

Журнал аналитической химии, 2022, T. 77, № 11, стр. 1016-1031
Особенности обработки и интерпретации аналитических данных с высокой степенью неопределенности
И. Г. Зенкевич a, *, Д. А. Никитина a, А. С. Кушакова b
a Санкт-Петербургский государственный университет, Институт химии
198504 Санкт-Петербург, Университетский просп., 26, Россия
b ЗАО “Биокад”
198515 Санкт-Петербург, ул. Связи, 34 лит. А, Россия
* E-mail: izenkevich@yandex.ru
Поступила в редакцию 08.12.2021
После доработки 08.01.2022
Принята к публикации 28.03.2022
- EDN: THEXHM
- DOI: 10.31857/S0044450222090146
Аннотация
Рассмотрены некоторые аналитические задачи, которые по объективным причинам часто считают некорректными. Главная из таких причин – аномально большой разброс исходных данных. Он может быть обусловлен либо низкой воспроизводимостью характеристик веществ, их количеств, интенсивностей аналитических сигналов, условий процессов и др., либо вариабельностью, обусловленной различиями природы самих объектов. В последнем случае на характер интерпретации данных влияют особенности принятых для их рассмотрения аналитических гипотез. В число рассмотренных задач входят вариации компонентного состава проявителей для черно-белых негативных фотоматериалов, сравнение параметров температурных режимов газохроматографического разделения различных органических соединений, характеристика токсичности (LD50) гомологов на примере 1-алканолов С3–С12 и возможности предсказания операций подготовки проб при определении лекарственных препаратов в плазме крови на основании их физико-химических характеристик. Выявлены основные особенности интерпретации данных, отличающихся высокой степенью неопределенности. Отмечено, что важные заключения могут быть получены на основании даже самих фактов низкой воспроизводимости (одномерные массивы) или неудовлетворительной корреляции переменных (двумерные массивы).
К числу важнейших требований к аналитическим методам (часто используемым термином является “валидация”) помимо точности, робастности, и др. традиционно относят такую характеристику, как прецизионность результатов измерений, подразделяемую на их повторяемость и воспроизводимость. Критерием оценок являются минимальные стандартные отклонения получаемых значений (или коэффициенты вариации). Двумерные массивы данных (прежде всего, градуировочные зависимости) характеризуют линейностью, критерием которой являются максимальные значения коэффициентов корреляции (R) или минимальные значения генеральных дисперсий (S0) [1]. Во многих практических руководствах указаны конкретные требования к характеристикам правильности. Часто это относят к биоаналитическим методам и контролю лекарственных препаратов [2–4]. Например, в соответствии с правилами [4] допустимые отклонения результатов для не менее 67% образцов должны находиться в пределах ±15% от их номинальных значений; в противном случае необходимо выяснение причин отклонений.
Однако в реальной аналитической практике встречается большое число примеров массивов данных, отличающихся низкой воспроизводимостью или плохой корреляцией переменных (иначе – высокой степенью неопределенности). Это может быть обусловлено либо большим разбросом непосредственных результатов измерений, либо вариабельностью, обусловленной различиями в природе объектов. Примером объективно большого разброса интенсивностей сигналов является электроискровая масс-спектрометрия [5, 6]; такие же особенности часто присущи определениям на уровне следов. Общим подходом к устранению подобных неопределенностей представляется совершенствование техники определений. Вариабельность второго типа иногда оказывается принципиально неустранимой, но зависит от принятых формулировок аналитических задач (иначе – от принимаемых аналитических гипотез). В качестве примера разброса данных, обусловленного особенностями выборки данных, можно привести интенсивности слабых сигналов так называемых масс-спектров ионных серий [7]11. Их стандартные отклонения могут превышать средние значения.
Еще один пример достаточно слабой корреляции величин – оценки индексов удерживания (RI) в обращенно-фазовой ВЭЖХ на основании факторов гидрофобности (lgP) аналитов [8, 9]. Коэффициенты корреляции зависимостей RI−lgP, как правило, оказываются на уровне 0.9, а S0 (средняя точность получаемых оценок) – не менее 50–70 ед. инд. Тем не менее, этот подход применяют до настоящего времени из-за относительной доступности значений lgP (преимущественно расчетных). Подобные корреляции часто расценивают как имеющие лишь ограниченное применение либо даже как неудовлетворительные. Однако многочисленность примеров большого разброса данных не только не позволяет ими пренебречь, но и заслуживает специального рассмотрения особенностей интерпретации подобной информации, что и является целью настоящей работы.
Для иллюстрации многообразия таких примеров подробно проанализированы четыре разнородные задачи. Все они относятся к вариабельности, обусловленной различиями в природе (свойствах, характеристиках) объектов и во всех проявляется сильная зависимость особенностей интерпретации данных от формулировок рассматриваемых задач. В качестве простейшего примера (одномерный массив данных) рассмотрены вариации состава проявителей для черно-белых негативных фотоматериалов. К более сложным задачам относятся вариации температурных режимов газохроматографического анализа, так как при объединении данных для разных аналитов задача становится двумерной. Еще одним примером большого разброса является токсичность гомологов. И, наконец, подробнее всего рассмотрена аналитически важная проблема выбора операций подготовки проб при определении следов лекарственных препаратов в плазме крови на основании физико-химических характеристик определяемых соединений.
ВЫБОР ИСХОДНЫХ ДАННЫХ И ПОДГОТОВКА ПРОБ К АНАЛИЗУ (ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ)
Общая характеристика четырех рассматриваемых примеров. Для исключения поиска и сравнения информации из недостаточно систематизированных источников разного времени издания сведения о составе проявителей для черно-белых негативных фотоматериалов (пример 1) заимствованы из руководства [10], относящегося к периоду большого разнообразия как самих негативных фотоматериалов, так и рецептов растворов для их обработки (1964 г.). В качестве источника сведений о режимах газохроматографического анализа с программированием температуры для пяти аналитов (нитробензол, бензонитрил, 2‑хлорфенол, 1,4-диметоксибензол и 1,3,5-трихлорбензол) выбрана база данных Национального института стандартов и технологии (NIST, США) (сайт http://webbook.nist.gov) [11], содержащая подробные сведения обо всех соединениях, индексы удерживания которых в нее включены (пример 2). Выбранные параметры включают начальную температуру (Т0, °С), продолжительность начального изотермического участка (t(T0), мин) и скорость нагрева (r, град/мин). Данные о токсичности (величины LD50, пример 3) 1-алканолов С3–С12 заимствованы из различных литературных источников [12–18]. Многие из них содержат дублирующие друг друга значения LD50, выявление и отбраковку которых не проводили. И, наконец, описания процедур подготовки проб лекарственных препаратов (51 соединение различной химической природы) резюмированы на основании оригинальных публикаций и данных, полученных в ЗАО “Биокад” (см. ниже) (пример 4). Экспериментальные данные, соответствующие каждому из рассматриваемых примеров, и необходимые ссылки приведены в табл. 1–4, номера которых согласованы с номерами примеров. Значения факторов гидрофобности (lgP) и степени связывания лекарственных препаратов с белками указаны на сайте Drugbank [62].
Таблица 1.
Содержание компонентов в некоторых метол-гидрохиноновых проявителях для негативных черно-белых фотоматериалов (по данным [10])
| Тип проявителя | Содержание компонентов, г/л | |||||
|---|---|---|---|---|---|---|
| метол | гидро-хинон | сульфит натрия (кристал-лический) |
карбонат натрия (безвод-ный)* |
бромид калия |
прочие добавки | |
| НП-1 | 2.5 | 2.5 | 60 | 5 | 0.5 | – |
| Подкрепляющий раствор к НП-1 | 7 | 10 | 60 | 20 | – | – |
| НП-2 | 1 | 5 | 52 | 20 | 1 | – |
| НП-3 | 5 | 6 | 80 | 40 | 3 | – |
| Контрастный | – | 30 | 120 | – | 20 | 19 (NaOH) |
| Контрастный КЦ-1 | 2 | 10 | 104 | 40 | 4 | – |
| Для высоких температур | 6 | – | 180 | – | 2 | 23 (бура), 100 (Na2SO4) |
| Мелкозернистый НП-15 | 2 | 5 | 200 | – | – | 2 (бура) |
| Подкрепляющий раствор к НП-15 | 3 | 7.5 | 200 | – | – | 20 (бура) |
| Стандартный | 8 | – | 250 | 5.75 | 2.5 | – |
| НП-19А | 10 | – | 120 | 90 | 2 | – |
| Мелкозернистый Д-23 | 7.5 | – | 200 | – | – | – |
| ДК-20 | 5 | – | 200 | – | 0.5 | 2 (бура), 1 (KNCS) |
| Для выравнивания контраста изображения | 5 | 6 | 40 | 25 | 4.5 | – |
| Для исправления недодержек | 14 | 14 | 50 | – | 9 | 9 (NaOH) |
| Для увеличения светочувствительности | 1 | 5 | 52 | 20 | 1 | 0.2 (сульфат гидразина), 0.01 (бензотриазол) |
| Для передержанных фотоматериалов | 7 | – | 50 | 12 | 5 | – |
| Для быстрого проявления | 5 | 45 | 180 | – | 10 | 40 (NaOH), 1 (бензотриазол) |
| Средние значения ± s | 5 ± 4 | 8 ± 12** | 120 ± 70 | 15 ± 23** | 4 ± 5** | – |
| Асимметрия выборок (А) | 1.6 | 3.5 | 1.5 | 2.3 | 2.9 | – |
Таблица 2.
Сравнение параметров температурных режимов газохроматографического анализа некоторых органических соединений (по данным [11]).
| Соединение | T0, °С | t(T0), мин | r, град/мин | Соединение | T0, °С | t(T0), мин | r, град/мин |
|---|---|---|---|---|---|---|---|
| Нитробензол | 40 | 0 | 5 | 1,4-Диметоксибензол | 60 | 2 | 10 |
| 60 | 1 | 5 | 60 | 5 | 3 | ||
| 75 | 0 | 6 | 35 | 3 | 4 | ||
| 50 | 2 | 8 | 60 | 1 | 6 | ||
| 40 | 1 | 10 | 40 | 7 | 6 | ||
| Средние значения | 55 ± 13 | 1 ± 1 | 7 ± 2 | 60 | 0 | 3 | |
| Бензонитрил | 0 | 12 | 12 | 40 | 5 | 3 | |
| 40 | 0 | 5 | 70 | 2 | 3 | ||
| 40 | 0 | 3 | 35 | 4 | 2 | ||
| 50 | 0 | 2.5 | 60 | 0 | 4 | ||
| 50 | 0 | 6 | 50 | 0 | 4 | ||
| 35 | 2 | 2 | 70 | 5 | 4 | ||
| Средние значения | 36 ± 19 | 2 ± 5 | 5 ± 4 | 80 | 0 | 3 | |
| 2-Хлорфенол | 40 | 0 | 3 | 50 | 2 | 3 | |
| 35 | 0 | 8 | 40 | 10 | 3 | ||
| 50 | 0 | 10 | Средние значения | 54 ± 14 | 3 ± 3 | 4 ± 2 | |
| 50 | 0 | 5 | 1,3,5-Трихлорбензол | 50 | 0 | 2 | |
| 50 | 4 | 6 | 50 | 0 | 2 | ||
| 100 | 0 | 10 | 100 | 0 | 10 | ||
| 100 | 0 | 2 | 100 | 0 | 5 | ||
| 100 | 0 | 6 | 50 | 0 | 10 | ||
| 100 | 0 | 10 | 50 | 0 | 5 | ||
| 100 | 0 | 5 | 30 | 0 | 1 | ||
| 50 | 0 | 10 | 80 | 0 | 2 | ||
| 50 | 0 | 5 | 80 | 0 | 3 | ||
| 50 | 3 | 3 | 40 | 3 | 2 | ||
| 80 | 0 | 2 | 80 | 0 | 2 | ||
| 50 | 2 | 8 | 50 | 5 | 5 | ||
| 40 | 3 | 8 | Средние значения | 63 ± 23 | 1 ± 2 | 4 ± 3 | |
| 50 | 4 | 6 | |||||
| Средние значения | 64 ± 25 | 1 ± 2 | 6 ± 3 |
Подготовка проб к анализу. Данные табл. 4, соответствующей последнему рассматриваемому примеру и содержащей сведения об операциях подготовки проб плазмы крови для ВЭЖХ-определения лекарственных препаратов, дополнены информацией для нескольких препаратов, методики анализа которых разработаны в ЗАО “Биокад” (Санкт-Петербург). Их стандартные растворы с концентрацией 1 мг/мл готовили растворением навесок анализируемых субстанций в подвижной фазе (состав растворителей соответствовал начальным составам элюентов в режимах градиентного элюирования). В качестве матрицы для приготовления модельных растворов использовали плазму крови здоровых добровольцев, хранившуюся в замороженном виде при температуре не выше –70°С.
Таблица 3.
Некоторые данные по токсичности 1-алканолов С3–С12 (LD50, мг/кг, перорально; отсутствие указания животного соответствует использованию крыс)
| 1-Алканол | LD50 | Источник |
|---|---|---|
| 1-Пропанол | 1870 | SDS* (Thermo Fisher Scientific, 2018) |
| 2160, 2800 (кролик), 1870 | PubChem | |
| 1870, 5700 (для человека) | SDS (ChemSupply, Australia) | |
| 4400, 7000 | SDS (Thermo Fisher Scientific) | |
| 2825 (кролик), 6800 (мышь), 1870, 2200 | SDS (АСС № 01356) | |
| Среднее значение (А) | 3400 ± 2000 (2.2) | |
| 1-Бутанол | 2680, 2510, 2020, 790, 1200 (хомяк), 2500 (птица), 3500 (кролик), 1782 (собака) | [12] |
| 4360, 2290, 2510, 2020, 790, 2680 (мышь), 3500 (кролик), 1200 (хомяк), 1782 (собака) | [13] | |
| 700, 790 | SDS (Thermo Fisher Scientific) | |
| 2292, 3430 (кролик) | SDS (Carl Roth, 2021) | |
| 790–4360 | PubChem | |
| 2500, 3400 (кролик), 790, 2680 (мышь), 3484, 2500 (птица) | SDS (Sigma-Aldrich) | |
| 1200 (хомяк), 2680 (мышь), 3400 (кролик), 3500 (кролик), 2100, 800–2000, 700 | [14] | |
| 2292 (крыса) | [15] | |
| 2290, 2510, 2680 (мышь), 3500 (кролик), 1200 (хомяк), 1782 (собака), <2000 | [16] | |
| Среднее значение (А) | 2200 ± 1000 (0.90) | |
| 1-Пентанол | 200 (мышь). 370 | SDS (Thermo Fisher Scientific) |
| >2000 | SDS (Carl Roth) | |
| >2000 | SDS (Acros Organics) | |
| 3645 | SDS (Agilent) | |
| 3670, 200 (мышь) | SDS (Sigma-Aldrich) | |
| >2000 | SDS (ChemSupply, Australia) | |
| 2200–3000 | SDS (Calderon Lab Chemicals) | |
| 2200, 3600, 140–4585 | [17] | |
| Среднее значение (А) | 2100 ± 1500 (0.97) | |
| 1-Гексанол | 1950 (мышь) | [12] |
| 720, >2000 | SDS (Carl Roth) | |
| 4590, 4870, 103–4870 | [17] | |
| 200–2000 | [18] | |
| 500–5000 | SDS (Cameo Chemicals) | |
| >2000 | ChemBook | |
| Среднее значение (А) | 2400 ± 1900 (1.8) | |
| 1-Гептанол | 3250, 6200, 5500, 500–6200 | [17] |
| 4300 (мышь) | PubChem | |
| 1870 | SDS (Cameo Chemicals) | |
| 500 (кролик), >2000 | ChemBook | |
| 500 | Green Chemistry | |
| 1-Октанол | >3200 | [12] |
| >5000, 1790 –>5000 | [11] | |
| >2000 | ChemBook | |
| 1-Нонанол | 3560, 800–6400 | [17] |
| 1-Деканол | 4720, 1000–5000 | [17] |
| 1-Ундеканол | 3000, 3000–>15800** | [17] |
| 1-Додеканол | >2000, 1500–26530** | [17] |
* SDS (Safety Data Sheet) или MSDS (Material Safety Data Sheet) – обозначение веб-сайтов различных фирм, содержащих токсикологические характеристики химических соединений. ** Аномально большие значения LD50, исключенные при графическом представлении данных (рис. 2).
Таблица 4.
Основные характеристики операций подготовки проб некоторых лекарственных препаратов в зависимости от их факторов гидрофобности (lgP) (оединения расположены по убыванию расчетных значений lgP (ChemAxon))
| Название | Мол. масса | CAS № | lgP | Связывание с белками плазмы, % | Вариант подготовки проб*, ссылка | Литера- тура |
|---|---|---|---|---|---|---|
| Everolimus | 958 | 159351-69-6 | 7.4 (ChemAxon) | ~74% | ЖЖЭ** | |
| Eltrombopag | 443 | 496775-61-2 | 6.03 (ChemAxon) | ~99% | Осаждение | [19] |
| Atorvastatin | 559 | 134523-00-5 | 5.39 (ChemAxon) 6.36 (эксп) |
~98% | ЖЖЭ | [20] |
| Bosentan | 552 | 147536-97-8; 157212-55-0 (моногидрат) |
4.94 (ChemAxon) 3.7 (эксп) |
~98% | Осаждение** | |
| Dapoxetine | 305 | 119356-77-3 | 4.67 (ChemAxon) | ~99% | ЖЖЭ | [21] |
| Vinorelbine | 779 | 71486-22-1 | 4.65 (ChemAxon) 4.0 (эксп) |
~90% | ЖЖЭ | [22] |
| Simeprevir | 750 | 923604-59-5 | 4.56 (ChemAxon) | ~99% | ЖЖЭ | [23] |
| Loratadine | 383 | 79794-75-5 | 4.55 ChemAxon | 99% | ЖЖЭ | [24] |
| Pyronaridine | 518 | 74847-35-1 | 4.22 (ChemAxon) | ~92% | ЖЖЭ | [25] |
| Nateglinide | 317 | 105816-04-4 | 4.03 ChemAxon | ~98% | Микро-ЖЖЭ | [26] |
| Abiraterone | 392 | 154229-18-2 | 3.97 (ChemAxon) | ~99% | ЖЖЭ** | |
| Chloroquine | 320 | 54-05-7 | 3.93 (ChemAxon) 4.63 (эксп) |
~74% | Осаждение | [25] |
| Gefitinib | 447 | 184475-35-2 | 3.75 (ChemAxon) 3.2 (эксп) |
~90% | ЖЖЭ** | |
| Crizotinib | 450 | 877399-52-5 | 3.57 (ChemAxon) | ~91% | Осаждение | [27, 28] |
| Pazopanib | 438 | 444731-52-6 635702-64-6 (гидрохлорид) |
3.55 (ChemAxon) 3.2 (эксп) |
~99% | Осаждение** | |
| Mycophenolic acid |
320 | 24280-93-1 | 3.53 (ChemAxon) 2.8 (эксп) |
~98% | ЖЖЭ | [29] |
| Canagliflozin | 444 | 842133-18-0 | 3.52 (ChemAxon) | ~99% | ЖЖЭ | [30] |
| Artemether | 298 | 71963-77-4 | 3.48 (ChemAxon) | ~95% | Осаждение | [25] |
| Olanzapine | 312 | 132539-06-1 | 3.39 (ChemAxon) | ~ 93% | ЖЖЭ | [31] |
| Asunaprevir | 748 | 630420-16-5 | 3.37 (ChemAxon) | ~ 99% | ЖЖЭ | [32] |
| Bupropion | 276 | 34911-55-2 | 3.27 (ChemAxon) 3.6 (эксп) |
~85% | ЖЖЭ | [33] |
| Erlotinib | 393 | 183321-74-6 | 3.2 (ChemAxon) | ~95% | Осаждение** | |
| Stiripentol | 234 | 49763-96-4 | 3.12 (ChemAxon) | ~99% | ЖЖЭ | [34] |
| Осаждение | [35] | |||||
| Diazepam | 285 | 439-14-5 | 3.08 (ChemAxon) | ~98% | ЖЖЭ | [36] |
| Sunitinib | 398.5 | 557795-19-4 | 2.93 (ChemAxon) | ~95% | ЖЖЭ | [37] |
| Docetaxel | 808 | 114977-28-5 | 2.92 (ChemAxon) 2.4 (эксп) |
~97% | ЖЖЭ | [38] |
| Fluvoxamine | 318 | 54739-18-3 | 2.8 (ChemAxon) 3.2 (эксп) |
~80% | ЖЖЭ | [39] |
| Gilteritinib | 553 | 1254053-43-4 | 2.79 ChemAxon | ~94% | ТФЭ | [40] |
| Retigabine | 303 | 150812-12-7 | 2.7 (ChemAxon) | ~80% | ЖЖЭ | [41] |
| Eslicarbazepine acetate |
296 | 236395-14-5 | 2.17 (ChemAxon) | ~40% | Осаждение + + высаливание | [42] |
| Teriflunomide | 270 | 163451-81-8 | 2.14 (ChemAxon) 1.536 (эксп) |
~ 99% | Осаждение** | |
| Dapagliflozin | 409 | 461432-26-8 | 2.11 (ChemAxon) 2.7 (эксп) |
~91% | ЖЖЭ | [30] |
| Oxcarbazepine | 252 | 28721-07-5 | 1.82 (ChemAxon) | ~40% | ЖЖЭ | [43] |
| Empagliflozin | 451 | 864070-44-0 | 1.66 (ChemAxon) | ~86% | ЖЖЭ | [30] |
| Dolutegravir | 419 | 1051375-16-6 | 1.1 (ChemAxon) | ~99% | Осаждение + ТФЭ | [44] |
| Doxorubicin | 544 | 23214-92-8 | 0.92 (ChemAxon) | ~74% | Осаждение | [45] |
| Monomethyl fumarate |
130 | 2756-87-8 | 0.34 (ChemAxon) | ~ 40% | Осаждение** | |
| Zonisamide | 212 | 68291-97-4 | 0.11 (ChemAxon) 0.5 (эксп) |
~40% | ЖЖЭ | [46, 47] |
| Cisplatin*** | 300 | 15663-27-1 | –2.19 (Hansch, 1995) –0.04 (ALOGPS) |
~95% | Дериватизация + + ЖЖЭ | [48] |
| Mercaptopurine | 152 | 50-44-2 | –0.12 (ChemAxon) | ~19% | Осаждение | [49] |
| Sulthiame | 290 | 61-56-3 | –0.27 (ChemAxon) | ~60% | Осаждение | [50] |
| Didanosine | 236 | 69655-05-6 | –0.35 (ChemAxon) | ~5% | ТФЭ | [51] |
| Metronidazole | 171 | 443-48-1 | –0.46 (ChemAxon) | ~20% | Осаждение | [52] |
| Isoniazid | 137 | 54-85-3 | –0.69 (ChemAxon) | ~10% | Осаждение | [53] |
| Lenalidomide | 259 | 191732-72-6 | –0.71 (ChemAxon) –0.4 (эксп) |
~30% | Осаждение** | |
| Entecavir | 277 | 142217-69-4 209216-23-9 (моногидрат) |
–1.4 (ChemAxon) –0.8 (эксп) |
~ 13% | Ультрафильтрация** | |
| Cytarabine | 243 | 147-94-4 | –2.8 (ChemAxon) –2.8 (эксп) |
~13% | Катионообменная ТФЭ | [54] |
| Azacitidine | 244 | 320-67-2 | –3.1 (ChemAxon) –3.5 (эксп) |
N/A | Ионообменная ТФЭ | [55] |
| Осаждение | [56] | |||||
| Tigecycline | 585.6 | 220620-09-7 | –3.9 (ChemAxon) 0.8 (эксп) | ~89% | Осаждение | [57] |
| Ceftazidime | 547 | 72558-82-8 | –4.1 (ChemAxon) | ~ 10% | Ультрафильтрация | [58] |
| Meropenem | 384 | 96036-03-2 | –4.4 (ChemAxon) –0.6 (эксп) |
~2% | Осаждение | [59, 60] |
| ТФЭ | [61] |
Образцы для ВЭЖХ-анализа готовили по одной из следующих схем. 1). Жидкостно-жидкостную экстракцию (ЖЖЭ) проводили путем добавления к аликвотам образцов экстрагента (1 : 3), полученные растворы перемешивали, центрифугировали, органические экстракты упаривали в токе азота, сухие остатки вновь растворяли в подвижной фазе. 2). Для осаждения белков плазмы крови в качестве осадителя использовали ацетонитрил в соотношении 1 : 3; полученные растворы перемешивали, центрифугировали, надосадочные слои переносили в виалы для хроматографического анализа. 3) Твердофазную экстракцию (ТФЭ) проводили с использованием картриджей (Oasis, Waters, США); наносили на них пробы и пропускали через картриджи различные растворители для элюирования примесей и целевых аналитов по отдельности. 4) Для фильтрации белков плазмы использовали центрифужные ультрафильтры Amicon® Ultra 3K (Millipore, США). В пробирки с фильтрами вносили заданные объемы образцов, центрифугировали; прошедшие через мембраны количества растворов отбирали для хроматографического анализа.
Во всех перечисленных примерах исходная информация отличалась значительной вариабельностью, а разброс числовых данных настолько велик, что какая-либо коррекция единичных значений в подобных массивах (выявление выбросов) [63] представляется невозможной. Для статистической обработки исходных данных и построения графиков использовали ПО ORIGIN (версии 4.1 и 8.1). Именно значительная вариабельность исходных данных ограничивает применение более сложных методов их интерпретации (факторный или кластерный анализ). Кроме того, во всех примерах число доступных значений недостаточно для корректного применения таких методов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Особенности компонентного состава метол-гидрохиноновых проявителей для негативных черно-белых фотоматериалов. Начать рассмотрение совокупностей данных с большим разбросом целесообразно с одномерных массивов, для которых применима обычная статистическая обработка. По современным представлениям наилучшие результаты обработки фотоматериалов обеспечивает строгое воспроизведение условий, в том числе состава растворов, продолжительности обработки и ее температурного режима (максимальная стандартизация). Однако 50–60 лет назад ситуация была принципиально иной: для многочисленных негативных фотоматериалов (с разной спектральной чувствительностью) использовали большое число проявителей различного назначения, в том числе нормальных (стандартных), мелкозернистых, контрастных, для снижения контраста, для увеличения светочувствительности, для передержанных фотоматериалов, для высоких температур обработки и т.д. Все такие проявители различались сочетаниями компонентов и их содержанием. Если ограничиться только метол-гидрохиноновыми проявителями22 для негативных фотоматериалов, то содержание компонентов в 19 из них по данным руководства [10] приведено в табл. 1.
Назначение такого многообразия составов в том, чтобы в зависимости от особенностей конкретного фотоматериала или условий съемки выбрать оптимальный вариант его обработки. Однако можно представить иную вполне реальную ситуацию: тип фотоматериала, особенности его экспозиции и, следовательно, априорные требования к обработке неизвестны. Иными словами, здесь мы имеем дело с иной формулировкой исходной гипотезы. В подобных случаях выбор конкретной рецептуры проявителя из числа ранее известных явно нерационален, и, следовательно, требуется изменение логической схемы действий. Из неопределенности формулировки задачи следует предпочтительное использование рецептуры, состав которой соответствует усредненным количествам всех компонентов (средние арифметические значения приведены в последней строке табл. 1).
Вариации содержания отдельных компонентов (размах) весьма велики и составляют 0–14 (метол), 0–45 (гидрохинон), 40–250 (сульфит натрия), 0–90 (карбонат натрия) и 0–20 г/л (бромид калия). Неудивительно, что для некоторых из них (гидрохинон, карбонат натрия и бромид калия) формальное вычисление стандартных отклонений приводит к величинам, превышающим их средние арифметические значения. Для двух остальных компонентов относительные стандартные отклонения (коэффициенты вариации) составляют 80% (метол) и 58% (сульфит натрия). Если стандартное отклонение некоторой величины больше ее среднего значения, то в практике математической обработки результатов измерений [64] это означает, что такая величина статистически незначима, и ее можно полагать равной нулю. Однако применительно к рассматриваемой задаче подобная интерпретация неприемлема и должна быть изменена: это подтверждает возможность отсутствия такого компонента в составе проявителя в отдельных случаях.
Если рассматриваемые наборы данных {xi} очевидно асимметричны (как в данном случае), то они могут быть охарактеризованы фактором асимметрии (А). Один из способов оценки этого параметра подразумевает независимое вычисление двух стандартных отклонений. Первое характеризует данные, превышающие среднее арифметическое значение, s(+), а второе соответственно меньшие, s(–). Их отношение А = s(+)/s(–) и является характеристикой асимметрии [65]. В нашем случае все значения А (указаны в последней строке табл. 1) значительно превышают единицу (от 1.5 до 3.5). Это типично для всех массивов данных, ограниченных “снизу” нулем, тогда как “сверху” таких ограничений нет.
Подобный разброс данных по составу проявителей заслуживает комментариев. Прежде всего, он соответствует некоторой среднестатистической рецептуре. Если рассмотреть 11 перечисленных в табл. 1 конкретных составов, то вычисленные средние значения лучше всего соответствуют проявителям НП-3 и “для выравнивания контраста изображения”. Кроме того, небезынтересно заметить, что если оценить точность задания требуемых количеств ингредиентов “на глаз” приблизительно как ±50 отн. %, то стандартные отклонения полученных средних значений превышают эту величину. Это означает, что при приготовлении подобных проявителей вполне можно исключить такую операцию, как взятие точных навесок компонентов, что тем не менее обеспечивает вполне приемлемый уровень качества обработки фотоматериалов.
Как отмечено выше, аналогичные примеры статистической обработки данных, характеризующихся заметным разбросом, встречаются при вычислении масс-спектров ионных серий [7], что определяется характером выбранной гипотезы (решение задач групповой масс-спектрометрической идентификации органических соединений).
Сравнение температурных режимов газохроматографического анализа различных органических соединений. Следующий по сложности пример совокупностей данных, характеризующихся большим разбросом, может быть представлен и как одномерный, и как двумерный массив. Это – параметры газохроматографического разделения различных аналитов с программированием температуры, а именно начальная температура (Т0, °С), продолжительность начального изотермического участка (t(T0), мин) и скорость нагрева (r, град/мин). Подобные сведения из разных источников информации доступны на сайте Национального института стандартов и технологии (NIST, США) (http://webbook.nist.gov) [11], где они приведены для всех соединений, охарактеризованных индексами удерживания. Для иллюстрации особенностей такой информации можно выбрать любые соединения, однако для сокращения объема обсуждения ограничимся пятью аналитами: нитробензолом, бензонитрилом, 2-хлорфенолом, 1,4-диметоксибензолом и 1,3,5-трихлорбензолом. Некоторые из значений Т0, t(T0) и r для них приведены в табл. 2.
Как и в предыдущем примере, обращают на себя внимание заметные вариации всех характеристик режимов программирования. При этом стандартные отклонения параметров Т0 и r не превышают соответствующих средних величин, чего нельзя сказать о продолжительности начального изотермического участка (r). Из общих положений можно было бы полагать, что начальные температуры T0 могли бы быть как-то связаны с химической природой аналитов, например, со значениями их индексов удерживания. Однако проверка зависимости RI−T0 показывает, что эти параметры слабо коррелируют между собой (рис. 1) и речь может идти всего лишь о некоторой слабо выраженной тенденции увеличения начальных температур режимов программирования для аналитов с большими индексами удерживания. Особенно это заметно, если предварительно не усреднять значения Т0, t(T0) и r для каждого соединения (рис. 1а), а нанести на график точки, соответствующие всем значениям этих параметров (рис. 1б). Коэффициент корреляции (R) в случае (б) составляет всего 0.075, что подтверждает отсутствие линейной связи переменных.
Рис. 1.
График, иллюстрирующий отсутствие линейной зависимости индексов удерживания (RI) некоторых аналитов от выбранных для их газохроматографического разделения начальных температур режима программирования (Т0); (а) – для средних значений Т0, (б) – по всем данным табл. 2. Параметры линейной регрессии RI = aT0 + b: a = 0.18 ± ± 0.45, b = 1047 ± 28, R = 0.053, S0 = 73.
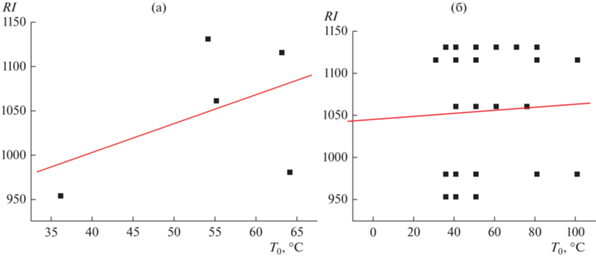
Если так, то вместо средних значений Т0, t(T0) и r для каждого из аналитов вполне можно вычислять и использовать общие средние значения и стандартные отклонения этих аналитических параметров, приведенные в последней строке табл. 2. Поскольку в интервалы ±2sRI должно попадать приблизительно 95% значений выборки, то эти данные соответствуют интервалу Т0 от 10 до 100°С (нет выбросов), t(T0) до 8 мин (один выброс, 10 мин) и r до 11 град/мин (нет выбросов). Коэффициенты асимметрии (А) всех указанных параметров по той же причине, как и в предыдущем случае составляет от 1.6 до 3.5 (последняя строка табл. 2). Из этого следует достаточно неожиданный вывод: при выборе режимов программирования температуры для различных соединений величины Т0, t(T0) и r можно задавать в широких интервалах, что не сказывается на разделении аналитов. Наибольшее, на что может повлиять неоптимальный выбор условий разделения (например, слишком малые значения T0, большие t(T0) и малые r) – это неоправданное увеличение продолжительности анализа. Иногда применительно к выбору режимов разделения можно встретить употребление выражения “разработка методики” (хроматографического анализа или, точнее, разделения). Однако с учетом сделанного вывода относительно больших вариаций параметров Т0, t(T0) и r следует признать, что правильнее было бы говорить о выборе некоторых значений из допустимых диапазонов.
Характеристика токсичности (LD50) гомологов на примере 1-алканолов С3–С12. Следующий пример также может рассматриваться и как одномерная, и как двумерная совокупность данных. Для значений LD50 конкретных соединений обычная статистическая обработка формально возможна (если это согласуется с принятой гипотезой), а сравнение таких данных для нескольких гомологов превращает массив в двумерный, когда для вычисления параметров регрессионных уравнений необходимо применение метода наименьших квадратов. Токсичность различных химических соединений представляют собой одну из их важнейших характеристик, однако ее определение (особенно in vivo) представляет собой весьма длительную, трудоемкую и дорогостоящую операцию. Известны многочисленные попытки теоретической оценки параметров токсичности (LC50 или LD50), однако поскольку это не является предметом рассмотрения настоящей работы, ограничимся лишь несколькими ссылками [66–70]. При этом важно заметить, что расчетные методы, так или иначе, базируются на экспериментальных данных, но если их разброс велик, то из этого неизбежно следует низкая точность всех получаемых оценок [63].
В табл. 3 приведены некоторые данные по токсичности (LD50, мг/кг, перорально) 1-алканолов С3–С12 для различных теплокровных животных (крыса, кролик, мышь, хомяк, собака, (без уточнения породы), птица (то же) и одно значение для человека (1-пропанол)). И здесь обращает на себя внимание значительный разброс данных. Если попытаться усреднить значения LD50 для спиртов С3–С6 (охарактеризованы наиболее подробно) по отдельности, то получаем оценки, для которых коэффициенты вариации составляют 45–80% (указаны в табл. 3). Кроме того, примечательно, что выборки данных для спиртов С4 и С5 практически симметричны: значения фактора асимметрии для них близки к единице и составляют 0.90 и 0.97 соответственно. Если ограничиться только одним видом животных (подробнее всего представлены крысы), то ситуация с разбросом данных принципиально не изменяется. По этой причине в дальнейшем можно рассматривать всю совокупность объектов без какого-либо дополнительного подразделения. При цитировании литературных источников в табл. 3 использован тот же принцип, что и в публикации [71]: указаны ссылки на конкретные работы, но иногда упомянуты доступные в Интернете SDS-страницы компаний, выпускающих те или иные химические реактивы.
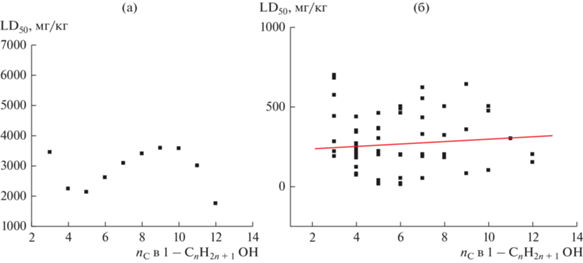
Этот пример позволяет проиллюстрировать, к каким парадоксам может приводить интерпретация данных, характеризуемых высокой вариабельностью. Если вычислить средние арифметические значения LD50 для каждого из спиртов С3‒С12, то на графике им соответствует гладкая синусоидальная кривая с двумя экстремумами (рис. 2а). Если же рассматривать всю совокупность данных без предварительной обработки (рис. 2б), то их графическое представление иллюстрирует значительный разброс. Авторы не могут не отметить тот факт, что мнения специалистов относительно возможности и невозможности характеристики токсичности гомологов средними значениями разделяются приблизительно в равной пропорции. Однако здесь важно подчеркнуть, что этот выбор также является следствием той или иной исходной гипотезы. Если учесть, что исходные значения LD50 для каждого из спиртов получены для разных животных, то их усреднение недопустимо. Если же пренебречь такой информацией, то приемлемо не только усреднение данных для каждого из гомологов, но и для всей их совокупности. В таком случае весь набор значений LD50 можно охарактеризовать общим средним значением и его стандартным отклонением: (2600 ± 1600) мг/кг и на основании этого заключить, что имеющиеся данные не подтверждают зависимость токсичности 1-алканолов C3–C12 от числа атомов углерода в молекуле. Этот вывод имеет общий характер: если для линейной регрессии y = ax + b стандартное отклонение коэффициента “а” превышает его абсолютную величину, sa > ∣a∣, то следует предпочесть обычное усреднение данных.
Рис. 2.
(а): Графическая иллюстрация зависимости средних арифметических значений LD50 каждого спирта от числа атомов углерода в молекуле 1-алканола; (б): то же для всех исходных данных табл. 3: параметры линейной регрессии (линия тренда) LD50 = anC + b: a = 78 ± 73, b = 2200 ± 400, R = 0.10, S0 = 1600.
Выбор операций подготовки проб при определении лекарственных препаратов в плазме крови в зависимости от их гидрофобно-гидрофильных свойств. Последний рассматриваемый пример обработки и интерпретации данных, характеризующихся большим разбросом, наиболее сложен. Во-первых, он относится к двумерным массивам, причем одну из переменных необходимо выбрать искусственно (ввести ранги операций подготовки проб). Второй фактор – актуальность рассматриваемой проблемы. Определение лекарственных препаратов в биологических жидкостях представляет собой весьма трудоемкую задачу, прежде всего, из-за сложности матрицы и, как следствие, стадии подготовки проб. Поскольку предпочтительным аналитическим методом при решении подобных задач является обращенно-фазовая ВЭЖХ, то на этой стадии необходимо обеспечить не только концентрирование целевых аналитов, но и удаление мешающих компонентов, в первую очередь – белков. Подготовка проб чаще всего включает следующие операции: жидкостно-жидкостную и твердофазную экстракцию, осаждение белковых компонентов, в том числе ультрафильтрацию, ионный обмен и, реже, другие. Выбор и оптимизацию операций подготовки проб (или их сочетаний) до настоящего времени проводят на основании общих представлений о природе определяемых соединений. Это приводит к большим затратам времени, которые можно было бы минимизировать, если бы удалось связать характер операций подготовки проб с физико-химическими характеристиками аналитов.
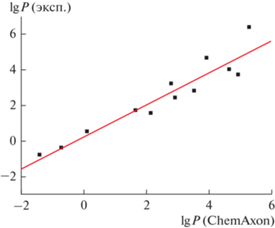
Из таких характеристик в первую очередь заслуживают внимания факторы гидрофобности, lgP. Однако экспериментальные значения lgP известны не для всех характеризуемых соединений. Их можно заменить расчетными оценками, в нашем случае вычисленными с использованием ПО ChemAxon [62]. При получении таких оценок из рассмотрения можно исключить значения lgP(ChemAxon) < –2, так как примеров применения ЖЖЭ для столь гидрофильных соединений нет. Проверка эквивалентности расчетных (указаны в табл. 4 для некоторых препаратов) и экспериментальных величин (13 пар значений), которую иллюстрирует рис. 3, показывает, что коэффициент “а” линейной регрессии lgP(эксп) = algP(ChemAxon) + b лишь незначительно меньше единицы (0.89 ± 0.09), а коэффициент “b” – статистически незначим (0.27 ± 0.31). Следовательно, экспериментальные и расчетные значения lgP в некотором приближении можно полагать эквивалентными. Однако средняя точность получаемых оценок невелика, так как значение S0 для представленной на рис. 3 линейной регрессии составляет 0.69. Кроме того, при использовании расчетных значений lgP нежелательно использовать значения lgP, вычисленные другими способами (например, с использованием ПО ACD).
Рис. 3.
Корреляция экспериментальных и вычисленных (ChemAxon) значений lgP. Параметры линейной регрессии lgP(эксп) = algP(ChemAxon) + b: a = 0.89 ± ± 0.09, b = 0.28 ± 0.31, R = 0.944, S0 = 0.69.
В табл. 4 сопоставлены важнейшие условия подготовки проб 51 лекарственного препарата, для каждого из которых указаны молекулярная масса, CAS №, значение lgP (ChemAxon) и доля препарата, связывающаяся с белками плазмы. Для каждого варианта подготовки проб приведена ссылка на оригинальную публикацию; отсутствие ссылки (10 препаратов) означает, что методика была разработана и валидирована в ЗАО “Биокад”.
Прежде всего, прокомментируем такую характеристику, как доли аналитов, связываемые с белками плазмы (варьируют от 2 до 99%), что непосредственно влияет на их пределы обнаружения. Проверка возможной взаимосвязи этой характеристики с такой операцией подготовки проб, как осаждение белков показывает, что значимой корреляции здесь не наблюдается. Среднее значение связываемой доли аналита при использовании осаждения составляет (89 ± 16)% , а при отсутствии этой операции – (70 ± 33)% (по данным табл. 4). Таким образом, речь идет лишь о слабо выраженной тенденции, которую можно не принимать во внимание.
Для последующей интерпретации данных различным операциям подготовки проб необходимо присвоить некоторые условные коды (ранги), что позволит применить метод, в некоторой степени сходный с ранговой корреляцией Спирмена. Условно нулевое значение цифрового кода можно приписать операции осаждения белков, так как она по своей сути менее всего имеет отношение к гидрофобно-гидрофильным свойствам целевых аналитов. Для концентрирования наиболее гидрофобных соединений используют ТФЭ (код +2), а в случае менее гидрофобных, когда допустимо использование ЖЖЭ (возможно варьирование растворителей), код можно приравнять +1. Такая операция, как ионный обмен применима только к соединениям, существующим в ионной форме (наиболее гидрофильные, код –2). Тогда процедура ультрафильтрации получает код (–1). Таким образом, получаем следующий набор переменных для ранговой корреляции:
Как и в предыдущих примерах, результаты проверки возможной связи значений lgP и предлагаемых цифровых кодов нагляднее всего представлять графически (рис. 4). Из рис. 4 следует, во-первых, что два варианта экстракции (ЖЖЭ и ТФЭ) – значительно более популярные способы подготовки проб по сравнению с остальными, что, правда, обусловлено преимущественно гидрофобными свойствами характеризуемых лекарственных препаратов. Однако даже при таком неравномерном распределении точек, как на рис. 4, можно заключить, что при значениях lgP > –1 примеров применения ультрафильтрации и ионного обмена нет или (альтернативная формулировка) на стадии подготовки проб используют только ЖЖЭ или ТФЭ. Операции ЖЖЭ и ТФЭ принципиально неразделимы и выбор между ними определяется, прежде всего, доступностью соответствующего оборудования, материалов и растворителей. Следует отметить, что ЖЖЭ при lgP < 0 принципиально возможна, но существенно сложнее, а примеров применения такой экстракции в области lgP < –1 среди рассматриваемых лекарственных препаратов нет.

* * *
Таким образом, примеры массивов данных, отличающихся большим разбросом, распространены достаточно широко. Самыми сложными представляются примеры вариабельности, обусловленной различиями в природе самих объектов. В таких случаях интерпретация данных осложняется влиянием характера принимаемых аналитических гипотез. Подобные массивы данных с высокими степенями неопределенности нередко исключают из рассмотрения, что не всегда оправданно. Обработке таких данных присущи свои особенности. Одномерные совокупности {xi}, как правило, отличаются высокой асимметрией. Большие значения относительных стандартных отклонений δi = sx/〈x〉 могут быть проинтерпретированы как отсутствие необходимости точного контроля значений переменных “х”, который можно заменить выбором из широких диапазонов их возможных значений. Условие sx > (〈x〉), в отличие от правил обычной статистической обработки, означает не равенство нулю средней величины 〈x〉, а то, что некоторые из значений {xi} выборки могут быть равными нулю. Если в результате обработки некоторых массивов данных методом наименьших квадратов для линейной регрессии y = ax + b стандартное отклонение коэффициента “а” превышает его величину (sa > ∣a∣), то следует предпочесть обычное усреднение данных с оценкой 〈x〉 ± sx. Вероятностный характер выводов, основанных на данных с высокими степенями неопределенности, как правило, выше, чем для данных с “нормальной” воспроизводимостью. При этом важные заключения могут быть получены даже из самих фактов высокой вариабельности переменных. Так, например, при характеристике зависимости индексов удерживания аналитов в обращенно-фазовой ВЭЖХ от содержания метанола в элюенте, dRI/dc [72], установлено, что эти параметры, в отличие от самих индексов удерживания, не коррелируют со значениями lgP, что позволяет в дальнейшем исключить подобную корреляцию из рассмотрения.
Список литературы
Аналитическая химия. Т. 3. Химический анализ / Под ред. Москвина Л.Н. Москва: Академия, 2010. 366 с.
Guideline on bioanalytical method validation. London: Eur. Med. Agency, 2011. 22 p.
The US Pharmacopeia 35. Section Chromatographia / Physical Tests / System Suitability. New York: The USP Convention, 2012. P. 262.
Об утверждении правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза. № 85. Астана: Евразийская экономическая комиссия, 2016. 161 с.
Adams F., Thomas J.M. Recent advances in analytical spark source mass spectrometry // Phil. Trans. R. Soc. Lond. A. 1981. V. 305. P. 509. https://doi.org/10.1098/rsta.1982.0048
Becker S., Dietze H.-J. Inorganic mass spectrometric methods for trace, ultratrace, isotope, and surface analysis // Int. J. Mass Spectrom. 2000. V. 197. № 1–3. P. 1. https://doi.org/10.1016/S1387-3806(99)00246-8
Зенкевич И.Г., Иоффе Б.В. Интерпретация масс-спектров органических соединений. Л.: Химия, 1986. 176 с.
Lochmuller C.H., Hui M. Calculated lgkOW as a guide for key set mobile phase selection in retention prediction // J. Chromatogr. Sci. 1998. V. 36. P. 11.
Hanai T., Koizumi K., Kinoshita T. Prediction of retention factors of phenolic and nitrogen containing compounds in reversed-phase liquid chromatography based on lgP and pKa obtained by computational chemical calculation // J. Liq. Chromatogr. Relat. Technol. 2000. V. 23. P. 363. https://doi.org/10.1081/JLC-100101457
Яштолд-Говорко В.А. Фотосъемка и обработка. М.: Искусство, 1964. 444 с.
NIST 20 (2020) Mass Spectral Library (NIST/EPA/NIH EI MS Library, 2020 Release). Software/Data Version; NIST Standard Reference Database, Number 69, May 2020. National Institute of Standards and Technology, Gaithersburg, MD 20899: http://webbook.nist.gov (дата обращения: декабрь 2021 г.).
Patochka J., Kuca K. Toxic alcohols: Aliphatic saturated alcohols // Mil. Med. Sci. Lett. 2012. V. 81. № 4. P. 142. https://doi.org/10.31482/mml.2012.022
n-Butyl Alcohol. CAS No: 71-36-3. SIDS Initial Assessment Report. Bern: UNEP Publ. 2001. 14 p.
Butanols: Four isomers. Environ. Health Criteria no. 65. New York: World Health Organization, 1987. 76 p.
ГОСТ 30333-2007. Паспорт безопасности химической продукции. Общие требования. М.: Изд. стандартов, 2008. 8 с.
Substance Evaluation Conclusion for Butan-1-ol. EC No. 200-751-6. Budapest: Ministry for Human Capacities, 2018. 33 p.
Case study on the use of integrated approaches for testing and assessment of 90-day rat oral repeated-dose toxicity for selected n-alkanols: read across. OECD Environ. Health and Safety Publ. No. 273. Paris: Organization for Economic Co-operation and Development, 2017. 44 p.
Wypych A., Wypych G. Databook of Solvents. 2nd Ed. Amsterdam: Elsevier, 2019. 800 p.
Yanagimachi N., Obara N., Sakata-Yanagimoto M., Chiba S., Doki K., Homma M. A simple HPLC assay for determining eltrombopag concentration in human serum // Biomed. Chromatogr. 2021. V. 35. № 5. Article e5049. https://doi.org/10.1002/bmc.5049
Bahrami G., Mohammadi B., Mirzaeei S., Kiani A. Determination of atorvastatin in human serum by reversed-phase high-performance liquid chromatography with UV detection // J. Chromatogr. B. 2005. V. 826. № 1–2. P. 41. https://doi.org/10.1016/j.jchromb.2005.08.008
Said R., Arafat B., Arafat T. High performance liquid chromatography – Mass spectrometric bioanalytical method for the determination of dapoxetine in human plasma: Application for bioequivalence study // J. Chromatogr. B. 2020. V. 1149. P. 122. https://doi.org/10.1016/j.jchromb.2020.122154
Qian J., Wang Y., Chang J., Zhang J., Wang J., Hu X. Rapid and sensitive determination of vinorelbine in human plasma by liquid chromatography–tandem mass spectrometry and its pharmacokinetic application // J. Chromatogr. B. 2011. V. 879. № 9. P. 662. https://doi.org/10.1016/j.jchromb.2011.01.039
Vanwelkenhuysen I., de Vries R., Timmerman P., Verhaeghe T. Determination of simeprevir: A novel, hepatitis C protease inhibitor in human plasma by high-performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2014. V. 958. P. 43. https://doi.org/10.1016/j.jchromb.2014.02.028
Patel B. N., Sharma N., Sanyal M., Shrivastav P. S. LC-MS-ESI for the determination of loratadine and descarboethoxyloratadine in human plasma // J. Chromatogr. Sci. 2010. V. 48. № 1. P. 35. https://doi.org/10.1093/chromsci.48.1.35
Casas M., Hansen M., Krogh K., Styrishave B., Björklund E. Analytical sample preparation strategies for the determination of antimalarial drugs in human whole blood, plasma and urine // J. Chromatogr. B. 2014. V. 962. P. 109. https://doi.org/10.1016/j.jchromb.2014.02.048
Hammad M., Kamal A., Kannouma R., Mansour F. Vortex-assisted dispersive liquid–liquid microextraction coupled with deproteinization for determination of nateglinide in human plasma using HPLC/UV // J. Chromatogr. Sci. 2021. V. 59 № 3. P. 297. https://doi.org/10.1093/chromsci/bmaa096
Qiu F., Gu Y., Wang T, Gao Y., Li X., Gao X., Cheng S. Quantification and pharmacokinetics of crizotinib in rats by liquid chromatography-tandem mass spectrometry // Biomed. Chromatogr. 2016. V. 30. P. 962. https://doi.org/10.3390/pharmaceutics10040221
Wong A., Xiang X., Ong P., Mitchell E., Syn N., Wee I., Kumar A., Yang W., Sethi G., Goh B., Ho P., Wang L.A review of liquid chromatography – tandem mass spectrometry methods for rapid quantification of oncology drugs // Pharmaceutics. 2018. V. 10. № 4. P. 221. https://doi.org/10.3390/pharmaceutics.10040221
Bahrami G., Mohammadi B. An isocratic high performance liquid chromatographic method for quantification of mycophenolic acid and its glucuronide metabolite in human serum using liquid–liquid extraction: Application to human pharmacokinetic studies // Clin. Chim. Acta. 2006. V. 370. № 1–2. P. 185. https://doi.org/10.1016/j.cca.2006.02.017
Mabrouk M., Soliman S., El-Agizy H., Mansour F. Ultrasound-assisted dispersive liquid–liquid microextraction for determination of three gliflozins in human plasma by HPLC/DAD // J. Chromatogr. B. 2020. V. 1136. https://doi.org/10.1016/j.jchromb.2019.121932
Aravagiri M., Ames D., Wirshing W.C., Marder S.R. Plasma level monitoring of olanzapine in patients with schizophrenia: Determination by high-performance liquid chromatography with electrochemical detection // Ther. Drug Monit. 1997. V. 19. № 3. P. 307. https://doi.org/10.1097/00007691-199706000-00011
Yuan L., Jiang H., Ouyang Z., Xia Y., Zeng J., Peng Q., Lange R., Deng Y., Arnold M., Aubry A. A rugged and accurate liquid chromatography–tandem mass spectrometry method for the determination of asunaprevir, an NS3 protease inhibitor, in plasma // J. Chromatogr. B. 2013. V. 921. P. 81. https://doi.org/10.1016/j.jchromb.2013.01.029
Loboz K, Gross A, Ray J, McLachlan A. HPLC assay for bupropion and its major metabolites in human plasma // J. Chromatogr. B. 2005. V. 823. № 2. P. 115. https://doi.org/10.1016/j.jchromb.2005.06.009
Peigné S., Chhun S., Tod M., Rey E., Rodrigues C., Chiron C., Pons G., Jullien V. Population pharmacokinetics of stiripentol in paediatric patients with dravet syndrome treated with stiripentol, valproate and clobazam combination therapy // Clin Pharmacokinet. 2018. V. 57. № 6. P. 739. https://doi.org/10.1007/s40262-017-0592-7
Takahashi R., Imai K., Yamamoto Y. Determination of stiripentol in plasma by high-performance Lliquid chromatography with fluorescence detection // Jpn. J. Pharm. Health Care Sci. 2015. V. 41. P. 643. https://doi.org/10.5649/jjphcs.41.643
Rouini M., Ardakani Y., Moghaddam K., Solatani F. An improved HPLC method for rapid quantitation of diazepam and its major metabolites in human plasma // Talanta. 2008. V. 75. № 3. P. 671. https://doi.org/10.1016/j.talanta.2007.11.060
Minkin P., Zhao M., Chen Z., Ouwerkerk J., Gelderblom H., Baker S.D. Quantification of sunitinib in human plasma by high-performance liquid chromatography-tandem mass spectrometry // J. Chromatogr. B. 2008. V. 874. P. 84. https://doi.org/10.1016/j.jchromb.2008.09.007
Wang L, Goh B, Grigg M, Lee S, Khoo Y, Lee H. A rapid and sensitive liquid chromatography/tandem mass spectrometry method for determination of docetaxel in human plasma // Rapid Commun. Mass Spectrom. 2003. V. 17. № 14. P. 1548. https://doi.org/10.1002/rcm.1091
Bahrami G., Mohammadi B. Rapid and sensitive bioanalytical method for measurement of fluvoxamine in human serum using 4-chloro-7-nitrobenzofurazan as precolumn derivatization agent: Application to a human pharmacokinetic study // J. Chromatogr. B. 2007. V. 857. № 2. P. 322. https://doi.org/10.1016/j.jchromb.2007.07.044
Yasu T., Sugi T., Momo K., Hagihara M., Yasui H. Determination of the concentration of gilteritinib in human plasma using HPLC // Biomed. Chromatogr. 2021. V. 35. № 4. Article e5028. https://doi.org/10.1002/bmc.5028
Perez H., Boram S., Evans C. Development and validation of a quantitative method for determination of retigabine and its N-acetyl metabolite; overcoming challenges associated with circulating labile N-glucuronide metabolites // Anal. Methods. 2015. V. 7. № 2. P. 723. https://doi.org/10.1039/C4AY02599G
Mohamed F., Ali M., Marwa F.B., Rageh A., Mostafa A. A highly sensitive HPTLC method for estimation of oxcarbazepine in two binary mixtures with two metabolically related antiepileptic drugs: Application to pharmaceutical and biological samples // Microchem. J. 2019. V. 146. P. 414. /https://doi.org/10.1016/j.microc.2019.01.031
Wang L., Wang J., Zhang J., Jiang Q., Zhao L., Zhang T. Simultaneous determination of topiramate, carbamazepine, oxcarbazepine and its major metabolite in human plasma by SFC-ESI-MS/MS with polarity switching: Application to therapeutic drug monitoring // Arab. J. Chem. 2019. V. 12. № 8. P. 4775. https://doi.org/10.1016/j.arabjc.2016.09.016
Charbe N., Baldelli S., Cozzi V., Castoldi S., Cattaneo D., Clementi E. Development of an HPLC–UV assay method for the simultaneous quantification of nine antiretroviral agents in the plasma of HIV-infected patients // J. Pharm. Analysis. 2016. V. 6. № 6. P. 396. https://doi.org/10.1016/J.JPHA.2016.05.008
Dharmalingam S.R., Ramamurthy S., Chidambaram K., Nadaraju S. A Simple HPLC bioanalytical method for the determination of doxorubicin hydrochloride in rat plasma: Application to pharmacokinetic studies // Tropical J. Pharm. Res. 2014. V. 13. № 3. P. 409. https://doi.org/10.4314/tjpr.v13i3.15
Lourenço D., Sarraguça M., Alves G., Coutinho P., Araujo A., Rodrigues M. A novel HPLC method for the determination of zonisamide in human plasma using microextraction by packed sorbent optimized by experimental design // Anal. Methods. 2017. V. 9. № 40. P. 5910. https://doi.org/10.1039/C7AY01912B
Majnooni M.B., Mohammadi B., Jalili R., Bahrami G.H. Rapid and sensitive high performance liquid chromatographic determination of zonisamide in human serum application to a pharmacokinetic study // Indian J. Pharm. Sci. 2012. V. 74. № 4. P. 360. https://doi.org/10.4103/0250-474X.107073
Kaushik K., Sripuram V., Bedada S., Reddy N., Priyadarshini G., Devarakonda K. A simple and sensitive validated HPLC method for quantitative determination of cisplatin in human plasma // Clin. Res. Reg. Affairs. 2010. V. 27. № 1. P. 1. https://doi.org/10.3109/10601330903490462
Naik K., Nandibewoor S. RP-HPLC Method for the Estimation of 6-mercaptopurine in spiked human plasma and pharmaceutical formulations // J. Anal. Chem. 2013. V. 68. № 12. P. 1212. https://doi.org/10.7868/S0044450213120049
Madej K., Paprotny Ł., Wianowska D., Kasprzyk J., Herman M., Piekoszewski W. A fully validated HPLC–UV method for determination of sulthiame in human serum/plasma samples // Biomed. Chromatogr. 2021. V. 35. № 3. Article e5002. https://doi.org/10.1002/bmc.5002
Hemasree S., Sumadhuri B., Murthy T. Quantization of didanosine in human plasma using high-performance liquid chromatography-tandem mass spectrometry // J. Adv. Pharm. Edu. Res. 2013. V. 3. № 3. P. 187
Galmier M., Frasey A., Bastide M., Beyssac E., Petit J., Aiache J., Lartigue-Mattei C. Simple and sensitive method for determination of metronidazole in human serum by high-performance liquid chromatography // J. Chromatogr. B: Biomed. Sci. App. 1998. V. 720. № 1–2. P. 239. https://doi.org/10.1016/s0378-4347(98)00443-5
Dasht Bozorg B., Goodarzi A., Fahimi F., Tabarsi P., Shahsavari N., Kobarfard F., Dastan F. Simultaneous determination of isoniazid, pyrazinamide and rifampin in human plasma by high-performance liquid chromatography and UV detection // Iran. J. Pharm. Res. 2019. V. 18. № 4. P. 1735. https://doi.org/10.22037/ijpr.2019.1100849
Hilhorst M., Hendriks G., van Hout M., Sillén H., van de Merbel N. HPLC–MS/MS method for the determination of cytarabine in human plasma // Bioanalysis. 2011. V. 3. № 14. P. 1603. https://doi.org/10.4155/bio.11.140
Anders N., Wanjiku T., He P., Azad N., Rudek M. A robust and rapid liquid chromatography tandem mass spectrometric method for the quantitative analysis of 5-azacytidine // Biomed. Chromatogr. 2016. V. 30. № 3. P. 494. https://doi.org/10.1002/bmc.3562
European Medicines Agency / Assessment report for vidaza (international nonproprietary name: azacytidine) // № 593162. 2008. P. 1.
D’Avolio A., Peila E., Simiele M., Pensi D., Baietto L., Cusato J., Cinnirella G., De Rosa F., Di Perri G. Ultra performance liquid chromatography PDA method for determination of tigecycline in human plasma // Ther. Drug Monit. 2013. V. 35. № 6. P. 853. https://doi.org/10.1097/FTD.0b013e31829403b1
Bergman J., Harvill L., Hawkins S., Sladky K., Cox S. Determination of ceftazidime in plasma by RP-HPLC and ultraviolet detection // Biomed. Chromatogr. 2021. V. 35. № 7. Article e5104. https://doi.org/10.1002/bmc.5104
Dincel D., Sagirli O., Topcu G. A high-performance liquid chromatographic method for the determination of meropenem in serum // J. Chromatogr. Sci. 2020. V. 58. № 2. P. 144. https://doi.org/10.1093/chromsci.bmz087
Paal M., Zoller M., Schuster C., Vogeser M., Schütze G. Simultaneous quantification of cefepime, meropenem, ciprofloxacin, moxifloxacin, linezolid and piperacillin in human serum using an isotope-dilution HPLC–MS/MS method // J. Pharm. Biomed. Anal. 2018. V. 152. P. 102. https://doi.org/10.1016/j.jpba.2018.01.031
Roth T., Fiedler S., Mihai S., Parsch H. Determination of meropenem levels in human serum by high-performance liquid chromatography with ultraviolet detection // Biomed. Chromatogr. 2017. V. 31. № 5. https://doi.org/10.1002/bmc.3880
Электронный ресурс: https://go.drugbank.com/drugs (дата обращения: ноябрь 2021 г.).
Зенкевич И.Г. Контроль, коррекция и восстановление значений физико-химических свойств органических соединений с использованием рекуррентных зависимостей // Журн. физ. химии. 2021. Т. 95. № 5. С. 700. https://doi.org/10.1134/S0036024421040294
Линник Ю.В. Метод наименьших квадратов и основы теории обработки наблюдений. М.: Физматгиз, 1958. 334 с.
Зенкевич И.Г. Модификация параметра “стандартное отклонение” для выявления и характеристики асимметричных выборок данных // Вестн. СПбГУ. Сер. физ.-хим. 1998. Вып. 2. С. 84.
Wu K., Wei G.-W. Quantitative toxicity prediction using topology based multitask deep neutral networks // J. Chem. Soc. Int. Model. 2018. V. 58. № 2. P. 520. https://doi.org/10.1021/acs.jvum.7b00558
Pu L., Naderi M., Liu T., Wu H.-C., Mukhopadhyay S., Brylinski M. eToxPred: A machine learning-based approach to estimate the toxicity of drug candidate // Pharmacol. Toxicol. Online version: https://doi.org/10.1186/s40360-018-0282-6
Semenova E., Lazic S.E. A Bayesian neural network for toxicity prediction // Computat. Toxicol. 2020. V. 16. P. 100. https://doi.org/10.1016/j.comtox.2020.100133
Xia X. Drug efficacy and toxicity prediction: An innovative application of transcriptomic data // Cell Biol. Toxicol. 2020. Online version: https://doi.org/10.1007/s10565-020-09552-2
Rim K.-T. In silico prediction of toxicity and its application for chemicals at work // Toxicol. Environ. Health Sci. 2020. V. 15. P. 1. https://doi.org/10.1007/s13530-020-00056-4
Зенкевич И.Г., Никитина Д.А., Деруиш А. Особенности образования и хроматографического детектирования гидратов органических соединений // Журн. аналит. химии. 2021. Т. 76. № 4. С. 331. https://doi.org/10.1134/S1061934821040146
Зенкевич И.Г., Деруиш А. Аналитические аспекты зависимости индексов удерживания органических соединений в обращенно-фазовой ВЭЖХ от содержания метанола в составе элюента // Аналитика и контроль. 2022. Т. 26. № 1. С. 41. https://doi.org/10.15826/analitika.2022.26.1.004
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии