

Журнал аналитической химии, 2022, T. 77, № 2, стр. 117-128
Ионселективные электроды на основе ионофоров: исследования механизма работы и возможности аналитического применения в условиях ненулевого тока
А. В. Бондарь a, В. М. Керестень a, К. Н. Михельсон a, *
a Институт химии Санкт-Петербургского государственного университетa
198504 Санкт-Петербург, Старый Петергоф, Университетский просп., 26, Россия
* E-mail: konst@km3241.spb.edu
Поступила в редакцию 02.06.2021
После доработки 21.07.2021
Принята к публикации 23.08.2021
- EDN: DNOZJM
- DOI: 10.31857/S0044450222020049
Аннотация
Кратко описаны данные фундаментальных исследований механизма потенциометрического отклика ионселективных электродов (ИСЭ) на основе ионофоров при ненулевом токе, а также данные об аналитических возможностях ИСЭ в режимах вольтамперометрии и хроноамперометрии/кулонометрии при постоянном потенциале, в частности определении содержания ионов K+ в сыворотке крови с чувствительностью 0.1%. Особое внимание уделено основам вольтамперометрии и хроноамперометрии/кулонометрии с ИСЭ на основе ионофоров и ответу на вопрос – чем и почему эти методы отличаются от классической вольтамперометрии и кулонометрии.
Ионселективные электроды (ИСЭ) с полимерными пластифицированными мембранами, содержащими нейтральные и заряженные ионофоры, уже в течение нескольких десятилетий используются в качестве потенциометрических сенсоров для определения широкого круга аналитов [1–8]. До сих пор измерения с ИСЭ в режиме ненулевого тока были главным образом направлены на фундаментальные исследования механизма отклика электродов и источника его селективности [9–15] либо на улучшение пределов функционирования [16–18] и/или чувствительности ИСЭ, особенно для определения полиионов [19–23]. До 2010 г. аналитические применения ИСЭ в токовом режиме были редкостью. Однако в последние годы ИСЭ в режимах вольтамперометрии, амперометрии и кулонометрии все чаще применяют для практических целей.
В настоящем обзоре мы сосредоточились на измерениях с ИСЭ c пластифицированными полимерными мембранами в режиме ненулевого тока. Не обсуждаются данные для чисто жидких систем – границ раздела несмешивающихся растворов электролитов, так как последние составляют отдельный раздел электрохимии (см. обзор [24]). Данный обзор посвящен краткому описанию исследований механизма работы ИСЭ, обсуждению применений ИСЭ в вольтамперометрическом, хроноамперометрическом и кулонометрическом режимах.
Исследования механизма потенциометрического отклика ионселективных с помощью токовых измерений. Большинство теоретических описаний мембранного потенциала и селективности ИСЭ использует равновесный подход, согласно которому коэффициенты селективности к основным ионам в присутствии мешающих пропорциональны отношениям констант устойчивости соответствующих комплексов ионов с ионофором [2, 5, 6, 25–27]. Камманн [9–11] отстаивал кинетический подход, согласно которому коэффициенты селективности пропорциональны отношениям стандартных плотностей токов обмена конкурирующих ионов.
Константы устойчивости комплексов ион–ионофор могут быть измерены потенциометрически с использованием мембран, содержащих интересующий ионофор и хромоионофор [28, 29], или (и этот метод более надежен) путем измерений потенциалов составных мембран [15, 27, 30–35]. Метод составных мембран позволяет также оценивать константы ассоциации ионных пар и ионных тройников в мембранах ИСЭ [36, 37].
Плотности токов обмена на границе сенсорной фазы (мембраны) и водного раствора оценивали главным образом путем измерений электрохимического импеданса [9–15, 38, 39]. Измерения плотностей токов обмена на ИСЭ представляют трудности. Фактически, измеряют величины сопротивлений межфазного переноса заряда. Последние на несколько порядков меньше объемного сопротивления мембран, поскольку удельное сопротивление мембран ИСЭ составляет порядка 105 Ом ⋅ м. Это затрудняет регистрацию сопротивления переноса заряда, соединенного последовательно с объемом мембраны, поэтому имеется только небольшое количество сообщений с надежными данными о токах обмена на ИСЭ [11, 13, 15, 39]. Нам известна только одна работа, в которой константы устойчивости комплексов ион–ионофор и плотности токов обмена параллельно измеряли с одними и теми же мембранами [15]. Исследование было проведено с Li+-селективными электродами на основе ионофора Li+. Константы устойчивости комплексов измеряли методом составных мембран, а плотности токов обмена – методом импеданса. Интересно, что коэффициенты селективности к ионам Li+ в присутствии ионов K+ и Na+ почти одинаково хорошо коррелируют как с равновесными, так и с кинетическими данными.
Импульсные методы (особенно метод импульса тока: хронопотенциометрия) пригодны как для измерения объемного сопротивления мембран, так и для регистрации поляризационных кривых η как функции времени [40]. Эти кривые обычно представляют собой суперпозицию затухающей экспоненты и концентрационной поляризации, пропорциональной квадратному корню из времени:
(1)
${{\eta }} = i\left[ {R\left( {1--{{{\text{e}}}^{{--~\frac{t}{{RC}}}}}} \right) + N\sqrt t } \right],$(2)
$N = \frac{{2RT}}{{{{F}^{2}}\sqrt {{\pi }} }}\left( {\frac{1}{{{{с}_{ + }}\sqrt {{{D}_{ + }}} }} + \frac{1}{{{{с}_{--}}\sqrt {{{D}_{--}}} }}} \right).$Здесь η – поляризация, i – плотность поляризующего тока, t – время, R – поляризационное сопротивление, C – параллельная ему емкость. Иногда эти параметры можно отнести к сопротивлению межфазного переноса заряда и емкости двойного электрического слоя на границе мембрана/раствор, но утверждать это в общем виде сложно. Фактор N связан с концентрацией заряженных частиц c+, c– и их коэффициентами диффузии D+, D– в мембранах. С помощью таких измерений иногда удается оценить степень ассоциации комплексов ион–ионофор с ионообменником в мембране, а также коэффициенты диффузии частиц [32, 37, 41]. Однако надежность этих оценок невелика вследствие большого числа упрощений при выводе уравнения (1).
Бубака [42] предложил хронопотенциометрический тест для быстрой оценки качества твердоконтактных ИСЭ. Поляризационная кривая позволяет предсказать, насколько выбранная комбинация из (1) электронопроводящего субстрата (например, стеклоуглерода), (2) переходного слоя (чаще всего электропроводящего полимера или углеродного материала с развитой поверхностью) и (3) сенсорной мембраны перспективна в смысле стабильности потенциала ИСЭ во времени. Это исследование сочетало академическую значимость с практической.
Согласно современным взглядам на механизм потенциометрического отклика ИСЭ, в пределах выполнения нернстовской функции объемное сопротивление мембран должно быть постоянным. Очевидно, что необменная сорбция электролита из раствора в мембрану приводит к снижению ее объемного сопротивления, но в этом случае потенциалы ИСЭ отклоняются от закона Нернста. Замещение основных ионов мешающими может вызвать изменения объемного сопротивления мембраны, если это замещение влечет за собой изменение степени ассоциации мембранного электролита. Опять-таки, это замещение приводит к отклонениям от нернстовского отклика вплоть до полной потери чувствительности к основному иону. Существуют, однако, признаки непостоянства объемного сопротивления мембран ИСЭ в диапазоне их нернстовского отклика [15, 43]. Более того, вариацию объемного сопротивления K+-селективной мембраны использовали для разработки кондуктометрического сенсора на ионы K+ [43]. Систематическое исследование объемного сопротивления мембран ИСЭ, селективных к ионам Ca2+, K+, Cd2+ и ${\text{NO}}_{3}^{ - },$ выполненное с помощью хронопотенциометрических и импедансных измерений, выявило следующий тренд: объемное сопротивление исследованных мембран приблизительно постоянно в диапазоне концентраций соответствующего электролита от 0.1 до 0.001 M и существенно возрастает при дальнейшем разбавлении [44–47]. Более того, основная часть изменений (90–95% от всего эффекта) происходит в течение нескольких минут после изменения концентрации [44, 45, 47]. Обнаружено, что объемное сопротивление мембран коррелирует с поглощением ими воды из раствора, которое регистрировали гравиметрически [47, 48]. Известно, что вода в мембранах образует капельки размером от ~16 нм, которые распределены по мембране неравномерно: приповерхностные слои обогащены водой [49–52]. Впервые эти данные были получены с использованием добавок окрашенных солей [49, 50] или хромоионофоров [51, 52]. В работе [48] эти сведения были подтверждены с помощью ATR-FTIR-имаджинга профилей поливинилхлорида (ПВХ), пластификатора о-нитрофенилоктилового эфира (о-НФОЭ) и воды в Cd2+-селективных мембранах. Показано [48], что основная часть воды локализована в пределах 50 мкм от поверхности мембраны, а на глубине более 150 мкм вода не обнаружена. Высказано предположение о том, что капельки воды в мембранах затрудняют диффузию липофильных комплексов ион–ионофор и ионообменников, поскольку липофильные частицы локализованы в связной органической фазе мембраны и вынуждены обходить капельки воды, поэтому их средняя длина пути возрастает. Таким образом, концентрация частиц в органической фазе мембраны постоянна, обеспечивая нернстовский отклик ИСЭ, тогда как сопротивление возрастает по мере увеличения сорбции воды [48, 53].
Может показаться, что эти данные представляют только академический интерес. Однако показано [53], что поглощение воды и объемное сопротивление мембран ИСЭ на самом деле зависят от ионной силы раствора или от общей концентрации ионов, а не от концентрации иона, к которому селективна мембрана. Это открывает возможность определения не только активностей, но также и концентраций ионов одним и тем же сенсором. ИСЭ в потенциометрическом (бестоковом) режиме измерений дает сведения об активности аналита, а измерения его сопротивления позволяет оценить ионную силу образца. Когда последняя известна, активность может быть пересчитана в концентрацию с помощью, например, теории Дебая−Хюккеля.
Ионселективные электроды в вольтамперометрическом режиме: фундаментальные исследования и возможности аналитического применения. Вольтамперометрия с электрохимически активными аналитами на электродах, изготовленных из электронного проводника (благородного металла или материала на основе углерода с немодифицированной или модифицированной поверхностью), предполагает наличие окислительно-восстановительной реакции на границе между электродом и водным раствором [54]. Объемные сопротивления электрода и раствора не затрудняют измерения. Мембраны ИСЭ, как правило, обладают высоким сопротивлением, и проведение вольтамперометрических измерений с такими объектами намного труднее. Из-за высокого омического сопротивления ИСЭ с мембранами толщиной 0.2–0.7 мм не дают пиков в режиме циклической вольтамперометрии (ЦВ) и ведут себя как высокоомные резисторы. Хорваи [55] смог получить пики на кривых ЦВ с мембранами из ПВХ, пластифицированного о-НФОЭ и содержащего хлориды тетрагексиламмония или тетрафениларсония, когда эти катионы присутствовали в водной фазе. По-видимому, мембраны в этом случае обладали сравнительно небольшим сопротивлением из-за высокой концентрации хлоридов тетрагексиламмония или тетрафениларсония в фазе мембраны. Группа Петрухина [56] сообщала о вольтамперометрическом определении ионов ${\text{NH}}_{4}^{ + }$ с использованием различных каликсаренов в качестве нейтральных ионофоров и тетра(п-хлорфенил)бората калия (KClTФБ) в качестве ионообменника в жидком о-НФОЭ. Сопротивление сенсорной фазы было низким из-за отсутствия ПВХ, так как система состояла из жидких фаз.
Несколько других примеров успешных вольтамперометрических измерений с относительно толстыми мембранами относятся к фундаментальным электрохимическим исследованиям. Методом квадратно-волновой вольтамперометрии с ПВХ-мембранами Серна [57] измерил стандартные потенциалы переноса ионов диалкилимидазолия и алкилпиридиния из воды в о-НФОЭ. Амемия и соавт. [58] получили важные результаты, относящиеся к механизму переноса ионов через границу раздела мембран ИСЭ и водного раствора. Путем ЦВ-измерений показано, что ионы Ag+, K+, Ca2+, Ba2+ и Pb2+ образуют комплексы с соответствующими нейтральными ионофорами непосредственно на границе мембрана/раствор, в отличие от термодинамически эквивалентного двухстадийного механизма, основанного на простом переносе иона из воды в мембрану с последующим комплексообразованием в ее объеме.
Использование тонких мембран облегчает вольтамперометрические измерения с ИСЭ и делает их более пригодными для аналитических применений. Невысокая механическая прочность тонких мембран из пластифицированного ПВХ требует применения так называемой “твердоконтактной конструкции”, в которой сенсорная мембрана наносится на твердый субстрат методом полива или спин-коатинга, а внутренний водный раствор отсутствует [2, 5, 6].
Вольтамперометрическое определение клинически важных электроактивных веществ, особенно в реальных объектах, например в крови, часто оказывается проблематичным из-за отравления электродов. Линднер и соавт. [59] систематически исследовали электроды из стеклоуглерода, модифицированные мембранами из пластифицированного ПВХ, подобными мембранам, используемым в потенциометрических ИСЭ. Показано, что покрытие из ПВХ резко снижает отравление сенсоров. Кроме того, улучшилась селективность из-за различия в коэффициентах распределения гидрофобных аналитов и гидрофильных примесей между образцами и ПВХ-мембраной. В числе успешно определяемых аналитов (в скобках даны пределы обнаружения, ПО, мкМ) были амитриптилин (0.03), арипипразон (0.009), циталопрам (0.008), пропофол (0.03), рапамицин (0.003), сертралин (0.13) и зортресс (0.01). Помимо практических результатов представлено также детальное теоретическое рассмотрение факторов, влияющих на селективность и чувствительность измерений.
Группа Бонда [60] разработала вольтамперометрические ИСЭ на Na+, K+ и Ca2+ с мембранами толщиной менее 1 мкм, содержащими тетрацианохинодиметан в качестве электрохимически активного агента. В случае сенсора на катионы перенос целевого иона из водной фазы в мембрану, облегченный ионофором, происходит в ходе восстановления электрохимически активного агента, присутствующего в мембране, что обеспечивает сохранение электронейтральности. В той же роли электроактивного агента для обеспечения электронейтральности функционализированные катионные [6]хелицены использовали для определения ионов Na+ с ИСЭ, содержащим Na+-селективный ионофор [61], и для определения ${\text{CO}}_{3}^{{2 - }}$ с ИСЭ на основе ионофора, селективного к анионам ${\text{CO}}_{3}^{{2 - }}$ [62].
Присутствие электроактивных агентов в мембране ИСЭ может вызвать чувствительность к редокс-агентам в образцах, что, очевидно, нежелательно. Устранение этого недостатка основано на применении проводящих полимеров (ПП) в качестве переходных слоев между электронопроводящим субстратом и ионопроводящей сенсорной мембраной. В таких вольтамперометрических ИСЭ, когда ион переходит из водного раствора в мембрану или обратно, электронейтральность поддерживается соответствующим окислением или восстановлением ПП с переходом также иона допанта между слоями ПП и мембраны. В этом случае необходимости в присутствии электроактивных агентов в мембране нет. В этих исследованиях решающие результаты были достигнуты научными группами Амемии и Баккера.
Амемия и соавт. [63–67] опубликовали ряд статей, посвященных субнаномолярному определению ионов методом инверсионной вольтамперометрии с твердоконтактными ИСЭ с мембранами толщиной ~0.7 мкм. На золотые электроды, модифицированные перхлоратом поли-3-октилтиофена (ПОТ-ClO4), наносили мембрану, содержащую ПВХ, о-НФОЭ и тетра(пентафторфенилборат) тетрадодециламмония (ТДДАТФФБ). Методом инверсионной вольтамперометрии ион ${\text{ClO}}_{4}^{ - }$ определяли с ПО 0.2–5 нМ на фоне деионизованной воды, коммерческой бутилированной воды и водопроводной воды [63]. Золотые электроды, модифицированные ПОТ-ClO4 или поли-3,4-этилендиокситиофеном, допированным ТФФБ (ПЭДОТ-ТФФБ), и покрытые тонкими мембранами, содержащими ПВХ, о-НФОЭ и ТДДАТФФБ, использовали для определения тетрапропиламмония и гексафторарсената с ПО 0.1 нМ [64]. При определении аниона гексафторарсената перенос аналита из раствора образца в мембрану сопровождается окислением ПОТ, и анион ТФФБ– компенсирует положительный заряд ПОТ+. Напротив, при определении катиона тетрапропиламмония перенос аналита из раствора в мембрану приводит к дедопированию ПЭДОТ, и ТФФБ– компенсирует заряд тетрапропиламмония в мембранном слое. В двух указанных случаях электроны соответственно переходят из слоя ПП на металл, или обратно. Инверсионную вольтамперометрию использовали также для определения ионов K+ и ${\text{NH}}_{4}^{ + }$ на наномолярном уровне с применением золотых электродов, модифицированных ПЭДОТ-ТФФБ, покрытых мембраной, содержащей валиномицин, методом спин-коатинга [65]. В случае протамина (антидот гепарина) показано, что сигнал ИСЭ вплоть до 0.038 мг/мл обусловлен обратимой адсорбцией протамина на границе мембрана/раствор [66]. Вольтамперометрический режим делает возможными мультиионные измерения: один и тот же ИСЭ используется для определения нескольких ионов. В ходе сканирования потенциала сначала в мембрану переходит ион, который селективно связывается ионофором. По мере расходования ионофора основной ион заменяется следующим по энергетической выгодности при дальнейшей развертке потенциала. Таким образом с ИСЭ, содержащим Na+-селективный ионофор, получены пики, отвечающие содержанию ионов Na+, а при более положительных потенциалах – пики, связанные с концентрациями ионов Ba2+ или Sr2+. С Li+-ионофором в мембране ИСЭ показал пики, обусловленные присутствием ионов Li+, а при более положительных потенциалах – ионов Ca2+ [67].
Возможности применения мультиионных измерений тщательно исследовали Баккер и соавт. [68, 69]. В отличие от подхода Амемии, мембраны ИСЭ содержали несколько ионофоров, каждый из которых облегчал перенос соответствующего иона из фазы образца в фазу мембраны ИСЭ. С мембранами толщиной ~300 нм, содержащими ионофоры на Li+ и Ca2+ и нанесенными на поверхность слоя ПОТ-ClO4 на стеклоуглероде, получены ЦВ-кривые с хорошо разрешенными пиками, обусловленными переносом ионов Li+ и Ca2+ [68]. Похожий ИСЭ с мембраной, содержащей ионофоры на ионы Li+, Na+ и K+, позволил одновременно определять эти три аналита в диапазоне от 0.1 до 100 мМ. Фактически хорошие пики получались и в случае ионов Mg2+ and Ca2+, если соответствующие ионофоры также присутствовали в мембране, но эти пики частично перекрывались пиками, отвечающими ионам Li+ и Na+ [69].
Важно, что закономерности вольтамперометрии с электродами (например, из стеклоуглерода), модифицированными проводящими полимерами, покрытыми в свою очередь тонкими мембранами на основе ионофоров, сильно отличаются от закономерностей классической вольтамперометрии с электродами без мембранных покрытий. В классической вольтамперометрии в идеале ток пика пропорционален концентрации аналита в растворе, а потенциал пика является характеристикой той или иной редокс-реакции, происходящей на границе электрода с раствором образца [54]. Напротив, в вольтамперометрии с электродами, модифицированными ПП и покрытыми ионопроводящей мембраной, ток пика не зависит от концентрации аналита в образце, тогда как потенциал пика меняется по мере изменения концентрации раствора. Более того, потенциал пика зависит от активности аналита, а не от его концентрации. Мы полагаем, что эти закономерности заслуживают специального рассмотрения, которое приведено ниже. Теория, объясняющая эти закономерности, была разработана Баккером и соавт. [68, 70, 71]. Суть теории в следующем. Рассмотрим электрод, например из стеклоуглерода, модифицированного слоем ПП, например, ПОТ, который присутствует в двух формах: нейтральной (восстановленной) ПОТ и окисленной ПОТ+. Слой ПП в свою очередь покрыт слоем ионселективной мембраны. Мембрана содержит ионофор, селективный к катиону M+, и ионообменник MR с липофильным анионом R–. Последний распределен между слоем ПП, компенсируя положительный заряд ПОТ+, и мембранным слоем, компенсируя заряд катиона M+. Предполагается, что диффузией частиц в мембране и в слое ПП можно пренебречь (осложнения, связанные с диффузией, рассмотрены в работах [70, 71]). Предполагается также, что процессы на границах между ПП и мембраной и между мембраной и раствором происходят быстро, поэтому обе границы находятся в состоянии электрохимического равновесия даже в условиях приложенного внешнего потенциала. Соответственно, межфазные потенциалы на границе ПП/мембрана (EP) и мембрана/раствор (EM) следуют закону Нернста с S ≅ 2.303RT/F:
(3)
${{E}_{{\text{P}}}} = E_{{\text{P}}}^{0}--S{\text{lg}}\frac{{{{c}_{{{\text{ПОТ}}}}}}}{{{{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}}},$(4)
${{E}_{{\text{M}}}} = E_{{\text{M}}}^{0} + S{\text{lg}}\frac{{{{a}_{{\text{M}}}}}}{{{{c}_{{\text{M}}}}}}.$Члены $E_{{\text{P}}}^{0},$ $E_{{\text{M}}}^{0}$ заданы стандартными химическим потенциалами частиц, вовлеченных в формирование межфазных электрических потенциалов и считаются постоянными. Величины ${{c}_{{{\text{ПОТ}}}}},$ ${{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}},$ ${{a}_{{\text{M}}}},$ и ${{c}_{{\text{M}}}}$ относятсяr к концентрациям ПОТ и ПОТ+ в слое ПП, активности M+ в водном растворе и концентрации M+ в мембране. При развертке приложенного потенциала в направлении более положительных значений, ПП окисляется и электрон с него переходит на стеклоуглерод. Положительный заряд ПП компенсируется переносом аниона обменника R– из мембраны в слой ПП. В свою очередь катион M+ выходит из мембраны в водную фазу. Именно таким образом ток протекает через такой ИСЭ. Для приложенного потенциала Eappl получим:
(5)
$\begin{gathered} {{E}_{{{\text{appl}}}}} = {\text{Const}} + {{E}_{{\text{P}}}} + {{E}_{{\text{M}}}} = E_{{\text{P}}}^{0} + E_{{\text{M}}}^{0}-- \\ - \,\,S\left( {{\text{lg}}\frac{{{{c}_{{{\text{ПОТ}}}}}}}{{{{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}}}--{\text{lg}}\frac{{{{a}_{{\text{M}}}}}}{{{{c}_{{\text{M}}}}}}} \right). \\ \end{gathered} $Член уравнения (5) ${\text{Const}}$ включает вклады от электрода сравнения, потенциала жидкостного соединения в области контакта электролитического ключа с раствором и потенциала на границе слоя ПП и стеклоуглерода, которые считаем постоянными. ПП не выходит за пределы своего слоя, поэтому общая концентрация ПП (независимо от формы) в этом слое постоянна:
(6)
$c_{{{\text{ПОТ}}}}^{{{\text{общ}}}} = {{c}_{{{\text{ПОТ}}}}} + {{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}.$В отличие от ПП, анион R– присутствует как в слое ПП, так и в мембране, постоянным является общее количество R– в ИСЭ как целом. Далее, согласно теории Баккера [68], мы полагаем, что либо объемы слоя ПП и слоя мембраны одинаковы, либо одинаковы их части, вовлеченные в электрохимический процесс. Тогда для общей концентрации R– можно записать:
(7)
$c_{{\text{R}}}^{{{\text{общ}}}} = {{c}_{{\text{M}}}} + {{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}}.$Теперь уравнение (5) можно переписать следующим образом:
(8)
$\begin{gathered} {{E}_{{{\text{appl}}}}}--S{\text{lg}}{{a}_{{\text{M}}}} = {\text{Const}} + E_{{\text{P}}}^{0} + E_{{\text{M}}}^{0}-- \\ - \,\,S\frac{{\left( {c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}--{{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}} \right)\left( {c_{{\text{R}}}^{{{\text{общ}}}}--{{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}} \right)}}{{{{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}}}. \\ \end{gathered} $Введя вспомогательный параметр:
и комбинируя уравнения (3)–(9) получим для ${{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}}{\text{:}}$(10)
$\begin{gathered} {{\alpha }}{{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}} = c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}c_{{\text{R}}}^{{{\text{общ}}}}--c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}{{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}}--{{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}}c_{{\text{R}}}^{{{\text{общ}}}} + \\ + \,\,c_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}^{2}. \\ \end{gathered} $Это квадратное уравнение имеет следующее решение:
(11)
$\begin{gathered} {{c}_{{{\text{ПО}}{{{\text{Т}}}^{ + }}}}} = \\ = \frac{{\left( {c_{{{\text{ПОТ}}}}^{{{\text{общ}}}} + c_{{\text{R}}}^{{{\text{общ}}}} + {{\alpha }}} \right)--\sqrt {{{{\left( {c_{{{\text{ПОТ}}}}^{{{\text{общ}}}} + c_{{\text{R}}}^{{{\text{общ}}}} + {{\alpha }}} \right)}}^{2}}--4c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}c_{{\text{R}}}^{{{\text{общ}}}}} }}{2}. \\ \end{gathered} $Наконец, примем во внимание, что приложенный потенциал меняется во времени со скоростью развертки ${v}$ от начального значения ${{E}_{{{\text{исх}}}}}$ в сторону более положительных значений:
Дальнейшие результаты представляют собой симуляцию линейной развертки потенциала в предположении ${\text{Const}} + E_{{\text{P}}}^{0} + E_{{\text{M}}}^{0} = 0~$ и ${{E}_{{{\text{исх}}}}} = --0.5\,\,{\text{В}},$ проведенную на основе теории Баккера. Кривые зависимости ${{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}}$ от приложенного потенциала приведены на рис. 1. Данные относятся к случаю $c_{{{\text{ПОТ}}}}^{{{\text{общ}}}} = 0.005\,\,{\text{М}}$ и $c_{{\text{R}}}^{{{\text{общ}}}} = 0.01\,\,{\text{М}}$ и трем значениям активности иона M+ в растворе: 1, 10 и 100 мМ. Видно, что кривые, по существу, представляют собой кривые титрования. В самом деле, когда приложен внешний потенциал и протекает ток, ПП окисляется. Постепенно, весь ПОТ превращается в ПОТ+. С точностью до постоянного слагаемого приложенный потенциал равен сумме ${{E}_{{\text{P}}}} + {{E}_{{\text{M}}}},$ а ${{E}_{{\text{M}}}}$ подчиняется закону Нернста, поэтому десятикратное изменение активности аналита в растворе приводит к изменению значения ${{E}_{{\text{M}}}}$ на ${{2.303RT} \mathord{\left/ {\vphantom {{2.303RT} F}} \right. \kern-0em} F}$ мВ. Таким образом, при одном и том же значении приложенного потенциала величина ${{E}_{{\text{P}}}}$ оказывается на ${{2.303RT} \mathord{\left/ {\vphantom {{2.303RT} F}} \right. \kern-0em} F}$ менее положительной, и окисление ПОТ в ходе развертки потенциала соответственно запаздывает. Поэтому кривая сдвигается в сторону более положительных потенциалов.
Рис. 1.
Кривые зависимости ${{c}_{{{\text{ПО}}{{{\text{Т}}}^{{\text{ + }}}}}}}$$~$ от приложенного потенциала при $c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}$ = 0.005 М, $c_{{\text{R}}}^{{{\text{общ}}}}$ = 0.01 М и ${{a}_{{\text{M}}}},$ равной 1, 10 или 100 мМ.

Ток представляет собой производную заряда по времени, поэтому кривые тока должны быть кривыми дифференциального титрования. Пики тока возникают вследствие того, что в точке перегиба кривой титрования ее производная проходит через максимум. Результаты симуляции той же системы, что и выше, приведены на рис. 2. Ток пика обусловлен скоростью окисления ПОТ в ПОТ+. Она зависит от скорости развертки потенциала, но не от состава раствора, поэтому и ток пика не зависит от состава образца.
Рис. 2.
Симуляция кривых тока при $c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}$ = 0.005 М, $c_{{\text{R}}}^{{{\text{общ}}}}$ = 0.01 М и ${{a}_{{\text{M}}}},$ равной 1, 10 или 100 мМ. Скорость развертки 100 мВ/с.
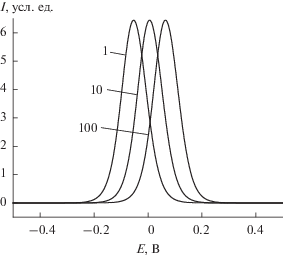
Можно задать вопрос: а куда же исчез фактор природы редокс-реакции? Он никуда не исчез: если мы заменим один ПП, например ПОТ, другим, например ПЭДОТ, кривые, показанные на рис. 1 и 2, сдвинутся.
Поскольку ток определяется скоростью окисления ПОТ в ПОТ+, т.е. скоростью развертки потенциала, следует ожидать линейной зависимости тока (в том числе тока пика) от скорости развертки, что иллюстрирует рис. 3. Здесь наблюдается аналогия с классической вольтамперометрией процессов, лимитированных скоростью реакции на электроде.
Рис. 3.
Зависимость тока от приложенного потенциала при скоростях развертки 10, 30, 50 и 100 мВ/с, случай $c_{{{\text{ПОТ}}}}^{{{\text{общ}}}}$ = 0.005 М, $c_{{\text{R}}}^{{{\text{общ}}}}$ = 0.01 М и ${{a}_{{\text{M}}}}$ = 10 мМ. Во вставке зависимость тока пика от скорости развертки.

Предсказания теории подтверждены экспериментальными данными [66–71].
Ионселективные электроды в режиме (хроно)амперометрии и кулонометрии при постоянном потенциале. Метод ионселективной кулонометрии предложен Бубакой и соавт. [72–76]. Фактически здесь измеряют ток, а заряд получают путем интегрирования тока во времени. Главное преимущество данного метода по сравнению с потенциометрией при нулевом токе – сильное повышение чувствительности: 0.1% вместо 1–5% [76]. Электроды представляют собой стержни из стеклоуглерода с электрохимически сформированным слоем ПП поли-3,4-этилендиокситиофена, допированного полистиролсульфонатом (ПЭДОТ-ПСС), покрытым нанесенной методом полива или спин-коатинга ПВХ-мембраной. Толщина мембран лежала в диапазоне ~(5–10) мкм, т.е. была больше, чем при вольтамперометрических измерениях, но намного меньше, чем при потенциометрии нулевого тока. Первоначально исследовали только K+-селективные ИСЭ [72–74, 76]. Позднее было показано, что этот метод может быть применен также для определения ионов Ca2+ и Ba2+ [77], Cl– [75] и ${\text{NO}}_{3}^{ - }$ [78].
Идея метода состоит в следующем. Потенциал ${{I}^{{{{z}_{I}}}}}$-селективного электрода как целого искусственно поддерживается постоянным с помощью потенциостата, тогда как межфазный потенциал на границе мембраны ИСЭ и раствора откликается на изменение активности иона ${{I}^{{{{z}_{I}}}}}$ в растворе по закону Нернста. Несоответствие между искусственно зафиксированным потенциалом ИСЭ как целого и изменениями межфазного потенциала компенсируется спонтанным протеканием тока через электрод. В первый момент после смены состава раствора соответствующее изменение межфазного потенциала на границе мембрана/раствор компенсируется омическим падением потенциала главным образом на объеме мембраны, так как сопротивление слоя ПП, стеклоуглеродного субстрата и других частей ячейки существенно ниже. Протекание тока приводит к окислению или восстановлению ПП и соответствующим изменениям потенциалов на границах мембрана/ПП и ПП/субстрат. Это изменение потенциала вносит вклад в компенсацию указанного выше несоответствия, и ток со временем затухает. В ходе этого процесса слой ПП перезаряжается, и величина заряда может быть получена интегрированием тока во времени.
Теория хроноамперометрического и кулонометрического отклика ИСЭ предполагает, что ИСЭ можно представить как резистор (мембрана) и конденсатор (слой ПП), соединенные последовательно [75]. Соответствующее уравнение для тока ${{i}_{t}},$ протекающего через ИСЭ в момент времени t после внезапной смены активности иона ${{I}^{{{{z}_{I}}}}}$ от начального значения $a_{I}^{{{\text{исх}}}}$ до конечного $a_{I}^{{{\text{кон}}}}$ может быть записано следующим образом:
(13)
${{i}_{t}} = \frac{{RT}}{{{{z}_{I}}F}}{\text{ln}}\frac{{a_{I}^{{{\text{исх}}}}}}{{a_{I}^{{{\text{кон}}}}}}\frac{1}{R}{{{\text{e}}}^{{--{\text{\;}}\frac{t}{{{{R}_{{{\text{мем}}}}}{{C}_{{{\text{пол}}}}}}}}}}.$Здесь ${{R}_{{{\text{мем}}}}}$ – это сопротивление мембраны, а ${{с}_{{{\text{пол}}}}}$ – емкость слоя ПП. Соответствующий заряд ${{Q}_{t}},$ накопленный за время t, описывается следующим образом:
(14)
${{Q}_{t}} = \frac{{RT}}{{{{z}_{I}}F}}{\text{ln}}\frac{{a_{I}^{{{\text{исх}}}}}}{{a_{I}^{{{\text{кон}}}}}}{{с}_{{{\text{пол}}}}}\left( {1--{{{\text{e}}}^{{--{\text{\;}}\frac{t}{{{\text{\;}}{{R}_{{{\text{мем}}}}}{{C}_{{{\text{пол}}}}}}}}}}} \right).$Движущей силой протекания тока и накопления заряда является изменение активности соответствующего иона в растворе. Поэтому такая кулонометрия дает информацию об активности, а не о концентрации ионов, в отличие от классической кулонометрии, при которой ток задан гальваностатом.
Выше отмечено, что главным преимуществом кулонометрического сигнала является повышение чувствительности анализа. Это достигается путем увеличения емкости слоя ПП между мембраной и субстратом, см. также уравнение (14) [76]. Продемонстрирована чувствительность к изменению концентрации на 0.1% по отношению к ее исходному значению. Кулонометрические измерения позволили увидеть изменения концентрации ионов K+ на 0.1 мМ в образце сыворотке крови, содержащем 4.4 мМ K+ [76]. Здесь речь идет о концентрации, а не об активности ввиду постоянства ионной силы образцов. С точки зрения чувствительности, кулонометрический режим измерений намного превосходит потенциометрию, где чувствительность отклика ИСЭ к ионам ${{I}^{{{{z}_{I}}}}}$ лимитирована фактором Нерста ${{RT} \mathord{\left/ {\vphantom {{RT} {{{z}_{I}}F}}} \right. \kern-0em} {{{z}_{I}}F}}.$
С другой стороны, кулонометрические измерения требуют относительно длительного времени для накопления сигнала. С этой точки зрения использование величины тока вместо заряда в качестве аналитического сигнала может иметь преимущество [79]. Однако смена состава раствора требует времени. Это создает неопределенность реального времени, отвечающего значению t = 0 в уравнениях (13), (14). Более того, сигнал, измеренный в ходе процедуры смены раствора, включает шумы, вызванные соответствующими манипуляциями и перемешиванием. Поэтому величина тока в момент t = 0 не может быть точно измерена. Напротив, чувствительность величины накопленного заряда к неопределенности значения t = 0 по очевидным причинам намного меньше. В связи с этим, в таких измерениях обычно используют заряд, а не ток в качестве аналитического сигнала.
Включение коммерчески доступного электронного конденсатора последовательно с ИСЭ позволяет резко сократить время отклика в кулонометрических измерениях [80], хотя и ценой снижения чувствительности. Использование конденсатора последовательно с ИСЭ позволяет проводить кулонометрические измерения с классическими ИСЭ, содержащими внутренний раствор и внутренний электрод, например Ag/AgCl [81].
Другой подход, направленный на сокращение времени отклика в кулонометрических измерениях, основан на фиттинге кривых ток–время и заряд–время к уравнениям (13), (14), ожидаемым из теории [82]. Это исследование выполнено с K+-ИСЭ на основе валиномицина в качестве модельной системы. Показано, что уравнения (13), (14) не всегда дают успешный фиттинг экспериментальных кривых. Сделан вывод о том, что протекающий через мембрану ИСЭ ток вызывает концентрационную поляризацию. Для учета этого обстоятельства уравнения (13), (14) модифицировали введением коттрелловского слагаемого, что дало следующие уравнения:
(15)
${{i}_{t}} = \frac{{RT}}{{{{z}_{I}}F}}{\text{ln}}\frac{{a_{I}^{{{\text{исх}}}}}}{{a_{I}^{{{\text{кон}}}}}}\left[ {\frac{1}{{{{R}_{{{\text{мем}}}}}}}{{{\text{e}}}^{{--~\frac{t}{{{{R}_{{{\text{мем}}}}}{{C}_{{{\text{пол}}}}}}}}}} + \left( {\frac{N}{2}} \right){{t}^{{{{ - 1} \mathord{\left/ {\vphantom {{ - 1} 2}} \right. \kern-0em} 2}}}}} \right],$(16)
${{Q}_{t}} = \frac{{RT}}{{{{z}_{I}}F}}{\text{ln}}\frac{{a_{I}^{{{\text{исх}}}}}}{{a_{I}^{{{\text{кон}}}}}}\left[ {{{c}_{{{\text{пол}}}}}\left( {1--{{{\text{e}}}^{{--~\frac{t}{{{{R}_{{{\text{мем}}}}}{{C}_{{{\text{пол}}}}}}}}}}} \right) + N{{t}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \right].$Фактор $N$ зависит от ${{A}_{E}}$ – площади сечения ИСЭ, ${{с}_{I}}$ – концентрации заряженных частиц в мембране и ${{D}_{I}}$ – их коэффициентов диффузии: $N = 2F{{A}_{E}}D_{I}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}{{с}_{I}}{{\left( {{{{{\pi }}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{t}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \right)}^{{--1}}}$ [83]. Экспериментально подтвердилось, что ток и заряд в произвольный момент времени t линейно зависят от логарифма активности иона в растворе (или его концентрации, если ионная сила постоянна) [82]. Этот результат согласуется с уравнениями (13)–(16) и поэтому служит дополнительным подтверждением теории. С практической точки зрения это означает, что линейные зависимости тока и заряда от активности иона аналита (относящиеся к одному и тому же времени) могут быть использованы как градуировочные. Это дает возможность останавливать измерения, не дожидаясь полного затухания тока и выхода кривой заряда на насыщение. Таким способом определена концентрация ионов K+ в образцах сыворотки крови – с использованием градуировки по модельным растворам с постоянной ионной силой 145 мМ [82].
Карякин и сотр. [84] показали, что амперометрические измерения при постоянном потенциале особенно перспективны в режиме проточно-инжекционного анализа. Печатные угольные электроды, модифицированные функционализированным боронатом полианилином или берлинской лазурью, успешно использованы для амперометрического определения лактата в человеческом поте. Строго говоря, эти исследования не относятся к мембранам на основе ионофоров. Однако перспективные результаты получены также с K+-селективными и Na+-селективными ИСЭ (стеклоуглерод/ПЭДОТ-ПСС/ПВХ-мембрана) в проточно-инжекционных амперометрических измерениях при постоянном потенциале [85].
* * *
Ионселективные мембраны на основе ионофоров традиционно применялись для анализа только в условиях равновесия или стационарного состояния в составе потенциометрических ИСЭ и оптодов. Измерения в условиях ненулевого тока использовались только в исследованиях механизма отклика таких сенсоров. Однако в настоящее время мембраны на основе ионофоров все шире применяются для анализа в нестационарных условиях, когда через сенсор протекает ток. Эти режимы измерений (вольтамперометрия, и хроноамперометрия и кулонометрия при постоянном потенциале) позволяют проводить многоцелевой анализ с одним и тем же сенсором и существенно улучшить чувствительность анализа. Вне сомнения, мы увидим дальнейшее развитие этих методов в ближайшем будущем.
Работа выполнена при поддержке РФФИ, проект 19-03-00259.
Список литературы
Stefanac Z., Simon W. In vitro verhalten von Macrotetraliden in Membranen als Grundlage für hochselektive kationenspezifische Elektrodensystem // Chimia. 1966. V. 20. № 12. P. 436.
Bakker E., Bühlmann P., Pretsch E. Carrier-based ion-selective electrodes and bulk optodes. 1. General characteristics // Chem. Rev. 1997. V. 97. № 8. P. 3083. https://doi.org/10.1021/cr940394a
Bühlmann P., Pretsch E., Bakker E. Carrier-based ion-selective electrodes and bulk optodes. 2. Ionophores for potentiometric and optical sensors // Chem. Rev. 1998. V. 98. № 4. P. 1593. https://doi.org/10.1021/cr970113+
Lewenstam A. Routines and challenges in clinical application of electrochemical ion-sensors // Electroanalysis. 2014. V. 26. P. 1171. https://doi.org/10.1002/elan.201400061
Mikhelson K.N. Ion-selective Electrodes (Lecture Notes in Chemistry. V. 81). Heidelberg–N.Y.–Dordrecht–London: Springer, 2013. 162 p. https://doi.org/10.1007/978-3-642-36886-8
Bobacka J., Ivaska A., Lewenstam A. Potentiometric ion sensors // Chem. Rev. 2008. V. 108. P. 329. https://doi.org/10.1021/cr068100w
Mikhelson K.N., Peshkova M.A. Advances and trends in ionophore-based chemical sensors // Russ. Chem. Rev. 2015. V. 84. P. 555. https://doi.org/10.1070/RCR4506
Zdrachek E., Bakker E. Potentiometric sensing // Anal. Chem. 2021. V. 93. P. 72. https://doi.org/10.1021/acs.analchem.0c04249
Cammann K., Rechnitz G.A. Exchange kinetics at ion-selective membrane electrodes // Anal. Chem. 1976. V. 48. P. 856.
Cammann K. Exchange kinetics at potassium-selective liquid membrane electrodes // Anal. Chem. 1978. V. 50. P. 936. https://doi.org/10.1021/ac50029a028
Xie S.-L., Cammann K. Apparent ion-exchange current densities at valinomycin-based potassium ion-selective PVC membranes obtained with an AC-impedance method // J. Electroanal. Chem. 1987. V. 229. P. 243.
Horvai G., Graf E., Toth K., Pungor E., Buck R.P. Plasticized poly(vinyl chloride) properties and characteristics of valinomycin electrodes. 1. High-frequency resistances and dielectric properties // Anal. Chem. 1986. V. 58. P. 2735. https://doi.org/10.1021/ac00126a034
Toth K., Graf E., Horvai G., Pungor E., Buck R.P. Plasticized poly(vinyl chloride) properties and characteristics of valinomycin electrodes. 2. Low-frequency, surface-rate, and Warburg impedance characteristics // Anal. Chem. 1986. V. 58. P. 2741. https://doi.org/10.1021/ac00126a035
Mikhelson K.N., Bobacka J., Lewenstam A., Ivaska A. Potentiometric performance and interfacial kinetics of neutral ionophore based ISE membranes in interfering ion solutions before and after contact with primary ions // Electroanalysis. 2001. V. 13. P. 876. https://doi.org/10.1002/15214109(200106)13:103.0.CO;2-#
Mikhelson K.N., Bobacka J., Ivaska A., Lewenstam A., Bochenska M. Selectivity of lithium electrodes: Correlation with ion-ionophore complex stability constants and with interfacial exchange current densities // Anal. Chem. 2002. V. 74. P. 518. https://doi.org/10.1021/ac0155660
Lindner E., Gyurcsanyi R.E., Buck R.P. Tailored transport through ion-selective membranes for improved detection limits and selectivity coefficients // Electroanalysis. 1999. V. 11 P. 695. https://doi.org/10.1002/(sici)1521-4109(199907)11:10/ 11%3c695::aid-elan695%3e3.0.co;2-g
Peshkova M.A., Sokalski T., Mikhelson K.N., Lewenstam A. Obtaining Nernstian response of Ca2+-selective electrode in a broad concentration range by tuned galvanostatic polarization // Anal. Chem. 2008. V. 80. P. 9181. https://doi.org/10.1021/ac8013143
Peshkova M.A., Koltashova E.S., Khripoun G.A., Mikhelson K.N. Improvement of the upper limit of the ISE Nernstian response by tuned galvanostatic polarization // Electrochim. Acta. 2015. V. 167. P. 187. https://doi.org/10.1016/j.electacta.2015.03.139
Mi Y., Mathison S., Bakker E. Polyion sensors as liquid junction-free reference electrodes // Electrochem. Solid-State Lett. 1999. V. 2. P. 198. https://doi.org/10.1149/1.1390782
Shvarev A., Bakker E. Reversible electrochemical detection of nonelectroactive polyions // J. Am. Chem. Soc. 2003. V. 125. P. 11192. https://doi.org/10.1021/ja037167n
Bakker E., Meyerhoff M.E. Ionophore-based membrane electrodes: new analytical concepts and non-classical response mechanisms // Anal. Chim. Acta. 2000. V. 466. P. 121. https://doi.org/10.1016/S0003-2670(00)00883-7
Gemene K.L., Shvarev A, Bakker E. Selectivity enhancement of anion-responsive electrodes by pulsed chronopotentiometry // Anal. Chim. Acta. 2007. V. 583. P. 190. https://doi.org/10.1016/j.aca.2006.09.042
Zdrachek E, Bakker E. Electrochemically switchable polymeric membrane ion-selective electrodes // Anal. Chem. 2018. V. 90. P. 7591. https://doi.org/10.1021/acs.analchem.8b01282
Izadyar A. Stripping voltammetry at the interface between two immiscible electrolyte solutions: a review paper // Electroanalysis. 2018. V. 30. P. 2210. https://doi.org/10.1002/elan.201800279
Морф В. Принципы работы ионоселективных электродов и мембранный транспорт. Пер. с англ. под ред. Петрухина О.М. М.: Мир, 1985. 289 с.
Mikhelson K.N., Smirnova A.L. A new equation for the electrical potential of liquid and PVC membranes containing both neutral carriers and ion-exchangers // Sens. Actuators B. 1992. V. 10. № 1. P. 47.
Mikhelson K.N. Ionselective electrodes in PVC matrix // Sens. Actuators B. 1994. V. 18. № 1–3. P. 31.
Bakker E., Willer M., Lerchi M., Seller K., Pretsch E. Determination of complex formation constants of neutral cation-selective ionophores in solvent polymeric membranes // Anal. Chem. 1994. V. 66. P. 516.
Bakker E., Pretsch E. Potentiometric determination of effective complex formation constants of lipophilic ion carriers within ion-selective electrode membranes // J. Electrochem. Soc. 1997. V. 144. № 5. P. 125.
Мокров С.Б., Стефанова О.К. Проявление в мембранном потенциале сопряженности потоков ионов и нейтрального комплексона на фоне содержащегося в мембране ионообменника. // Электрохимия. 1985. Т. 21. № 4. С. 540.
Мокров С.Б., Стефанова О.К. Оценка констант нестойкости комплексов нейтральных ионофоров и ионов в пленочных мембранах методом э.д.с. // Электрохимия. 1990. Т. 26. № 3. С. 294.
Lutov V.M., Mikhelson K.N. A new pH sensor with PVC-membrane: the analytical evaluation and mechanistic aspects // Sens. Actuators B. 1994. V. 19. № 1–3. P. 400. https://doi.org/10.1016/0925-4005(93)01010-2
Mi Y., Bakker E. Determination of complex formation constants of lipophilic neutral ionophores in solvent polymeric membranes with segmented sandwich membranes // Anal. Chem. 1999. V. 71. P. 5279. https://doi.org/10.1021/ac9905930
Qin Y., Mi Y., Bakker E. Determination of complex formation constants of 18 neutral alkali and alkaline earth metal ionophores in poly(vinyl chloride) sensing membranes plasticized with bis(2-ethylhexyl)sebacate and o-nitrophenyloctyl ether // Anal. Chim. Acta. 2000. V. 421. № 2. P. 207. https://doi.org/10.1016/S0003-2670(00)01038-2
Shultz M.M., Stefanova O.K., Mokrov S.B., Mikhelson K.N. Potentiometric estimation of the stability constants of ion-ionophore complexes in ion-selective membranes by sandwich membrane method: theory, advantages, and limitations // Anal. Chem. 2002. V. 74. P. 510. https://doi.org/10.1021/ac015564f
Mikhelson K.N. Numeric Simulation of Ion-site Association Effects in Ion-selective Electrode Response // Electroanalysis. 2003. V. 15. № 15–16. P. 1236. https://doi.org/10.1002/elan.200302804
Peshkova M.A., Korobeynikov A.I., Mikhelson K.N. Estimation of Ion-Site Association Constants in Ion-selective Electrode Membranes by Modified Segmented Sandwich Membrane Method // Electrochim. Acta. 2008. V. 53. P. 5819. https://doi.org/10.1016/j.electacta.2008.03.030
Zhang W., Spichiger U.E. An impedance study of Mg2+-selective membranes // Electrochim. Acta. 2000. V. 45. P. 2259.
Mikhelson K.N. AC-impedance studies of ion transfer across ionophore-based ion-selective membranes // Chem. Anal. 2006. V. 51. P. 853.
Ivanova N.M., Podeshvo I.V., Goikhman M.Ya., Yakimanskii A.V., Mikhelson K.N. Potassium-selective solid contact electrodes with poly(amidoacid) Cu(I) complex, electron-ion exchanging resin and different sorts of carbon black in the transducer layer // Sens. Actuators B. 2013. V. 186. P. 589. https://doi.org/10.1016/j.snb.2013.06.072
Михельсон К.Н., Лутов В.М., Сулко К., Стефанова О.К. Исследование валиномицинсодержащих мембран одноимпульсным гальваностатическим методом // Электрохимия. 1988. Т. 24. № 11. С. 1487.
Bobacka J. Potential stability of all-solid-state ion-selective electrodes using conducting polymers as ion-to-electron transducers // Anal. Chem. 1999. V. 71. P. 4932. https://doi.org/10.1021/ac990497z
Shvarev A.E., Rantsan D.A., Mikhelson K.N. Potassium-selective conductometric sensor // Sens. Actuators B. 2001. V. 76. № 3. P. 500.
Kondratyeva Ye.O., Solovyeva E.V., Khripoun G.A., Mikhelson K.N. Non-constancy of the bulk resistance of ionophore-based ion-selective electrode: A result of electrolyte co-extraction or of something else? // Electrochim. Acta. 2018. V. 259. P. 458. https://doi.org/10.1016/j.electacta.2017.10.176
Кондратьева Е.О., Соловьева Е.В., Хрипун Г.А., Михельсон К.Н. Парадокс вариации объемного сопротивления мембран калиевых ионоселективных электродов в области нернстовского потенциометрического отклика // Электрохимия. 2019. Т. 55. № 11. С. 1371. (Kondratyeva Ye.O., Solovyeva E.V., Khripoun G.A., Mikhelson K.N. Paradox of the variation of the bulk resistance of potassium ion-selective electrode membranes within nernstian potentiometric response range // Russ. J. Electrochem. 2019. V. 55. № 11. P. 1118. https://doi.org/10.1134/S102319351911009010.1134/S1023193519110090)https://doi.org/10.1134/S0424857019110100
Ivanova A., Mikhelson K. Electrochemical properties of nitrate-selective electrodes: The dependence of resistance on the solution concentration // Sensors. 2018. V. 18. P. 2062. https://doi.org/10.3390/s18072062
Kalinichev A.V., Solovyeva E.V., Ivanova A.R., Khripoun G.A., Mikhelson K.N. Non-constancy of the bulk resistance of ionophore-based Cd2+-selective electrode: a correlation with the water uptake by the electrode membrane // Electrochim. Acta. 2020. V. 334. Article 135541. https://doi.org/10.1016/j.electacta.2019.135541
Solovyeva E.V., Lu H., Khripoun G.A., Mikhelson K.N., Kazarian S.G. In situ ATR-FTIR spectroscopic imaging of PVC, plasticizer and water in solvent-polymeric ion-selective membrane containing Cd2+-selective neutral ionophore // J. Membr. Sci. 2020. V. 619. Article 118798. https://doi.org/10.1016/j.memsci.2020.118798
Harrison J.D., Li X. Measurement of concentration profiles inside a nitrite ion selective electrode membrane // Anal. Chem. 1991. V. 63. P. 2168.
Chan A.D.C., Li X., Harrison J.D. Evidence for a water-rich surface region in poli(vinyl chloride)-based ion-selective electrode membranes // Anal. Chem. 1992. V. 64. P. 2512.
Schneider B., Zwickl T., Federer B., Pretsch E. Spectropotentiometry: A new method for in situ imaing of concentration profiles in ion-selective membranes with simultaneous recording of potential-time transients // Anal. Chem. 1996. V. 68. P. 4342. https://doi.org/10.1021/ac9604245
Zwickl T., Schneider B., Lindner E., Sokalski T., Schaller U., Pretsch E. Chromoionophore-mediated imaging of water transport in ion-selective membranes // Anal. Sci. 1998. V. 14. P. 57. https://doi.org/10.2116/analsci.14.57
Keresten V., Solovyeva E., Mikhelson K. The Origin of the Non-Constancy of the Bulk Resistance of Ion-Selective Electrode Membranes within the Nernstian Response Range // Membranes. 2021. V. 11. Article 344. https://doi.org/10.3390/membranes11050344
Compton R.G., Banks C.E. Understanding Voltammetry. 2nd Ed. London: Imperial College Press, 2010. 444 p. ISBN-13: 978-1848165861, ISBN-10: 1848165862.
Horvath V., Horvai G. Cyclic voltammetric experiments with plasticized PVC membranes // Anal. Chim. Acta. 1993. V. 273. P. 145. https://doi.org/10.1016/0003-2670(93)80153-C
Золотов С.А., Владимирова Е.В., Дунаева А.А., Шипуло Е.В., Петрухин О.М., Вацуро И.М., Ковалев В.В. Определение иона аммония вольтамперометрией на границе двух жидкостей с использованием каликсаренов в качестве нейтральных переносчиков // Электрохимия. 2014. Т. 50. № 10. С. 1045.
Ortuno J.A., Serna C., Molina A., Torralba E. Ion transfer square wave voltammetry of ionic liquid cations with a solvent polymeric membrane ion sensor // Electroanalysis. 2009. V. 21. P. 2297. https://doi.org/10.1002/elan.200904684
Ishimatsu R, Izadyar A., Kabagambe B., Kim Y., Kim J., Amemiya S. Electrochemical mechanism of ion_ionophore recognition at plasticized polymer membrane/water interfaces // J. Am. Chem. Soc. 2011. V. 133. P. 16300. https://doi.org/10.1021/ja207297q
Lindner E, Guzinski M., Pendley B., Chaum E. Plasticized PVC membrane modified electrodes: Voltammetry of highly hydrophobic compounds // Membranes. 2020. V. 10. P. 202. https://doi.org/10.3390/membranes10090202
Zhang J., Harris A.R., Cattrall R.W., Bond A.M. Voltammetric ion-selective electrodes for the selective determination of cations and anions // Anal. Chem. 2010. V. 82. P. 1624. https://doi.org/10.1021/ac902296r
Jarolımova Z., Bosson J., Labrador G.M., Lacour J., Bakker E. Ion transfer voltammetry at thin films based on functionalized cationic helicenes // Electroanalysis. 2018. V. 30. P. 650. https://doi.org/10.1002/elan.201700669
Jarolımova Z., Bosson J., Labrador G.M., Lacour J., Bakker E. Ion transfer voltammetry in polyurethane thin films based on functionalised cationic helicenes for carbonate detection // Electroanalysis. 2018. V. 30. P. 1378. https://doi.org/10.1002/elan.201800080
Kim Y., Amemiya S. stripping analysis of nanomolar perchlorate in drinking water with a voltammetric ion-selective electrode based on thin-layer liquid membrane // Anal. Chem. 2008. V. 80. P. 6056. https://doi.org/10.1021/ac8008687
Kim Y., Rodgers P.J., Ishimatsu R., Amemiya S. Subnanomolar ion detection by stripping voltammetry with solid-supported thin polymeric membrane // Anal. Chem. 2009. V. 81. P. 7262. https://doi.org/10.1021/ac900995a
Kabagambe B., Izadyar A., Amemiya S. Stripping voltammetry of nanomolar potassium and ammonium ions using a valinomycin-doped double-polymer electrode // Anal. Chem. 2012. V. 84. P. 7979. https://doi.org/10.1021/ac301773w
Garada M.B., Kabagambe B., Amemiya S. Extraction or adsorption? Voltammetric assessment of protamine transfer at ionophore-based polymeric membranes // Anal. Chem. 2015. V. 87. P. 5348. https://doi.org/10.1021/acs.analchem.5b00644
Greenawalt P.J., Amemiya S. Voltammetric mechanism of multiion detection with thin ionophore-based polymeric membrane // Anal. Chem. 2016. V. 88. P. 5827. https://doi.org/10.1021/acs.analchem.6b00397
Crespo G.A., Cuartero M., Bakker E. Thin layer ionophore-based membrane for multianalyte ion activity detection // Anal. Chem. 2015. V. 87. P. 7729. https://doi.org/10.1021/acs.analchem.5b01459
Cuartero M., Crespo G.A., Bakker E. Ionophore-based voltammetric ion activity sensing with thin layer membranes // Anal. Chem. 2016. V. 88. P. 1654. https://doi.org/10.1021/acs.analchem.5b03611
Yuan D., Cuartero M., Crespo G.A., Bakker E. Voltammetric thin-layer ionophore-based films: Part 1. Experimental evidence and numerical simulations // Anal. Chem. 2017.V. 89. P. 586. https://doi.org/10.1021/acs.analchem.6b03354
Yuan D., Cuartero M., Crespo G.A., Bakker E. Voltammetric thin-layer ionophore-based films: Part 2. Semi-empirical treatment // Anal. Chem. 2017. V. 89. P. 595. https://doi.org/10.1021/acs.analchem.6b03355
Hupa E., Vanamo U., Bobacka J. Novel ion-to-electron transduction principle for solid-contact ISEs // Electroanalysis. 2015. V. 27. P. 591. https://doi.org/10.1002/elan.201400596
Vanamo U., Hupa E., Yrjänä V., Bobacka J. New signal readout principle for solid-contact ion-selective electrodes // Anal. Chem. 2016. V. 88. P. 4369. https://doi.org/10.1021/acs.analchem.5b04800
Han T., Vanamo U., Bobacka J. Influence of electrode geometry on the response of solid-contact ion-selective electrodes when utilizing a new coulometric signal readout method // ChemElectroChem. 2016. V. 3. P. 2071. https://doi.org/10.1002/celc.201600575
Jarolímová Z., Han T., Mattinen U., Bobacka J., Bak-ker E. Capacitive model for coulometric readout of ion-selective electrodes // Anal. Chem. 2018. V. 90. P. 8700. https://doi.org/10.1021/acs.analchem.8b02145
Han T., Mattinen U., Bobacka J. Improving the sensitivity of solid-contact ion-selective electrodes by using coulometric signal transduction // ACS Sens. 2019. V. 4. P. 900. https://doi.org/10.1021/acssensors.8b01649
Han T., Mousavi Z., Mattinen U., Bobacka J. Coulometric response characteristics of solid contact ion-selective electrodes for divalent cations // J. Sol. St. Electrochem. 2020. V. 24. P. 2975. https://doi.org/10.1007/s10008-020-04718-8
Han T., Mattinen U., Mousavi Z., Bobacka J. Coulometric response of solid-contact anion-sensitive electrodes // Electrochim. Acta. 2021. V. 367. Article 13756. https://doi.org/10.1016/j.electacta.2020.137566
Jaworska E., Pawłowski P., Michalska A., Maksymiuk K. Advantages of amperometric readout mode of ion-selective electrodes under potentiostatic conditions // Electroanalysis. 2019. V. 31. P. 343. https://doi.org/10.1002/elan.201800649
Kraikaew P., Sailapu S.K., Bakker E. Rapid constant potential capacitive measurements with solid contact ion-selective electrodes coupled to electronic capacitor // Anal. Chem. 2020. V. 92. P. 14174. https://doi.org/10.1021/acs.analchem.0c03254
Kraikaew P., Jeanneret S., Soda Y., Cherubini T., Bakker E. Ultrasensitive seawater pH measurement by capacitive readout of potentiometric sensors // ACS Sens. 2020. V. 5. P. 650. https://doi.org/10.1021/acssensors.0c00031
Kondratyeva Ye.O., Tolstopjatova E.G., Kirsanov D.O., Mikhelson K.N. Chronoamperometric and coulometric analysis with ionophore-based ionselective electrodes: A modified theory and the potassium ion assay in serum samples // Sens. Actuators B. 2020. V. 310. Article 127894. https://doi.org/10.1016/j.snb.2020.127894
Bard L.J., Faulkner L.R. Electrochemical Methods. Fundamentals and Applications. 2nd Ed. New York-Chichester-Weinheim-Brisbane-Singapore-Toronto: John Wiley & Sons Inc., 2001. 864 p. ISBN: 978-0-471-04372-0.
Zavolskova M.D., Nikitina V.N., Maksimova E.D., Karyakina E.E., Karyakin A.A. Constant potential amperometric flow-injection analysis of ions and neutral molecules transduced by electroactive (conductive) polymers // Anal. Chem. 2019. V. 91. P. 7495. https://doi.org/10.1021/acs.analchem.9b00934
Nikitina V.N., Maksimova E.D., Zavolskova M.D., Karyakin A.A. Flow injection amperometry as an alternative to potentiometry for solid contact ion-selective membrane-based electrodes // Electrochim. Acta. 2021. V. 377. Article 138074. https://doi.org/10.1016/j.electacta.2021.138074
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии