемых содержаний. “Диапазон определяемого содержания (аналита) – это область значений содержания аналита в пробе вещества или материала объекта аналитического контроля, которые могут быть определены по данной методике анализа вещества или материала” [3]. Чаще всего в спектральном анализе используют прямолинейные градуировочные графики, построенные для определенного диапазона определяемых содержаний.
Предложенный нами подход позволяет оценить динамический диапазон определяемых содержаний элементов. Для оценки динамического диапазона предложено анализировать серию из растворов с концентрациями, последовательно отличающимися друг от друга на порядок величины. Далее необходимо провести расчет сопр. эксп с использованием предложенной формулы и по полученным данным оценить динамический диапазон определяемых содержаний.
В табл. 6 приведены интенсивности аналитического сигнала и значения сопр. эксп, полученные для растворов кадмия в концентрационном диапазоне от 0.001 до 100 мкг/мл методом АЭС-ИСП. Для наглядности построена зависимость lg(сопр. эксп) от концентрации кадмия (рис. 1). Видно, что плато графика соответствует диапазону концентраций 0.1–10 мкг/мл. Другими словами, градуировочный график для кадмия при длине волны 214.4 нм прямолинеен только в диапазоне от 0.1 до 10 мкг/мл. Если необходимы концентрационные диапазоны выше или ниже области прямолинейной зависимости, то можно предложить построение градуировочной зависимости в пределах одного порядка концентрации, например от 0.01 до 0.1 мкг/мл по нескольким точкам. В качестве альтернативы могут быть также рассмотрены концентрирование для низких содержаний или разбавление – для высоких.
Таблица 6.
Значения сопр. эксп кадмия (214.4 нм), полученные методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой
| Содержание Cd, мкг/мл | I, имп/с | сопр. эксп, мкг/мл |
|---|---|---|
| Холостой р-р (1%-ная HNO3) | 1.4 | |
| 0.001 | 5.4 | 0.0003 |
| 0.01 | 15.4 | 0.0009 |
| 0.1 | 82.5 | 0.0017 |
| 1.0 | 808 | 0.0017 |
| 10.0 | 5871 | 0.0024 |
| 100.0 | 14120 | 0.01 |
Рис. 1.
Нахождение динамического диапазона определяемых содержаний кадмия (214.4 нм) методом АЭС-ИСП.

Способ выявления матричных и межэлементных влияний. Аргоновая индуктивно связанная плазма является высокотемпературной (8000–10 000 К) и характеризуется высокой концентрацией электронов и ионов аргона (~1021–1022 см−3). В связи с этим происходит практически полная атомизация/ионизация легкоионизируемых элементов и частичная атомизация/ионизация остальных элементов. С одной стороны, это позволяет определять аналиты, с другой стороны, способствует возникновению межэлементных и матричных влияний, повышающих значения собн/сопр аналитов. В АЭС-ИСП такие помехи возникают из-за большого количества спектральных линий, излучаемых каждым элементом, плазменным газом, поддерживающим плазму, компонентами матрицы образца и компонентами атмосферы, попадающими в плазму. В МС-ИСП помехи чаще возникают из-за перекрывания пиков интересующих изотопов и пиков от полиатомных частиц, полученных из материала образца или за счет комбинации атомов образца и аргона.
Дополнительным фактором, осложняющим ИСП-анализ, является изменение степени атомизации/ионизации при варьировании состава образцов и при переходе от стандартов к образцам. В связи с этим для обеспечения качества получаемых результатов необходимо выявить все источники межэлементных и матричных влияний, правильно выбрать оптимально-компромиссные инструментальные параметры регистрации получаемых сигналов в условиях многоэлементного анализа методами АЭС-ИСП и МС-ИСП и использовать стандартные образцы состава или “синтезировать” образцы сравнения, по возможности полностью имитирующие состав анализируемых образцов. В качестве инструмента для решения проблем, связанных с выявлением спектральных влияний, может быть рассмотрен предложенный нами способ нахождения сопр. эксп.
В качестве примера спектральных влияний можно использовать результаты анализа растворов, содержащих 1 мг/мл циркония (табл. 2). Цирконий имеет богатый спектр и оказывает матричные и межэлементные влияния практически на все выбранные спектральные линии определяемых элементов, что дополнительно подтверждает предложенный способ нахождения сопр. эксп (табл. 2).
Рассмотрим значения сопр. эксп для спектральных линий Cu 224.7 нм и Cu 327.3 нм в присутствии циркония. Из табл. 2 видно, что значения сопр. эксп изменяются при увеличении концентрации меди. Используя предложенный подход, можно говорить о наличии спектральных влияний.
Увеличение значений сопр. эксп при наличии спектральных влияний можно объяснить с помощью расчетов по уравнению Саха (вычисления проведены с использованием ресурса [22]), которое позволяет вычислить степень ионизации газа, находящегося в тепловом равновесии:
В табл. 7 и на рис. 2 приведены расчетные значения интенсивностей спектральных линий меди при 224.7 и 327.3 нм. Из табл. 7 видно, что зависимость суммарной интенсивности спектральных линий меди и циркония от концентрации меди становится нелинейной при изменении соотношения меди и циркония, что дополнительно иллюстрирует рис. 2.
Таблица 7.
Теоретические значения интенсивностей спектральных линий меди и циркония, рассчитанные с помощью уравнения Саха (Те = 1 эВ, Ne = 1 × 1017e/см3)
| Соотношение Сu : Zr, % | Интенсивность линии 224.7 нм, а. е. | ∑I | Интенсивность линии 327.3 нм, а. е. | |||
|---|---|---|---|---|---|---|
| Сu II | Zr III | Сu II + Zr III | Сu I | Zr III | Сu I + Zr III | |
| 1 : 99 | 4752 | 3.2 | 4755 | 1736 | 45.5 | 1782 |
| 0.1 : 99.9 | 478 | 3.9 | 482 | 173.6 | 72.6 | 246.4 |
| 0.01 : 99.99 | 47.8 | 4.3 | 52 | 16.9 | 57.3 | 74.2 |
| 0.001 : 99.999 | 4.8 | 3.7 | 8.5 | 1.6 | 48.5 | 50.3 |
| 0.0001 : 99.9999 | 0.48 | 3.2 | 3.7 | |||
Рис. 2.
Зависимости ∑(I Сu II + I Zr III) (1) и I Cu II (2) (224.7 нм) от концентрации меди в присутствии циркония.

На рис. 3 представлены спектры вблизи аналитической линии Cu 224.7 нм, полученные с помощью уравнения Саха, при варьировании соотношения концентраций Cu и Zr. Видно, что увеличение концентрации циркония значительно влияет на интенсивность аналитической линии меди. Причем, начиная с соотношения концентраций Cu : Zr (0.001 : 99.999) регистрация аналитического сигнала (интенсивности) меди при выбранной длине волны становится невозможной.
Рис. 3.
Спектры вблизи аналитической линии Cu 224.7 нм при варьировании соотношения концентраций Cu и Zr: (а) 0.0001 : 99.9999; (б) 0.001 : 99.999; (в) 0.01 : 99.99; (г) 0.1 : 99.9.
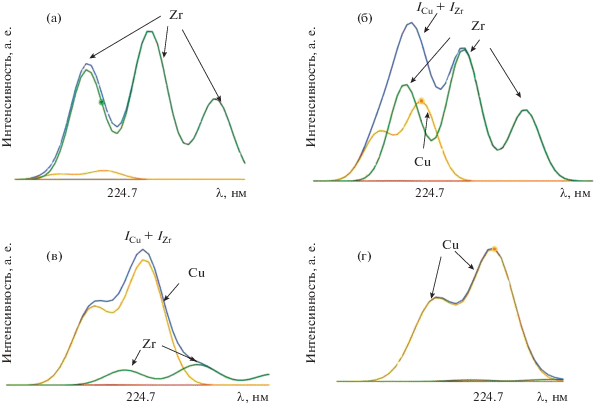
Расчеты, представленные в табл. 2 и 7 (на рис. 2 и 3), свидетельствуют о том, что любые коррекции межэлементного и матричного влияния применимы в пределах одного порядка величины, а при более высоких концентрациях матричных элементов они не могут быть использованы. В качестве решения данной проблемы можно предложить отделение матрицы, полное или частично контролируемое. Данная процедура позволит проводить корректное определение примесей в таких образцах, как циркониевые сплавы, оксид циркония, природные и технологические объекты с высоким содержанием циркония.
* * *
Таким образом, предложен подход к экспериментальному нахождению предела определения (сопр. эксп). Использование полученных значений сопр. эксп позволяет: 1) рассчитывать реальные пределы определения аналитов в образцах; 2) оценивать динамический диапазон определяемых содержаний; 3) выявлять спектральные и неспектральные влияния и выбирать для анализа линии или изотопы, свободные от наложений; 4) оперативно выбирать оптимально-компромиссные условия анализа, в том числе и при многоэлементном определении; 5) оценивать качество получаемых результатов при использовании в расчетах коэффициентов межэлементной коррекции; 6) делать выводы о пригодности спектрального оборудования для конкретных аналитических задач; 7) контролировать чистоту реактивов и химической посуды.
Автор выражает благодарность д. х. н. И.В. Куб-раковой за ценные замечания, С.Ю. Лапшину, О.А. Тютюнник, М.С. Киселевой и И.Н. Громяк за помощь в проведении экспериментальной работы.
Список литературы
Пупышев А.А. Атомно-абсорбционный спектральный анализ. М.: Техносфера, 2009. 784 с.
Дворкин В.И. Метрология и обеспечение качества химического анализа. М.: Техносфера, 2020. 317 с.
ГОСТ Р 52361-2018. Контроль объекта аналитический. Термины и определения. М.: Стандартинформ, 2018. 12 с.
Mermet J.-M. Limit of quantitation in atomic spectrometry: An unambiguous concept? // Spectrochim. Acta B. 2008. V. 63. P. 166.
Mermet J.-M., Granier G., Fichet P. A logical way through the limits of quantitation in inductively coupled plasma spectrochemistry // Spectrochim. Acta B. 2012. V. 76. P. 221.
Badocco D., Lavagnini I., Mondin A., Pastore P. Estimation of the uncertainty of the quantification limit // Spectrochim. Acta B. 2014. V. 96. P. 8.
Badocco D., Lavagnini I., Mondin A., Tapparo A., Pastore P. Limit of detection in the presence of instrumental and non-instrumental errors: study of the possible sources of error and application to the analysis of 41 elements at trace levels by ICP−MS technique // Spectrochim. Acta B. 2015. V. 107. P. 178.
Badocco D., Lavagnini I., Mondin A., Favaro G., Pastore P. Definition of the limit of quantification in the presence of instrumental and non-instrumental errors. Comparison among various definitions applied to the calibration of zinc by inductively coupled plasma–mass spectrometry // Spectrochim. Acta B. 2015. V. 114. P. 81.
Voigtman E., Abraham K.T. Statistical behavior of ten million experimental detection limits // Spectrochim. Acta B. 2011. V. 66. P. 105.
Voigtman E., Abraham K. T. True detection limits in an experimental linearly heteroscedastic system. Part 1 // Spectrochim. Acta B. 2011. V. 66. P. 822.
Voigtman E., Abraham K.T. True detection limits in an experimental linearly heteroscedastic system. Part 2 // Spectrochim. Acta B. 2011. V. 66. P. 828.
Carlson J., Wysoczanski A., Voigtman E. Limits of quantitation – yet another suggestion // Spectrochim. Acta B. 2014. V. 69. P. 69.
Prudnikov E.D. Detection limits and error estimation in analysis // Fresenius J. Anal. Chem. 1990. V. 337. P. 412.
Prudnikov E.D., Elgersma J.W., Smit H.C. Theoretical calculation of the standard deviation and detection limit in lnductively coupled plasma atomic emission spectrometry // J. Anal. Atom. Spectrom. 1994. V. 9. P. 619.
Prudnikov E.D., Barnes R.M. Estimation of detection limits in inductively coupled plasma mass spectrometry // Fresenius J. Anal. Chem. 1998. V. 362. P. 465.
Prudnikov E.D., Barnes R.M. Theoretical calculation of the standard deviation in inductively coupled plasma mass spectrometry // J. Anal. Atom. Spectrom. 1999. V. 14. P. 27.
Guide to Quality in Analytical Chemistry. An Aid to Accreditation. 3rd Ed., 2016. www.eurachem.org (07.08.2021).
Гребнева-Балюк О.Н., Кубракова И.В., Тютюнник О.А., Лапшин С.Ю., Пряжников Д.В. Многоэлементный анализ нефти методами АЭС-ИСП и МС-ИСП с использованием микроволновой пробоподготовки // Журн. аналит. химии. 2021. Т. 76. № 3. С. 1.
Inductively Coupled Plasma Spectrometry and Its Applications / Ed Hill S.J. Blackwell, 2007. 420 p.
Dean J.R. Practical Inductively Coupled Plasma Spectroscopy. England: John Wiley & Sons Ltd., 2005. 197 p.
Rodushkin I., Engström E., Baxter D.C. Sources of contamination and remedial strategies in the multi-elemental trace analysis laboratory // Anal. Bioanal. Chem. 2010. V. 396. P. 365.
www.physics.nist.gov (01.08.2021).
Чен Ф. Введение в физику плазмы. М.: Мир, 1987. 396 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии