

Журнал аналитической химии, 2022, T. 77, № 11, стр. 969-1015
Определение остаточных количеств антибиотиков в объектах окружающей среды и пищевых продуктах
О. И. Лаврухина a, b, В. Г. Амелин a, b, *, Л. К. Киш a, А. В. Третьяков a, Т. Д. Пеньков a
a Всероссийский государственный центр качества и стандартизации
лекарственных средств для животных и кормов
123022 Москва, Звенигородское шоссе, 5, Россия
b Владимирский государственный университет
имени Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 24.03.2022
После доработки 14.04.2022
Принята к публикации 14.04.2022
- EDN: EHJXDO
- DOI: 10.31857/S004445022211007X
Аннотация
Представлен обзор методов пробоподготовки и определения остаточных количеств антибиотиков в объектах окружающей среды и продуктах питания: аминогликозидов, амфениколов, гликопептидов, диаминопиримидинов, ионофоров, линкозамидов, макролидов, нитроимидазолов, нитрофуранов, пенициллинов, плевромутилинов, полипептидов, сульфаниламидов, тетрациклинов, хинолонов, хиноксалинов и цефалоспоринов. Рассмотрены способы их индивидуального и группового определения. Показано, что для группового определения остаточных количеств антибиотиков различных классов используют в основном высокоэффективную и ультравысокоэффективную жидкостную хроматографию с масс-спектрометрическим детектированием. Перспективными способами пробоподговки в настоящее время являются QuEChERS, дисперсионная жидкостно-жидкостная микроэкстракция и твердофазная экстракция.
Антибиотики широко применяются в ветеринарии. В организме животных они метаболизируются и частично выводятся в неизменном виде, что приводит к их попаданию в окружающую среду и биоаккумуляции в продукции животноводства. Потребление человеком продуктов, содержащих антибиотики, представляет угрозу для здоровья и обусловливает развитие антибиотикорезистентности. Для обеспечения “здоровья человека через здоровье животных” необходим контроль их остаточных содержаний в объектах окружающей среды и пищевой продукции.
Как минимум 70% антибиотиков, имеющих жизненно важное значение для человека, реализуется на рынке для ветеринарного применения, среди них наиболее значимы аминогликозиды, амфениколы, гликопептиды, диаминопиримидины, ионофоры, линкозамиды, макролиды, нитроимидазолы, нитрофураны, пенициллины, плевромутилины, полипептиды, сульфаниламиды, тетрациклины, хинолоны, хиноксалины и цефалоспорины. Они используются не только для лечения инфекционных заболеваний животных, но и в профилактических целях при выращивании крупного (КРС) и мелкого рогатого скота (МРС), птицы, объектов аквакультуры и др. Антибактериальные препараты могут быть введены инъекционно (внутривенно, внутримышечно или подкожно), перорально, местно (на кожу), и другими способами. Несоблюдение дозировки, а также сроков выведения из организма живых животных, а кроме того, использование антибиотиков в качестве стимуляторов роста приводит к их попаданию в животноводческую продукцию, сточные воды и почву [1–5].
В странах Европейского Союза наиболее часто используемыми в ветеринарии антибиотиками являются тетрациклины (32.4%), пенициллины (25.8%) и сульфаниламиды (11.5%) [6]. Далее следуют макролиды (7.0%), аминогликозиды (5.1%), полимиксины (5.1%), линкозамиды (3.0%), плевромутилины (2.8%) и фторхинолоны (2.2%). На другие группы приходится 5.1% от общего объема продаж этих веществ. Согласно отчету Европейского Медицинского Агентства 2018 г., охватывающего 30 стран (27 стран ЕС, Исландия, Норвегия и Швейцария), больше всего ветеринарных антибактериальных препаратов использовалось в Испании, Италии и Германии, меньше всего – в Исландии, Люксембурге и Словении. В США также чаще всего используются тетрациклины (71%) [4].
После потребления животными антибиотики метаболизируются и частично выводятся из организма в неизменном виде: 75–80% доз тетрациклинов (согласно некоторым источникам 90% [5]), 50–90% – эритромицина и 60% – линкомицина [3]. Биоаккумуляция антибиотиков и их метаболитов в органах и тканях животных, их перенос в молоко, яйца, мед и другую продукцию животноводства обуславливают риск для здоровья потребителей и усугубляют проблему развития антибиотикорезистентности [3, 7].
Нарушения в технологических процессах утилизации неиспользованных терапевтических препаратов, очистки сточных вод фармацевтических и сельскохозяйственных предприятий, а также использование в качестве удобрения навоза влекут за собой загрязнение таких объектов окружающей среды, как почва, поверхностные и грунтовые воды, растения [3, 6, 8–10] и, соответственно, корма [7]. В качестве удобрения используются и биосолиды (твердые органические материалы, богатые питательными веществами и полученные фильтрацией бытовых сточных вод), которые также могут содержать экскретируемые антибиотики. Все это в итоге приводит к загрязнению продукции животноводства. Кроме того, попадание антибиотиков в почву влияет на активность и разнообразие почвенных микробных сообществ [3].
Загрязнение воды и почвы антибиотиками зависит в основном от уровня антропогенной нагрузки в конкретном регионе. Отмечено значительно большее содержание антибиотиков в почве пахотных земель, чем в лесной и садовой [2]. Их содержание в сточных водах колеблется от нг/мл до мкг/мл [3].
ОПРЕДЕЛЕНИЕ АНТИБИОТИКОВ В ОБЪЕКТАХ ОКРУЖАЮЩЕЙ СРЕДЫ
Предложено большое количество методик определения антибиотиков и их метаболитов в почве, навозе, биосолидах, сточных и природных водах, а также в донных отложениях (табл. 1). Перечисленные объекты не столь сложны для анализа, как, например, биологические жидкости, пищевые продукты и продовольственное сырье, в особенности животного происхождения.
Таблица 1.
Определение антибиотиков в почве, навозе, биосолидах, сточных и природных водах
| Антибиотик | Метод, колонка | Подвижная фаза | Предел обнаружения | Предел определения | Объект анализа, пробоподготовка, степень извлечения (%) |
Литера-тура |
|---|---|---|---|---|---|---|
| 3 ТЦ и их эпимеры | УВЭЖХ-МС/МС, ACQUITY UPLC BEH C18 | Градиент 0.1% МК – АЦН | 0.6–2.5 мкг/кг | 2.0–10.0 мкг/кг | Почва с добавлением свиного навоза; ЖЖЭ, ТФЭ-очистка, 23.3–159.2 |
[11] |
| 4 НФ и 4 их метаболита | ВЭЖХ-ДМД, Mightysil RP-18 (250 × 4.6 мм,
5 мкм), λ = 362–369 нм ВЭЖХ-МС/МС, Zorbax Eclipse XDB-C18 (250 × 4.6 мм, 5 мкм) |
АЦН + 25 мМ NaH2PO4 0.05 мМ АА + метанол | 0.05–0.6 мг/л 0.2–0.6 мкг/л |
0.08–1.0 мг/л 0.3–1.0 мкг/л |
Прудовая вода и донные отложения;
фильтрация и
ЖЖЭ + фильтрация, 72.9–102.3 |
[12] |
| 13 фармацевтических препаратов, в т.ч. ХЛ, ТЦ, СА, МЛ | УВЭЖХ-МС/МС, BEH C8 (2.1 × 100 мм, 1.7 мкм) | Градиент 0.1% УК – АЦН + 0.1% УК | – | 4.0–272.0 нг/л | Сточные воды, навоз и донные отложения; QuEChERS, онлайн-ТФЭ (рН 2.6), >75.0 | [13] |
| 6 ТЦ и 2 эпимера | ВЭЖХ-МС-ВР, Hypersil GOLDTM C18 (150 × 2.1 мм, 3 мкм) | Градиент 0.1% МК – метанол + + 0.1% МК | 1.5–3.6 нг/г | 6.6–40.0 нг/г | Свиной навоз; ЖЖЭ, ТФЭ-очистка, 40.0–103.0 |
[14] |
| 26 препаратов, в т.ч. ТЦ, СА, МЛ, ХЛ, ЛА, ПМ | ВЭЖХ-МС/МС, Eclipse XDB-C18 (150 × 2.1 мм, 3.5 мкм) | Градиент АЦН – 0.1% МК | 0.01–1.86 мкг/кг | 0.05–5.91 мкг/кг | Свиной навоз;
QuEChERS, 61.39–105.65 |
[15] |
| Ципрофлоксацин | ВЭЖХ-ФЛД, LaChrom C18 (250 × 4.6 мм, 5 мкм), λ = 280/452 нм | 25 мМ H3PO4 + + триэтиламин/ АЦН (82/18) | 0.2 мкг/л | 0.67 мкг/л | Морская вода; МИ-ТФЭ, 75.2–112.4 | [16] |
| 82 препарата, в т.ч. СА, НИ, ТЦ, ХЛ, БЛ, МЛ | УВЭЖХ-–МС/МС, Acquity BEH C18 (50 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – метанол/АЦН (20/80) + 0.1% МК | 0.3–2.0 мкг/кг | 1.0–10.0 мкг/кг | Донные отложения отстойника свинофермы; ТФЭ, 60.0–110.0 | [17] |
| 67 соединений, в т.ч. ХЛ, МЛ, ТЦ, СА, ДП | УВЭЖХ-МС/МС, ZORBAX Eclipse XDB C18 (50 × 4.6 мм, 1.8 мкм) | Градиент 0.1% МК–АЦН | 1–357 нг/л | 10–500 нг/л | Очищенные сточные воды; фильтрация с добавлением АЦН, 70.0–120.0 | [18] |
| 3 МЛ | ВЭЖХ-МС/МС, Kinetex XB-C18 (100 × 3 мм, 2.6 мкм) | Градиент АЦН–0.1% МК | 11.5–26.0 нг/л | 34.0–77.7 нг/л | Вода; магнитная ТФЭ, >84.0 | [19] |
| 4 АФ и их метаболиты | УВЭЖХ-МС/МС, ACQUITY UPLC®BEH C18 (2.1 × 100 мм, 1.7 мкм) | Изократическое элюирование 0.05% NH3/АЦН (40/60) | 0.05–0.15 мкг/л | 0.17–0.49 мкг/л | Вода для аквакультуры; ДТФЭ, 70.1–109.2 | [20] |
| Окситетрациклин, флорфеникол, флумеквин | ВЭЖХ-МС-ВП, Luna Omega Polar C18 (100 Å, 100 × 2.1 мм, 5 мкм) | Градиент 0.1% МК – метанол | – | 0.1–0.5 мкг/л, 1–500 мкг/кг |
Морская вода, донные отложения, биологи-ческие образцы; ТФЭ, 63.0–120.0 | [21] |
| 3 ТЦ | УВЭЖХ-УФ, BEH C18 (50 × 2.1 мм, 1.7 мкм), λ = 314 нм | 0.02 М ЩК/метанол/АЦН (7/1/2) | 0.027–0.107 мкг/л | 0.133–0.267 мкг/л | Речная вода; магнитная ТФЭ, 91.0–104.6 | [22] |
| 28 антибиотиков, в т.ч. СА, ХЛ, ЛА, МЛ, ТЦ | УВЭЖХ-МС/МС, Kinetex C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК–АЦН | 0.002–0.200 нг/мл | 0.005–0.500 нг/мл | Сточные воды свино-водческих хозяйств; QuEChERS, 50.0–100.0 | [23] |
| 4 СА и 3 метаболита | ВЭЖХ-МС/МС, Phenomenex Kinetex C18 (100 × 2.1 мм, 2.6 мкм) | Градиент метанол – вода | 0.01–0.23 нг/л | 0.03–0.78 нг/л | Сточные воды; ТФЭ, 77.7–148.1 | [24] |
| Триметоприм и 6 СА | ВЭЖХ-УФ, Kinetex XB C18 50 × 3.0 мм, 2.6 мкм), λ = 265 нм | Градиент 10 мМ ФА–АЦН/ метанол (8/92) | 2.15–7.64 нг/мл | 36.8–119.2 нг/мл | Природные воды; ДЖЖМЭ, – | [25] |
| 62 фарм. препарата, в т.ч. ТЦ, СА, БЛ, МЛ, ПМ, ДП, ХЛ | УВЭЖХ-МС/МС, ACQUITY BEH C18 (100 × 3.0 мм, 1.7 мкм) | Градиент 0.1% МК – АЦН – АЦН/метанол (2/1) | 0.00119–0.623 нг/л | 0.00475–2.49 нг/л | Питьевая вода; онлайн-ТФЭ, 80.7–119.9 |
[26] |
| 37 пестицидов и 33 антибиотика, в т.ч. АФ, МЛ, СА и НИ | УВЭЖХ-МС/МС, ACQUITY BEH C18 UPLC (2.1 × 100 мм, 1.7 мкм) | Градиент 2 мМ ФА + + 0.1% МК | 0.2–3.5 мкг/кг | 0.5–11.5 мкг/кг | Навоз домашнего скота и птицы; ASE, магнитная очистка, 60.3–110.0 | [27] |
| 40 антибиотиков, в т.ч. ЦС, ЛА, МЛ, НИ, ХЛ, СА, ТЦ | ВЭЖХ-МС/МС, Kinetex C18 (100 × 2.1 мм, 2.6 мкм) | Градиент 0.001% МК – метанол | 0.01–34.3 мкг/кг | 0.03–115.0 мкг/кг | Донные отложения; ЖЖЭ, ДТФЭ, 24.0–162.0 |
[28] |
| Пенициллин G | ВЭЖХ-УФ, Ultimate XB C18 (4.6 × 250 мм, 5 мкм), λ = 220 нм | Метанол + фосфатный буферный раствор (рН 4.5) | 0.493 мкг/л | 1.638 мкг/л | Сточные и природные воды; ТФЭ, 91.31–123.27 | [29] |
| 10 СА | УВЭЖХ-МС/МС, Hypersil gold C18 (2.1 × 100 мм, 1.9 мкм) | Градиент метанол – 0.1% МК | 0.54–4.5 нг/л | 1.9–15.0 нг/л | Природные воды; ДТФЭ, 82.0–105.4 | [30] |
| 7 СА | ВЭЖХ-УФ, InertSustain C18 (4.6 × 250 мм, 5 мкм), λ = 270 нм | Метанол/ 0.01 М ЩК + вода (32/68) | 0.01–0.03 мкг/л | – | Вода; ДТФЭ, 90.5–101.9 |
[31] |
| 168 фарм. препаратов и их метаболитов, в т.ч. ХЛ, ТЦ, СА, МЛ, БЛ | ВЭЖХ-МС/МС, Phenomenex C18 (50 × 3.0 мм, 2.6 мкм) | Градиент 0.1% МК–0.1% МК + АЦН | – | <0.1 нг/л | Сточные и природные воды; ТФЭ, 77.0–117.0 | [32] |
| 11 ХЛ | ВЭЖХ-МС/МС, Poroshell 120 SB-C18 (100 × 2.1 мм, 2.7 мкм) | Градиент 0.1% МК – метанол + 0.1% МК | 6–150 нг/л | 33–500 нг/л | Сточные воды; МИ-ТФЭ, >75.0 | [33] |
| 45 антибиотиков, в т.ч. АФ, ЦС, ДП, ЛА, МЛ, НФ, ПЦ, ПМ, ХЛ, СА, ТЦ | УВЭЖХ-МС-ВР, Hypersil Gold C18 (100 × 2.1 мм, 1.9 мкм) | Градиент 0.1% МК – 0.1% МК + метанол | 10–50 нг/л | 50 нг/л | Прудовая вода; ТФЭ, 65.7–118.3 | [34] |
| 8 антибиотиков, в т.ч. ЦС, СА, ДП, МЛ, ХЛ |
ВЭЖХ-МС/МС, Shim-pack XR-ODSIII C18 (150 × 2.1 мм, 2.6 мкм | Градиент 0.1% МК – 0.1% МК + АЦН | 18.54–78.49 нг/л | 58.96–249.59 нг/л | Сточные воды; низкотемпературная разделительная экстракция, 15.6–57.2 | [35] |
| 13 МЛ и их метаболиты | ВЭЖХ-МС/МС, ACE C18 PFP | Градиент 0.1% МК–АЦН | 0.13–1.1 нг/г | 0.39–3.2 нг/г | Донные отложения и глубинный водоносный горизонт; ЖЭ под давлением, ТФЭ (для воды), 54.0–99.0 | [36] |
| 107 соединений, в т.ч. 2 ЛА |
ВЭЖХ-МС/МС, Kinetex C18 (150 × 4.6 мм, 2.6 мкм) | Градиент 0.1% МК – метанол | – | 5–500 нг/л | Очищенные городские сточные воды, QuEChERS, 70.0–120.0 | [37] |
| 172 антропогенных загрязнителя, в т.ч. ЛА, ПЦ, ЦС, СА, ДП | ВЭЖХ-МС-ВР, Thermo Hypersil GOLD aQ column (50 × 2.1 мм, 1.9 мкм) | Градиент 0.1% МК – 0.1% МК + метанол | – | <1 нг/л | Сточная и водопроводная вода; ТФЭ, >70.0 | [38] |
| 3 ТЦ | ВЭЖХ-УФ, C18 (250 × 4.6 мм, 5 мкм), λ = 224 нм | Градиент АЦН/метанол (50/50) – 0.01 М ЩК | 1.37–4.38 мкг/л | 4.58–14.60 мкг/л | Вода колодезная, дождевая из леса, прибрежная морская, садовая, минеральная; ДЖЖМЭ, 74.0–113.0 | [39] |
| 3 ХЛ | ВЭЖХ-УФ, ZORBAX SB-C18 (150 мм × 4.6 мкм, 5 мкм), λ = 293 нм | Градиент АЦН – 0.01 М H3PO4 | 6.4–9.9 нг/мл | 17.8–34.5 нг/мл | Речная вода; магнитная МИ-ТФЭ, 84.1–91.9 | [40] |
Обозначения: АА – ацетат аммония; АФ – амфениколы; АЦН – ацетонитрил; БЛ – β-лактамы; ДП – диаминопиримидины; ДТФЭ – дисперсионная твердофазная экстракция; ЖЖЭ – жидкостно-жидкостная экстракция; ЛА – линкозамиды; МИ-ТФЭ – твердофазная экстракция с использованием молекулярно-импринтированных полимеров; МК – муравьиная кислота; МЛ – макролиды; МС-ВП – времяпролётная масс-спектрометрия; МС-ВР – масс-спектрометрия высокого разрешения; НИ – нитроимидазолы; НФ – нитрофураны; ПМ – плевромутилины; ПЦ – пенициллины; СА – сульфаниламиды; СПМР – супрамолекулярные растворители; ТЦ – тетрациклины; УК – уксусная кислота; ФА – формиат аммония; ХЛ – хинолоны; ЦС – цефалоспорины; ЩК – щавелевая кислота.
Благодаря развитию аналитической химии, инструментария и методических подходов к подготовке проб и последующему анализу, многие фармацевтические соединения удается обнаружить в сточных и природных водах на уровне нг/л [41]. Их определение в основном предполагает предварительную твердофазную экстракцию (ТФЭ). Наиболее часто используемым сорбентом для ТФЭ в случае анализа водных сред на содержание в них антибактериальных препаратов является гидрофильно-липофильный балансный сорбент (hydrophilic-lipophilic balance, HLB), поскольку он селективен и эффективнен при выделении полярных соединений [6].
На сегодняшний день, на наш взгляд, одним из перспективных способов пробоподготовки, соответствующих принципам “зеленой химии”, является дисперсионная жидкостно-жидкостная микроэкстракция (ДЖЖМЭ). С момента внедрения в 2006 г. этот подход стал популярным экологически безопасным методом подготовки образцов [42, 43]. Преимуществами ДЖЖМЭ являются предельно малые объемы экстрагента 50–500 мкл, экспрессность и высокие (до нескольких тысяч) коэффициенты концентрирования. Пределы обнаружения примесей, достигаемые при сочетании хроматографического анализа и микроэкcтракционного концентрирования, составляют 10–7–10–4 мг/л.
Метод ДЖЖМЭ основан на использовании трехкомпонентных систем растворителей: проба−экстрагент−диспергент, он включает два этапа. На первом этапе смесь экстракционного и диспергирующего растворителей быстро вводится в водный раствор пробы и диспергируется в виде очень мелких капель. В связи с большой площадью поверхности контакта между экстрагентом и водным образцом равновесное состояние достигается быстро, и извлечение не зависит от времени, что является наиболее важным преимуществом метода. Второй стадией является центрифугирование мутного раствора. После центрифугирования фаза, содержащая определяемые вещества, отбирается микрошприцем. Органические растворители выбирают на основании их более высокой плотности по сравнению с водой и экстракционной возможности интересующих компонентов [42]. В качестве экстрагирующего растворителя обычно выбирают галогенированные углеводороды, такие как хлорбензол, хлороформ, четыреххлористый углерод и тетрахлорэтилен, из-за их высокой плотности.
В настоящее время ДЖЖМЭ применяется для определения достаточно широкого круга органических соединений, в том числе антибиотиков в объектах окружающей среды и продуктах питания. Благодаря сочетанию ДЖЖМЭ и высокоэффективной жидкостной хроматографии с ультрафиолетовым (ВЭЖХ-УФ) [25, 39, 44–47], диодноматричным (ВЭЖХ-ДМД) [48–51] и флуоресцентным (ВЭЖХ-ФЛД) детектированием [51–53], ВЭЖХ- и ультравысокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием (УВЭЖХ-МС/МС) [54–56] становится возможным определение ряда антибиотиков в природных, минеральных и грунтовых водах, молоке, яйцах, курице, говядине, рыбе и детских молочных порошковых смесях с достаточно высокой степенью извлечения и низкими пределами определения.
Несмотря на все явные преимущества Д-ЖЖМЭ, метод редко применяется для определения антибиотиков различных классов одновременно, за исключением использования дорогостоящего метода УВЭЖХ-МС/МС [54]. Особенностью ДЖЖМЭ является ее наиболее частое использование для анализа жидких образцов (табл. 1, 2). Кроме того, в процессе пробоподготовки ДЖЖМЭ может сочетаться с ТФЭ. Предложена УВЭЖХ-МС/МС-методика определения 10 антибиотиков в воде (питьевой, речной, сточной) [58]. В качестве экстракционного растворителя выбран дихлорметан, а в качестве диспергирующего – смесь метанол−ацетонитрил (1 : 1). Максимальные значения предела обнаружения и предела определения составили 1.67 и 5.57 нг/мл соответственно. Степень извлечения находилась в диапазоне от 64.16 до 99.80%, относительное стандартное отклонение − от 0.7 до 8.4%.
Таблица 2.
Дисперсионная жидкостно-жидкостная микроэкстракция при определении антибактериальных препаратов
| Антибиотик | Матрица | Экстрагент (объем, мкл) |
Диспергент (объем, мл) | Объем пробы, мл (навеска, г) | Степень извлечения, % (фактор концентри-рования) | Метод анализа | Предел обнаружения | Лите-ратура |
|---|---|---|---|---|---|---|---|---|
| 5 СА | Молоко | [C4MIM-TEMPO]Cl (50 мг) |
0.1 г/мл KPF6 (0.26) | 40.0 (4.0) | 97.2–101.6 | ВЭЖХ-УФ, Amethyst C18-H (4.6 × 250 мм, 5 мкм) | 0.534–0.891 мкг/л | [44] |
| 2 СА | Курица | Тетрабутил-аммония бромид, малоновая и гексановая кислоты (1 : 1 : 1) (2 г) | Вода (10) | 1.0 | 45.0–75.0 | ВЭЖХ-УФ, Phenomenex C18 (250 × 4.6 мм, 5 мкм) | 3.0–7.0 мкг/кг | [45] |
| 3 СА | Молоко | Тимол + октановая кислота (3.40 г + 1.65 г) (500) | Вода (25) | 10.0 | 64.0–100.0 | ВЭЖХ-УФ, Luna C18(2) (250 × 4.6 мм, 5 мкм), λ = 280 нм | 1.0–5.0 мкг/л | [46] |
| 4 ТЦ | Молоко, яйца | [C6MIM][PF6] (130 мкл) | [C2MIM][BF4]/ [C4MIM][NPA] (120/45) (165 мкл) |
3.0 (молоко) 1.0 (яйца) |
94.1–102.1 | ВЭЖХ-УФ, Zorbax Eclipse SB-C18 (4.6 × 250 мм, 5 мкм), λ = 355 нм | 0.08–1.12 мкг/кг | [47] |
| 6 ХЛ | Куриный ливер | Трихлорметан (200) | АЦН-экстракт (1.0) | 5.0 | 83.0–102.0 | ВЭЖХ-ДМД, λ = 280 нм | 5.0–19.0 мкг/кг | [48] |
| 11 СА и 14 ХЛ | Вода | CHCl3 | АЦН (1.25) | 5.0 | 78.0–117.0 | УВЭЖХ-ДМД (260–300 нм) | 0.35–10.5 нг/мл | [49] |
| 6 СА | Детские молочные смеси | [C6MIM][PF6] (70) |
Ультразвук | 0.5 | 90.4–114.8 | ВЭЖХ-ДМД (270 нм) |
2.94–6.66 мкг/кг | [50] |
| Альбендазол, хлорамфеникол, триметоприм, энрофлоксацин, окситетрациклин, никарбазин |
Яйца | Дихлорметан (160) | АЦН (1.84) | 1.0 | 24.1–98.3 | ВЭЖХ-ДМД/ФЛД | – | [51] |
| 8 ХЛ | Грунтовые воды | [C8MIM][PF6] (65 мг) |
Метанол (0.4), ультразвук |
10.0 | 85.0–107.0 (122.0–205.0) |
ВЭЖХ-ФЛД, λ = 278–324/ 366– 514 нм |
0.8–13.0 нг/л | [52] |
| 6 ХЛ | Молоко | Хлороформ (200) | Сухой остаток после QuEChERS в АЦН + + 10% УК (0.1) | 2.0 QuEChERS | 69.2–104.8 | ВЭЖХ-ФЛД, λ =278–294/ 466–514 нм |
0.8–5.0 мкг/кг | [53] |
| 17 ХЛ, 6 ЦС, 8 ПЦ | Сырое коровье молоко | Трихлорметан (570, 440) | АЦН (1.070, 1.500) | 1.0 | 72.0–110.0 | УВЭЖХ-МС/МС (экстракты объединены) | 0.1–1.3 нг/г | [54] |
| 6 ТЦ | Говядина | Дихлорметан (200) | Метанол (1.0) | 1.0 | 80.0–105.0 | ВЭЖХ-МС/МС | 2.22–3.59 мкг/кг | [55] |
| 2 НИ, 2 АФ, 2 красителя | Рыба | Дихлорметан (500) |
АЦН (0.7) | 2.0 | Микровол-новая экстракция и ТФЭ очистка; > 87.0 | ВЭЖХ-МС/МС | 4.52–101.3 пг/кг | [56] |
| 3 ТЦ | Молоко | АЦН (1000) | Тимол/октано-вая кислота (150 мкл) | 1.0 | 70.0–113.0 | ВЭЖХ-УФ, C18 (250 × 4.6 мм, 5 мкм), λ = 369 нм | 1.5–8.5 мкг/л | [57] |
Обозначения: АФ – амфениколы;АЦН – ацетонитрил; НИ – нитроимидазолы; ПЦ – пенициллины; СА – сульфаниламиды; ТЦ – тетрациклины; ХЛ – хинолоны; ЦС – цефалоспорины; УК – уксусная кислота; C2MIM – 1-этил-3-метилимидазолий; C4MIM – 1-бутил-3-метилимидазолий; C6MIM – 1-гексил-3-метилимидазолий; C4MIM-TEMPO – условное обозначение синтезированного молекулярно-импринтированного полимера; C8MIM – 1-октил-3-метилимидазолий; NPA – соль нафтоевой кислоты.
Процедура определения антибиотиков в почвах и донных отложениях водоемов сложнее в связи с более трудозатратной пробоподготовкой, но в настоящее время предложена масса решений этой проблемы: ускоренная экстракция растворителем (accelerated solvent extraction, ASE), жидкостная экстракция с использованием ультразвука (УЗ-ЖЭ), дисперсионная ТФЭ (ДТФЭ) [41, 59, 60]. При необходимости процесс пробоподготовки завершается дополнительной ТФЭ-очисткой и фильтрацией. Доступные технологии позволяют определять соединения, в том числе антибиотики, на уровне следовых количеств (несколько нг/л и менее). Так как молекулы большинства антибактериальных препаратов полярны, для анализа водных сред и донных отложений предпочтителен метод высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС) благодаря его высокой селективности и чувствительности (табл. 1). Определение соединений в основном проводится в режиме мониторинга множественных реакций (MRM) – используются как минимум два наиболее характерных ионных перехода ион-предшественник/продукт и надежность результатов обеспечивается не только определением времени удерживания, но и использованием характеристических ионов. Вместе с тем в последнее время повышен интерес и к УВЭЖХ – она обеспечивает еще более высокое разрешение и скорость выполнения анализа [41].
Для снижения и оценки матричных эффектов, в том числе при анализе морской воды и донных отложений, используются метод разбавления после экстракции и изотопно-меченные стандарты, хотя это приводит к увеличению стоимости и/или продолжительности анализа [41]. Однако для получения надежных результатов применение указанных приемов необходимо, поэтому потребность в разработке более селективных, чувствительных, но при этом быстрых и доступных методик сохраняется.
Распределение антибиотиков в почве обусловлено, во-первых, структурой самих соединений и физико-химическими свойствами представителей различных классов (в первую очередь полярностью). Во-вторых – особенностями почвы, так как сорбционные свойства и связующая способность частиц определяются ее составом, рН среды и содержанием органических веществ [5, 8, 10]. Максимальные содержания в почве (мг/кг) определены для окситетрациклина – 50, хлортетрациклина – 11, ципрофлоксацина – 5.6, сульфаметазина – от 0.2 до 25 и тилозина – 1.3 [3, 61]. Для тетрациклинов установлен наибольший риск попадания в пищевую цепь при низком содержании в почве органических веществ, так как в таких условиях их сорбция существенно снижена [5].
Основным источником поступления тетрациклинов, фторхинолонов, сульфаниламидов и макролидов в почву является применение навоза в качестве удобрения. Фторхинолоны отличаются высокой специфичностью взаимодействия с почвами: коэффициент сорбции в почве более чем в 600 раз выше, чем в курином помете [9]. Разработана методика определения ветеринарных антибиотиков в навозе бройлеров, почве и компосте методом ВЭЖХ-МС/МС, предел определения 2–16 мкг/кг [62]. Пробоподготовка, сочетающая жидкостную экстракцию и последующую ТФЭ-очистку на HLB-картриджах позволяет достичь степени извлечения для девяти антибиотиков 63.0–121.0%, максимальные содержания в реальных образцах куриного навоза и почвы установлены для доксициклина и флумеквина. В жидких фракциях свиного навоза содержится до 20.4 мг/л линкомицина – это один из самых высоких показателей остаточного содержания неметаболизированных антибиотиков, экскретируемых животными [63]. Твердофазная экстракция с использованием HLB-картриджей также предложена в качестве пробоподготовки для определения β-лактамных антибиотиков и полиэфирных ионофоров методом ВЭЖХ-МС/МС в навозе КРС, сточных и прудовых водах [64]. Пределы обнаружения цефапирина, пенициллина G, клоксациллина, монензина, салиномицина и наразина составили 0.15–2.13 мкг/л в воде, 0.34–2.94 мкг/кг в навозе, соответствующие степени извлечения (%) – 74.4–91.0 и 71.7–94.2. Цефапирин, пенициллин G и клоксациллин обнаружены в пробах воды отстойника в концентрациях 0.97–43.31 мкг/л. Из ионофоров обнаружен только монензин (94–1077 мкг/л) в образцах прудовой воды со стоками ферм КРС. В образцах навоза молочного скота обнаружено 8.09–45.20 мкг/кг клоксациллина. Согласно литературным данным биосолиды содержат гораздо меньшее количество антибиотиков [3].
Сорбционная емкость почв по отношению к антибиотикам различна [8]. Вместе с тем отличается и способность их аккумуляции разными видами сельскохозяйственных культур. Как отмечено в работе [65], бóльшее внимание следует уделять региону, в котором выращиваются овощи с более высокой способностью к накоплению антибиотиков. Существенное значение имеют также сезонные различия биодеградации антибиотиков [63]. Все это необходимо учитывать при оценке риска контаминации растительного сырья, произрастающего в различных регионах.
Антибиотики могут присутствовать и в донных отложениях вблизи хозяйств, где их добавляют непосредственно в воду для лечения бактериальных инфекций объектов аквакультуры [7]. Связывание антибиотиков с частицами почвы и отложений может затруднять биодеградацию, повышая их устойчивость в окружающей среде. Отмечено, что уровень загрязнения воды и почвы антибиотиками близок к уровню загрязнения пестицидами, остаточные содержания в сточных водах варьируются от нескольких нг/л до мкг/л, а в твердых веществах, в том числе в почве, от мкг/кг до мг/кг [66].
Нельзя исключать и естественный путь образования антибиотиков. В работе [67] показано, что существует возможность образования хлорамфеникола (ХАФ) почвенными бактериями в их естественной среде с дальнейшим его поглощением растениями. Механизм образования запрещенного к применению в ветеринарной практике ХАФ в почве на данный момент глубоко не изучен, хотя его естественное (фоновое) содержание в растительной продукции может быть значимо (до 32 мкг/кг), что более чем в 200 раз превышает контрольное значение 0.15 мкг/кг, регламентируемое Европейским законодательством [68].
ОПРЕДЕЛЕНИЕ АНТИБИОТИКОВ В ПИЩЕВЫХ ПРОДУКТАХ
Пробоподготовка. Значительные трудности анализа продуктов питания животного происхождения обусловлены наличием большого количества сопутствующих компонентов в экстракте при содержании антибиотиков на уровне остаточных количеств. Кроме того, они могут быть преобразованы микроорганизмами, а также другими физическими и химическими способами в метаболиты, в результате чего образуется смесь экотоксикантов, представляющих еще больший риск для здоровья человека, чем отдельные соединения. Таким образом, растет интерес к доступным многокомпонентным методам анализа антимикробных смесей в пищевых продуктах (табл. 3). Немаловажным этапом при этом является стадия пробоподготовки. От выбора способа подготовки образцов и его осуществления во многом зависят конечные результаты исследования. Из-за сложности матриц для достижения требуемой чувствительности кроме стадии экстракции требуются дополнительная очистка и концентрирование экстракта. Наиболее распространенными анализируемыми пищевыми матрицами являются молоко, мед, ткани животных и яйца. Пробоподготовка включает в себя процедуры удаления белков, обезжиривания и гидролиза сахаров (для меда). Удаление белков обычно достигается с помощью органических растворителей, таких как ацетонитрил или метанол, и при необходимости образец далее обезжиривают гексаном [112].
Таблица 3.
Определение остаточных количеств антибиотиков различных классов в пищевых продуктах
| Антибиотик | Метод, колонка | Подвижная фаза | Предел обнаружения | Предел определения | Объект анализа, пробоподготовка, степень извлечения (%) |
Лите-ратура |
|---|---|---|---|---|---|---|
| 10 антибиотиков, в т.ч. ТЦ, ХЛ, АФ, СА, ЦС | УВЭЖХ-МС/МС, Waters ACQUITY UPLC® BEH Phenyl (50 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – АЦН | 0.11–1.67 нг/мл | 0.27–5.57 нг/мл | Вода; ТФЭ + ДЖЖМЭ дихлорметан/метанол/АЦН (1/1) УЗ; 64.16–99.80 | [58] |
| 11 препаратов, в т.ч. ЦС, ХЛ, СА |
ВЭЖХ-ФДЛ, ВЭЖХ-УФ, ВЭЖХ-МС/МС, 250 × 4.6 мм, 5 мкм), λЦС = 280 нм, λХЛ = 280/ 440 нм, λСА = 405/495 нм | Градиент 2% УК – АЦН | 8.0–20.0 нг/мл | 10.0–32.0 нг/мл | Молоко; ЖЖЭ, предколоночная дериватизация, >98.0 | [69] |
| 17 препаратов, в т.ч. МЛ | ВЭЖХ-МС/МС, Hypersil Gold C18 (100 × 2.1 мм, 5 мкм) | Градиент метанол – формиатный буфер | 0.21–0.61 мкг/кг | 5.0–10.0 мкг/кг | Мясо; ASE, ≈75.0 | [70] |
| 34 препарата, в т.ч. СА, ХЛ, МЛ, ТЦ, ПЦ | УВЭЖХ-МС-ВР, Hypersil GOLD aQ C18 (100 × × 2.1 мм, 1.7 мкм) | Градиент 4 мМ ФА + + 0.1% МК – метанол + + 0.1% МК | 0.1–50 мкг/кг | 5.0–50 мкг/кг | Мeд; ЖЖЭ, автоматизированная ТФЭ, 68.0–121.0 | [71] |
| 8 СА и триметоприм | ВЭЖХ-МС/МС, Xterra MS C18 (2.1 × 150 мм, 3.5 мкм) | Градиент 0.1% МК – АЦН + 0.1% МК | 0.06−0.18 мкг/кг | – | Мeд; ТФЭ, 70–106 | [72] |
| 3 СА, 2 ХН | ВЭЖХ-УФ, Diamonsil C18 (250 × 4.6 мм, 5 мкм), λ = 265 нм | АЦН/вода (0.2% УК) | 0.01–0.014 мкгг/л | 0.039-0.096 мкг/л | МИ-онлайн-ТФЭ, 71.0–108.2 |
[73] |
| 3 ЛА и 10 МЛ | ВЭЖХ-МС/МС, Kinetex XB-C18 (2.1 × 100 мм, 2.6 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | 0.5–3.0 мкг/кг | 0.5–5.0 мкг/кг | Рыба, креветки; ЖЖЭ,
ТФЭ-очистка, 47.0–99.0 |
[74] |
| 26 препаратов, в т.ч. СА, ФХ, ХЛ, триметоприм | ВЭЖХ-МС/МС, YMC ODS-AQ (2 × 100 мм, 3 мкм) | Градиент 0.1% МК – АЦН | – | 0.40–10.0 нг/г | Рыба и креветки; ЖЖЭ,
фильтрация, 88.0–112.0 |
[75] |
| 38 препаратов, в т.ч. СА, ХЛ | УВЭЖХ-МС/МС, Acquity BHE C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК + + 5 мМ АА – метанол | 1–125 мкг/кг | – | Молоко; ЖЖЭ, ТФЭ-очистка, 87.0–119.0 | [76] |
| 2 ТЦ, хлорамфеникол | ВЭЖХ-УФ, Zorbax Eclipse XDB-C18 column (4.6 × 150 мм, 3.5 мкм), λТЦ = 270 нм, λАФ = 272 нм |
Градиент АЦН – 0.8% МК | 4.2–8.2 мкг/кг | 13.8–27.4 мкг/кг | Мeд; ИЖ-ЖЖЭ, 85.5–110.9 |
[77] |
| 39 антибиотиков, в т.ч. СА, ТЦ, МЛ, ХЛ, ПЦ, АФ, ДП | УВЭЖХ-МС/МС, Acquity HSS T3 (2.1 × 100 мм, 1.8 мкм) | Градиент 0.1% МК – АЦН | 0.48−1048.0 мкг/кг | – | Печень; ЖЖЭ, ТФЭ-очистка, 80.0–110.0 | [78] |
| 45 антибиотиков, в т.ч. ПЦ, ЦС, ХЛ, ТЦ, МЛ, СА, ЛА, ДП | ВЭЖХ-МС/МС, Luna C18 (50 × 3.0 мм, 3 мкм) | Градиент АЦН – ГФМК | 0.01–3.79 мкг/л | 0.02–10 мкг/л | Вода; ТФЭ, 84.3–109.3 |
[79] |
| НФ и их метаболиты, АФ | УВЭЖХ-МС-ВР, Acquity UHPLC BEH C18 (2.1 × 50 мм, 1.7 мкм) | Градиент 10 мМ АА + метанол + NH3 – метанол | 0.05–0.5 мкг/кг | – | Мышечная ткань, почки, ливер, рыба, Мeд, яйца, молоко; гидролиз, ЖЖЭ, ОФ-ТФЭ, 22–237 | [80] |
| 78 препаратов, в т.ч. БЛ, ЛА, МЛ, АФ, ХЛ | ВЭЖХ-МС/МС, Acquity CSH C18, (2.1 × 150 мм, 1.7 мкм) | Градиент метанол/АЦН (3/1) + 0.1 мМ ФА + + 0.5 мМ МК – 0.1 мМ ФА + 0.5 мМ МК | – | 0.1–10.0 мкг/кг | Молоко, яйца, мясо; ЖЖЭ, ДТФЭ-очистка, 70.0–120.0 | [81] |
| 391 соединение, в т.ч. антибиотики, применяемые в ветеринарии | УВЭЖХ-МС-ВР, Hypersil GOLD aQ C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК + 4 мМ ФА – 0.1% МК + 4 мМ ФА + метанол | 0.5–50.0 мкг/кг | 10.0–100.0 мкг/кг | Детское питание; ЖЖЭ, ТФЭ-очистка, 70.0–120.0 | [82] |
| 88 препаратов, в т.ч. МЛ, ХЛ, СА, ТЦ, ПЦ | ВЭЖХ-МС/МС, CAP-CELL PAK C18 MG III (2.1 × 150 мм, 5 мкм) | Градиент АЦН + 0.1% МК | 0.2–2.0 мкг/кг | 0.5–10.0 мкг/кг | Молоко; онлайн-ТФЭ, 63.1–117.4 | [83] |
| 76 препаратов, в т.ч. МЛ, ПЦ, СА, ДП, АГ | ВЭЖХ-МС/МС, ACQUITY UPLC BEH HILIC (100 × 2.1 мм, 1.7 мкм) | Градиент АЦН – 1 мМ ФА + 0.1% МК – метанол | – | 0.37–178.0 мкг/кг | Мышечная ткань КРС; ЖЖЭ (2 стадии), ТФЭ-очистка, 60.0–109.0 | [84] |
| 62 антибиотика: АФ, БЛ, ДП, ЛА, МЛ, ПМ, ХЛ, СА, ТЦ | ВЭЖХ-МС-ВР, Poroshell 120 EC-C18 (100 × 3.0 мм, 2.7 мкм) | Градиент метанол – 0.1% МК | 1–5 мкг/кг | 3.3–10 мкг/кг | Мясо; ЖЖЭ, 59.0–93.0 | [85] |
| 61 препарат, в т.ч. БЛ, МЛ, СА, ТЦ, ХЛ | УВЭЖХ-МС/МС, HSS T3 (2.1 × 100 мм, 1.8 мкм) | Градиент АЦН – 0.1% МК | 0.003–1.57 мкг/кг | 0.01–5.18 мкг/кг | Молоко; ТФЭ, 61.5–118.6 | [86] |
| 120 препаратов, в т.ч. ХЛ, МЛ, БЛ, НИ, СА, ЛА, АФ, ХН, ТЦ | ВЭЖХ-МС/МС, Hypersil Gold C18 (150 × 2.1 мм, 5 мкм) | Градиент АЦН – 0.1% МК | 0.5–3.0 мкг/кг | 1.5–10.0 мкг/кг | Мясо, молоко, яйца; УЗ-ЖЖЭ, ТФЭ-очистка; >60 | [87] |
| 138 препаратов и метаболитов | УВЭЖХ-МС/МС, Hypersil Gold aQ C-18 (100×2.1 мм, 1.9 мкм) + + Accucore C-18 aQ (10 × × 2.1 мм, 1.9 мкм) | Градиент вода + 1 мл/л МК+ 4 мМ ФА – метанол + 1 мл/л МК+ 4 мМ ФА | 0.01–4.73 мкг/кг (возможность обнаружения) | – | Рыба; ЖЖЭ, ДТФЭ, 81.0–111.0 | [88] |
| 8 ХЛ, 8 СА, 4 ТЦ | УВЭЖХ-МС/МС, BEH C18 (2.1 × 50 мм, 1.7 мкм) | Градиент 0.2% МК – АЦН/метанол (1/1) | 0.5–3.0 нг/г | 1.5–6.0 нг/г | Мясо; МИ-ТФЭ, 74.5–102.7 | [89] |
| 80 препаратов, в т.ч. БЛ, ЛА, МЛ, ХЛ, НИ, СА, ДП, АФ | ВЭЖХ-МС-ВР, Acquity UPLC BEH C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – метанол | – | 0.25–25 мкг/кг | Рыба; ЖЖЭ, фильтрация, 60.74–109.85 |
[90] |
| 54 препарата, в т.ч. МЛ, СА, ХЛ | ВЭЖХ-МС-ВР, C18 (150 × 2.1 мм, 2.7 мкм) | Градиент 0.1% МК – 0.1% МК + метанол | 0.31–1.61 мкг/кг | 1.05–6.94 мкг/кг | Рыба; микро-ДТФЭ, 56.3–119.9 |
[91] |
| 36 антибиотиков, в т.ч. СА, ТЦ, АФ, ФХ, ПЦ и ДП |
ВЭЖХ-МС/МС, C18 PerfectSil Target ODS-3 HD (150 × 3.0 мм, 3 мкм) | Градиент 0.1% МК – метанол | 0.14–2.91 нг/г | 0.50–9.70 нг/г | Мeд; ЖЖЭ, 65.0–116.1 |
[92] |
| Эритромицин, тетрациклин, хлорамфеникол | ВЭЖХ-ИРС, C18 (4.6 × 250 мм, 5 мкм) | АЦН + 0.05% АА | 10–20 мкг/кг | – | Молоко; МИ-ТФЭ, 72.94–88.17 | [93] |
| 25 антибиотиков, в т.ч. ПЦ, ТЦ, СА, ХЛ, МЛ, ЛА и ДП | ВЭЖХ-МС/МС, Synergi Hydro RP C18 (150 × 2 мм, 4 мкм) | Градиент 0.1% МК – метанол | 1 мкг/кг | 2 мкг/кг | Рыба; ЖЖЭ, 91.1–105.6 |
[94] |
| 24 фарм. субстанции, в т.ч. ХЛ, СА, ТЦ | УВЭЖХ-МС/МС, XBridge BEH C18 (2.1 × 100 мм, 2.5 мкм) | Градиент метанол – 0.1% МК | – | 5–10 мкг/кг | Креветки; Двухступенчатая ЖЖЭ, 83.0–100.0 | [95] |
| 39 препаратов, в т.ч. НИ, ЛА, ХЛ, СА, АФ, МЛ, ТЦ, ПЦ и ЦС | УВЭЖХ-МС/МС, Poroshell 120 EC-C18 (150 × 2.1 мм, 2.7 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | – | 1–5 мкг/кг | Мясо, печень, почки; ЖЖЭ, ТФЭ-очистка, 60.0–120.0 | [96] |
| 25 препаратов, в т.ч. БЛ, ХЛ, АФ, НФ | УВЭЖХ-МС/МС, XBridge® MS C18 (2.1 × 100 мм, 5 мкм) | Градиент вода – АЦН + + 0.1% МК | – | 0.1–12.0 нг/г |
Молоко; ЖЖЭ, 65.9–123.5 |
[97] |
| СА, ТЦ, МЛ | ВЭЖХ-МС/МС, XTerra® C18 3.5 мм, 125 Å (100 × 2.1 мм) | Градиент 0.1% МК – 0.1% МК + АЦН | – | 0.0025–0.005 мкг/кг | Мeд; ЖЖЭ, 36.0−139.0 | [98] |
| 4 ХЛ, 3 ТЦ, 7 СА, 3 МЛ, линкомицин |
ВЭЖХ-МС/МС, Kinetex XB-C18 (100 × 3 мм, 2.6 мкм) | Градиент АЦН – 0.1% муравьиная кислота | 0.30–1.8 нг/г | 0.8–5.9 нг/г | Зерно; УЗ-ЖЖЭ, ДТФЭ, 74.0–127.0 | [99] |
| 2 ГП, 5 ПП | ВЭЖХ-МС/МС, Kinetex Biphenyl (50 × 2.1 мм, 2.6 мкм) | Градиент 0.1% МК + + АЦН – 0.1% МК | 5-20 мкг/кг | 15–30 мкг/кг | Корма; ДТФЭ, 63.1–107.5 |
[100] |
| 45 антибиотиков, в т.ч. ЦС, ДП, ФХ, ЛА, МЛ, ПЦ, ПМ, СА, ТЦ | ВЭЖХ-МС/МС, InfinityLab Poroshell 120 (50 × 2.1 мм, 1.9 мкм) | 0.1% МК+ АЦН/ метанол (80/20) |
0.3–3.0 мкг/кг | 1.0–10.0 мкг/кг | Шампиньоны; 73.0–118.0 |
[101] |
| 55 антибиотиков, в т.ч. ХЛ, СА, ТЦ, МЛ, ЛА, ПЦ, НИ, АФ | УВЭЖХ-МС/МС, ZORBAX RRHD Eclipse plus C18 (100 × 2.1 мм, 1.8 мкм) | Градиент 0.1% МК – АЦН | 1.0–5.0 нг/г | 3.0–10.0 нг/г | Креветки; ЖЖЭ, 74.3–113.3 |
[102] |
| 77 препаратов, в т.ч. СА, БЛ и ХЛ | УВЭЖХ-МС/МС, UPLC HSS T3 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | – | На уровне МДУ Канады и США | Курица; автоматизированная ТФЭ, 60.0–140.0 | [103] |
| 164 фарм. субстанций, в т.ч. АФ, ЦС, ИФ, ЛА, МЛ, НИ, ПЦ, ХЛ, СА, ТЦ | ВЭЖХ-МС-ВР, Phenomenex Luna Omega (100 × 2.1 мм, 1.6 мкм) | Градиент 0.1% МК – АЦН + 0.1% МК – метанол + 0.1% МК | – | От 0.9 нг/кг | Мясо; ЖЖЭ, 70.0–120.0 | [104] |
| 141 препарат, в т.ч. АФ, ИФ, ЛА, МЛ, НИ, ХЛ, СА, ТЦ | ВЭЖХ-МС-ВП, Poroshell 120 EC–C18 (150 × 2.1 мм, 2.7 мкм) | Градиент 0.1% МК + + 10 мМ АА – АЦН | 0.1–5.0 мкг/кг | 0.1–10.0 мкг/кг | Свинина; ЖЖЭ, ТФЭ-очистка, > 70 | [105] |
| 45 антибиотиков, в т.ч. ПЦ, ЦС, ТЦ, СА, МЛ, ХЛ | УВЭЖХ-МС-ВП, Acquity HSS T3 (2.1 × 100 мм, 1.8 мкм) | Градиент 0.1% МК – АЦН | 0.20–30.93 мкг/кг | – | Свиная печень; ЖЖЭ,
ТФЭ-очистка, 82.0–120.0 |
[106] |
| 62 препарата, в т.ч. АФ, ЛА, МЛ, ХЛ, ХН, ДП, СА, ТЦ, БЛ | ВЭЖХ-МС/МС, Hypersil Gold HILIC (150 × 3.0 мм, 5 мкм) | Градиент 5 мМ АА – АЦН + 0.1% УК | 25 мкг/кг | 75 мкг/кг | Пищевые продукты и корма; ASE, >57 | [107] |
| 291 загрязнитель, в т.ч. НИ и их метаболиты, БЛ, ЛА, МЛ, ХН, ХЛ, СА, ДП, ИФ | УВЭЖХ-МС/МС, Acquity HSS-T3 (100 × 2.1 мм, 1.8 мкм) | Градиент 0.1% МК + + 0.5 мМ АА – Метанол + + 0.1% МК; 2 мМ АА – метанол | – | 0.1–50.0 мкг/кг | Протеиновый порошок; ЖЖЭ, ТФЭ-очистка, низкотемпературная фильтрация, 65.6–142.2 |
[108] |
| 78 препаратов, в т.ч. СА, ХЛ, БЛ | УВЭЖХ-МС/МС, Acquity UPLC BEH C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН/ метанол (8/2) | 0.03–0.33 мкг/кг | 0.1–1.0 мкг/кг | Яйца; ДТФЭ, 70.5–119.2 |
[109] |
| 54 препарата, в т.ч. СА, ХЛ, МЛ, АФ, ЦС, ЛА | УВЭЖХ-МС/МС, EclipsePlusC18 RRHD (100 × 2.1 мм, 1.8 мкм) | Градиент Вода/метанол (95/5) + 0.1% МК + + 5 мМ АА – метанол | 0.3~10.9 мкг/кг | 0.1~3.8 мкг/кг | Яйца; ТФЭ, 61.5–97.0 |
[110] |
| 18 препаратов, в т.ч. ПЦ, МЛ, ЛА, ХЛ, ТЦ, СА, АФ | УВЭЖХ-МС/МС, Acquity UPLC BEH C18 (2.1 × 50 мм, 1.7 мкм) | Градиент АЦН–0.1% МК | 0.1–3.2 мкг/кг | 1.0–10.0 мкг/кг | Мясо, рыба, яйца, мед; ТФЭ, 92.59–102.86 | [111] |
Обозначения: АГ – аминогликозиды; АЦН – ацетонитрил; ГП – гликопептиды; ГФМК – гептафтормасляная кислота; ЖЖЭ – жидкостно-жидкостная экстракция; ИЖ – ионные жидкости; ИФ – ионофоры; МДУ – максимально допустимый уровень; МИ – молекулянрно-импринтированная; МС-ВР – масс-спектрометрия высокого разрешения; ОФ-ТФЭ – обращённо-фазовая твердофазная экстракция; ПП – полипептиды; ТФМЭ – твердофазная микроэкстракция; ФА – формиат аммония; ФХ – фторхинолоны; ХН – хиноксалины.
Экстракция и разбавление – самый простой способ пробоподготовки для многокомпонентных методов с высокочувствительным оборудованием. При разбавлении экстрактов могут незначительно снижаться матричные эффекты, однако для поддержания воспроизводимости в масс-спектрометрии необходимы регенерация колонки и очистка ионного источника. Сочетание жидкостной экстракции с использованием ультразвука и дополнительной очистки экстрактов дисперсионной твердофазной экстракцией позволяет снизить матричный эффект и повысить степень извлечения аналитов [99], хотя отмечены случаи когда дополнительная очистка добавлением сорбента не влияла на матричный эффект и пробоподготовка ограничивалась ЖЭ с добавлением высаливателей [113].
Тем не менее большинство методов подготовки образцов для многокомпонентного анализа антибиотиков в пищевых матрицах используют ТФЭ для достижения чувствительности на уровне нг/кг. Типичные сорбенты для ТФЭ – Oasis HLB и полимерные сорбенты Strata X. Картриджи Oasis HLB предпочтительны из-за достигаемой высокой воспроизводимости для широкого спектра соединений, как полярных, так и неполярных. Strata X картриджи имеют схожую с Oasis HLB функциональность, обеспечивая сопоставимые результаты.
Среди многокомпонентных методик описана процедура, которая включает растворение образца в смеси ацетонитрил−ЭДТА в слабокислой среде (pH 4.0) с последующей твердофазной очисткой на картриджах Oasis HLB. Этот способ применен для одновременного определения 30 антибиотиков (макролидов, тетрациклинов, хинолонов, сульфаниламидов, пенициллинов, амфениколов и диаминопиримидинов) в печени КРС, свиней, овец и домашней птицы. Степень извлечения варьировалась от 80 до 110% [78]. Очевидно, что данный способ пробоподготовки трудоемок, а анализ в целом занимает слишком много времени, что не позволяет анализировать множество образцов в короткие сроки. Кроме того, предполагается использование большого количества токсичных органических растворителей, в том числе и для восстановления картриджей.
Твердофазная экстракция продолжает широко применяться в практике анализа (табл. 1, 3, 4). Ее развитие связано в первую очередь с применением новых материалов в качестве твердой фазы, например молекулярно-импринтированных полимеров, появлением метода твердофазной микроэкстракции, что позволяет существенно повысить селективность очистки [115–117, 128, 143, 194, 195]. Молекулярный импринтинг впервые исследовали в 1949 г. В связи с необходимостью разработки более селективных сорбентов интерес к ТФЭ с использованием молекулярно-импринтированных полимеров (МИ-ТФЭ) возрос в последние десятилетия. Кроме того, появилась возможность синтеза и использования в пробоподготовке сложных матриц магнитных наночастиц на основе молекулярно-импринтированных полимеров [195]. В основном с их применением предложены методики определения отдельных представителей аминогликозидов, нитроимидазолов, сульфаниламидов, тетрациклинов, фторхинолонов и β-лактамов (табл. 1, 4). Ограничение использования МИ-ТФЭ в многокомпонентном анализе обусловлено ее основным преимуществом – селективностью.
Таблица 4.
Характеристика методик определения антибиотиков различных классов
| Антибиотик | Метод определения, детектор, колонка | Подвижная фаза | Предел обнаружения | Предел определения | Объект анализа; пробоподготовка, степень извлечения, % |
Лите-ратура |
|---|---|---|---|---|---|---|
| Аминогликозиды | ||||||
| 6 АГ | ВЭЖХ-МС/МС, X-Terras C18 (100 × 2.1 мм, 3.5 мкм) | Градиент 10 мМ НФПК – АЦН + + 10 мМ НФПК | 5–100 нг/г | 12.5–250 нг/г | Молоко, мышечная ткань; ЖЖЭ 36.8–98.0 |
[114] |
| 3 АГ | ВЭЖХ-ИРС, Syncronis C18 (250 мм × 4.6 мм, 5 мкм) | Вода + 0.2% ТФА – АЦН (9/1) | 3.0–5.2 мкг/кг | – | Рыба; ТФЭ, 82.1–96.7 |
[115] |
| 10 АГ | ВЭЖХ-МС/МС, Kinetex HILIC (100 × 2.1 мм, 1.7 мкм) | Градиент 150 мМ АА + 1% МК– АЦН | 2.3–14.7 мкг/кг | 4.2–49 мкг/кг | Молоко и молочная продукция; МИ-ТФЭ, 70.0–106.0 | [116] |
| 11 АГ | ВЭЖХ-МС/МС, ZIC-HILIC (50 × 2.1 мм, 3.5 мкм) | Градиент 175 мМ ФА + 0.3% МК– метанол + 0.3% МК | 2–30 мкг/кг | 7–100 мкг/кг | Мeд, молоко, свинина; МИ-ТФЭ, 78.2–94.8 | [117] |
| 3 АГ | ВЭЖХ-МС/МС, Click Xion-C18 (3.0 × 150 мм, 5 мкм) | Градиент АЦН/вода/МК (80/19/1) + 30 мМ ФА – вода/МК (99/1) + 30 мМ ФА | 0.913–1.23 мкг/кг | – | Мeд; ДТФЭ, 82.9–100.7 | [118] |
| Амфениколы | ||||||
| Хлорамфеникол | ВЭЖХ-МС/МС, Luna C18 (150 × 4.6 мм, 5 мкм) | Градиент вода – АЦН | 0.02 мкг/г | 0.04–0.09 мкг/г | Мeд, рыба, креветки; ЖЖЭ; 85.5–115.6 | [119] |
| 3 АФ | ВЭЖХ-ДМД, Perfectsil-120 ODS-2 (250 × 4 мм, 5 мкм) | АЦН/0.05 АА (25/75) | 55.9–58.99 мкг/кг | – | Молоко; волоконная ТФЭ, 44.0–81.4 | [120] |
| 4 АФ | ВЭЖХ-МС/МС, Acquity BEH C18 (100 × 2.1 мм, 1.7 мкм) | Градиент метанол – вода | 0.03–0.5 мкг/кг | 0.1–2.0 мкг/кг | Курица; ЖЖЭ (субкритическая вода), 86.8–101.5 | [121] |
| 3 АФ | ВЭЖХ-УФ, ODS-AP
(4.6 × 250 мм, 5 мкм), λ = 224–277 нм |
АЦН/вода (30/70) | 0.08–0.16 мкг/кг | 0.27–0.5 мкг/кг | Вода, кровь, яйца; магнитная МИ-ТФЭ, 88.3–99.1 | [122] |
| Хлорамфеникол | ВЭЖХ-УФ, PerfectSil ODS-2 (250 × 4.0 мм, 5 мкм), ВЭЖХ-МС, λ = 280 нм |
АЦН/вода (30/70) Метанол/вода (40/60) |
17 мкг/кг 0.1 мкг/кг |
50 мкг/кг 0.3 мкг/кг |
Молоко; МИ-ТФЭ, 85.0–106.0 |
[123] |
| Флорфеникол | ВЭЖХ-МС/МС, XBridge BEH C18 XP (100 × 2.1 мм, 2.5 мкм) | АЦН/вода (20/80) | 0.98 мкг/кг | 3.2 мкг/кг | Курица; ДТФЭ, ∼94.6 | [124] |
| Гликопептиды | ||||||
| 5 ГП | ВЭЖХ-МС/МС, BEH-C18 (100 × 2.1 мм, 1.7 мкм) | – | ≥ 0.33 мкг/кг | 1 мкг/кг | Яйца, курица, молоко;
ЖЖЭ + ТФЭ-очистка, 83.0–102.0 |
[125] |
| 3 ГП | ВЭЖХ-АД, циано-капиллярная колонка (50 × 375 мкм, 3 мкм) | Градиент 0.1% МК– АЦН | 1.0–8.0 мкг/л | – | Корма; ЖЖЭ + капиллярная микроэкстракция, 80.0–120.0 | [126] |
| 3 ГП | ВЭЖХ-МС, GOLD C18 (150 × 2.1 мм, 5 мкм) | Градиент 0.1% МК – АЦН | 5.02–10 мкг/л | – | Корма, свинина; ЖЖЭ + + капиллярная микроэкстракция, 76.0–109.7 | [127] |
| 2 ГП | ВЭЖХ-МС/МС, Phenomenex Kinetex Biphenyl (50 × 2.1 мм, 5 мкм) | Градиент 0.1% МК + + АЦН – 0.1% МК | 0.5 мкг/кг | 1 мкг/кг | Молоко; МИ-ТФЭ, 83.3–92.1 |
[128] |
| Диаминопиримидины | ||||||
| 3 ДП | ВЭЖХ-МС/МС, Eclipse Plus C18 (3.0 × 100 мм, 1.8 мкм) | Изократическое элюирование 0.2% МК/метанол (80/20) | 20 мкг/кг | 40 мкг/кг | Корма; ЖЖЭ, фильтрация, 74.4–105.2 | [129] |
| Ионофоры | ||||||
| 5 ИФ | ВЭЖХ-МС/МС, Zorbax Eclipse XDB-C8 (3 × 150 мм, 3.5 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | – | 0.01–1.0 мкг/кг | Молочная продукция;
ЖЖЭ + ТФЭ-очистка, 86.8–111.2 |
[130] |
| 20 кокци- диостатиков, в т.ч. 7 ИФ | УВЭЖХ-МС/МС, Acquity BEH C8 (2.1 × 50 мм, 1.7 мкм) | Градиент 0.1% МК – 0.1% МК + метанол | – | 0.13–0.42 мг/кг | Мышечная ткань; ЖЖЭ + + фильтрация, 80.0–125.0; Молоко; QuEChERS, 84.0–120.0 | [131] |
| 4 ИФ | ВЭЖХ-МС/МС, Hypersil Gold (2.1 × 150 мм, 5 мкм) | Градиент 0.1% МК + 1 мМ АА – АЦН + 0.1% МК | 0.01-0.3 мкг/кг | 0.4–1 мкг/кг | Молоко, мышечная ткань, яйца, печень; ЖЖЭ, ТФЭ-очистка 68.2–113.8 | [132] |
| 5 ИФ | ВЭЖХ-МС/МС, ACQUITY UPLC BEH HILIC (100 × 2.1 мм, 1.7 мкм | Градиент АЦН – 1 мМ ФА + 0.1% МК | 0.004–0.56 мкг/кг | 0.021–0.13 мкг/кг | Мышечная ткань, яйца; ЖЖЭ, ТФЭ-очистка, 92.0–114.0 | [133] |
| 6 ИФ | ВЭЖХ-МС/МС, C18 Phenomenex Luna (100 × 2 мм, 3 мкм) | Градиент 0.1% МК – 0.1% МК + метанол | 0.004−0.07 мкг/кг | – | Печень, молоко, курица, говядина, почки, яйца, жир; ЖЖЭ (СПМР), ДТФЭ, 71.0–112.0 | [134] |
| Линкозамиды | ||||||
| Линкомицин | ВЭЖХ-УФ, ODSA C18 (4.6 × 150 мм, 5 мкм), λ = 208 нм | Градиент фосфатный буферный раствор (рН 6) – АЦН | 0.02 мкг/мл | 0.08 мкг/мл | Молоко; МИ-ТФЭ, 80.0–89.0 |
[135] |
| Макролиды | ||||||
| Натамицин | ВЭЖХ-ДМД, Kromasil ODS (C18) (150 × 3.2 мм, 5 мкм), λ = 304 нм | Градиент АЦН – вода | 0.01 мкг/мл | 0.05 мкг/мл | Сыр, сосиски; ЖЖЭ, фильтрация, 90.3 ± 11.4, 92.4 ± 6.5 | [136] |
| 6 МЛ | ВЭЖХ-УФ, SunFireTM C18 (250 × 4.6 мм, 5 мкм), λ = 210 нм | Градиент АЦН – 25 мМ KH2PO4 (pH 3) | 0.015–0.075 мкг/кг | 0.075-0.5 мкг/кг |
Свинина, рыба, креветки; МИ-ТФЭ, >89.1 | [137] |
| Азитромицин | ВЭЖХ-МС/МС, Ecosil C8-SH (250 × 4.6 мм, 5 мкм) | Градиент АЦН – 0.1% МК |
0.03 мкг/кг | 0.1 мкг/кг | Свинина; МИ-ТФЭ, 85.8–96.5 | [138] |
| 7 МЛ | ВЭЖХ-МС/МС, XCharger-C18 (100 × 2.1 мм, 5 мкм) | Градиент 0.2% МК + + АЦН – 0.2% МК | 0.003–0.017 мкг/кг | 0.012–0.057 мкг/кг | Мeд; МИ-ТФЭ, 88.0–117.0 |
[139] |
| 11 МЛ | УВЭЖХ-МС/МС, Acquity CSH C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 0.2% МК – 0.2% МК + АЦН | 0.1–2.0 мкг/кг | 2.0–5.0 мкг/кг | Мышечная ткань, печень, почки, жир, яйца; ДТФЭ, 83.5–111.4 | [140] |
| Нитроимидазолы | ||||||
| 9 НИ и 3 метаболита | ВЭЖХ-МС/МС, C18 Zorbax Eclipse Plus (50 × 2.1 мм, 1.8 мкм) | Градиент 0.1% МК – метанол | 0.04–0.11 мкг/кг | 0.11–2.79 мкг/кг | Икра; ЖЖЭ с добавлением высаливателей, ≥68.9 | [113] |
| 3 НИ и 2 метаболита | ВЭЖХ-МС/МС, Eclipse C18 HPLC (150 × 4.6 мм, 3 мкм) | Градиент МК – АЦН | 0.08–0.25 мкг/кг | – | Мышечная ткань КРС; ЖЖЭ, фильтрация, – | [141] |
| 4 НИ и 3 метаболита | ВЭЖХ-МС/МС, Kinetex XB (150 × 2.1 мм, 2.6 мкм) | Градиент 0.1% МК + + АЦН – 0.1% МК | – | <3 мкг/кг | Молоко; ЖЖЭ, ТФЭ-очистка, 96.6–105.2 | [142] |
| 8 НИ и 3 метаболита | ВЭЖХ-УФ, Zorbax XDB-C18 (150 мм × 0.5 мм, 5 мкм) | Градиент вода – АЦН | 0.9–3.2 мкг/кг | – | Аквакультура; МИ-ТФЭ, >67.0 | [143] |
| 3 НИ | ВЭЖХ-ДРР, Kinetex® C18 100 Å (250 × 4.60 мм, 5 мкм) | АЦН, фосфатный буферный раствор рН 3 | 0.013–0.27 мкг/мл | – | Мeд; ТФЭ, 96.8–102.2 |
[144] |
| Нитрофураны и их метаболиты | ||||||
| 4 метаболита НФ | УВЭЖХ-МС/МС, Kinetex C18 (50 × 2.1 мм, 2.6 мкм) | Градиент метанол – 10 мМ ФА | 0.5–0.8 мкг/кг | 1 мкг/кг | Морепродукты; гидролиз, дериватизация, ТФЭ, фильтрация, 73.0–103.0 | [145] |
| Семикарбазид | ВЭЖХ-УФ, BDS HYPERSIL C18 (4.6 × 150 мм, 5 мкм) | Градиент метанол – 25 мМ фосфатный буферный раствор | 0.59 нг/мл | 50 нг/мл | Рыба; МИ-ТФЭ, дериватизация, 96.2–105.1 | [146] |
| 4 метаболита НФ | УВЭЖХ-МС/МС, Eclipse Plus C18 (2.1 × 50 мм, 5 мкм) |
Градиент 0.04% МК + + АЦН/вода (1/99) – АЦН/вода (99/1) | – | 0.4 мкг/кг | Рыба; гидролиз, дериватизация, 55.0–75.8 | [147] |
| 2 НФ и 4 метаболита | УВЭЖХ-МС/МС, Atlantis C18 (150 × 2.1 мм, 5 мкм) | Градиент 10 мМ АА + АЦН + гидроксид аммония – АЦН | 0.01–0.2 мкг/кг |
0.04–0.5 мкг/кг | Моллюски и рыба; гидролиз, деривати-зация, ЖЖЭ (все процедуры без доступа света), 91.6–107.3 | [148] |
| 4 метаболита НФ | ВЭЖХ-МС/МС, ACCLAIM 120C18 (2.1 × 100 мм, 3 мкм) | Градиент 0.5% МК – 0.5% МК + + АЦН/метанол (50/50) | 0.1–0.3 мкг/кг | 0.3–1.0 мкг/кг | Мeд; гидролиз, дериватизация, магнитная ТФЭ, >85.0 | [149] |
| 5 метаболитов НФ | ВЭЖХ-МС/МС, Symmetry C18 (100 × 2.1 мм, 3.5 мкм) | Градиент 2 мМ АА + + 0.01% УК – метанол | 0.1–0.2 мкг/кг | – | Мясо и аквакультура; гидролиз, дериватизация, ЖЖЭ, фильтрация, 86.5–108.2 | [150] |
| Полипептиды | ||||||
| 7 ПП | УВЭЖХ-МС/МС, Poroshell 120 (100 × 2.1 мм, 2.7 мкм) | Градиент 0.1% МК – метанол | 9–22 мкг/мкг | 30–74 мкг/кг | Мышечная ткань курицы; ЖЖЭ, ТФЭ-очистка, 41.0–52.0 (эффективность экстракции) | 151] |
| 5 ПП | ВЭЖХ-МС/МС, Hypersil GOLD C18 (100 × 2.1 мм, 5 мкм) | Градиент 0.1% МК – 0.1% муравьиная кислота+ АЦН | – | 25 мкг/кг | Корма; ДТФЭ, 75.9–87.9 | [152] |
| Колистин Б | УВЭЖХ-МС/МС, Hypersil GOLD C18 (100 × 2.1 мм, 1.9 мкм) | Градиент 1% МК – 1% МК + АЦН | – | 5–10 мкг/кг | Мышечная ткань курицы, яйца; ЖЖЭ, фильтрация, 70.0–107.0 | [153] |
| Плевромутилины | ||||||
| Вальнемулин | УВЭЖХ-МС/МС, Acquity BEH C18 (50 × 2.1 мм, 1.7 мкм) | Градиент 0.1% вода – 0.1% АЦН | 0.2 мкг/кг | 1.0 мкг/кг | Мышечная ткань, печень, почки КРС и свиней; ТФЭ, 93.4–104.3 | [112] |
| Вальнемулин | ВЭЖХ-УФ, Diamonsil TM C18 (250 × 4.6 мм, 5 мкм), λ = 210 нм | Градиент фосфатный буферный раствор (рН 2.5) – АЦН | 0.1 мг/кг | 0.25 мг/кг | Свинина; МИ-ТФЭ, 80.5–94.8 | [154] |
| Сульфаниламиды | ||||||
| 17 СА и 4 их метаболита | ВЭЖХ-МС/МС, ZORBAX Eclipse AAA (150 × 4.6 мм, 3.5 мкм) | Градиент 0.02% МК – 0.02% МК + АЦН | – | ≤7.5 нг/г | Свинина, курица, рыба, Мeд, молоко; ЖЖЭ, ДТФЭ, фильтрация, 71.0–109.0 | [155] |
| 22 СА | ВЭЖХ-МС/МС, Shim-pack XR-ODS II (100 × 2 мм, 2.2 мкм) | Градиент 0.01% МК – 0.01% МК + АЦН | 0.004–0.028 нг/г | 0.013–0.099 нг/г | Курица; дисперсионная МИ-ТФЭ, 85.0–112.2 | [156] |
| 5 СА | ВЭЖХ-УФ, ZORBAX SB-C18 (150 × 4.6 мм, 5 мкм), λ = 272 нм | АЦН + 0.5% УК | 0.71–0.98 нг/г | – | Яйца, Мeд; ЖЖЭ, мини-ТФЭ, 75.3–105.2 | [157] |
| 9 СА | ВЭЖХ-МС, ZORВAX C18 (50 × 2.1 мм, 3.5 мкм) | Градиент 0.5% УК + + 5% метанол – метанол | 5.04–6.59 мкг/кг | – | Мeд; гидролиз, ТФЭ, 89.0–118.0 | [158] |
| 27 СА и метаболитов | УВЭЖХ-МС-ВР, Hypersil Gold aQ C18 (100 × 2.1 мм, 1.9 мкм) | Градиент вода + МК + 4 мМ ФА – метанол + МК + 4 мМ ФА | 0.07–2.33 мкг/кг | – | Рыба; онлайн-ТФЭ, 83.0–109.0 | [159] |
| 5 СА | ВЭЖХ-УФ, Ultimate AQ-C18 (250 × 4.6 мм, 5 мкм), λ = 270 нм | АЦН/вода (30/70) | 0.32–1.7 нг/мл | – | Курица, свинина, рыба, печень; ТФЭ, 76.0–102.0 | [160] |
| 8 СА | ВЭЖХ-МС-ВП, Poroshell 120 HILIC (150 × 3 мм, 2.7 мкм) | Градиент 20 мМ ФА + метанол – АЦН | š | 5.0–20.0 мкг/кг | Детская смесь; преципитация, ДТФЭ, 72.9–109.2 | [161] |
| 4 СА | ВЭЖХ-УФ, C18 (4.6 × 150 мм, 5 мкм), λ = 270 нм | Вода/АЦН (75/25) | 0.11–2.24 мкг/кг | 0.37–7.47 мкг/кг | Молоко, мясо; магнитная ДТФЭ, 77.2–118.0 |
[162] |
| 18 СА | УВЭЖХ-МС/МС, Phenomenex Torrance F5 (50 × 3.0 мм, 2.6 мкм) | Градиент 0.1% МК – АЦН + 0.1% МК | 1.46–15.5 нг/кг | 4.9–51.6 нг/кг | Рыба, креветки, крабы; онлайн-ТФЭ, 71.5–102.0 | [163] |
| 10 СА | ВЭЖХ-МС/МС, Phenomenex Kinetex C18 LC (100 × 3.0 мм, 2.6 мкм) | Градиент АЦН – вода | 0.31–2.3 нг/л | 1.0–7.6 нг/л | Мeд, природные воды; микро-ТФЭ, 83.5–119.0 | [164] |
| 4 СА | ВЭЖХ-УФ, C18 column (4.6 × 150 мм, 5.0 мкм), λ = 270 нм | Градиент 0.2% УК – АЦН | 2.5–5.0 мкг/кг | 5.0–10.0 мкг/кг | Молоко; магнитная ДТФЭ, 83.0–99.2 | [165] |
| 2 СА | ВЭЖХ-УФ, – | – | 0.9–1.3 нг/мл | 3.0 нг/мл | Молоко; магнитная МИ-ТФЭ, >90.0 | [166] |
| Тетрациклины | ||||||
| 4 ТЦ | ВЭЖХ-МС/МС, Symmetry C18 (150 × 2.1 мм, 3.5 мкм) | Градиент 0.05% УК – 0.05% УК + АЦН | 5.45–9.24 мкг/кг | – | Мeд; гидролиз, фильтрация, ТФЭ, фильтрация, 83.5–109.7 | [167] |
| 7 ТЦ | ВЭЖХ-УФ, ZORBAX SB-C18 (150 × 4.6 мм, 5 мкм), λ = 355 нм | Градиент метанол/АЦН/ 0.01 М ЩК | 5.0–10.0 мкг/кг | 10.0–15.0 мкг/кг | Яйца, рыба, креветки; ASE, 75.6–103.5 | [168] |
| 5 ТЦ | УВЭЖХ-МС/МС, Zorbax RRHD Eclipse Plus C18 (2.1 × 50 мм, 1.8 мкм) | Градиент 0.05% МК – метанол | 0.05–0.14 мкг/кг | 0.16–0.48 мкг/кг | Детское питание; ЖЖЭ, 89.2–96.8 | [169] |
| 4 ТЦ | ВЭЖХ-УФ, VP-ODS C18 (150 × 4.6 мм, 5 мкм), λ=350 нм | Метанол/АЦН/ 0.01 М ЩК | 1.02–1.21 мкг/л | 3.56–4.32 мкг/л | Молоко; магнитная МИ-ТФЭ, 84.1–95.8 | [170] |
| Тетрациклин | ВЭЖХ-ДМД, Luna Omega C18 (250 × 4.6 мм, 5 мкм), λ = 276 нм, λ = 358 нм | Изократическое элюирование pH 4.0 оксалатный буферный раствор/ метанол/АЦН (70/10/20) | 9.56–21.11 нг/мл | 28.8 нг/мл – 1.01 мкг/мл | Молоко, печень, куриное мясо; ЖЖЭ, фильтрация, 93.9–104.8 | [171] |
| 4 ТЦ | ВЭЖХ-УФ, ZORBAX SB-C18 (4.6 × 150 мм, 5 мкм), λ = 360 нм | Градиент 0.01 М ЩК – АЦН/метанол (1/1) | 8.0–16.8 мкг/кг | 22.6–55.9 мкг/кг | Молоко, яйца, куриная печень; ТФЭ, 81.3–98.7 | [172] |
| 3 ТЦ | ВЭЖХ-ФДД, C18 (250 × × 4.6 мм, 3.5 мкм) | 0.05 М ЩК/АЦН/ метанол (70/20/10) | 0.015–0.062 мкг/г | 0.125–0.175 мкг/г | Морепродукты; преципитация, ЖЖЭ, фильтрация, 95.0–105.0 | [173] |
| 4 ТЦ | ВЭЖХ-МС/МС, ACQUITY UPLC® HSS T3 (1.0 × 150 мм, 1.8 мкм) | 0.4% МК/АЦН (72/28) | 0.073–0.435 нг/г | 0.239–1.449 нг/г | Мeд; ДТФЭ, 88.1–126.2 | [174] |
| Хиноксалины | ||||||
| 2 ХН и 2 метаболита | ВЭЖХ-МС/МС, Hypersil ODS (150 × 4.6 мм, 5 мкм) | Градиент 1% УК – метанол + 1% УК | 0.12–0.41 мкг/кг | – | Мышечная ткань и печень КРС и свиней; гидролиз, ТФЭ, 92.0–101.0, 60.0–62.0 (метаболиты) | [175] |
| 2 ХН и 11 метаболитов | УВЭЖХ-МС/МС, Acquity BEH C18 (50 × 2.1 мм, 1.7 мкм) | Градиент 0.1% МК – АЦН + градиент 0.1% МК | 0.05–5.2 мкг/кг | 0.15–16.0 мкг/кг | Курица, свинина; ЖЖЭ, ТФЭ-очистка, 69.1–113.3 |
[176] |
| 2 ХН | ВЭЖХ-МС/МС, Lichrospher C18 (3 × 250 мм, 5 мкм) | Градиент метанол – АЦН – вода | 9.0–80.0 мкг/кг | 12.0–110.0 мкг/кг | Корма для кур и свиней; ЖЖЭ, ДТФЭ-очистка, 99.41–104.62 | [177] |
| 3-Метил-хиноксалин-2-карбоновая кислота | ВЭЖХ-УФ, Zorbax Eclipse XDB C18 (250 × 4.6 мм, 5 мкм), λ = 320 нм | 0.1% МК/АЦН (40/60) | 1.0–3.0 мкг/кг | 4.0–10.0 мкг/кг | Съедобные ткани животных; УЗ-ЖЭ, ТФЭ, 80.1–87.7 | [178] |
| Метил-3-хиноксалин-2-карбоновая кислота | УВЭЖХ-МС/МС, BEH C18 (2.1 × 50 мм, 1.7 мкм) | Градиент 0.1% МК – метанол | 0.2 мкг/кг | 0.5 мкг/кг | Ткани животных; гидролиз, ЖЖЭ, иммуноаффинная колоночная ТФЭ, 90.2–103.5 | [179] |
| Хинолоны и фторхинолоны | ||||||
| 6 ХЛ | ВЭЖХ-ФЛД, Kinetex C18 (150 × 4.6 мм, 2.6 мкм), λ = 280–297/450–507 нм | Градиент 0.01 М ЩК pH 4.0 – АЦН | – | 0.08–0.13 | Корма для животных; ЖЖЭ, 80.0–105.0 | [180] |
| 6 ХЛ | ВЭЖХ-ДМД, Zorbax SB-C18 (150 × 0.5 мм,
5 мкм), λ = 250–300 нм, ВЭЖХ-МС |
Градиент АЦН – 5 мМ ФА | 6.0–30.0 мкг/кг, 2.4–6.0 мкг/кг |
22.0–110.0 мкг/кг, 8.0–20.0 мкг/кг |
Молоко; депротеинизация, фильтрация, 64.0–96.0 |
[181] |
| 7 ХЛ | ВЭЖХ-УФ, Perfectsil ODS-2 (250 × 4 мм, 5 мкм), λ = 255–275 нм | Градиент 0.1% ТФК – АЦН – метанол | 1.9–11.6 мкг/кг | 5.7–35.0 мкг/кг | Рыба; ЖЖЭ, ТФЭ-очистка, 95.7–102.7 | [182] |
| 9 ХЛ | ВЭЖХ-ФЛД, Zorbax Eclipse XDB C18 (150 × 3 мм, 5 мкм), λ = 280–312/360–450 нм | Градиент 0.1% МК– АЦН – метанол | 3–50 мкг/кг | 7.5–100.0 мкг/кг | Яйца, молоко, рыба, мясо, почки; ЖЖЭ, ТФЭ-очистка, 77.0–120.0 |
[183] |
| 9 ХЛ | ВЭЖХ-МС/МС, XTerra C18 (100 × 2.1мм, 3.5 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | 5 мкг/кг | 10 мкг/кг | Говядина, свинина, курица, рыба; ЖЖЭ, низкотемпературная очистка, 79.0–115.0 | [184] |
| Флумеквин | ВЭЖХ-ФЛД, монолитная колонка, λ = 325/360 нм | Метанол/0.01 М H3PO4 (65/35) | <0.32 нг/г | – | Рыба; МИ-ТФЭ, >95.2 | [185] |
| β-Лактамы | ||||||
| 3 БЛ | ВЭЖХ-УФ, XbridgeTM C18 (250 × 4.6 мм, 5 мкм), λ = 215 нм | Изократическое элюирование фосфатный буферный раствор (рН 6.6)/АЦН (75/25) | 1.0–2.0 нг/мл | 3.0-7.0 нг/мл | Говядина и молоко; ЖЖЭ, >85.0 | [186] |
| 3 БЛ | ВЭЖХ-МС/МС, Ultimate XB-C18 (100 × 2.1 мм, 5 мкм) | Градиент 0.1% МК – 0.1% МК + АЦН | 0.06–0.09 нг/мл | 0.23–0.26 нг/мл | Молоко; магнитная ТФЭ, 87.55–88.38 | [187] |
| Бензилпеницил-лин | УВЭЖХ-МС/МС, UPLC BEH C18, 100 × 2.1 мм, 1.7 мкм | Градиент вода/АЦН (95/5) + 0.3% УК – АЦН/вода (95/5) + + 0.3% УК | 6.2–14.4 мкг/кг | 12.3–28.8 мкг/кг | Мясо; МИ-ТФЭ, 94.63–108.2 |
[188] |
| 22 БЛ | ВЭЖХ-МС/МС, ZORBAX Eclipse XDB-C18 (150 × 4.6 мм, 5 мкм) | Градиент 0.1% УК – 0.1% УК + АЦН | 0.04–3.0 мкг/л | 0.06–10.0 мкг/л | Свинина; ЖЖЭ, фильтрация, 83.6–113.0 | [189] |
| 15 БЛ | УВЭЖХ-МС/МС, ACQUITY UPLC HSS T3 C18 (2.1 × 100 мм, 1.7 мкм) | Градиент 0.01% МК – 0.01% МК + АЦН | 0.02–0.63 мкг/кг | 0.07–0.7 мкг/кг | Свинина; ДТФЭ, 92.0–111.0 |
[190] |
| 15 БЛ | УВЭЖХ-МС/МС, Kinetex Biphenyl Core- Shell (50 × 2.1 мм, 1.7 мкм) | Градиент 0.05% УК – метанол | 00.2–2.7 мкг/кг | <9.0 мкг/кг | Детская молочная продукция; ЖЖЭ, 79.0–93.0 |
[191] |
| Клоксациллин | ВЭЖХ-УФ, Kromasil ODS (150 × 4.6 мм, 5 мкм), λ = 225 нм | АЦН + ацетатный буферный раствор (рН 6) | 0.03 мкг/г | 0.1 мкг/г | Креветки; МИ-ТФЭ, 76.0–84.3 | [192] |
| 5 БЛ | УВЭЖХ-МС/МС, AcquityTM BEH Shield C18 (100 × 2.1 мм, 1.7 мкм) | Градиент 50 мМ ФА – метанол | 0.03–0.2 мкг/кг | 0.17–0.68 мкг/кг | Молоко; микро-ДТФЭ, 87.0–107.0 | [193] |
| Цефтиофур | ВЭЖХ-УФ, МИ-ТФЭ колонка (4.6 × 150 мм), λ = 292 нм | АЦН/0.2% УК (30/70) | 0.0015 мг/л | 0.005 мг/л | Молоко, курица, свинина, говядина; МИ-ТФЭ, 91.9–106.8 | [194] |
Обозначения: АА – ацетат аммония; АГ – аминогликозиды; АЦН – ацетонитрил; ВЭЖХ-АД – высокоэффективная жидкостная хроматография с амперометрическим детектированием; ГП – гликопептиды; ДРР – детектор рассеяния Рэлея; ЖЖЭ – жидкостно-жидкостная экстракция; ИРС – испарительный детектор рассеяния света; ИФ – ионофоры; МК – муравьиная кислота; НФПК – нонафторпентановая кислот; ПП – полипептиды; СПМР – супрамолекулярные растворители; УК – уксусная кислота; ФА – формиат аммония; ХН – хиноксалины; ЩК – щавелевая кислота
Для сокращения продолжительности анализа и необходимого объема пробы предложена онлайн-ТФЭ [196], и в настоящее время разработано множество методик с ее применением [13, 26, 73, 83, 159, 163]. Автоматизация пробоподготовки для дальнейшего исследования сложных образцов, а именно объектов окружающей среды и продуктов питания, изучается с конца восьмидесятых годов. Онлайн-методы имеют ряд преимуществ: сокращение потребления растворителей и меньший контакт с ними исполнителя, возможность регенерации ТФЭ-колонок, экономия времени.
Жидкостная и твердофазная экстракция совершенствуются и активно используются на этапе подготовки образцов в рутинном анализе пищевых продуктов. При этом неавтоматизированные процедуры трудоемки и требуют применения больших количеств токсичных органических растворителей. В то же время в аналитическую методологию уже более 30 лет внедряется концепция “зеленой химии”. Изначально она была ориентирована на методы органического синтеза, но позже адаптирована и к другим областям химии. Кроме экологической безопасности, применение принципов “зеленой химии” позволяет снизить стоимость анализа, повысить его скорость и безопасность для исполнителей [197]. С этой точки зрения предложен такой способ пробоподготовки как дисперсионная твердофазная экстракция QuEChERS (Quick, Easy, Cheap, Effective, Rugged and Safe – быстрый, простой, дешевый, эффективный, точный и надежный), известный с 2003 г. и изначально применявшийся для быстрого извлечения пестицидов [198]. Эффективность метода настолько существенна, что два его варианта в настоящее время являются официальными методами анализа международных организаций по стандартизации ЕС и AOAC International (Association of Official Agricultural Chemists International – Международная Ассоциация Официальных Агрохимиков, США) при определении остаточных содержаний пестицидов во фруктах и овощах [197]. Экстракцию целевых компонентов проводят ацетонитрилом в присутствии буферирующих солей. Очистку экстрактов от липидов, жиров и белков осуществляют насыпными сорбентами Bondesil-PSA, C18, графитированной сажей, ионообменными смолами и их комбинациями. В последнее время метод применяется в многокомпонентном определении лекарственных средств для ветеринарного применения в продуктах питания [198]. QuEChERS позволяет сократить продолжительность пробоподготовки; нет необходимости применения дополнительных способов очистки, что уменьшает риск ошибки; характеризуется высокой степенью извлечения для широкого спектра антибиотиков; позволяет использовать меньшее количество органических растворителей. Метод показал хорошие результаты и в анализе объектов окружающей среды (табл. 1). Его простота обеспечивает высокую надежность и воспроизводимость. Применение QuEChERS для определения антибиотиков иллюстрирует табл. 5.
Таблица 5.
Пробоподготовка QuEChERS при определении остаточных количеств антибиотиков
| Антибиотик | Матрица | Степень извлечения, % | Метод определения, детектор, колонка | Предел обнаружения | Предел определения | Лите- ратура |
|---|---|---|---|---|---|---|
| 6 ХЛ и хлорамфеникол | Мясо, рыба, корма, яйца, молоко | 62.0–100.0 | ВЭЖХ-ДМД, XTerra RP18 (150 × 3.9 мм, 3 мкм) | 0.002–0.04 мг/кг | 0.008–0.12 мг/кг | [199] |
| 7 СА, 8 ХЛ, 4 ТЦ | Мясо | 19.0–29.0 | ВЭЖХ-МС/МС, Symmetry C18 (75 × 4.6 мм, 3.5 мкм) | – | 25–50 мкг/кг | [200] |
| 8 ХЛ | Продукты пчеловодства | 40.0–100.0 | УВЭЖХ-МС/МС, Zorbax Eclipse Plus C18 (50 × 2.1 мм, 1.8 мкм) | 0.2–4.1 мкг/г | 0.8–13 мкг/г | [201] |
| 31 препарат, в т.ч. ХЛ, ПЦ, СА, ДП, ТЦ, МЛ | Рыба садкового содержания | 69.0–125.0 | УВЭЖХ-МС/МС, Acquity UHPLC BEH C18 (100 × 2.1 мм, 1.7 мкм) |
7.5–15.0 мкг/кг | 25–50 мкг/кг | [202] |
| Более 50 препаратов, в т.ч. АГ, ХЛ, ИФ, ПЦ, МЛ, АФ, СА, ТЦ | Молоко, мeд | 50.0–120.0 | УВЭЖХ-МС-ВП, Acquity UPLC BEH C18, (100 × 2.1 мм, 1.7 мкм) | <1 мкг/кг | – | [203] |
| 20 препаратов, в т.ч. ХЛ, СА, МЛ, ДП | Курица | 65.6–120.0 | УВЭЖХ-МС/МС, Acquity UPLC BEH C18, (100 × 2.1 мм, 1.7 мкм) | 3.0–6.0 мкг/кг | 10.0–20.0 мкг/кг | [204] |
| 20 запрещенных препаратов, в т.ч. ХН | Корма | 56.7–103.0 | ВЭЖХ-МС/МС, Zorbax SB-Aq C18 (150 × 2.1 мм, 3.5 мкм) | 5.70–9.81 мкг/кг | – | [205] |
| 55 препаратов, в т.ч. ХЛ, СА, МЛ | Мышечная ткань (КРС и МРС) | 70.0–120.0 | ВЭЖХ-МС/МС, ZORBAX SB-C18 | – | 0.1–18.4 мкг/кг | [206] |
| 22 СА | Съедобные ткани животных | 88.0–112.0 | ВЭЖХ-МС-ВР, Zorbax Eclipse XDB-C8 (2.1 × 100 мм, 3.5 мкм) | 3–26 мкг/кг | 11–88 мкг/кг | [207] |
| 50 препаратов, в т.ч. МЛ, ХЛ, СА, ТЦ |
Сыр | 70.0–120.0 | ВЭЖХ-МС/МС, ZORBAX-SB-C18 |
– | 0.05–20.0 мкг/кг | [208] |
| 90 препаратов, в т.ч. ЛА, МЛ, СА, ХЛ, ТЦ, БЛ, НИ, НФ |
Молоко | 72.62–122.2 | УВЭЖХ-МС-ВП, BEH C18 (100 × 2.1 мм, 1.7 мкм) | 0.03–5.20 мкг/кг | 0.1–17.3 мкг/кг | [209] |
| 492 загрязнителя, в т.ч. АГ, АФ, ИФ, ЛА, МЛ, ХЛ, ХН и метаболиты, НИ, СА, ТЦ, БЛ, НФ и метаболиты, ПМ, ПП | Молоко, мясо, яйца, рыба, корма и зерно, овощи, фрукты | – | ВЭЖХ-МС-ВР, Acclaim 120 C18 (150 × 2.1 мм, 2.2 мкм) | 0.002–50.0 нг/мл | 0.006–250.0 нг/мл | [210] |
| 90 препаратов, в т.ч. ЛА, МЛ, СА, ХЛ, ТЦ, БЛ, НИ и НФ |
Маточное молочко | 70.21–121.0 | УВЭЖХ-МС/МС, ACQUITY® UPLC BEH C18 (100 × 2.1 мм, 1.7 мкм) | 0.13–6.0 мкг/кг | 0.21–20.0 мкг/кг | [211] |
| 4 метаболита НФ, 2 НИ | Мeд | 90.96–104.80 | ВЭЖХ-МС/МС, ZORBAX Eclipse XDB C-18 (4.6 × 150 мм, 5 мкм) | 0.21–1.27 мкг/кг | – | [212] |
| 8 СА | Курица, яйца | 65.9–88.1 | ВЭЖХ-ФЛД, постколоноч-ная дериватизация, λ = 240/350 нм | 4.1–19.9 мкг/кг | 14–85 мкг/кг | [213] |
| 23 антибиотика, в т.ч. МЛ, ТЦ, СА, НИ, ДП, АФ | Морепродукты | 28.0–71.0 | УВЭЖХ-МС/МС, Acquity HSS T3 (50 × 2.1 мм, 1.8 мкм) | 0.01–0.31 нг/г | 0.02–1.03 нг/г | [214] |
| 26 препаратов, в т.ч. НИ | Мышечная ткань КРС | 84.3–111.6 | УВЭЖХ-МС/МС, Acquity UPLC® BEH C18 (50 × 2.1 мм, 1.7 мкм) | 0.007– 66.715 мкг/кг | 0.011– 113.674 мкг/кг | [215] |
| 11 СА и 5 метаболитов | Детское питание | 60.9–85.9 | УВЭЖХ-МС-ВР, Hypersil Gold aQ (100 × 2.1 мм, 1.9 мкм) | 0.03–0.17 мкг/кг | 0.10–0.55 мкг/кг | [216] |
| 5 ТЦ | Рыба | >80.0 | УВЭЖХ-МС/МС, Zorbax Eclipse plus RRHD (50 × 2.1 мм, 1.8 мкм) | 0.5–1.2 мкг/кг | 1.7–4.4 мкг/кг | [217] |
| 13 препаратов, в т.ч. ПЦ, АФ, СА, ДП | Молоко | 62.0–125.0 | ВЭЖХ-МС/МС, XTerra C18 (50 × 3 мм, 3.5 мкм) | 0.3–16.6 мкг/кг | 1.0–50.0 мкг/кг | [218] |
| 13 пестицидов,
13 микотоксинов и 48 антибиотиков в т.ч. СА, БЛ, МЛ, НИ, ЛА, АФ |
Яйца | 60.5–114.6 | УВЭЖХ-МС/МС, Shim-pack XR-ODS III (150 × 2.0 мм, 2.2 мкм) | – | 0.1–17.3 мкг/кг | [219] |
| 238 пестицидов и 78 препаратов, в т.ч. ПЦ, ХЛ, ТЦ, МЛ, НИ, БЛ, СА, АФ, ИФ | Молоко | 70.0–120.0 | УБЖХ-МС/МС, Atlantis T3
(100 × 2.1 мм, 5 мкм), ГХ-МС/МС, HP-5 MS (30 м × 250 мкм, 0.25 мкм) |
– | 0.02–25.0 нг/г | [220] |
| 26 препаратов, в т.ч. СА, ХЛ, ТЦ, МЛ, ЛА, НФ, НИ, АФ |
Морепродукты | 56.0–108.0 (<19.0 для ТЦ) | УВЭЖХ-МС/МС, Hypersil Gold C18 (100 × 2.1 мм, 1.9 мкм) | 0.002–3.00 мкг/кг | – | [221] |
| 30 микотоксинов и 24 фарм. активные субстанции, в т.ч. ЦС, ИФ, СА, ХЛ | Молоко | 79.0–86.0 | УВЭЖХ-МС-ВР, Luna Omega (50 × 2.1 мм, 1.6 мкм) | – | 0.01−0.5 нг/мл | [222] |
| 65 пестицидов, 41 препарат, в т.ч. СА, БЛ, МЛ, ЛА, ХЛ | Рыба | 56.0–115.0 | УВЭЖХ-МС/МС, Eclipse Plus C18 (2.1 × 100 мм, 1.8 мкм) | – | 0.02–3.7 мкг/кг | [223] |
| 2 ГП | Рыба | 86.7–98.6 | УВЭЖХ-МС/МС, C18 (50 × 2.1 мм, 1.7 мкм) | 0.51 мкг/кг | 1.73 мкг/кг | [224] |
Обозначения: АГ – аминогликозиды; АФ – амфениколы; БЛ – β-лактамы; ГП – гликопептиды; ДП – диаминопиримидины; ИФ – ионофоры; ЛА – линкозамиды; МЛ – макродиды; НИ – нитроимидазолы; НФ – нитрофураны; ПМ – плевромутилины; ПП – полипептиды; ПЦ – пенициллины; СА – сульфаниламиды; ТЦ – тетрациклины; ХЛ – хинолоны; ХН – хиноксалины; ЦС – цефалоспорины; ЭДМК − этилендиметакрилат
Надежный и быстрый УВЭЖХ-МС/МС-способ определения восьми антибиотиков хинолонового ряда (марбофлоксацина, ципрофлоксацина, данофлоксацина, энрофлоксацина, сарафлоксацина, дифлоксацина, флумеквина и оксолиновой кислоты) разработан для анализа продуктов пчеловодства [201]. Образец меда, маточного молочка или прополиса помещали в центрифужную пробирку и растворяли в среде 30 мМ NaH2PO4 буферного раствора с рН 7.0. Затем экстрагировали 5%-ной муравьиной кислотой в ацетонитриле с добавлением солей и сорбентов QuEChERS. Аликвоту надосадочной жидкости переносили в пробирку и высушивали в токе азота. Остаток повторно растворяли в смеси вода−ацетонитрил−муравьиная кислота (88 : 10 : 2), фильтровали и анализировали методом УВЭЖХ-МС/МС. Степени извлечения антибиотиков составили 40.0–100.0%, пределы обнаружения 0.2–4.1 мкг/кг, пределы определения 0.8–13 мкг/кг. Предложена высокочувствительная УВЭЖХ-МС/МС-методика определения гликопептидов ванкомицина и норванкомицина в рыбе [224]. В работе использовали сорбент из катионообменной смолы (степень извлечения составила 86.7–98.6%) и 96-луночный планшет для реализации способа пробоподготовки QuEChERS. Предел обнаружения – 0.51 мкг/кг, предел определения – 1.73 мкг/кг.
Разработана методика определения 23 сульфаниламидов и их метаболитов (сульфагуанидина, сульфацетамида, дапсона, сульфадиазина, сульфисомидина, сульфатиазола, сульфапиридина, сульфамеразина, сульфаметра (сульфаметоксидиазина), сульфаметизола, сульфаметазина, сульфаметоксипиридазина, сульфахлоропиридазина, сульфаметоксазола, сульфамонометоксина, сульфадоксина, сульфисоксазола, сульфабензамида, сульфафеназола, сульфадиметоксина, сульфахиноксалина, сульфаклозина и сульфанитрана) в тканях животных, которая включает экстракцию метанолом с уксусной кислотой, добавление буферирующих солей, очистку экстракта первичными вторичными аминами (QuEChERS) и фильтрацию [207]. Степени извлечения антибиотиков составили 88.0–112.0%. Последующие идентификацию, подтверждение и количественный анализ проводили методом ВЭЖХ с масс-спектрометрическим детектированием высокого разрешения (ВЭЖХ-МС-ВР) на колонке с фазой C8 (размер частиц 3.5 мкм). Предел обнаружения 3.0–26.0 мкг/кг. Оптимизированная методика успешно применена к анализу 30 образцов тканей животных. Сульфаниламиды обнаружены в восьми образцах.
Простая, селективная, экспрессная и многокомпонентная УВЭЖХ-МС/МС-методика разработана для определения 32 остатков лекарственных препаратов, применяемых в ветеринарии, в том числе антибиотиков пяти разных классов, в рыбе садкового содержания [202]. Степени извлечения варьировались от 69.0 до 125.0%. Относительное стандартное отклонение – 20–30%. Пределы обнаружения составили 7.5–15.0 мкг/кг, пределы определения – 25–50 мкг/кг.
Методика, совмещающая пробоподготовку QuEChERS и времяпролетную (ВП) УВЭЖХ-МС, разработана для определения 90 препаратов 20 различных классов (включая антибиотики: линкозамиды, макролиды, сульфаниламиды, хинолоны, тетрациклины, β-лактамы, нитроимидазолы, нитрофураны) в молоке [209]. Экстракцию проводили 1%-ной уксусной кислотой в ацетонитриле с добавлением буферирующих солей с последующей очисткой экстракта насыпными сорбентами. Упаривали в токе азота досуха. Остаток повторно растворяли в 25%-ном ацетонитриле. Восстановленный экстракт фильтровали через мембранный фильтр в микрофлакон и хроматографировали. Эта методика обеспечила пределы обнаружения в диапазоне 0.03–5.20 мкг/кг. Степени извлечения антибиотиков в зависимости от матрицы составили от 72.62 до 122.20%.
Разработан способ одновременного ВЭЖХ-МС/МС-определения 65 пестицидов и 41 препарата в лососе [223]. В работе адаптирован официальный метод AOAC 2007.01 определения пестицидов в жировых матрицах. В качестве привычных для QuEChERS насыпных сорбентов использовали их улучшенные варианты для очистки липидов. Сульфагуанидин, как и в других подобных исследованиях, показал низкую степень извлечения. Наиболее полярные соединения, включая и сульфагуанидин, оставались в водной фазе и не переходили в ацетонитрильную, для остальных степень извлечения составила 70.0–120.0%. Пределы определения находились в диапазоне 0.02–3.7 мкг/кг.
Методики, описанные в табл. 5, зачастую характеризуются низкими степенями извлечения антибиотиков из мяса [200] и морепродуктов [214], высокими пределами обнаружения и определения аналитов в мясе [200, 204, 207, 213], рыбе [202] и яйцах [213]. Кроме того, эти способы не всегда дают возможность совместного определения антибиотиков различных классов [201], а также не являются универсальными для нескольких типов матриц [200, 202]. При этом метод QuEChERS продолжает развиваться, и не стоит забывать, что появляются более современные материалы и для решения перечисленных проблем исследуются возможности использования различных сочетаний растворителей, буферирующих солей и новых сорбентов [198]. Например, при одновременном ВЭЖХ-МС/МС-определении в молоке 57 препаратов для ветеринарного применения (в том числе сульфаниламидов, хинолонов, макролидов, нитроимидазолов, амфениколов) экстракт очищали с испльзованием меламиновой губки [225]. Хитозан из отходов панцирей креветок предложен в качестве сорбента при одновременном определении в молоке остаточных содержаний 13 препаратов, включая амоксициллин, кларитромицин, хлорамфеникол, эритромицин, пенициллин G, сульфадиазин, сульфаметазин, сульфаметоксазол и триметоприм с пределом определения 1.0–50.0 мкг/кг и степенью извлечения 62.0–125.0% [218]. Хитозан обеспечивает хорошее извлечение аналитов, снижает матричный эффект, при этом является нетоксичным, возобновляемым биополимером и более дешевой и экологичной альтернативой традиционным сорбентам. Таким образом, востребованность и перспективность дальнейшего использования QuEChERS в рутинном анализе продуктов питания и продовольственного сырья сохраняется.
Методы ЖЭ и ТФЭ эффективны для концентрирования аналитов и подавления матричных эффектов, но большой расход токсичных органических растворителей является их основным недостатком. “Зеленая химия” предлагает варианты их замены на более экологичные, такие как субкритическая вода и сверхкритический диоксид углерода, супрамолекулярные растворители (СПМР), ионные жидкости (ИЖ) и глубокие эвтектические растворители (ГЭР) [10]. СПМР – фаза, обогащенная наноструктурным поверхностно-активным веществом (ПАВ), образующаяся путем последовательной самосборки на нано- и молекулярном уровнях. В зависимости от типа, состояния и концентрации ПАВ могут быть получены водные неионные, ионные и смешанные, а также обратные мицеллы и везикулы (пузырьки).
Ионные жидкости представляют собой соли, состоящие из органических катионов и органических или неорганических анионов, которые остаются жидкими при температуре ниже 100°С (и даже при комнатной) [10]. Ионные жидкости считаются экологически чистыми растворителями из-за низкого давления пара, нелетучести, настраиваемой кислотности/основности, гидрофобности/гидрофильности, (их называют “дизайнерскими растворителями” в связи с возможностью изменять физические и химические свойства, выбирая катионы и анионы).
Применению СПМР для извлечения антибиотиков из пищевых продуктов посвящено несколько исследований, хотя с момента их внедрения прошло много времени. Везикулярный супрамолекулярный растворитель (смесь катионного ПАВ с добавлением для коацервации NaCl) использовали для извлечения тетрациклиновых антибиотиков из образцов молока, яиц и меда, при этом образцы молока и яиц депротеинизировали и обезжиривали добавлением ацетонитрила и трифторуксусной кислоты, а образцы меда просто разбавляли водой и фильтровали [226]. Достигнуты высокий коэффициент предварительного концентрирования (48–198) и низкие пределы обнаружения (0.7–3.4 мкг/л). Сочетание СПМР (индуцирован добавлением воды к коллоидной системе гексанол–тетрагидрофуран) и дисперсионной ТФЭ обеспечило хорошее извлечение ионофоров из образцов яиц, молока, жира, печени, почек и мышечной ткани (71.0–112.0%) без дополнительных процедур, а пределы обнаружения находились в диапазоне 0.004–0.07 мкг/кг [227].
Функционализированные адсорбенты с иммобилизированными на поверхности магнитных твердых материалов ИЖ использованы в магнитной ТФЭ, но в основном при определении антибиотиков в природных водах [228]. Глубокие эвтектические растворители более успешно применяются в анализе продуктов питания. Предложено несколько методик с их использованием на стадии пробоподготовки при определении пенициллина G, стрептомицина и эритромицина в прополисе [229], окситетрациклина, пенициллина G и тилмикозина в колбасных изделиях [230], гамбургерах и печени КРС [231].
Таким образом, хотя ИЖ считаются “зелеными растворителями”, процедура синтеза, устойчивость к биоразложению и, более того, токсичность многих из них, противоречат концепции “зеленой химии”. А вот СПМР и ГЭР обладают уникальными свойствами и могут быть идеальной заменой обычным органическим растворителям.
Методы определения антибиотиков в пищевых продуктах. Для определения остаточных количеств антибиотиков в пищевых продуктах животного происхождения предложен ряд методов, в том числе микробиологические [232], иммуноферментный анализ [233], тонкослойная хроматография [234], капиллярный электрофорез [235], газовая хромато-масс-спектрометрия (ГХ-МС) [236], ВЭЖХ и ВЭЖХ-МС (табл. 1–5). Методы ГХ не получили широкого распространения, несмотря на то что они очень чувствительны и специфичны. Сложность их использования заключается в необходимости предварительной дериватизации для подготовки летучего соединения перед ГХ-анализом. Методы капиллярного электрофореза имеют ряд преимуществ перед ВЭЖХ: малый объем органического растворителя в рабочем буферном растворе, короткое время разделения, высокая эффективность разделения. Однако капиллярный электрофорез ограниченно применим для определения остаточных количеств препаратов, так как некоторые из них не могут быть разделены, поскольку являются незаряженными или незначительно отличающимися по электрофоретической подвижности. ВЭЖХ стала наиболее широко используемым методом для определения остаточных количеств антибиотиков.
При определении аминогликозидов чаще всего для подготовки образцов используют ТФЭ [115–117], являющуюся наиболее эффективным способом очистки и концентрирования экстрактов, но требующую относительно больших затрат времени. Жидкостно-жидкостная экстракция (ЖЖЭ) в данном случае дает слишком низкую степень извлечения аналитов (36.8–98.0%) [114], поскольку молекулы аминогликозидов достаточно крупные, с двумя и более аминосахарами, связанными с центральной гексозой/пентозой гликозидными связями. Они крайне высокополярны из-за наличия нескольких амино- и многочисленных гидроксильных групп, поэтому их определение в рамках многокомпонентного анализа проблематично – резко отличаются физико-химические свойства, а соответственно и необходимые условия пробоподготовки [84]. Определение следовых количеств аминогликозидов одновременно с другими группами антибиотиков методом обращенно-фазовой (ОФ) ВЭЖХ-МС/МС практически невозможно – они не задерживаются на ОФ-колонках и имеют асимметричную форму пика из-за сильных ионных взаимодействий с остаточными группами силанолов на поверхности неподвижной фазы. Единственный способ добиться разделения в данном случае – это добавление агентов для ионного сопряжения, например фторированных кислот, или дериватизация, но в случае сочетания с МС-детектированием использование ион-парных реагентов приводит к загрязнению источника. Среди методов определения аминогликозидов наиболее распространена ВЭЖХ-МС/МС (табл. 4). В предложенных методиках одновременного определения аминогликозидов и других препаратов (табл. 3) проблема разделения решается использованием ион-парных реагентов, например гептафтормасляной кислоты, добавляемой в подвижную фазу, либо применением гидрофильного разделения (HILIC) [84].
Для определения остаточных количеств амфениколов в настоящее время используют два основных метода пробоподготовки: жидкостную [119, 121, 140, 236] и твердофазную экстракцию [120, 124], а в последнее время МИ-ТФЭ [122, 123]. Наиболее подходящими экстрагентами являются этилацетат, ацетонитрил, гексан и хлороформ. Остаточные содержания амфениколов часто определяют методами жидкостной хроматографии с УФ- [122] и ДМД-детектированием [120] (табл. 4), хотя также используют ГХ с электронно-захватным детектированием и ГХ-МС [236]. Максимум поглощения хлорамфеникола 278 нм, тогда как тиамфеникола и флорфеникола он лежит в диапазоне 225–230 нм. Однако чувствительности данных методов не хватает для достижения необходимых пределов определения амфениколов в связи с их низкими максимально допустимыми уровнями (МДУ), поэтому в данном случае необходимо использование ВЭЖХ-МС/МС [119, 121].
Ограниченное количество методик по сравнению с другими группами антибиотиков предложено для определения гликопептидов в пищевых продуктах и кормах для животных [125–128], в том числе совместно с другими циклопептидами [100], а также в сыворотке и плазме крови [237]. Пробоподготовка в основном предполагает жидкостную экстракцию в сочетании с ТФЭ, кроме того, для очистки предложена капиллярная микроэкстракция (полимерная монолитная колонка) [126, 127]. Гликопептиды запрещены для использования в ветеринарии на территории ЕС, США, Китая, однако в связи с их высокой эффективностью они нелегально используются в качестве кормовых добавок для лечения бактериальных инфекций животных и обнаруживаются в продукции животноводства [100, 128, 224].
Диаминопиримидины, а чаще всего триметоприм, определяют в растительной и животноводческой продукции, а также в сточных и природных водах в основном совместно с другими антибиотиками методом ВЭЖХ-МС/МС [51, 79, 88, 94, 238]. Они являются потенциаторами сульфаниламидов и используются в комбинированных препаратах. Предложена методика определения диаверидина, триметоприма и орметоприма в кормах [129].
Ионофорные антибиотики широко используются в ветеринарии для борьбы с кокцидиозами. В продукции животноводства их в основном определяют методом ВЭЖХ-МС/МС после жидкостной экстракции и ТФЭ-очистки; разработанные методики отличаются высокой чувствительностью: предел обнаружения на уровне сотых долей мкг/кг, а в самых поздних работах – тысячных [130–134]. Совершенствование предлагаемых методик связано с модификацией пробоподготовки: от трудоемкой жидкостной экстракции, дополненной стадией ТФЭ-очистки, а в некоторых случаях еще и обезжириванием образца и испарением растворителя, до простой в исполнении и высокоэффективной СПМР-ЖЖЭ в сочетании с ДТФЭ [231].
Основными представителями группы линкозамидов являются линкомицин – природный антибиотик − и клиндамицин – его полусинтетический аналог [239]. Однако их индивидуальное определение в пищевой продукции хроматографическими методами ограничено в последнее десятилетие лишь несколькими работами, в том числе с использованием на стадии пробоподготовки МИ-полимеров для ТФЭ [135]. В основном же предложены многокомпонентные методики их определения совместно с макролидами [74] и представителями других групп (табл. 1, 3, 5).
При определении макролидов преобладают методики ВЭЖХ-УФ и ВЭЖХ-МС/МС (табл. 4). Макролиды и линкозамиды являются липофильными соединениями. Как правило, для их извлечения из образцов пищевой продукции используют различные варианты ТФЭ, хотя в последнее время при их определении большое внимание уделяют ДЖЖМЭ, позволяющей добиться высокого концентрирования аналитов при минимальных затратах растворителей и времени [1]. Для совместного определения с другими препаратами в основном предложены ВЭЖХ- и УВЭЖХ-МС/МС, в том числе высокого разрешения (табл. 1, 3, 5).
Для определения нитроимидазолов одновременно с сульфаниламидами, фторхинолонами, тетрациклинами, макролидами, нитрофуранами и другими антибиотиками предложено несколько УВЭЖХ-МС/МС-методик с пробоподготовкой QuEChERS [209, 211, 214, 219]. В большинстве же случаев используют либо жидкостно-жидкостную экстракцию либо ее сочетание с последующей ТФЭ-очисткой [87, 90, 102, 104, 105]. При групповом определении нитроимидазолов наиболее распространена ТФЭ с последующим ВЭЖХ-УФ- и ВЭЖХ-МС/МС-определением [113, 143, 144].
Нитрофураны быстро трансформируются, образуя канцерогенные и мутагенные метаболиты 3-амино-5-морфолинометил-1,3-оксазолидинон (АМОЗ), 3-амино-2-оксазолидинон (АОЗ), 1‑аминогидантоин (АГД) и семикарбазид (СЕМ), сохраняющиеся в органах и тканях от нескольких недель до нескольких месяцев, в связи с чем нитрофураны запрещены к применению в животноводстве многих стран [145]. Метаболиты, ковалентно связанные с белками, сложно разрушить, поэтому пробоподготовка для их определения является одной из самых сложных и многостадийных и в основном предполагает гидролиз и дериватизацию [145, 147, 150, 240]. В случае нитрофуранов методик для определения исходных соединений крайне мало в связи с их быстрой метаболизацией. Дериватизация необходима при использовании жидкостной хроматографии, так как гидролизованные метаболиты нитрофуранов высокополярны, слабо удерживаются на обращенно-фазовых колонках и обладают плохими ионизационными характеристиками [147]. При ВЭЖХ-МС/МС-определении нитрофуранов и их метаболитов в большинстве случаев используют изотопно-меченные стандарты, что позволяет учесть влияние матрицы и обеспечить необходимую специфичность и правильность анализа. Использование МИ-ТФЭ позволяет миновать стадию гидролиза при определении метаболитов нитрофуранов и при этом обеспечить хорошее извлечение аналита (>96%), но стадия дериватизации сохраняется и в случае использования В-ЭЖХ-УФ [146].
В работе [149] в качестве дериватизирующего агента предложен 5-нитро-2-фуральдегид. АОЗ, АМОЗ, АГД и СЕМ взаимодействуют с этим альдегидом, образуя исходные нитрофураны, что существенно ускоряет анализ и снижает его стоимость, поскольку позволяет выбрать условия пробоподготовки и определения с использованием доступных и недорогих стандартов нитрофуранов. Разработано ограниченное количество методик определения нитрофуранов и их метаболитов одновременно с другими группами препаратов [80, 97, 209, 211, 221], что связано с необходимостью гидролиза и дериватизации образцов, которое может привести к разрушению остальных аналитов. Возможны и альтернативные пути попадания метаболитов нитрофуранов в продукты питания. В случае СЕМ не до конца изучен вопрос его естественного образования в организме живых животных, а также в результате разложения азодикарбонамида, используемого в качестве вспенивающего агента при производстве прокладок для крышек пищевых банок. В связи с этим возможно обнаружение его фоновых концентраций, но исследований в этой области недостаточно [240].
Пенициллины и цефалоспорины относятся к β-лактамным антибиотикам, уникальной структурной особенностью которых является наличие четырехчленного β-лактамного (2-азетидинонового) кольца [241]. Они довольно полярны, объемны и умеренно чувствительными к нагреванию [242]. β-Лактамы, как правило, демонстрируют значительно меньшую по сравнению с другими антибиотиками стабильность в условиях пробоподготовки. Органические растворители и кислоты гидролизуют β-лактамное кольцо и приводят к образованию неактивной формы, что усложняет их определение [242]. Для решения этой проблемы используют дериватизацию. Результаты показали, что дериватизация пиперидином снижает деградацию пенициллинов и повышает точность и анализа. В связи с этими особенностями наиболее частыми методами определения пенициллинов и цефалоспоринов в пищевой продукции являются ВЭЖХ-УФ [154, 186, 192, 194], ВЭЖХ-МС/МС [187, 189] и УВЭЖХ-МС/МС [190, 191, 193]. Для повышения степени извлечения и селективности используют ионные жидкости [186, 193], а также широко представлена МИ-ТФЭ [192, 194].
Бацитрацин, колистин и вирджиниамицин относятся к группе полипептидных антибиотиков, их часто добавляют в корма для животных в качестве стимуляторов роста [116]. Хотя бацитрацин и вирджиниамицин запрещены для применения в продуктивном животноводстве ЕС, они разрешены в Китае [100]. С 2016 г. правительство Китая запретило использовать колистин в связи с появлением колистин-резистентных штаммов бактерий, однако при этом Китай – лидирующий производитель и экспортер колистина во Вьетнам, Индию и Южную Корею [153]. Для определения полипептидов предложены ВЭЖХ- и УВЭЖХ-МС/МС-методики [151–153].
Всего несколько работ за последнее десятилетие посвящено индивидуальному определению плевромутилинов [112, 154], хотя они широко используются в ветеринарии, в том числе в качестве стимуляторов роста. Большинство методик применяют для их одновременного определения с фторхинолонами, сульфаниламидами, макролидами, линкозамидами, тетрациклинами и β-лактамами [18, 34, 85, 101, 109].
Сульфаниламиды − самая обширная группа антибиотиков, для определения которых предложено очень большое количество методик (табл. 4). Методы ВЭЖХ с УФ-, ФЛД- и МС-детектированием успешно применяют при определении сульфаниламидов в продуктах животного происхождения и кормах, однако среди разработанных преобладают ВЭЖХ-УФ-методики. В качестве пробоподготовки широко используется ТФЭ, в том числе магнитная и дисперсионная. Сульфаниламиды положительно заряжены в кислой среде при рН < 2, нейтральны при рН 3–5 и отрицательно заряжены при рН > 5. Легко извлекаются молекулярные формы сульфаниламидов, поэтому важно поддерживать определенное значение рН при экстракции [158]. Для извлечения сульфаниламидов из мяса, рыбы, меда используют ацетонитрил, метанол, этилацетат или дихлорметан, а также супрамолекулярные растворители на основе обратных мицелл [243]. Стадия кислотного гидролиза требуется, как правило, при их определении в образцах пищевых продуктов богатых сахарами, таких как мед [242]. При определении сульфаниламидов с помощью ВЭЖХ-ФЛД необходимым этапом подготовки является дериватизация [69, 213], что также неизбежно ведет к усложнению анализа. Предложено большое количество методик определения сульфаниламидов одновременно с другими антибактериальными препаратами (табл. 3). ВЭЖХ-МС/МС широко применяется как в групповом, так и в многокомпонентном анализе.
Тетрациклины обладают высокой хелатирующей способностью по отношению к ионам переходных и щелочноземельных металлов, поэтому использование солей ЭДТА с буфером Макилвейна (или без него) повышает эффективность экстракции [242]. Экстракция тетрациклинов улучшается в кислых средах с добавлением трихлоруксусной, муравьиной и щавелевой кислот, подкисленного метанола и при увеличении времени встряхивания [242]. Кроме того, в своей структуре они содержат несколько хромофорных групп и интенсивно поглощают свет в ультрафиолетовой области между 270 и 360 нм как в кислой, так и в нейтральной среде. Чаще всего при определении тетрациклинов используют ВЭЖХ. ВЭЖХ-УФ является официальным методом AOAC для определения остаточного содержания тетрациклинов в молоке и тканях свиней и КРС, и большинство методик, разработанных в последние 10 лет, основано именно на его применении (табл. 4). ВЭЖХ-МС/МС-методики в основном предложены для одновременного определения тетрациклинов и других антибиотиков (табл. 3).
Хиноксалины используют в качестве стимуляторов роста, в организме животных они очень быстро трансформируются. В 1998 г. в ЕС отозваны лицензии на карбадокс и олаквиндокс в связи с возможным канцерогенным, мутагенным и фотоаллергенным действием препаратов и их дезоксиметаболитов [241]. Карбадокс быстро метаболизируется через моно- и дезоксисоединения до хиноксалин-2-карбоновой кислоты (ХКК), которая длительно сохраняется в тканях и поэтому является маркером для карбадокса. Путь метаболизма олаквиндокса аналогичен, при этом образуется 3-метилхиноксалин-2-карбоновая кислота, структура которой схожа с ХКК. Для определения в кормах для животных карбадокса (запрещенного в ЕС, Китае и Японии) и олаквиндокса (запрещенного в ЕС) [241] разработана ВЭЖХ-МС/МС-методика с экстракцией смесью вода–ацетонитрил (1 : 1) и последующей очисткой экстракта С18 [177]. Определение метаболитов, как правило, сложнее, чем исходных соединений. Наиболее эффективны в данном случае ВЭЖХ-МС/МС [175] и УВЭ-ЖХ-МС/МС [176, 179] (табл. 4). Пробоподготовка для большинства разработанных методик – ЖЖЭ с последующей ТФЭ-очисткой. ВЭЖХ-МС/МС предложена для одновременного определения хиноксалинов с другими препаратами [87, 107], при этом использовали УЗ-ЖЭ с последующей ТФЭ и экстракцию под давлением (табл. 3).
Хинолоны широко используют в ветеринарии и аквакультуре. Они имеют амфотерную природу, поскольку содержат и кислотные, и основные группы [242], нестабильны в сильно кислой среде. Хинолоны хорошо растворимы в полярных органических растворителях, поэтому в большинстве случаев проводят общую экстракцию растворителем с последующей ТФЭ-очисткой [183, 242]. Как правило, хинолоны в пищевой матрице связаны с белками, и необходима депротеинизация во время подготовки образца [181]. При экстракции используют ацетонитрил, поскольку он демонстрирует высокую эффективность осаждения белков [183]. Большинство методов позволяют проводить одновременное определение только нескольких хинолонов, а при использовании ВЭЖХ с УФ- и ФЛД-детектированием зачастую требуется сложная пробоподготовка, сочетающая ЖЖЭ и ТФЭ [182, 183]. Процедуры перед хроматографическим анализом предполагают использование больших количеств органических растворителей, трудоемки и финансово затратны. ВЭЖХ-МС/МС применяется преимущественно при одновременном определении в пищевых продуктах хинолонов и других антибиотиков (табл. 3).
* * *
Методики одновременного определения антибиотиков, включая метаболиты, представляют наибольший интерес в случае мониторинга остаточных содержаний препаратов в пищевой продукции и продовольственном сырье [88, 244]. Разнообразие и количество органических загрязнителей в объектах окружающей среды и продуктах питания неуклонно растет, и в этом контексте разработка УВЭЖХ-методик позволяет сократить продолжительность анализа, а при совмещении с МС/МС разработать простые, недорогие методики, характеризующиеся высокой точностью [245]. Принимая во внимание различия в физико-химических свойствах, необходимо использовать общие условия экстракции, хроматографического разделения и обнаружения. Твердофазная экстракция и QuEChERS − наиболее распространeнные подходы к пробоподготовке в многокомпонентном анализе [244]. Анализ литературы за последнее десятилетие позволил проследить основные тенденции в определении остаточных количеств антибиотиков в объектах окружающей среды и пищевых продуктах. В пробоподготовке для индивидуального и группового анализа лидируют ЖЖЭ (18%), ТФЭ (15%) и МИ--ТФЭ (15%), однако не менее востребованы ЖЖЭ с ТФЭ-очисткой, ДЖЖМЭ и ДТФЭ (14, 13 и 12% соответственно) (рис. 1а). Разработано наибольшее количество ВЭЖХ-МС/МС- и ВЭЖХ-ДМД-методик (35 и 31%) (рис. 2а). При одновременном определении препаратов разных групп в пробоподготовке преобладают QuEChERS (30%), ЖЖЭ в сочетании с последующей ТФЭ-очисткой (19%) и ЖЖЭ (15%) (рис. 1б), предложено больше УВЭ-ЖХ- и ВЭЖХ-МС/МС-методик (36 и 35%) (рис. 2б). Выбор методов пробоподготовки и определения антибиотиков обусловлен, во-первых, особенностями их физико-химических свойств: большинство соединений полярны и гидрофильны, некоторые группы неустойчивы в агрессивных средах или быстро метаболизируются, поэтому предпочтительны ЖЖЭ и ТФЭ (в различных вариантах исполнения) с последующим использованием ВЭЖХ. Во-вторых, имеет значение сложность матриц и необходимость одновременного многокомпонентного анализа: данные задачи предполагают универсальность пробоподготовки (QuEChERS) и высокую специфичность метода (ВЭЖХ- и УВЭЖХ-МС/МС). В-третьих, следует учитывать необходимость определения остаточных содержаний антибактериальных препаратов на уровне МДУ, значения которых могут быть крайне низкими: для решения данной проблемы требуются высокочувствительные методы в основном с масс-спектрометрическим детектированием, в том числе МС-ВР.
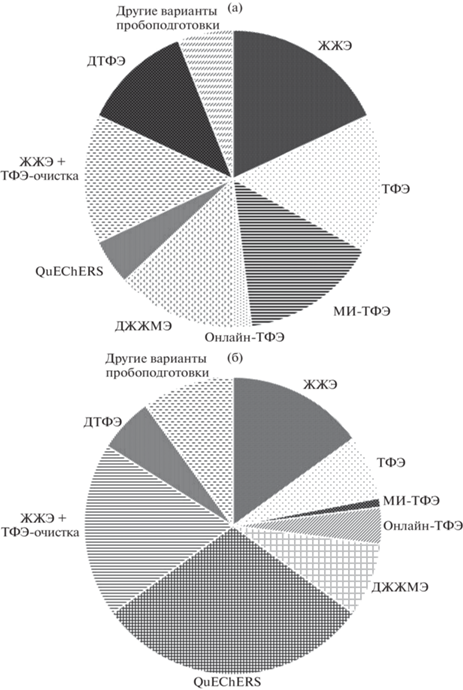

Список литературы
Hu C., Zhang Y., Zhou Y., Liu Z.F., Meng Q., Feng X.S. A review of pretreatment and analysis of macrolides in food (Update Since 2010) // J. Chromatogr. A. 2020. V. 1634. Article 461662.
Zhao F., Chen L., Yang L., Li S., Sun L., Yu X. Distribution, dynamics and determinants of antibiotics in soils in a peri-urban area of Yangtze River Delta, Eastern China // Chemosphere. 2018. V. 211. P. 261.
Cycoń M., Mrozik A., Piotrowska-Seget Z. Antibiotics in the soil environment – Degradation and their impact on microbial activity and diversity // Front. Microbiol. 2019. V. 10. Article 338.
Ronquillo M.G., Hernandez J.C.A. Antibiotic and synthetic growth promoters in animal diets: Review of impact and analytical methods // Food Control. 2017. V. 72. Part B. P. 255.
Conde-Cid M., Ferreira-Coelho G., Núñez-Delgado A., Fernández-Calviño D., Arias-Estévez M., Álvarez-Rodríguez E., Fernández-Sanjurjo M.J. Competitive adsorption of tetracycline, oxytetracycline and chlortetracycline on soils with different pH value and organic matter content // Environ. Res. 2019. V. 178. Article 108669.
Szymańska U., Wiergowski M., Sołtyszewski I., Kuzemko J., Wiergowska G., Woźniak M.K. Presence of antibiotics in the aquatic environment in Europe and their analytical monitoring: Recent trends and perspectives // Microchem. J. 2019. V. 147. P. 729.
Lyu J., Yang L., Zhang L., Ye B., Wang L. Antibiotics in soil and water in China – A systematic review and source analysis // Environ. Pollut. 2020. V. 266. Part 1. Article 115147.
Wegst-Uhrich S., Navarro D., Zimmerman L., Aga D. Assessing antibiotic sorption in soil: A literature review and new case studies on sulfonamides and macrolides // Chem. Cent. J. 2014. V. 8. P. 5.
Leal R.M., Figueira R.F., Tornisielo V.L., Regitano J.B. Occurrence and sorption of fluoroquinolones in poultry litters and soils from São Paulo State, Brazil // Brazil. Sci. Total Environ. 2012. V. 432. P. 344.
Khataei M.M., Epi S.B.H., Lood R., Spégel S.P., Yamini Y., Turner C. A review of green solvent extraction techniques and their use in antibiotic residue analysis // J. Pharm. Biomed. Anal. 2021. V. 209. Article 114487.
Zheng W.-L., Zhang L.-F., Zhang K.-Y., Wang X.-Y., Xue F.-Q. Determination of tetracyclines and their epimers in agricultural soil fertilized with swine manure by ultra-high-performance liquid chromatography tandem mass spectrometry // J. Integr. Agric. 2012. V. 11. № 7. P. 1189.
Yu W.-H., Chin T.-S., Lai H.-T. Detection of nitrofurans and their metabolites in pond water and sediments by liquid chromatography (LC)-photodiode array detection and LC-ion spray tandem mass spectrometry // Int. Biodeterior. Biodegradation. 2013. V. 85. P. 517.
Bourdat-Deschamps M., Leang S., Bernet N., Daudin J.-J., Nélieu S. Multi-residue analysis of pharmaceuticals in aqueous environmental samples by online solid-phase extraction–ultra-high-performance liquid chromatography-tandem mass spectrometry: Optimisation and matrix effects reduction by quick, easy, cheap, effective, rugged and safe extraction // J. Chromatogr. A. 2014. V. 1349. P. 11.
Solliec M., Roy-Lachapelle A., Sauvé S. Quantitative performance of liquid chromatography coupled to Q-Exactive high resolution mass spectrometry (HRMS) for the analysis of tetracyclines in a complex matrix // Anal. Chim. Acta. 2015. V. 853. P. 415.
Guo C., Wang M., Xiao H., Huai B., Wang F., Pan G., Liao X., Liu Y. Development of a modified QuEChERS method for the determination of veterinary antibiotics in swine manure by liquid chromatography tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1027. P. 110.
Lian Z., Wang J. Determination of ciprofloxacin in Jiaozhou Bay using molecularly imprinted solid-phase extraction followed by high-performance liquid chromatography with fluorescence detection // Mar. Pollut. Bull. 2016. V. 111. № 1–2. P. 411.
Li X., Guo P., Shan Y., Ke Y., Li H., Fu Q., Wang Y., Liu T., Xia X. Determination of 82 veterinary drugs in swine waste lagoon sludge by ultra-high performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2017. V. 1499. P. 57.
Campos-Mañas M.C., Plaza-Bolaños P., Sánchez-Pérez J.A., Malato S., Agüera A. Fast determination of pesticides and other contaminants of emerging concern in treated wastewater using direct injection coupled to highly sensitive ultra-high performance liquid chromatography-tandem mass spectrometry // J. Chromatogr. A. 2017. V. 1507. P. 84.
Pérez R.A., Albero B., Férriz M., Tadeo J.L. Analysis of macrolide antibiotics in water by magnetic solid-phase extraction and liquid chromatography–tandem mass spectrometry // J. Pharm. Biomed. Anal. 2017. V. 146. P. 79.
Cai Q., Zhang L., Zhao P., Lun X., Li W., Guo Y., Hou X. A joint experimental-computational investigation: Metal organic framework as a vortex assisted dispersive micro-solid-phase extraction sorbent coupled with UPLC-MS/MS for the simultaneous determination of amphenicols and their metabolite in aquaculture water // Microchem. J. 2017. V. 130. P. 263.
González-Gaya B., Cherta L., Nozal L., Rico A. An optimized sample treatment method for the determination of antibiotics in seawater, marine sediments and biological samples using LC-TOF/MS // Sci. Total Environ. 2018. V. 643. P. 994.
Lian L., Lv J., Wang X., Lou D. Magnetic solid–phase extraction of tetracyclines using ferrous oxide coated magnetic silica microspheres from water samples // J. Chromatogr. A. 2018. V. 1534. P. 1.
Wang Z., Wang X.-Y., Tian H., Wei Q.-H., Liu B.-S., Bao G.-M., Liao M.-L., Peng J.-L., Huang X.-Q., Wang L.-Q. High through-put determination of 28 veterinary antibiotic residues in swine wastewater by one-step dispersive solid phase extraction sample cleanup coupled with ultra-performance liquid chromatography-tandem mass spectrometry // Chemosphere. 2019. V. 230. P. 337.
Yuan S.-F., Liu Z.-H., Yin H., Dang Z., Wu P.-X., Zhu N.-W, Lin Z. Trace determination of sulfonamide antibiotics and their acetylated metabolites via SPE-LC-MS/MS in wastewater and insights from their occurrence in a municipal wastewater treatment plant // Sci. Total Environ. 2019. V. 653. P. 815.
Mokhtar H.I., Abdel-Salam R.A., Hadad G.M. Tolerance intervals modeling for design space of a salt assisted liquid-liquid microextraction of trimethoprim and six common sulfonamide antibiotics in environmental water samples // J. Chromatogr. A. 2019. V. 1586. P. 18.
Liang Y., Liu J., Zhong Q., Yu D., Yao J., Huang T., Zhu M., Zhou T. A fully automatic cross used solid-phase extraction online coupled with ultra-high performance liquid chromatography–tandem mass spectrometry system for the trace analysis of multi-class pharmaceuticals in water samples // J. Pharm. Biomed. Anal. 2019. V. 174. P. 330.
Wang J., Xu J., Ji X., Wu H., Yang H., Zhang H., Zhang X., Li Z., Ni X., Qian M. Determination of veterinary drug/pesticide residues in livestock and poultry excrement using selective accelerated solvent extraction and magnetic material purification combined with ultra-high-performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2020. V. 1617. Article 460808.
Rashid A., Mazhar S.H., Zeng Q., Kiki C., Yu C.-P., Sun Q. Simultaneous analysis of multiclass antibiotic residues in complex environmental matrices by liquid chromatography with tandem quadrupole mass spectrometry // J. Chromatogr. B. 2020. V. 1145. Article 122103.
Mahmoudian M.H., Fazlzadeh M., Niari M.H., Azari A., Lima E.C. A novel silica supported chitosan/glutaraldehyde as an efficient sorbent in solid phase extraction coupling with HPLC for the determination of Penicillin G from water and wastewater samples // Arab. J. Chem. 2020. V. 13. № 9. P. 7147.
Duan R., Sun L., Yang H.-Y., Ma Y.-R., Deng X.-Y., Peng C., Zheng C., Dong L.-Y., Wang X.-H. Preparation of phenyl–boronic acid polymeric monolith by initiator-free ring-opening polymerization for microextraction of sulfonamides prior to their determination by ultra-performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2020. V. 1609. Article 460510.
Lu D., Liu C., Qin M., Deng J., Shi G., Zhou T. Functionalized ionic liquids-supported metal organic frameworks for dispersive solid phase extraction of sulfonamide antibiotics in water samples // Anal. Chim. Acta. 2020. V. 1133. P. 88.
Zhang Y., Duan L., Wang B., Liu C.S., Jia Y., Zhai N., Blaney L., Yu G. Efficient multiresidue determination method for 168 pharmaceuticals and metabolites: Optimization and application to raw wastewater, wastewater effluent, and surface water in Beijing, China // Environ. Pollut. 2020. V. 261. Article 114113.
Yu R., Chen L., Shen R., Li P., Shi N. Quantification of ultratrace levels of fluoroquinolones in wastewater by molecularly imprinted solid phase extraction and liquid chromatography triple quadrupole mass // Environ. Technol. Innov. 2020. V. 19. Article 100919.
Goessens T., Huysman S., De Troyer N., Deknock A., Goethals P., Lens L, Vanhaecke L., Croubels S. Multi-class analysis of 46 antimicrobial drug residues in pond water using UHPLC-Orbitrap-HRMS and application to freshwater ponds in Flanders, Belgium // Talanta. 2020. V. 220. Article 121326.
Da Cunha C.C.R.F., Freitas M.G., da Silva Rodrigues D.A., de Barros A.L.C., Ribeiro M.C., Sanson A.L., de Cássia Franco Afonso R.J. Low-temperature partitioning extraction followed by liquid chromatography tandem mass spectrometry determination of multiclass antibiotics in solid and soluble wastewater fractions // J. Chromatogr. A. 2021. V. 1650. Article 462256.
Senta I., Terzic S., Ahel M. Analysis and occurrence of macrolide residues in stream sediments and underlying alluvial aquifer downstream from a pharmaceutical plant // Environ. Pollut. 2021. V. 273. Article 116433.
Martínez-Piernas A.B., Plaza-Bolaños P., Gilabert A., Agüera A. Application of a fast and sensitive method for the determination of contaminants of emerging concern in wastewater using a quick, easy, cheap, effective, rugged and safe-based extraction and liquid chromatography coupled to mass spectrometry // J. Chromatogr. A. 2021. V. 1653. Article 462396.
Ofrydopoulou A., Nannou C., Evgenidou E., Lambropoulou D. Sample preparation optimization by central composite design for multi class determination of 172 emerging contaminants in wastewaters and tap water using liquid chromatography high-resolution mass spectrometry // J. Chromatogr. A. 2021. V. 1652. Article 462369.
Sereshti H., Karami F., Nouri N. A green dispersive liquid-liquid microextraction based on deep eutectic solvents doped with β-cyclodextrin: Application for determination of tetracyclines in water samples // Microchem. J. 2021. V. 163. Article 105914.
Zhao G., Ouyang X.-K., Yang L.-Y., Jin M.-C. Facile fabrication of surface molecularly imprinted magnetic polydopamine for selective adsorption of fluoroquinolone from aqueous solutions // J. Mol. Struct. 2021. V. 1243. Article 130894.
Białk-Bielińska A., Kumirska J., Borecka M., Caban M., Paszkiewicz M., Pazdro K., Stepnowski P. Selected analytical challenges in the determination of pharmaceuticals in drinking/marine waters and soil/sediment samples // J. Pharm. Biomed. Anal. 2016. V. 121. P. 271.
Rykowska I., Ziemblińska J., Nowak I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: A review // J. Mol. Liq. 2018. V. 259. P. 319.
Mousavi L., Tamiji Z., Khoshayand M.R. Applications and opportunities of experimental design for the dispersive liquid–liquid microextraction method – A review // Talanta. 2018. V. 190. P. 335.
Yao T., Du K. Simultaneous determination of sulfonamides in milk: In-situ magnetic ionic liquid dispersive liquid-liquid microextraction coupled with HPLC // Food Chem. 2020. V. 331. Article 127342.
Shishov A., Gorbunov A., Baranovskii E., Bulatov A. Microextraction of sulfonamides from chicken meat samples in three-component deep eutectic solvent // Microchem. J. 2020. V. 158. Article 105274.
Pochivalov A., Cherkashina K., Shishov A., Bulatov A. Microextraction of sulfonamides from milk samples based on hydrophobic deep eutectic solvent formation by pH adjusting // J. Mol. Liq. 2021. V. 339. Article 116827.
Gao J., Wang H., Qu J., Wang H., Wang X. Development and optimization of a naphthoic acid-based ionic liquid as a “non-organic solvent microextraction” for the determination of tetracycline antibiotics in milk and chicken eggs // Food Chem. 2017. V. 215. P. 138.
Moema D., Nindi M.M., Dube S. Development of a dispersive liquid–liquid microextraction method for the determination of fluoroquinolones in chicken liver by high performance liquid chromatography // Anal. Chim. Acta. 2012. V. 730. P. 80.
Herrera-Herrera A.V., Hernández-Borges J., Borges-Miquel T.M., Rodríguez-Delgado M.Á. Dispersive liquid–liquid microextraction combined with ultra-high performance liquid chromatography for the simultaneous determination of 25 sulfonamide and quinolone antibiotics in water samples // J. Pharm. Biomed. Anal. 2013. V. 75. P. 130.
Gao S., Yang X.,Yu W., Liu Z., Zhang H. Ultrasound-assisted ionic liquid/ionic liquid-dispersive liquid–liquid microextraction for the determination of sulfonamides in infant formula milk powder using high-performance liquid chromatography // Talanta. 2012. V. 99. P. 875.
Teglia C.M., Gonzalo L., M.J. Culzoni, Goicoechea H.C. Determination of six veterinary pharmaceuticals in egg by liquid chromatography: Chemometric optimization of a novel air assisted-dispersive liquid-liquid microextraction by solid floating organic drop// Food Chem. 2019. V. 273. P. 194.
Parrilla Vázquez M.M., Parrilla Vázquez P., Martínez Galera M., Gil García M.D. Determination of eight fluoroquinolones in groundwater samples with ultrasound-assisted ionic liquid dispersive liquid–liquid microextraction prior to high-performance liquid chromatography and fluorescence detection // Anal. Chim. Acta. 2012. V. 748. P. 20.
Karami-Osboo R., Shojaee M.H., Miri R., Kobarfard F., Javidnia K. Simultaneous determination of six fluoroquinolones by validated QuEChERS-DLLME HPLC-FLD in milk // Anal. Methods. 2014. V. 6. № 15. P. 1.
Dorival-Garcíac N., Junza A., Zafra-Gómez A., Barrón D., Ballesteros O., Barbosa J., Navalónc A. Multiclass method for the determination of quinolones and β-lactams, in raw cow milk using dispersive liquid–liquid microextraction and ultra high performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2014. V. 1356. P. 10.
Mookantsa S.O., Dube S., Nindi M.M. Development and application of a dispersive liquid-liquid microextraction method for the determination of tetracyclines in beef by liquid chromatography mass spectrometry // Talanta. 2016. V. 148. P. 321.
Huang P., Zhao P., Dai X., Hou X., Zhao L., Liang N. Trace determination of antibacterial pharmaceuticals in fishes by microwave-assisted extraction and solid-phase purification combined with dispersive liquid-liquid microextraction followed by ultra-high performance liquid chromatography-tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1011. P. 136.
Sereshti H., Jazani S.S., Nouri N., Shams G. Dispersive liquid–liquid microextraction based on hydrophobic deep eutectic solvents: Application for tetracyclines monitoring in milk // Microchem. J. 2020. V. 158. Article 105269.
Liang N., Huang P., Hou X., Li Z., Tao L., Zhao L. Solid-phase extraction in combination with dispersive liquid-liquid microextraction and ultra-high performance liquid chromatography-tandem mass spectrometry analysis: The ultra-trace determination of 10 antibiotics in water samples// Anal. Bioanal. Chem. 2016. V. 408. № 6. P. 1701
Крылов В.А., Волкова В.В., Савельева О.А. Микроэкстракционное концентрирование примесей из воды с ультразвуковым диспергированием экстрагента // Аналитика и контроль. 2013. Т. 17. № 1. С. 82.
Bottoni P., Caroli S. Presence of residues and metabolites of pharmaceuticals in environmental compartments, food commodities and workplaces: A review spanning the three-year period 2014–2016 // Microchem. J. 2018. V. 136. P. 2.
Conde-Cid M., Núñez-Delgado A., Fernández-Sanjurjo M.J., Álvarez-Rodríguez E., Fernández-Calviño D., Arias-Estévez M. Tetracycline and sulfonamide antibiotics in soils: Presence, fate and environmental risks // Processes. 2020. V. 8. № 11. Article 1479.
Ho Y.B., Zakaria M.P., Latif P.A., Saari N. Simultaneous determination of veterinary antibiotics and hormone in broiler manure, soil and manure compost by liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2012. V. 1262. P. 160.
Mehrtens A., Licha T., Burke V. Occurrence, effects and behaviour of the antibiotic lincomycin in the agricultural and aquatic environment – A review // Sci. Total Environ. 2021. V. 778. Article 146306.
Cha J., Carlson K.H. Occurrence of β-lactam and polyether ionophore antibiotics in lagoon water and animal manure // Sci. Total Environ. 2018. V. 640–641. P. 1346.
Li X.W., Xie Y.F., Li C.L., Zhao H.N., Zhao H., Wang N., Wang J.F. Investigation of residual fluoroquinolones in a soil-vegetable system in an intensive vegetable cultivation area in Northern China // Sci. Total Environ. 2014. V. 468-469. P. 258.
Mejías C., Martín J., Santos J.L., Aparicio I., Alonso E. Occurrence of pharmaceuticals and their metabolites in sewage sludge and soil: A review on their distribution and environmental risk assessment // Trends Environ. Anal. Chem. 2021. V. 30. Article e00125.
Berendsen B., Pikkemaat M., Römkens P., Wegh R., van Sisseren M., Stolker L., Nielen M. Occurrence of chloramphenicol in crops through natural production by bacteria in soil // J. Agr. Food Chem. 2013. V. 61. № 17. P. 4004.
Commission Regulation (EU) 2019/1871 of 7 November 2019 on reference points for action for non-allowed pharmacologically active substances present in food of animal origin and repealing Decision 2005/34/EC (Text with EEA relevance) C/2019/7908. URL: https:// eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX %3A32019R1871 (дата обращения 22.02.2022).
Toaldo I.M., Gamba G.Z., Picinin L.A., Rubensam G., Hoff R., Bordignon-Luiz M. Multiclass analysis of antibacterial residues in milk using RP-liquid chromatography with photodiode array and fluorescence detection and tandem mass spectrometer confirmation // Talanta. 2012. V. 99. P. 616.
Tao Y., Yu G., Chen D., Pan Y., Liu Z., Wei H., Peng D., Huang L., Wang Y., Yuan Z. Determination of 17 macrolide antibiotics and avermectins residues in meat with accelerated solvent extraction by liquid chromatography-tandem mass spectrometry // J. Chromatogr. B. 2012. V. 897. P. 64.
Aguilera-Luiz M.M., Romero-Gonza R., Plaza-Bolan P., Mart J.L., Frenich A.G. Rapid and semiautomated method for the analysis of veterinary drug residues in honey based on turbulent-flow liquid chromatography coupled to ultrahigh-performance liquid chromatography−orbitrap mass spectrometry (TFC-UHPLCOrbitrap-MS)// Agric. Food Chem. 2013. V. 61. P. 829.
Economou A., Petraki O., Tsipi D., Botitsi E. Development of a liquid chromatography–tandem mass spectrometry method for the determination of sulfonamides, trimethoprim and dapsone in honey and validation according to Commission Decision 2002/657/EC for banned compounds [corrected] // Talanta. 2012. V. 97. P. 32.
Zhang Q., Xiao X., Li G. Porous molecularly imprinted monolithic capillary column for on-line extraction coupled to high-performance liquid chromatography for trace analysis of antimicrobials in food samples // Talanta. 2014. V. 123. P. 63.
Dickson L.C. Performance characterization of a quantitative liquid chromatography–tandem mass spectrometric method for 12 macrolide and lincosamide antibiotics in salmon, shrimp and tilapia // J. Chromatogr. B. 2014. V. 967. P. 203.
Storey J.M., Clark S.B., Johnson A.S., Andersen W.C., Turnipseed S.B., Lohne J.J., Burger R.J., Ayres P.R., Carr J.R., Madson M.R. Analysis of sulfonamides, trimethoprim, fluoroquinolones, quinolones, triphenylmethane dyes and methyltestosterone in fish and shrimp using liquid chromatography-mass spectrometry // J. Chromatogr. B. 2014. V. 972. P. 38.
Hou X.-L., Chen G., Zhu L., Yang T., Zhao J., Wang L., Wu Y.-L. Development and validation of an ultra-high-performance liquid chromatography tandem mass spectrometry method for simultaneous determination of sulfonamides, quinolones and benzimidazoles in bovine milk // J. Chromatogr. B. 2014. V. 962. P. 20.
Yang X., Zhang S., Yu W., Liu Z., Lei L., Li N., Zhang H., Yu Y. Ionic liquid-anionic surfactant based aqueo-us two-phase extraction for determination of antibiotics in honey by high-performance liquid chromatography // Talanta. 2014. V. 124. P. 1.
Freitas A., Barbosa J., Ramos F. Multidetection of antibiotics in liver tissue by ultra-high-pressure-liquid-chromatography–tandem mass spectrometry // J. Chromatogr. B. 2015. V. 976–977. P. 49.
Gbylik-Sikorska M., Posyniak A., Sniegocki T., Zmudzki J. Liquid chromatography–tandem mass spectrometry multiclass method for the determination of antibiotics residues in water samples from water supply systems in food-producing animal farms // Chemosphere. 2015. V. 119. P. 8.
Kaufmann A., Butcher P., Maden K., Walker S., Widmer M. Determination of nitrofuran and chloramphenicol residues by high resolution mass spectrometry versus tandem quadrupole mass spectrometry // Anal. Chim. Acta. 2015. V. 862. P. 41.
Chung S.W.C., Lam C.-H. Development of a 15-class multiresidue method for analyzing 78 hydrophilic and hydrophobic veterinary drugs in milk, egg and meat by liquid chromatography−tandem mass spectrometry // Anal. Methods. 2015. V. 7. P. 6764.
Gómez-Pérez M.L., Romero-González R., Luis Martínez V.J., Frenich A.G. Analysis of pesticide and veterinary drug residues in baby food by liquid chromatography coupled to Orbitrap high resolution mass spectrometry // Talanta. 2015. V. 131. P. 1.
Zhu W.-X., Yang J.-Z., Wang Z.-X., Wang C.-J., Liu Y.-F., Zhang L. Rapid determination of 88 veterinary drug residues in milk using automated TurborFlow online clean-up mode coupled to liquid chromatography−tandem mass spectrometry // Talanta. 2016. V. 148. P. 401.
Dasenaki M.E., Michali C.S., Thomaidis N.S. Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2016. V. 1452. P. 67.
Moretti S., Dusi G., Giusepponi D., Pellicciotti S., Rossi R., Saluti G., Cruciani G., Galarini R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat // J. Chromatogr. A. 2016. V. 1429. P. 175.
Tian H., Wang J., Zhang Y., Li S., Jiang J., Tao D., Zheng N. Quantitative multiresidue analysis of antibiotics in milk and milk powder by ultra-performance liquid chromatography coupled to tandem quadrupole mass spectrometry // J. Chromatogr. B. 2016. V. 1033–1034. P. 172.
Chen D., Yu J., Tao Y., Pan Y., Xie S., Huang L., Peng D., Wang X., Wang Y., Liu Z., Yuan Z. Qualitative screening of veterinary anti-microbial agents in tissues, milk, and eggs of food-producing animals using liquid chromatography coupled with tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1017–1018. P. 82.
Jia W., Chu X., Chang J., Wang P.G., Chen Y., Zhang F. High-throughput untargeted screening of veterinary drug residues and metabolites in tilapia using high resolution orbitrap mass spectrometry // Anal. Chim. Acta. 2017. V. 957. P. 29.
Wang G.N., Zhang L., Song Y.P., Liu J.X., Wang J.P. Application of molecularly imprinted polymer based matrix solid phase dispersion for determination of fluoroquinolones, tetracyclines and sulfonamides in meat // J. Chromatogr. B. 2017. V. 1065–1066. P. 104.
Zhao F., Gao X., Tang Z., Luo X., Wu M., Xu J., Fu X. Development of a simple multi-residue determination method of 80 veterinary drugs in Oplegnathus punctatus by liquid chromatography coupled to quadrupole orbitrap mass spectrometry // J. Chromatogr. B. 2017. V. 1065–1066. P. 20.
Zhang Y., Guo W., Yue Z., Lin L., Zhao F., Chen P., Wu W., Zhu H., Yang B., Kuang Y., Wang J. Rapid determination of 54 pharmaceutical and personal care products in fish samples using microwave-assisted extraction-hollow fiber-liquid/solid phase microextraction // J. Chromatogr. B. 2017. V. 1051. P. 41.
Louppis A.P., Kontominas M.G., Papastephanou C. Determination of antibiotic residues in honey by high-performance liquid chromatography with electronspray ionization tandem mass spectrometry // Food Anal. Methods. 2017. V. 10. P. 3385.
Xie Y., Hu Q., Zhao M., Cheng Y., Guo Y., Qian H., Yao W. Simultaneous determination of erythromycin, tetracycline, and chloramphenicol residue in raw milk by molecularly imprinted polymer mixed with solid-phase extraction // Food Anal. Methods. 2017. V. 11. P. 374.
Chiesa L., Panseri S., Pasquale E., Malandra R., Pavlovic R., Arioli F. Validated multiclass targeted determination of antibiotics in fish with high performance liquid chromatography–benchtop quadrupole orbitrap hybrid mass spectrometry // Food Chem. 2018. V. 258. P. 222.
Saxena S.K., Rangasamy R., Krishnan A.A., Singh D.P., Uke S.P., Malekadi P.K., Sengar A.S., Mohamed D.P., Gupta A. Simultaneous determination of multi-residue and multi-class antibiotics in aquaculture shrimps by UPLC-MS/MS // Food Chem. 2018. V. 260. P. 336.
Zhao L., Lucas D., Long D., Richter B., Stevens J. Multi-class multi-residue analysis of veterinary drugs in meat using enhanced matrix removal lipid cleanup and liquid chromatography−tandem mass spectrometry // J. Chromatogr. A. 2018. V. 1549. P. 14.
Li J., Ren X., Diao Y., Chen Y., Wang Q., Jin W., Zhou P., Fan Q., Zhang Y., Liu H. Multiclass analysis of 25 veterinary drugs in milk by ultra-high performance liquid chromatography−tandem mass spectrometry // Food Chem. 2018. V. 257. P. 259.
Ribeiro C.B., Martins M.T., Jank L., Barreto F., Hoff R.B., Arsand J.B. Development and validation of a simple and fast method for sulfonamides, tetracyclines and macrolides in honey using LC-MS/MS // Drug Anal. Res. 2018. № 2. P. 21.
Albero B., Tadeo J.L., Miguel E., Pérez R.A. Rapid determination of antibiotic residues in cereals by liquid chromatography triple mass spectrometry // Anal. Bioanal. Chem. 2019. V. 411. № 23. P. 6129.
Song X., Huang Q., Zhang Y., Zhang M., Xie J., He L. Rapid multiresidue analysis of authorized/banned cyclopolypeptide antibiotics in feed by liquid chromatography–tandem mass spectrometry based on dispersive solid-phase extraction // J. Pharm. Biomed. Anal. 2019. V. 170. P. 234.
Gbylik-Sikorska M., Gajda A., Nowacka-Kozak E., Posyniak A. Simultaneous determination of 45 antibacterial compounds in mushrooms – Agaricus bisporus by ultra-high performance liquid chromatography−tandem mass spectrometry // J. Chromatogr. A. 2019. V. 1587. P. 111.
Susakate S., Poapolathep S., Chokejaroenrat C., Tanhan P., Hajslova J., Giorgi M., Saimek K., Zhang Z., Poapolathep A. Multiclass analysis of antimicrobial drugs in shrimp muscle by ultra-high performance liquid chromatography−tandem mass spectrometry // J. Food Drug Anal. 2019. V. 27. № 1. P. 118.
Khaled A., Gionfriddo E., Acquaro V., Singh V., Pawliszyn J. Development and validation of a fully automated solid phase microextraction high throughput method for quantitative analysis of multiresidue veterinary drugs in chicken tissue // Anal. Chim. Acta. 2019. V. 1056. P. 34.
Pugajeva, I., Ikkere L.E., Judjallo E., Bartkevics V. Determination of residues and metabolites of more than 140 pharmacologically active substances in meat by liquid chromatography coupled to high resolution orbitrap mass spectrometry // J. Pharm. Biomed. Anal. 2019. V. 166. P. 252.
Li X., Chi Q., Xia S., Pan Y., Chen Y., Wang K. Untargeted multi-residue method for the simultaneous determination of 141 veterinary drugs and their metabolites in pork by high-performance liquid chromatography time-of-flight mass spectrometry // J. Chromatogr. A. 2020. V. 1634. Article 461671.
Magalhães D., Freitas A., Pouca A.S.V., Barbosa J., Ramos F. The use of ultra-high-pressure-liquid-chromatography tandem time-of-flight mass spectrometry as a confirmatory method in drug residue analysis: Application to the determination of antibiotics in piglet liver // J. Chromatogr. B. 2020. V. 1153. Article 122264.
Hoff R.B., Molognoni L., Deolindo C.T.P., Vargas M.O., Kleemann C.R., Daguer H. Determination of 62 veterinary drugs in feedingstuffs by novel pressurized liquid extraction methods and LC-MS/MS // J. Chromatogr. B. 2020. V. 1152. Article 122232.
Zhan J., Shi X.-Z., Xu X.-W., Cao G.-Z., Chen X.-F. Generic and rapid determination of low molecular weight organic chemical contaminants in protein powder by using ultra-performance liquid chromatography−tandem mass spectrometry // J. Chromatogr. B. 2020. V. 1138. Article 121967.
Wang C., Li X., Yu F., Wang Y., Ye D., Hu X., Zhou L., Du J., Xia X. Multi-class analysis of veterinary drugs in eggs using dispersive-solid phase extraction and ultra-high performance liquid chromatography−tandem mass spectrometry // Food Chem. 2021. V. 334. Article 127598.
Xu X., Zhao W., Ji B., Han Y., Xu G., Jie M., Wu N., Wu Y., Li J., Li K., Zhao D., Bai Y. Application of silanized melamine sponges in matrix purification for rapid multi-residue analysis of veterinary drugs in eggs by UPLC-MS/MS // Food Chem. 2022. V. 369. Article 130894.
Liang S., Dai H., Wang C., Zhang H., Li J., Xu Q., Zhang Q. Application of polydopamine fibers mat for simultaneous detection of multi-class drug residues in various animal-original foods // Food Control. 2022. V. 132. Article 108523.
Li H., Wang Y., Li X., Fu Q., Shan Y., Liu T., Xia X. Determination of valnemulin in swine and bovine tissues by ultra-high performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1014. P. 102.
Hernández-Mesa M., Cruces-Blanco C., García-Campaña A.M. Simple and rapid determination of 5-nitroimidazoles and metabolites in fish roe samples by salting-out assisted liquid liquid extraction and UHPLC-MS/MS // Food Chem. 2018.V. 252. P. 294.
Arsand J.B., Jank L., Martins M.T., Hoff R.B., Barreto F., Pizzolato T.M., Sirtori C. Determination of aminoglycoside residues in milk and muscle based on a simple and fast extraction procedure followed by liquid chromatography coupled to tandem mass spectrometry and time of flight mass spectrometry// Talanta. 2016. V. 154. P. 38.
Wang J., Zhao Q., Jiang N., Li W., Chen L., Lin X., Xie Z., You L., Zhang Q. Urea−formaldehyde monolithic column for hydrophilic in-tube solid-phase microextraction of aminoglycosides // J. Chromatogr. A. 2017. V. 1485. P. 24.
Moreno-González D., Hamed A.M., García-Campaña A.M., Gámiz-Gracia L. Evaluation of hydrophilic interaction liquid chromatography tandem mass spectrometry and extraction with molecularly imprinted polymers for determination of aminoglycosides in milk and milk-based functional foods // Talanta. 2017. V. 171. P. 74.
Yang B., Wang L., Luo C., Wang X., Sun C. Simultaneous determination of 11 aminoglycoside residues in honey, milk, and pork by liquid chromatography with tandem mass spectrometry and molecularly imprinted polymer solid phase extraction // J. AOAC Int. 2017. V. 100. P. 1869.
Li D., Li T., Wang L., Ji S. A polyvinyl alcohol-coated core-shell magnetic nanoparticle for the extraction of aminoglycoside antibiotics residues from honey samples // J. Chromatogr. A. 2018. V. 1581–1582. P. 1.
Barreto F., Ribeiro C., Hoff R.B., Costa T.D. Determination and confirmation of chloramphenicol in honey, fish and prawns by liquid chromatography–tandem mass spectrometry with minimum sample preparation: Validation according to 2002/657/EC Directive // Food Addit. Contam. 2012. V. 29. № 4. P. 550.
Samanidou V., Galanopoulos L.-D., Kabir A., Furton K.G. Fast extraction of amphenicols residues from raw milk using novel fabric phase sorptive extraction followed by high-performance liquid chromatography-diode array detection // Anal. Chim. Acta. 2015. V. 855. P. 41.
Xiao Z., Song R., Rao Z., Wei S., Jia Z., Suo D., Fan X. Development of a subcritical water extraction approach for trace analysis of chloramphenicol, thiamphenicol, florfenicol, and florfenicol amine in poultry tissue // J. Chromatogr. A. 2015. V. 1418. P. 29.
Wei S., Li J., Liu Y., Ma J. Development of magnetic molecularly imprinted polymers with double templates for the rapid and selective determination of am-phenicol antibiotics in water, blood, and egg samples // J. Chromatogr. A. 2016. V. 1473. P. 19.
Samanidou V., Kehagia M., Kabir A., Furton K.G. Matrix molecularly imprinted mesoporous sol–gel sorbent for efficient solid-phase extraction of chloramphenicol from milk // Anal. Chim. Acta. 2016. V. 914. P. 62.
Imran M., Fazal-e-Habib, Tawab A., Rauf W., Rahman M., Khan Q.M., Asi M.R., Iqbal M. LC–MS/MS based method development for the analysis of florfenicol and its application to estimate relative distribution in various tissues of broiler chicken // J. Chromatogr. B. 2017. V. 1063. P. 163.
Deng F., Yu H., Pan X., Hu G., Wang Q., Peng R., Tan L., Yang Z. Ultra-high performance liquid chromatography tandem mass spectrometry for the determination of five glycopeptide antibiotics in food and biological samples using solid-phase extraction // J. Chromatogr. A. 2018. V. 1538. P. 54.
Tang Y., Zhang N., Zhang B., Lei X., Wu X. On-line coupling of hydrophilic ionic liquids-based polymer monolith microextraction to capillary liquid chromatography with amperometric detection: An ultrasensitive residue analysis method for glycopeptide antibiotics // J. Chromatogr. A. 2018. V. 1556. P. 10.
Lei X., Cui J., Wang S., Huang T., Wu X. Preparation of a biomimetic ionic liquids hybrid polyphosphorylcholine monolithic column for the high efficient capillary microextraction of glycopeptide antibiotics // J. Chromatogr. A. 2020. V. 1623. Article 461175.
Zhou H., Liu R., Chen Q., Zheng X., Qiu J., Ding T., He L. Surface molecularly imprinted solid-phase extraction for the determination of vancomycin and norvancomycin in milk by liquid chromatography coupled to tandem mass spectrometry // Food Chem. 2022. V. 369. Article 130886.
Yang Y.-J., Liu X.-W., Li B., Li S.-H., Kong X.-J., Qin Z., Li J.-Y. Simultaneous determination of diaveridine, trimethoprim and ormetoprim in feed using high performance liquid chromatography tandem mass spectrometry // Food Chem. 2016. V. 212. P. 358.
Nász S., Károlyné E.M. Determination of coccidiostats in milk products by LC–MS and its application to a fermentation experiment // Chromatographia. 2012. V. 75. № 11–12. P. 645.
Clarke L., Moloney M., O’Mahony J., O’Kennedy R., Danaher M. Determination of 20 coccidiostats in milk, duck muscle and non-avian muscle tissue using UHPLC-MS/MS // Food Addit. Contam. Part A. 2013. V. 30. № 6. P. 958.
Jing H., Ge S., Lian-Feng A., Jian-Chen L. Determination of six polyether antibiotic residues in foods of animal origin by solid phase extraction combined with liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1017. P. 187.
Dasenaki M.E., Thomaidis N.S. Multi-residue methodology for the determination of 16 coccidiostats in animal tissues and eggs by hydrophilic interaction liquid chromatography–tandem mass spectrometry // Food Chem. 2019. V. 275. P. 668.
González-Rubio S., García-Gómez D., Ballesteros-Gómez A., Rubio S. A new sample treatment strategy based on simultaneous supramolecular solvent and dispersive solid-phase extraction for the determination of ionophore coccidiostats in all legislated foodstuffs // Food Chem. 2020. V. 326. Article 126987.
Negarian M., Mohammadinejad A., Mohajeri S.A. Preparation, evaluation and application of core-shell molecularly imprinted particles as the sorbent in solid-phase extraction and analysis of lincomycin residue in pasteurized milk // Food Chem. 2019. V. 288. P. 29.
Paseiro-Cerrato R., Otero-Pazos P., Rodríguez-Bernaldo de Quirós A., Sendón R., Angulo I., Paseiro-Losada P. Rapid method to determine natamycin by HPLC-DAD in food samples for compliance with EU food legislation // Food Control. 2013. V. 33. № 1. P. 262.
Zhou Y., Zhou T., Jin H., Jing T., Song B., Zhou Y., Mei S., Lee Y.I. Rapid and selective extraction of multiple macrolide antibiotics in foodstuff samples based on magnetic molecularly imprinted polymers // Talanta. 2015. V. 137. P. 1.
Zhou T., Yang H., Jin Z., Liu Q., Song X., He L., Fang B., Meng C. Determination of azithromycin residue in pork using a molecularly imprinted monolithic microcolumn coupled to liquid chromatography with tandem mass spectrometry // J. Sep. Sci. 2016. V. 39. P. 1339.
Ji S., Li T., Yang W., Shu C., Li D., Wang Y., Ding L. A hollow porous molecularly imprinted polymer as a sorbent for the extraction of 7 macrolide antibiotics prior to their determination by HPLC-MS/MS // Mikrochim Acta. 2018. V. 185. P. 203.
Du J., Li X., Tian L., Li J., Wang C., Ye D., Zhao L., Liu S., Xu J., Xia X. Determination of macrolides in animal tissues and egg by multi-walled carbon nanotube-based dispersive solid-phase extraction and ultra-high performance liquid chromatography–tandem mass spectrometry // Food Chem. 2021. V. 365. Article 130502.
Granja R.H.M.M., Nino A.M.M., Reche K.V.G., Giannotti F.M., Lima A.C., Wanschel A.C.B.A., Salerno A.G. Determination and confirmation of metronidazole, dimetridazole, ronidazole and their metabolites in bovine muscle by LC-MS/MS // Food Addit. Contam. Part A: Chem. 2013. V. 30. P. 970.
Mitrowska K., Antczak M., Posyniak A. Confirmatory method for the determination of nitroimidazoles in milk by liquid chromatography–tandem mass spectrometry // Bull. Vet. Inst. Pulawy. 2014. V. 58. P. 581.
Hernández-Mesa M., Moreno-González D., Cruces-Blanco C., García-Campaña A.M. Evaluation of a selective approach for the determination of 5-nitroimidazoles in aquaculture products by capillary liquid chromatography using molecularly imprinted solid-phase extraction // Food Anal. Methods. 2017. V. 10. P. 3647.
Chen F., Li S., Peng J., Wang X., Peng H., Chen Y., He Y. Study on simultaneous determination of three nitroimidazole residues in honey by high performance liquid chromatography–resonance Rayleigh scattering spectra // Microchem. J. 2018. V. 141. P. 423.
Valera-Tarifa N.M., Plaza-Bolaños P., Romero-González R., Martínez-Vidal J.L., Garrido-Frenich A. Determination of nitrofuran metabolites in seafood by ultra-high performance liquid chromatography coupled to triple quadrupole tandem mass spectrometry // J. Food Compost. Anal. 2013. V. 30. № 2. P. 86.
Tang T., Wei F., Wang X., Ma Y., Song Y., Ma Y., Song Q., Xu G., Cen Y., Hu Q. Determination of semicarbazide in fish by molecularly imprinted stir bar sorptive extraction coupled with high performance liquid chromatography // J. Chromatogr. B. 2018. V. 1076. P. 8.
Wu S., Yang B., Yu H., Li Y. A rapid derivatization method for analyzing nitrofuran metabolites in fish using ultra-performance liquid chromatography−tandem mass spectrometry // Food Chem. 2020. V. 310. Article 125814.
Yuan G., Zhu Z., Yang P., Lu S., Liu H., Liu W., Liu G. Simultaneous determination of eight nitrofuran residues in shellfish and fish using ultra-high performance liquid chromatography–tandem mass spectrometry // J. Food Compost. Anal. 2010. V. 92. Article 103540.
Melekhin A.O., Tolmacheva V.V., Shubina E.G., Dmitrienko S.G., Apyari V.V., Grudev A.I. Determination of nitrofuran metabolites in honey using a new derivatization reagent, magnetic solid-phase extraction and LC–MS/MS // Talanta. 2021. V. 230. Article 122310.
Guichard P., Laurentie M., Hurtaud-Pessel D., Verdon E. Confirmation of five nitrofuran metabolites including nifursol metabolite in meat and aquaculture products by liquid chromatography-tandem mass spectrometry: Validation according to European Union Decision 2002/657/EC // Food Chem. 2021. V. 342. Article 128389.
Boison J.O., Lee S., Matus J. A multi-residue method for the determination of seven polypeptide drug residues in chicken muscle tissues by LC-MS/MS // Anal. Bioanal. Chem. 2015. V. 407. № 14. P. 4065.
Tao Y., Xie S., Zhu Y., Chen D., Pan Y., Wang X., Liu Z., Huang L., Peng D., Yuan Z. Analysis of major components of bacitracin, colistin and virginiamycin in feed using matrix solid-phase dispersion extraction by liquid chromatography–electrospray ionization tandem mass spectrometry // J. Chromatogr. Sci. 2018. V. 56. № 3. P. 285.
Kumar H., Kumar D., Nepovimova E., Oulkar D., Kumar A., Azad R.M.R., Budakoti S.K., Upadhyay N.K., Verma R., Kuča K. Determination of colistin B in chicken muscle and egg using ultra-high-performance liquid chromatography–tandem mass spectrometry // Int. J. Environ. Res. Public Health. 2021. V. 18. Article 2651.
Guo H., Liu Y., Fang B., Ding H., Wang M., He L. Determination of valnemulin residues in porcine tissues by molecularly imprinted solid-phase extraction coupling with high-performance liquid chromatography // J. Liq. Chromatogr. Relat. Technol. 2014. V. 37. P. 1597.
Huang C., Guo B., Wang X., Li J., Zhu W., Chen B., Ouyang S., Yao S. A generic approach for expanding homolog-targeted residue screening of sulfonamides using a fast matrix separation and class-specific fragmentation-dependent acquisition with a hybrid quadrupole-linear ion trap mass spectrometer // Anal. Chim. Acta. 2012. V. 737. P. 83.
Zhao Y.-G., Zhou L.-X., Pan S.-D., Zhan P.-P., Chen X.-H., Jin M.-C. Fast determination of 22 sulfonamides from chicken breast muscle using core–shell nanoring amino-functionalized superparamagnetic molecularly imprinted polymer followed by liquid chromatography-tandem mass spectrometry // J. Chromatogr. A. 2014. V. 1345. P. 17.
Li Y., Li Z., Wang W., Zhong S., Chen J., Wang A.J. Miniaturization of self-assembled solid phase extraction based on graphene oxide/chitosan coupled with liquid chromatography for the determination of sulfonamide residues in egg and honey // J. Chromatogr. A. 2016. V. 1447. P. 17.
Guillén I., Guardiola L., Almela L., Núñez-Delicado E., Gabaldón J.A. Simultaneous determination of nine sulphonamides by LC-MS for routine control of raw honey samples // Food Anal. Methods. 2017. V. 10. P. 1430.
Jia W., Shi L., Chu X. Untargeted screening of sulfonamides and their metabolites in salmon using liquid chromatography coupled to quadrupole orbitrap mass spectrometry // Food Chem. 2018. V. 239. P. 427.
Li Y., Zhu N., Chen T., Ma Y., Li Q. A green cyclodextrin metal-organic framework as solid-phase extraction medium for enrichment of sulfonamides before their HPLC determination // Microchem. J. 2018. V. 138. P. 401.
Petrarca M.H., Braga P.A.C., Reyes F.G.R., Bragotto A.P.A. Hydrophilic interaction liquid chromatography coupled to quadrupole time-of-flight mass spectrometry as a potential combination for the determination of sulfonamide residues in complex infant formula matrices // J. Chromatogr. A. 2020. V. 1633. Article 461606.
Liu X., Tong Y., Zhang L. Tailorable yolk-shell Fe3O4@graphitic carbon submicroboxes as efficient extraction materials for highly sensitive determination of trace sulfonamides in food samples // Food Chem. 2020. V. 303. Article 125369.
Li T., Wang C., Xu Z., Chakraborty A. A coupled method of on-line solid phase extraction with the UHPLC‒MS/MS for detection of sulfonamides antibiotics residues in aquaculture // Chemosphere. 2020. V. 254. Article 126765.
Wu J., Li Y., Li W., Gong Z., Huang X. Preparation of a novel monolith-based adsorbent for solid-phase microextraction of sulfonamides in complex samples prior to HPLC-MS/MS analysis // Anal. Chim. Acta. 2020. V. 1118. P. 9.
Jullakan S., Bunkoed O. A nanocomposite adsorbent of metallic copper, polypyrrole, halloysite nanotubes and magnetite nanoparticles for the extraction and enrichment of sulfonamides in milk // J. Chromatogr. B. 2021. V. 1180. Article 122900.
Dil E.A., Ghaedi M., Mehrabi F., Tayebi L. Highly selective magnetic dual template molecularly imprinted polymer for simultaneous enrichment of sulfadiazine and sulfathiazole from milk samples based on syringe–to–syringe magnetic solid–phase microextraction // Talanta. 2021. V. 232. Article 122449.
Tarapoulouzi M., Papachrysostomou C., Constantinou S., Kanari P., Hadjigeorgiou M. Determinative and confirmatory method for residues of tetracyclines in honey by LC-MS/MS // Food Addit. Contam. Part A. 2013. V. 30. № 10. P. 1728.
Liu Y., Yang H., Yang S., Hu Q., Cheng H., Liu H., Qiu Y. High-performance liquid chromatography using pressurized liquid extraction for the determination of seven tetracyclines in egg, fish and shrimp // J. Chromatogr. B. 2013. V. 917–918. P. 11.
Moreno-González D., García-Campaña A.M. Salting-out assisted liquid–liquid extraction coupled to ultra-high performance liquid chromatography–tandem mass spectrometry for the determination of tetracycline residues in infant foods // Food Chem. 2017. V. 221. P. 1763.
Xu Y., Tang Y., Zhao Y., Gao R., Zhang J., Fu D., Li Z., Li H., Tang X. Bifunctional monomer magnetic imprinted nanomaterials for selective separation of tetracyclines directly from milk samples // J. Colloid Interface Sci. 2018. V. 515. P. 18.
Saridal K., Ulusoy H.İ. A simple methodology based on cloud point extraction prior to HPLC-PDA analysis for tetracycline residues in food samples // Microchem. J. 2019. V. 150. Article 104170.
Zhao W., Zuo H., Guo Y., Liu K., Wang S., He L., Jiang X., Xiang G., Zhang S. Porous covalent triazine-terphenyl polymer as hydrophilic–lipophilic balanced sorbent for solid phase extraction of tetracyclines in animal derived foods // Talanta. 2019. V. 201. P. 426.
Alanazi F., Almugbel R., Maher H.M., Alodaib F.M., Alzoman N.Z. Determination of tetracycline, oxytetracycline and chlortetracycline residues in seafood products of Saudi Arabia using high performance liquid chromatography–photo diode array detection // Saudi Pharm. J. 2021.V. 29. № 6. P. 566.
Pang Y.-H., Lv Z.-Y., Sun J.-C., Yang C., Shen X.-F. Collaborative compounding of metal–organic frameworks for dispersive solid-phase extraction HPLC–MS/MS determination of tetracyclines in honey // Food Chem. 2021. V. 355. Article 129411.
Merou A., Kaklamanos G., Theodoridis G. Determination of carbadox and metabolites of carbadox and olaquindox in muscle tissue using high performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2012. V. 881–882. P. 90.
Li Y., Liu K., Beier R.C., Cao X., Shen J., Zhang S. Simultaneous determination of mequindox, quinocetone, and their major metabolites in chicken and pork by UPLC–MS/MS // Food Chem. 2014. V. 160. P. 171.
Lutero W., Dibai S., de Alkimin Filho J.F., da Silva Oliveira F.A., de Assis D.C.S., Lara L.J.C., de Figueiredo T.C., de Vasconcelos Cançado S. HPLC-MS/MS method validation for the detection of carbadox and olaquindox in poultry and swine feedingstuffs // Talanta. 2015. V. 144. P. 740.
Peng D., Zhang X., Wang Y., Pan Y., Liu Z., Chen D., Sheng F., Yuan Z. An immunoaffinity column for the selective purification of 3-methyl-quinoxaline-2-carboxylic acid from swine tissues and its determination by high-performance liquid chromatography with ultraviolet detection and a colloidal gold-based immunochromatographic assay // Food Chem. 2017. V. 237. P. 290.
Li P., Zhang X., Zhang J., Yan Z., Zhang S., Chen S., Fang Y. A sensitive and selective immunoaffinity column clean up coupled to UPLC-MS/MS for determination of trace methyl-3-quinoxaline-2-carboxylic acid in animal tissues // J. Chromatogr. B. 2018. V. 1074–1075. P. 39.
Borras S., Gabriel J., Companyo R., Prat M.-D. Analysis of fluoroquinolones in animal feeds by liquid chromatography with fluorescence detection // J. Sep. Sci. 2012. V. 35. № 1. P. 1.
Ruiz-Viceo J.A., Rosales-Conrado N., Guillén-Casla V., Pérez-Arribas L.V., León-González M.E., Polo-Díez L.M. Fluoroquinolone antibiotic determination in bovine milk using capillary liquid chromatography with diode array and mass spectrometry detection // J. Food Compost. Anal. 2012. V. 28. № 2. P. 99.
Evaggelopoulou E.N., Samanidou V.F. HPLC confirmatory method development for the determination of seven quinolones in salmon tissue (Salmo salar L.) validated according to the European Union Decision 2002/657/EC // Food Chem. 2013. V. 136. № 2. P. 479.
Stoilova N.A., Surleva A.R., Stoev G. Simultaneous determination of nine quinolones in food by liquid chromatography with fluorescence detection // Food Anal. Methods. 2013. V. 6. № 3. P. 803.
Barreto F., Ribeiro C.B.D., Hoff R.B., Costa T.D. Development and validation of a high-throughput method for determination of nine fluoroquinolones residues in muscle of different animal species by liquid chromatography coupled to tandem mass spectrometry with low temperature clean up // J. Chromatogr. A. 2017. V. 1521. P. 131.
Zhai H., Liang G., Guo X., Chen Z., Yu J., Lin H., Zhou Q. Novel coordination imprinted polymer monolithic column applied to the solid-phase extraction of flumequine from fish samples // J. Chromatogr. B. 2019. V. 1118–1119. P. 55.
Kukusamude C., Burakham R., Chailapakul O., Srijaranai S. High performance liquid chromatography for the simultaneous analysis of penicillin residues in beef and milk using ion-paired extraction and binary water–acetonitrile mixture // Talanta. 2012. V. 92. P. 38.
Liu X., Yu Y., Zhao M., Zhang H., Li Y., Duan G. Solid phase extraction using magnetic core mesoporous shell microspheres with C18-modified interior pore-walls for residue analysis of cephalosporins in milk by LC–MS/MS // Food Chem. 2014. V. 150. P. 206.
Van Royen G., Dubruel P., Van Weyenberg S., Daeseleire E. Evaluation and validation of the use of a molecularly imprinted polymer coupled to LC–MS for benzylpenicillin determination in meat samples // J. Chromatogr. B. 2015. V. 1025. P. 48.
Li W., Shen H., Hong Y., Zhang Y., Yuan F., Zhang F. Simultaneous determination of 22 cephalosporins drug residues in pork muscle using liquid chromatography–tandem mass spectrometry // J. Chromatogr. B. 2016. V. 1022. P. 298.
Huang Z., Pan X.-D., Huang B.-F., Xu J.-J., Wang M.-L., Ren Y.-P. Determination of 15 β-lactam antibiotics in pork muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry // Food Control. 2016. V. 66. P. 145.
Moreno-González D., Rodríguez-Ramírez R., del Olmo-Iruela M., García-Campaña A.M. Validation of a new method based on salting-out assisted liquid-liquid extraction and UHPLC-MS/MS for the determination of betalactam antibiotics in infant dairy products // Talanta. 2017. V. 167. P. 493.
Du W., Sun M., Guo P., Chang C., Fu Q. Molecularly imprinted membrane extraction combined with high-performance liquid chromatography for selective analysis of cloxacillin from shrimp samples // Food Chem. 2018. V. 259. P. 73.
Sahebi H., Konoz E., Ezabadi A., Niazi A., Ahmadi S.H. Simultaneous determination of five penicillins in milk using a new ionic liquid-modified magnetic nanoparticle based dispersive micro-solid phase extraction followed by ultra-performance liquid chromatography-tandem mass spectrometry// Microchem. J. 2020. V. 154. Article 104605.
Cheng G., Zhao J., Wang X., Yang C., Li S., Lu T., Li X., Wang X., Zhu G. A highly sensitive and selective method for the determination of ceftiofur sodium in milk and animal-origin food based on molecularly imprinted solid-phase extraction coupled with HPLC-UV // Food Chem. 2021. V. 347. Article 129013.
Ansari S., Karimi M. Novel developments and trends of analytical methods for drug analysis in biological and environmental samples by molecularly imprinted polymers // Trends Anal. Chem. 2017. V. 89. P. 146.
Valsecchi S., Polesello S., Mazzoni M., Rusconi M., Petrovic M. On-line sample extraction and purification for the LC–MS determination of emerging contaminants in environmental samples // Trends Environ. Anal. Chem. 2015. V. 8. P. 27.
Socas-Rodríguez B., González-Sálamo J., Herrera-Herrera A.V., Hernández-Borges J., Rodríguez-Delgado M.Á. Chapter eleven – Recent advances and developments in the QuEChERS method / Eds. Ibáñez E., Cifuentes A. // Compr. Anal. Chem. 2017. V. 76. P. 319.
Perestrelo R., Silva P., Porto-Figueira P., Pereira J.A.M., Silva C., Medina S., Câmara J.S. QuEChERS – Fundamentals, relevant improvements, applications and future trends // Anal. Chim. Acta. 2019. V. 1070. P. 1.
Волкова Н.М., Амелин В.Г., Третьяков А.В., Абраменкова О.И., Тимофеев А.А. Пробоподготовка QuEChERS при одновременном определении остаточных количеств антибиотиков хинолонового ряда и хлорамфинекола в пищевых продуктах методом ВЭЖХ-ДМД // Изв. Сарат. ун-та Новая сер. Сер. Химия. Биология. Экология. 2013. № 2. С. 30.
Bittencourt M.S., Martinsa M.T., de Albuquerque F.G.S., Barreto F., Hoff R. High-throughput multiclass screening method for antibiotic residue analysis in meat us-ing liquid chromatography-tandem mass spectrometry: A novel minimum sample preparation procedure // Food Addit. Contam. 2012. V. 29. P. 508.
Lombardo-Agui M., Garcia-Campaña A.M., Gamiz-Gracia L., Cruces-Blanco C. Determination of quinolones of veterinary use in bee products by ultra-high performance liquid chromatography–tandem mass spectrometry using a QuEChERS extraction procedure // Talanta. 2012. V. 93. P. 193.
Pereira-Lopes R., Cazorla-Reyes R., Romero-González R., Martínez-Vidal J.L., Garrido-Frenich A. Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra high performance liquid chromatography coupled to tandem mass spectrometry // J. Chromatogr. B. 2012. V. 895–896. P. 39.
Wang J., Leung D. The challenges of developing a generic extraction procedure to analyze multi-class veterinary drug residues in milk and honey using ultra-high pressure liquid chromatography quadrupole time-of-flight mass spectrometry // Drug Test. Anal. 2012. V. 4. P. 103.
Lopes R.P., Reyes R.C., Romero-González R., Frenich A.G., Vidal J.L.M. Development and validation of a multiclass method for the determination of veterinary drug residues in chicken by ultra-high performance liquid chromatography–tandem mass spectrometry // Talanta. 2012. V. 89. P. 201.
Zhang G.-J., Fang B.-H., Liu Y.-H., Wang X.-F., Xu L.-X., Zhang Y.-P., He L.-M. Development of a multi-residue method for fast screening and confirmation of 20 prohibited veterinary drugs in feedstuffs by liquid chromatography tandem mass spectrometry // J. Chromatogr. B. 2013. V. 936. P. 10.
Kang J., Fan C.-L., Chang Q.-Y., Bu M.-N., Zhao Z.-Y., Wang W., Pang G.-F. Simultaneous determination of multi-class veterinary drug residues in different muscle tissues by modified QuEChERS combined with HPLC-MS/MS // Anal. Methods. 2014. V. 6. Article 6285.
Abdallah H., Arnaudguilhem C., Jaber F., Lobinski R. Multiresidue analysis of 22 sulfonamides and their metabolites in animal tissues using quick, easy, cheap, effective, rugged, and safe extraction and high resolution mass spectrometry (hybrid linear ion trap-Orbitrap) // J. Chromatogr. A. 2014. V. 1355. P. 61.
Cao Y., Kang J., Chang Q., Hu X., Wang Z., Fan C., Wang M. Multi-residue determination of veterinary drugs in cheese by QuEChERS and liquid chromatography−tandem mass spectrometry // Se Pu. 2015. V. 33. P. 132.
Zhang Y., Li X., Liu X., Zhang J., Cao Y., Shi Z., Sun H. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography−quadrupole time-of-flight mass spectrometry // J. Dairy Sci. 2015. V. 98. P. 8433.
Amelin V., Korotkov A., Andoralov A. Identification and determination of 492 contaminants of different classes in food and feed by high-resolution mass spectrometry using the standard addition method // J. AOAC Int. 2016. V. 99. P. 1600.
Zhang Y., Liu X., Li X., Zhang J., Cao Y., Su M., Shi Z., Sun H. Rapid screening and quantification of multi-class multi-residue veterinary drugs in royal jelly by ultra performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry // Food Control. 2016. V. 60. P. 667.
Shendy A.H., Al-Ghobashy M.A., Gad Alla S.A., Lotfy H.M. Development and validation of a modified QuEChERS protocol coupled to LC-MS/MS for simultaneous determination of multi-class antibiotic residues in honey // Food Chem. 2016. V. 190. P. 982.
Huertas-Pérez J.F., Arroyo-Manzanares N., Havlíková L., Gámiz-Gracia L., Solich P., García-Campaña A.M. Method optimization and validation for the determination of eight sulfonamides in chicken muscle and eggs by modified QuEChERS and liquid chromatography with fluorescence detection // J. Pharm. Biomed. Anal. 2016. V. 124. P. 261.
Serra-Compte A., Álvarez-Muñoz D., Rodríguez-Mozaz S., Barceló D. Multi-residue method for the determination of antibiotics and some of their metabolites in seafood // Food Chem. Toxicol. 2017. V. 104. P. 3.
Silva G.R., Lima J.A., Souza L.F., Santos F.A., Lana M.A.G., Assis D.C.S., Cançado S.V. Multiresidue method for identification and quantification of avermectins, benzimidazoles and nitroimidazoles residues in bovine muscle tissue by ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) using a QuEChERS approach // Talanta. 2017. V. 171. P. 307.
Konak Ü.İ., Certel M., Şık B., Tongur T. Development of an analysis method for determination of sulfonamides and their five acetylated metabolites in baby foods by ultra-high performance liquid chromatography coupled to high-resolution mass spectrometry (orbitrap-MS) // J. Chromatogr. B. 2017. V. 1057. P. 81.
Grande-Martínez Á., Moreno-González D., Arrebola-Liébanas F.J., Garrido-Frenich A., García-Campaña A.M. Optimization of a modified QuEChERS method for the determination of tetracyclines in fish muscle by UHPLC–MS/MS // J. Pharm. Biomed. Anal. 2018. V. 155. P. 27.
De Oliveira Arias J.L., Schneider A., Batista-Andrade J.A., Vieira A.A., Caldas S.S., Primel E.G. Chitosan from shrimp shells: A renewable sorbent applied to the clean-up step of the QuEChERS method in order to determine multi-residues of veterinary drugs in different types of milk // Food Chem. 2018. V. 240. P. 1243.
Xu X., Xu X., Han M., Qiu S., Hou X. Development of a modified QuEChERS method based on magnetic multiwalled carbon nanotubes for the simultaneous determination of veterinary drugs, pesticides and mycotoxins in eggs by UPLC-MS/MS // Food Chem. 2019. V. 276. P. 419.
Jadhav M.R., Pudale A., Raut P., Utture S., Shabeer T.P.A., Banerjee K. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC–MS/MS // Food Chem. 2019. V. 272. P. 292.
Dinh Q.T., Munoz G., Duy S.V., Do D.T., Bayen S., Sauvé S. Analysis of sulfonamides, fluoroquinolones, tetracyclines, triphenylmethane dyes and other veterinary drug residues in cultured and wild seafood sold in Montreal, Canada // J. Food Compost. Anal. 2020. V. 94. Article 103630.
Izzo L., Rodríguez-Carrasco Y., Tolosa J., Graziani G., Gaspari A., Ritieni A. Target analysis and retrospective screening of mycotoxins and pharmacologically active substances in milk using an ultra-high-performance liquid chromatography/high-resolution mass spectrometry approach // J. Dairy Sci. 2020. V. 103. № 2. P. 1250.
Castilla-Fernández D., Moreno-González D., Bouza M., Saez-Gómez A., Ballesteros E., García-Reyes J.F., Molina-Díaz A. Assessment of a specific sample cleanup for the multiresidue determination of veterinary drugs and pesticides in salmon using liquid chromatography/tandem mass spectrometry // Food Control. 2021. V. 130. Article 108311.
Shen Q., Zhu X., Zhao Q., Li S., Wang Y., Xue J., Wang P. QuEChERS and 96-well plate solid phase extraction for determination of vancomycin and norvancomycin in fish meat by UPLC–MS/MS // Food Chem. 2021. V. 342. Article 128326.
Ji B., Zhao W., Xu X., Han Y., Jie M., Xu G., Bai Y. Development of a modified quick, easy, cheap, effective, rugged, and safe method based on melamine sponge for multi-residue analysis of veterinary drugs in milks by ultra-performance liquid chromatography tandem mass spectrometry // J. Chromatogr. A. 2021. V. 1651. Article 462333.
Gissawong N., Boonchiangma S., Mukdasai S., Srijaranai S. Vesicular supramolecular solvent-based microextraction followed by high performance liquid chromatographic analysis of tetracyclines // Talanta. 2019. V. 200. P. 203.
González-Rubio S., García-Gómez D., Ballesteros-Gómez A., Rubio S. A new sample treatment strategy based on simultaneous supramolecular solvent and dispersive solid-phase extraction for the determination of ionophore coccidiostats in all legislated foodstuffs // Food Chem. 2020. V. 326. 1 Article 26987.
Yao T., Yao S. Magnetic ionic liquid aqueous two-phase system coupled with high performance liquid chromatography: A rapid approach for determination of chloramphenicol in water environment // J. Chromatogr. A. 2017. V. 1481. P. 12.
Shahi M., Javadi A., Mogaddam M.R.A., Mirzaei H., Nemati M. Extraction of some antibiotics from propolis samples using homogenous liquid–liquid extraction coupled with deep eutectic solvent–based hollow fibre protected preconcentration // Int. J. Environ. Anal. Chem. 2020. P. 1.
Saei A., Javadi A., Mogaddam M.R.A., Mirzaei H., Nemati M. Development of homogeneous liquid–liquid extraction combined with dispersive liquid–liquid microextraction based on solidification of floating droplets of a ternary component deep eutectic solvent for the analysis of antibiotic residues in sausage samples prior to ion mobility spectrometry // Anal. Methods. 2020. V. 12. № 34. P. 4220.
Saei A., Javadi A., Mogaddam M.R.A., Mirzaei H., Nemati M. Determination of three antibiotic residues in hamburger and cow liver samples using deep eutectic solvents-based pretreatment method coupled with ion mobility spectrometry // Int. J. Environ. Anal. Chem. 2020. P. 1.
Wu Q., Shabbir M.A.B., Peng D., Yuan Z., Wang Y. Microbiological inhibition-based method for screening and identifying of antibiotic residues in milk, chicken egg and honey // Food Chem. 2021. V. 363. Article 130074.
Xiao X., Hu S., Lai X., Peng J., Lai W. Developmental trend of immunoassays for monitoring hazards in food samples: A review // Trends Food Sci. Technol. 2021. V. 111. P. 68.
Anyakudo F., Adams E., Van Schepdael A. Analysis of amikacin, gentamicin and tobramycin by thin layer chromatography-flame ionization detection // Microchem. J. 2020. V. 157. Article 105032.
Semail N.-F., Keyon A.S.A., Saad B., Kamaruzaman S., Zain N.N.M., Lim V., Miskam M., Abdullah W.N.W., Yahaya N., Chen D.D.Y. Simultaneous preconcentration and determination of sulfonamide antibiotics in milk and yoghurt by dynamic pH junction focusing coupled with capillary electrophoresis // Talanta. 2022. V. 236. Article 122833.
Guidi L.R., Tette P.A.S., Fernandes C., Silva L.H.M., Gloria M.B.A. Advances on the chromatographic determination of amphenicols in food // Talanta. 2017. V. 162. P. 324.
Tan L., Deng F., Luo X., Pan X., Zhang L., Marina M.L., Jiang Z. Glycosyl imprinted mesoporous microspheres for the determination of glycopeptide antibiotics using ultra-high performance liquid chromatography coupled with tandem mass spectrometry // J. Chromatogr. A. 2021. V. 1659. Article 462630.
Vardanyan R., Hruby V. Chapter 30 – Antibiotics / Eds. Vardanyan R., Hruby V. / Synthesis of Best-Seller Drugs. Academic Press, 2016. 868 p.
Jank L., Martins M.T., Arsand J.B., Motta T.M.C., Hoff R.B., Barreto F., Pizzolato T.M. High-throughput method for macrolides and lincosamides antibiotics residues analysis in milk and muscle using a simple liquid-liquid extraction technique and liquid chromatography-electrospray-tandem mass spectrometry analysis (LC–MS/MS) // Talanta. 2015. V. 144. P. 686.
Kwon J.-W. Semicarbazide: Natural occurrence and uncertain evidence of its formation from food processing // Food Control. 2017. V. 72. Part B. P. 268.
Pang G.-F. Analytical Methods for Food Safety by Mass Spectrometry / Ed. Pang G.-F. Academic Press, 2018. 880 p.
Manimekalai M., Rawson A., Sengar A.S., Kumar K.S. Development, optimization, and validation of methods for quantification of veterinary drug residues in complex food matrices using liquid-chromatography – A review // Food Anal. Methods. 2019. V. 12. P. 1823.
Dmitrienko S.G., Kochuk E.V., Apyari V.V., Tolmacheva V.V., Zolotov Y.A. Recent advances in sample preparation techniques and methods of sulfonamides detection – A review // Anal. Chim. Acta. 2014. V. 850. P. 6.
Regal P., Lamas A., Franco C.M., Cepeda A. Veterinary drugs: Progress in multiresidue technique / Eds. Melton L., Shahidi F., Varelis P. / Encyclopedia of Food Chemistry. Academic Press, 2019. 2194 p.
Frenich A.G., Romero-González R., del Mar Aguilera-Luiz M. Comprehensive analysis of toxics (pesticides, veterinary drugs and mycotoxins) in food by UHPLC-MS // Trends Anal. Chem. 2014. V. 63. P. 158.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии