

Журнал аналитической химии, 2022, T. 77, № 6, стр. 490-513
Биоэлектрохимия как область анализа: исторические аспекты и современное состояние
Е. В. Супрун a, *, Г. К. Будников b
a Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича
119121 Москва, ул. Погодинская, 10, стр. 8, Россия
b Казанский (Приволжский) федеральный университет, Химический институт им. А.М. Бутлерова
420008 Казань, ул. Кремлевская, 29/1, Россия
* E-mail: lenasuprun@mail.ru
Поступила в редакцию 01.09.2021
После доработки 19.11.2021
Принята к публикации 19.11.2021
- EDN: EUDITH
- DOI: 10.31857/S0044450222060184
Аннотация
В обзоре рассмотрены исторические аспекты становления и развития биоэлектрохимии и некоторые вопросы аналитического применении электрохимических методов (преимущественно вольтамперометрии и родственных методов) в исследовании свойств биологически активных соединений и биополимеров в контексте интересов биохимии и медицины. Отмечена важность учета процессов на поверхности электродов, генерирующих аналитический сигнал. Обзор затрагивает принципы прямого электрохимического анализа биополимеров (белков и нуклеиновых кислот) за счет электроактивности аминокислотных остатков, редокс активных центров, остатков нуклеиновых оснований на немодифицированных электродах. Обсуждается электрохимическое поведение белков, пептидов, олигонуклеотидов, одно- и двунитевых молекул нуклеиновых кислот с точки зрения их пространственной структуры. Сделан акцент на направлениях развития биоэлектрохимии.
ТЕРМИН “БИОЭЛЕКТРОХИМИЯ”
Применение методов и подходов электрохимии к изучению биомолекул стало выделяться в отдельную область знаний около полувека тому назад, хотя первые работы были опубликованы несколько ранее. Междисциплинарный характер данной области формировался постепенно, и термин “биоэлектрохимия” появился не сразу. К настоящему моменту он не имеет четких границ, поскольку в его дефиницию включены несколько концепций и познавательных подходов, объединенных приставкой “био-”. Тем не менее представляют интерес исторические аспекты становления и развития биоэлектрохимии в формате движения идей, направленных на решение аналитических задач.
В 1979 г. по инициативе Джулио Милаццо было создано международное Биоэлектрохимическое общество, официально зарегистрированное в 1981 г. во Франции (Bioelectrochemical Society, BES, http://www.bioelectrochemical-soc.org). Интересно, что за несколько лет до этого Милаццо был участником VI Дискуссии Гейровского (1972 г., замок Либлице, недалеко от Праги), посвященной вопросам электроаналитичекой химии применительно к биомедицине. В настоящее время продолжается серия симпозиумов Биоэлектрохимического общества под названием “Biannual Symposium series of the Society on Bioelectrochemistry and Bioenergetics”, начатая еще в 1971 г., а выпускаемый с 1974 г. международный журнал “Bioelectrochemistry and Bioenergetics” c 2000 г. сменил свое название на “Bioelectrochemistry” (официальный сайт: https://www.journals.elsevier.com/bioelectrochemistry). Журнал посвящен электрохимическим принципам в биологии и биологическим аспектам электрохимии. В год 40-летия Общества, в мае 2021 г. Биоэлектрохимический симпозиум прошел в двадцать шестой раз, впервые в онлайн формате (XXVI International Symposium on Bioelectrochemistry and Bioenergetics of the Bioelectrochemical Society, http://www.bes2021.org). Следует упомянуть, что чешская традиция проводить Дискуссии Гейровского также сохранилась (http://www.physchem.cz), и почти полвека спустя конференция была посвящена электрохимии биологически активных соединений, включая биополимеры.
Интерес представляет сама история появления в науке электрохимии термина с приставкой “био-”. Хотя биология и химия возникли независимо друг от друга, связь между соответствующими науками была осознана лишь в ХIХ в., когда естествоиспытатели стали целенаправленно проводить эксперименты с живыми организмами при воздействии на них различных физических факторов. Электрические эксперименты, в своем названии с приставкой “био-”, впервые были проведены еще в конце XVIII века. В 1771 г. итальянский анатом и физиолог Луиджи Гальвани открыл и исследовал феномен сокращения мышц препарированной лягушки под влиянием электрического тока [1]. Наблюдая сокращение мышц при соединении их металлом с нервами или спинным мозгом, он обратил внимание на то, что мышца сокращается при одновременном прикосновении к ней двух разных металлов. Гальвани объяснил эти явления существованием “животного электричества”, благодаря которому мышцы заряжаются подобно лейденской банке. Сами явления, открытые Гальвани, долгое время называли “гальванизмом”. В 1790-х гг., заинтересовавшись “животным электричеством”, итальянский физик и физиолог Алессандро Вольта провел ряд опытов и показал, что наблюдаемые Гальвани явления были связаны с наличием замкнутой цепи, состоящей из двух разнородных металлов и жидкости. Вольта считал причины “гальванизма” физическими, а физиологические действия – одними из проявлений этого физического процесса. Проведя опыты с разными парами электродов, Вольта установил, что физиологическое раздражение нервов тем сильнее, чем дальше отстоят друг от друга два металла в следующем ряду: цинк, оловянная фольга, олово, свинец, железо, латунь и т.д., до серебра, ртути, графита. Этот знаменитый ряд напряжений (активностей) Вольта и составлял ядро эффекта; мышца лягушки была лишь пассивным, хотя и очень чувствительным электрометром, а активными звеньями являлись металлы, от контакта которых и происходила их взаимная электризация. Проводя многочисленные сравнительно-физиологические опыты, Вольта наблюдал у животных бóльшую электрическую возбудимость нервов по сравнению с мышцами, а также гладкой мускулатуры кишечника и желудка по сравнению со скелетной. Он обнаружил электрическую раздражимость органов зрения и вкуса у человека [1]. Сегодня изучением гальванических эффектов в биологии занимается электрофизиология, отцом которой можно по праву считать Гальвани. Отдавая дань истории, традиционно раз в два года Биоэлектрохимическое общество вручает приз имени Луиджи Гальвани молодому ученому, внесшему наиболее весомый вклад в область биоэлектрохимии (http://www.bioelectrochemical-soc.org/awards-LuigiGalvani.php).
Под приставкой “био-” в термине “биоэлектрохимия” подразумевают биологически активные вещества, биополимеры (белки, нуклеиновые кислоты), целые клетки и живые организмы. Первые работы, посвященные электрохимическому поведению белков на ртутном электроде, появились в начале 1930-х гг., т.е. менее чем через десять лет после открытия полярографии Ярославом Гейровским. Если период практического использования электрохимических методов, начиная с полярографии, в анализе биомедицинских объектов охватывает сто лет, то собственно биоэлектрохимия как междисциплинарный раздел науки появился сравнительно недавно. В настоящее время термин “биоэлектрохимия” включает в себя достаточно широкий спектр направлений, так или иначе связанных с живыми системами и регистрацией переноса электронов. Биоэлектрохимия изучает электрохимические закономерности, лежащие в основе биологических процессов (в частности, передачи информации по нервным волокнам, преобразования энергии, фотосинтеза, рецепции), а также воздействие внешних электрических и магнитных полей на биологические системы [2]. Общая стадия всех упомянутых процессов – разделение зарядов (ионов и электронов), реализующееся в ходе окислительно-восстановительной реакции или при транспорте ионов через мембраны. Это приводит к возникновению мембранного потенциала и градиентов концентрации ионов между внутренней частью клетки и окружающей средой. Свободная энергия, накопленная в виде мембранного потенциала или концентрационных градиентов, обеспечивает генерацию и передачу нервных импульсов, синтез АТФ, некоторые виды механического движения и т.п. Согласно Березовчуку [3], биоэлектрохимия изучает структуру и свойства мембран живых клеток, механизм переноса ионов через мембрану, природу скачка потенциала на мембране живой клетки, механизм передачи потенциала вдоль нервного волокна. Знание механизма работы клеточной мембраны позволит сконструировать различные приборы, работающие по принципу живой клетки. Добавим, что, с другой стороны, процессы переноса ионов в живых системах могут быть смоделированы с использованием знаний ионометрии – одного из разделов электрохимии. Таким образом, биоэлектрохимия – это научное направление, предмет которого – электрохимические аспекты работы живых систем [4]. Под термином “биоэлектрохимия” в настоящее время также понимают область электрохимии, связанную с изучением электрохимических свойств биомолекул и разработкой способов их электрохимического определения в биообразцах.
Новое “дыхание” биоэлектрохимии дало появление биосенсоров: в 1962 г. Кларк и Лайонс [5] предложили концепцию “ферментных мембранных электродов”. Раствор фермента (например, глюкозооксидазы или уреазы) должен был быть помещен между двумя мембранами на поверхности электрода, регистрирующего убыль субстрата или прирост продукта ферментативной реакции. В качестве сенсорного элемента были использованы pH- и pO2-электроды. Идея авторов заключалась в использовании свойств ферментов для определения концентрации аналита при проведении сердечно-сосудистых операций и для послеоперационного прогноза состояния пациента [5]. Таким образом, биосенсоры – это химические сенсоры, в которых распознающая система основана на биохимическом механизме. Согласно определению, принятому в 1999 г. Международным союзом теоретической и прикладной химии (ИЮПАК) [6], химический сенсор представляет собой устройство, трансформирующее химическую информацию, начиная с концентрации компонента и заканчивая композиционным составом, в аналитический сигнал. Химические сенсоры обычно состоят из двух основных частей: химической (молекулярной) распознающей системы (рецептора) и физико-химического трансдьюсера. Биологическая распознающая система передает информацию от биохимического рецептора, обычно о концентрации определяемого вещества, в виде химического или физического выходящего сигнала с определенной чувствительностью. Согласно рекомендациям ИЮПАК [6], термин “биосенсор” следует отделять от аналитических систем, которые включают в себя дополнительные стадии разделения (такие как высокоэффективная жидкостная хроматография), вспомогательные устройства и/или обработку образца введением специальных реагентов (например, проточно-инжекционный анализ). Таким образом, биосенсор должен представлять собой аналитическое устройство, не требующее дополнительных реагентов помимо природных косубстратов, необходимых для определения аналита. В то же время существует и альтернативная точка зрения: Энтони Тернер [7] подразделяет современные биосенсоры на (1) сложное, высокопроизводительное лабораторное оборудование, способное быстро, легко и точно детектировать биологические взаимодействия и определять интересующие компоненты; и (2) простые в использовании, портативные устройства, рассчитанные на широкий круг потребителей, для децентрализованного, in situ или домашнего анализа. Первые – дорогие, последние – массово производимые и доступные. Семейства биосенсоров – это иммунодатчики, биосенсоры на основе ферментов, а также биосенсоры на основе целых клеток. Биосенсоры были применены к широкому спектру аналитических проблем, включая использование в медицине, биомедицинских исследованиях, фармакологии, экологии, пищевой промышленности, перерабатывающих отраслях, сфере безопасности. Самыми известным примером аналитического устройства на основе электрохимического биосенсора служит коммерчески доступный глюкометр. Дизайн и изучение молекулярных и супрамолекулярных структур с молекулярными биорецепторными и биомиметическими свойствами для использования в аналитических устройствах также входит в область интересов биоэлектрохимии. Здесь основное внимание уделяется комплементарному пересечению молекулярного распознавания, нанотехнологии, молекулярного импринтинга и супрамолекулярной химии для улучшения аналитических характеристик и надежности устройств. Параллельно с биосенсорикой примерно в то же время возникла область биоэлектроники, которая стремится использовать биологию в сочетании с электроникой в более широком контексте, охватывая, например, биологические топливные элементы, бионики и биоматериалы для обработки и хранения информации. Ключевым аспектом здесь является интерфейс между биологическими материалами и микро- и наноэлектроникой.
Относительно недавно вышли рекомендации ИЮПАК [8], касающиеся терминологии методов биоаналитической химии, анализа и исследования биомакромолекул. В них вводятся четкие определения, связанные с биоаналитическими образцами, ферментативными методами, иммуноаналитическими методами, методами, используемыми в геномике и анализе нуклеиновых кислот, протеомике, метаболомике, гликомике, липидомике и интерактомике [8]. Согласно рекомендациям 2018 г., в область биоаналитической химии входят выявление (идентификация, расшифровка последовательности), характеристика (полярность и заряд, структура, межмолекулярные взаимодействия), а также количественная оценка и мониторинг (стабильность, динамика, фрагментация, деградация, метаболизм и др.). Под термином “биосенсор” понимается измерительный инструмент (устройство, средство), не требующий дополнительных реагентов и обеспечивающий получение селективной качественной и/или количественной аналитической информации с использованием элемента биологического распознавания, который удерживается в непосредственном пространственном контакте с элементом трансдукции (преобразователем) [8].
БИОЭЛЕКТРОХИМИЯ: ИСТОРИЧЕСКАЯ РЕТРОСПЕКТИВА
История электрохимических методов анализа прослеживается с середины ХIХ в. [9]. Открытие в 1922 г. Ярославом Гейровским метода полярографии означало появление новой области электрохимии, в которой основным инструментом становится полярограф, позволяющий регистрировать поляризационные кривые (полярограммы) ртутного капельного электрода в испытуемых растворах. Первое время объектами исследования служили, как правило, неорганические соединения, и развитие теории метода было связано с интерпретацией их полярографического поведения. В 1925 г. Шиката в лаборатории Гейровского в Праге зарегистрировал полярограмму первого органического соединения – нитробензола [10]. Если принять во внимание, что нитрогруппа входит в структуру многих органических соединений с биологической активностью, то можно принять эту дату началом аналитической биоэлектрохимии. Таким образом, все началось с полярографии низкомолекулярных органических соединений. Из личного опыта: один из авторов настоящего обзора в 1960 г. поступил в аспирантуру Химического института Казанского филиала Академии наук СССР (КФ АН СССР, Казань) по специальности “полярография органических соединений”. Объектами исследования были семи- и тиосемикарбазоны алдегидов и кетонов, из которых ряд веществ имел противотуберкулезную активность. В период обучения в аспирантуре была пройдена стажировка в Полярографическом институте Чехословацкой академии наук, инициированная Арбузовым и Китаевым. Стажировка проходила под патронажем Гейровского, получившего накануне Нобелевскую премию “за открытие и развитие полярографических методов анализа” (1959 г.). Обстановка в институте способствовала глубокому знакомству с работами в области полярографии в биомедицине и фармации. Таким образом, вначале полярографические методы успешно развивались как эффективные по тому времени средства исследования строения и реакционной способности органических соединений в процессах с участием электронов. Постепенно обширные сведения об электрохимических свойствах органических соединений в растворах становились востребованными при разработке способов их определения в различных объектах.
Потребовалось почти полвека исследований, чтобы показать, что в качестве материала для поляризуемого электрода в вольтамперометрии можно использовать не только ртуть, но и благородные металлы, их оксиды, различные углеродные материалы, пасты и композиты на их основе; помимо обычных стали использовать и так называемые модифицированные электроды. Появление стационарного твердого электрода означало переход от катодной (восстановление) к анодной (окисление) вольтамперометрии, что для органического анализа оказалось крайне перспективным. Размеры самих электродов уменьшились в десятки и даже сотни раз, а для достижения воспроизводимости их характеристик стали применять унифицированные способы изготовления, например, трафаретную печать. Одновременно велись исследования по аппаратурной тематике, т.е. по совершенствованию приборов, обеспечивающих надежную регистрацию полезного сигнала по отношению к шуму (помехе) на принципах микроэлектроники. Современный вольтамперограф (потенциостат) мало напоминает набор реостатов и батарей Гейровского. Как правило, это небольшой блок, а иногда и плата, вставляемая в компьютер или смартфон. Например, такие мини-приборы успешно выпускает фирма “PalmSens” (Нидерланды, официальный сайт: www.palmsens.com), в частности, совместимый со смартфоном потенциостат “Sensit Smart”. Миниатюризация коснулась аппаратуры, электродов и электрохимических ячеек. Это связано с тем, что электроды стали использовать в качестве преобразователей сигнала в интегрированных устройствах, таких как проточные тонкослойные ячейки, микрофлюидные ячейки и “лаборатории на чипе”. Миниатюризация подобных систем позволила добиться выдающихся успехов в области снижения как определяемых концентраций аналитов, так и объема потребляемой пробы. Тем самым созданы предпосылки для очередного качественного скачка и перехода к имплантируемым устройствам, сочетающим функции врача-диагноста и лечебного средства. При этом определяемые количества аналита вполне могут спуститься на уровень пико- и фемтограммов в образце.
Отметим, что создание устройств, которые применяют в электрохимическом анализе, в том числе и в мобильном исполнении, возможно лишь на основе глубокого понимания как задач биоаналитической химии, так и природы процессов, происходящих на поверхности электрода. Аналитический отклик на изучаемое соединение формируется в ходе протекания на электроде процесса, часто состоящего из нескольких стадий [11]. Обычно собственно механизм электродной реакции остается вне интересов аналитика. Важно, чтобы отклик был повторяем в требуемых пределах. Тем не менее полезно иметь общие представления о механизме. Под механизмом понимают всю совокупность гетерогенных реакций переноса электронов на границе раздела электрод/раствор, а также химических гомогенных реакций вблизи электрода и в объеме раствора. Изучение механизма включает в себя определение последовательности этих электрохимических (Е) и химических (С) стадий, констант их скоростей, что позволяет выделить лимитирующую стадию переноса, а также чисел электронов и протонов, переносимых на одну молекулу, участвующую в реакции, установление других фундаментальных электрохимических характеристик отдельных стадий, определение природы образующихся частиц и так далее. Следует иметь в виду, что каждый электрохимический и химический процесс должен включать в себя только один элементарный акт [11]. В большинстве случаев лимитирующей стадией процесса является одна из химических стадий, предшествующая, последующая или параллельная переносу электрона. Для установления природы продуктов реакций, образующихся на электроде, проводят электролиз при контролируемом потенциале, и затем продукты идентифицируют с помощью соответствующих физико-химических методов.
Необходимо отметить мировой тренд в электроаналитике в отношении объектов биомедицины. Из низкомолекулярных органических соединений в моде остаются различные флавоноиды, включая антиоксиданты [12]. Успехи наблюдаются и в области биосенсорики, в которой используют как природные, так и искусственные материалы для организации селективного отклика, включая “синтетические рецепторы” и “нанозимы” [13, 14]. Повторимся, сказав, что мы живем в век серьезных преобразований в областях биомедицины, а отсюда и в биоэлектрохимии. Здесь несомненный интерес для биоэлектроаналитиков представляют области с междисциплинарными взаимодействиями. Это – проблемы ранней диагностики заболеваний, функционирования клеток, контроля эффективности и токсичности лекарственных средств, контроля содержания пищевых красителей и т.д.
Белки. Началом электрохимии белков можно считать работу Гейровского и Бабички 1930 г. [15], в которой при добавлении белка в полярографическую ячейку наблюдали появление на вольтамперограмме новой волны при потенциале –1.6 В (на 0.2 В более положительном, чем восстановление ${\text{NH}}_{4}^{ + }$-ионов аммиачного буферного раствора) [16]. Волну отнесли к процессу электрохимического восстановления водорода на поверхности ртути, катализируемого белком (рис. 1). Следом, в 1932 г., Герлз и Ванкура, два врача, изучающие по приглашению Гейровского полярографическую активность различных биологических жидкостей человека, зарегистрировали другой, менее выраженный сигнал: внесение белка в раствор хлорида натрия вызвало рост тока при потенциале, на 0.3 В более положительном, чем восстановление натриевых ионов фона [17]. Сигнал был назван “преднатриевая волна” (рис. 1). Дальнейшие исследования показали, что процессы, протекающие в полярографической ячейке с участием белка, могут быть выражены следующими уравнениями реакций [18]:
(1)
${\text{P}} + {\text{DH\;}} \rightleftarrows {\text{P}}{{{\text{H}}}^{ + }} + {{{\text{D}}}^{--}},$(4)
$E = \frac{{2RT}}{F}\ln \left[ {{{{\text{H}}}^{ + }}} \right]--\frac{{3RT}}{{2F}}{\text{ln}}I + a,$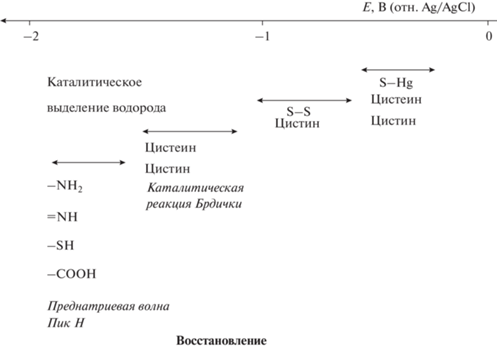
Подобный эффект каталитического выделения водорода наблюдал и Брдичка в 1933 г. [23], когда пытался подавить сигнал кобальта на фоне аммиачного буферного раствора сывороткой крови: сигнал был подавлен, но при потенциалах, более положительных, чем для “преднатриевой волны”, появилась новая четко выраженная двойная волна. Данное явление было названо “каталитическая реакция Брдички” (рис. 1). Из аминокислот только цистин (Цис-Цис) и Цис давали рост этой волны. Высота новой волны была в сотни раз больше, чем сигнал восстановления SS-групп Цис-Цис, что говорило о ее каталитической природе. Так как высота каталитической волны для Цис была в два раза ниже, чем для Цис-Цис при той же концентрации, Брдичка заключил [24], что происходит восстановление SS-групп Цис-Цис до SH-групп, а SH-группы в свою очередь служат источниками ионов водорода для регистрируемого сигнала. С помощью данного метода был проведен анализ более 250 образцов сыворотки крови онкологических больных и здоровых доноров, который выявил существенное различие между ними [25]. Так, в случае сыворотки крови онкобольных высота характеристической волны была всегда ниже, чем для сыворотки здоровых добровольцев. Отвечая на вопрос, какие из групп – SH или SS – ответственны за полярографическую реакцию, был сделан вывод, что наблюдаемый эффект относится главным образом к SS-группам [25]. Более глубокое исследование, проведенное на образцах сыворотки крови от 386 человек (онкологических больных, больных другими заболеваниями и здоровых доноров) выявило влияние ряда факторов (возраста пациента, вида опухоли) на равновесие ${\text{Цис - Цис}} \rightleftarrows {\text{Цис}}$ [26]. С этого времени полярографическое изучение белков строилось главным образом на каталитических волнах в присутствии кобальта [27–29]. Каталитическая реакция Брдички нашла свое применение в биоанализе для определения Цис-содержащих белков, таких как металлотионеины [30, 31] и фитохелатины [32]. Считается, что катализаторами реакции выступают Цис- и/или Цис-Цис-содержащие белки, однако ее механизм до сих пор до конца не изучен.
В 1950–1960-е гг. расшифровка аминокислотной последовательности инсулина (Сенгер, Нобелевская премия по химии 1958 г.) и получение пространственых структур миоглобина и гемоглобина рентгеноструктурным анализом (Перутц и Кендрю, Нобелевская премия по химии 1962 г.) вывели исследования в области белков и аминокислот, включая электрохимические, на новый уровень. В 1960-х гг. было открыто полярографическое восстановление белков за счет дисульфидных связей Цис-Цис (RSSR) [33, 34]. Суммарное уравнение процессов, протекающих при этом на поверхности ртутного электрода, можно записать как [35, 36]:
Согласно механизму реакции, предложенному для восстановления Цис-Цис [35], сначала происходит образование цистеината ртути(II) (уравнение (6)), а затем его восстановление (уравнение (7)):
(7)
${\text{Hg}}{{\left( {{\text{SR}}} \right)}_{2}} + 2\bar {e} + 2{{{\text{H}}}^{ + }} \rightleftarrows {\text{Hg}} + 2{\text{RSH}}{\text{.}}$Другой механизм был предложен для реакции восстановления окисленной формы глутатиона (GSSG) (уравнения (8)–(9)) [36]:
(8)
${\text{GSSG}} + \bar {e} + {{{\text{H}}}^{ + }} \rightleftarrows {\text{G}}{{{\text{S}}}^{ \bullet }} + {\text{GSH,}}$(9)
${\text{G}}{{{\text{S}}}^{ \bullet }} + \bar {e} + {{{\text{H}}}^{ + }} \rightleftarrows {\text{GSH}}{\text{.}}$Отметим, что для каталитического электровосстановления водорода в присутствии белка и ранее требовалось восстановление SS-групп остатков Цис-Цис (рис. 1), но сообщения такого рода не были опубликованы. Объяснением этому противоречию может служить значительная разница высот двух типов волн. Токи восстановления SS-групп Цис-Цис белков составляют порядка 0.1 мкА, тогда как токи каталитических волн выделения водорода – 10–100 мкА. Таким образом, было необходимо достичь более высокой чувствительности измерения сигнала, чтобы зарегистрировать восстановление SS-мостиков, что впервые продемонстрировали Сесил и Вейцман [33] в 1964 г. на примере инсулина и ряда других белков. Известно, что SH-группы Цис (RSH) после предварительного полярографического окисления также образуют цистеинаты ртути(I) и ртути(II) и адсорбируются на поверхности электрода (уравнения (10)–(11)) [37, 38]:
(10)
$2{\text{RSH}} + {\text{Hg}} \rightleftarrows {\text{Hg}}{{\left( {{\text{SR}}} \right)}_{2}} + 2\bar {e} + 2{{{\text{H}}}^{ + }}{\text{\;,}}$(11)
$2{\text{RSH}} + 2{\text{Hg}} \rightleftarrows {\text{H}}{{{\text{g}}}_{2}}{{\left( {{\text{SR}}} \right)}_{2}} + {\text{\;}}2\bar {e} + 2{{{\text{H}}}^{ + }}.$Определение концентрации белковых молекул возможно и через восстановление образующейся связи Hg–S (уравнение (7), рис. 1). Позже, в 1970-х гг., изучение полярографического восстановления гемопротеинов – белков, содержащих ион Fe(II)/Fe(III) в активном центре, выявило прямой перенос электрона между электродом и простетической группой, дав старт электрохимии содержащих кофактор белков [39–41].
В 1970-х гг. было показано, что адсорбция молекул белка на поверхности электрода – ключ к регистрации прямого переноса электрона между электродом и активным центром белка. Термин “прямой перенос электрона” означает обмен электронами между кофактором фермента (белка) и электродом в отсутствие каких-либо медиаторов [42]. Прямой перенос электрона описан для небольшого числа ферментов и белков, которые принято называть “редокс-активными”. В 1972 г. Бетсо с соавт. [39] описали восстановление иона Fe(III) гема цитохрома с (Цит c), находящегося в растворе, на ртутном, платиновом и золотом электродах без необратимой денатурации белка. Авторы заключили: “Адсорбция белка на поверхности электрода значительно влияет на наблюдаемый электрохимический сигнал, но не вызывает загрязнения электрода или потерю способности электрода переносить электроны” [39]. Позже, в 1977 г., был зарегистрирован обратимый прямой перенос электрона между Цит c, находящимся в растворе, и электродом [43, 44]:
(12)
${\text{Fe}}\left( {{\text{III}}} \right){\text{ - гем}} + \bar {e} \rightleftarrows {\text{Fe}}\left( {{\text{II}}} \right){\text{ - гем}}{\text{.}}$На электроде из смешанных оксидов индия и олова методом циклической вольтамперометрии в области от 0.45 до –0.30 B (отн. Ag/AgCl) зарегистрировали четкую волну, характерную для контролируемого диффузией прямого переноса электрона между Цит c и электродом [43]. В то же время на золотом дисковом электроде перенос электрона между Цит c и электродной поверхностью удалось зарегистрировать только после внесения в ячейку 4,4'-бипиридила [44]. Присутствие 4,4'-бипиридила (который в данной области потенциалов неэлектроактивен) стимулировало перенос электрона между Цит c и электродом. Авторы предположили, что 4,4'-бипиридил взаимодействует с белком и/или с электродом, изменяя двойной электрический слой [44]. Позже было экспериментально подтверждено, что 4,4'-бипиридил образует адсорбционный слой на поверхности электрода, с которым Цит c связывается через остатки лизина перед переносом электрона [45]. Таким образом, 4,4'-бипиридил – первое соединение, которое послужило “мостиком” между белком и электродом, ориентируя белок на поверхности электрода и стимулируя прямой перенос электрона. Более поздние работы подтвердили важность наличия некого связующего звена между активным центром фермента и поверхностью электрода для эффективного переноса электрона. В частности, подобный эффект наблюдали для гемопротеинов при использовании пленок поверхностно-активных веществ [46].
Дальнейшие исследования в области электрохимии содержащих кофактор белков привели к иммобилизации ферментов на поверхности электрода и открытию явления “биоэлектрокатализа”. Термин “биоэлектрокатализ” определяется как ускорение электрохимических реакций с помощью биологических катализаторов [47]. “Биоэлектрокатализом” также можно назвать биокатализ с заменой биологических систем доставки электронов на электрохимические системы. Прямой биоэлектрокатализ предполагает отсутствие каких-либо свободно диффундирующих или иммобилизованных медиаторов; электроны беспрепятственно переходят между активным центром фермента и электродом [47]. Пионерские работы в области биоэлектрокатализа были выполнены в конце 1970–1980-х гг. советскими учеными во главе с Березиным. Первый кислородный ферментный электрод был изготовлен иммобилизацией лакказы на электроде из сажи [48]. Молекулярный кислород восстанавливался в активном центре фермента, при этом электроны поступали из материала электрода (уравнение (13)):
(13)
${{{\text{O}}}_{2}} + 4\bar {e} + 4{{{\text{H}}}^{ + }} \to 2{{{\text{H}}}_{2}}{\text{O}}{\text{.}}$Лакказа относится к семейству голубых медьсодержащих оксидаз, к которому также принадлежат билирубиноксидаза, аскорбатоксидаза и церулоплазмин. Медьсодержащие оксидазы обладают способностью к прямому восстановлению кислорода до воды (уравнение (13)) без образования высокореакционноспособных токсичных промежуточных соединений кислорода, таких как супероксид анион-радикал $({\text{O}}_{2}^{{-- \bullet }}),$ гидроксил радикал (•OH) и пероксид водорода (H2O2). Еще один большой класс белков, содержащих редокс-активные кофакторы, – это гемопротеины. Первая работа, описывающая биоэлектрокатализ с участием гемопротеина пероксидазы, иммобилизованной на электроде из сажи [49], была опубликована в 1979 г., а пять лет спустя был открыт прямой биоэлектрокатализ с участием другого гемопротеина – гидрогеназы [50]. Очевидно, что принципы электрохимии содержащих кофактор редокс-активных белков основаны на биологических реакциях переноса электронов. Природа использует относительно ограниченную палитру редокс-активных центров: гем, хиноны, флавины, железо-серные кластеры. Понимание биологических принципов, определяющих эффективный перенос электрона, важно при использовании природных переносчиков электрона (оксидоредуктаз, НАДH-зависимых дегидрогеназ и редокс-активных белков) в биосенсорах, биотопливных элементах и биоэлектросинтезе [51].
Параллельно в 1960-х гг. было продемонстрировано и детально изучено электрохимическое окисление серосодержащих аминокислот Цис и Цис-Цис на электродах из платины и золота [52–54]. Спустя два десятилетия, в 1980 г., две независимые группы под руководством Брабека [55] и Рейно [56] описали окисление белков за счет аминокислотных остатков тирозина (Тир) и триптофана (Трп) на электродах из углеродных материалов. Сигнал окисления рибонуклеазы, альбумина, лизоцима и инсулина наблюдали на импрегнированном графитовом электроде при потенциалах около 0.7–0.8 В (отн. насыщенного каломельного электрода, НКЭ) [55]. На угольно-пастовом электроде при потенциалах 0.8–0.9 В (отн. НКЭ) удалось зарегистрировать сигналы электроокисления РНКазы (рибонуклеазы), альбумина и конканавалина А, в то время как на золотом, платиновом и стеклоуглеродном электродах четких пиков окисления этих белков получено не было [56]. С тех пор электрохимия белков и пептидов развивается преимущественно с использованием твердых электродов.
Нуклеиновые кислоты. У истоков электрохимии нуклеиновых кислот стоял Эмиль Палечек (1930–2018, Брно, Чехия). Свою жизнь он посвятил исследованию свойств ДНК и других биополимеров и оставил после себя огромное число работ (книг, обзоров и оригинальных статей). Активное изучение электрохимических свойств нуклеиновых кислот началось в конце 1950-х гг., когда было показано окисление и восстановление молекул ДНК и РНК на ртутном электроде [57, 58]. Ученик Гейровского, Палечек в 1958 г., еще будучи аспирантом, наблюдал восстановление аденина (Аде) и цитозина (Цит) на ртутном капающем электроде, причем восстановление последнего протекало не только в кислой, но и в нейтральной среде; гуанин (Гуа) давал характерный анодный сигнал, обусловленный окислением продукта восстановления ДНК при высоких отрицательных потенциалах [57, 59–61] (рис. 2). До этого считалось, что из компонентов нуклеиновых кислот только Аде способен электрохимически восcтанавливаться на ртути в сильнокислых средах [62]. Любопытно, что в то же самое время, в 1957 г., Берг [63], определяя следы белка в образцах ДНК и РНК в присутствии ионов кобальта, подтвердил электрохимическую инертность нуклеиновых кислот. В 1960 г. Миллер [64, 65] впервые доказал способность ДНК адсорбироваться на поверхности поляризованного ртутного электрода. Эмиль Палечек так вспоминал о своей работе в те годы [57]: “Вскоре после публикации моей статьи об осциллографической полярографии ДНК из тимуса теленка и продуктов ее деградации в журнале Nature (1960 г.) [60] я был приглашен Юлиусом Мармуром из Гарвардского университета пройти стажировку в его лаборатории в качестве докторанта. Прошло около двух лет, прежде чем мне разрешили покинуть коммунистическую Чехословакию, но для меня это было похоже на чудо. В 1962 г. оборудование для осцилографической полярографии “Polaroskop P 524” производили только в Чехословакии. Мармур, который к этому времени перешел из Гарварда в университет Брандейса, посоветовал мне привезти прибор с собой. Таким образом, я отправил прибор авиагрузом. Вооружившись рекомендательным письмом от Гейровского, в ноябре 1962 г. я поехал в Соединенные Штаты без каких-либо сомнений о пригодности моего метода в исследовании нуклеиновых кислот. К сожалению, прибор прибыл только через девять месяцев, полностью сломанный. В лаборатории преобладало мнение, что он был тщательно “обыскан” секретными службами по обеим сторонам “железного занавеса”. В этом месте заканчиваются воспоминания, процитированные по вышестоящей ссылке [57]. В Соединенных Штатах Палечек приобрел необходимые микробиологические знания и практический опыт в молекулярной биологии. В то время Мармур был одним из ведущих специалистов в области ДНК. Мармур первым обнаружил ренатурацию и гибридизацию ДНК, т.е. ее способность восстанавливать двойную спираль из комплементарных одиночных нитей [66, 67]. Эта способность ДНК используется сегодня в целом ряде молекулярно-биологических методов, также как и в биотехнологии, включая электрохимические ДНК-датчики. Мармур также предложил метод выделения ДНК [68], который применялся с некоторыми поправками в течение нескольких десятилетий как классический метод извлечения ДНК из бактерий и других организмов. Вернувшись из США, Палечек продолжил активную работу в области электроанализа нуклеиновых кислот. Открытие Палечека заложило фундамент для дальнейшего изучения электрохимического поведения молекул нуклеиновых кислот и родственных им соединений.

В конце 1970-х гг. Каммингс и Элвинг осуществили одноэлектронное восстановление на ртути тимина (Тим, El/2 = –2.4 В) [69] и урацила (Ура, El/2 = –2.3 В) [70] с формированием соответствующих анион-радикалов и переносом протонов в среде из диметилсульфоксида (рис. 2). При этом, анионы Ура и Тим образовывали с ртутью нерастворимые соли, давая последующие волны окисления продуктов восстановления (El/2 от –0.1 до –0.3 В) [69, 70]. В отличие от Ура, в случае Тим, содержащего в пятом положении метильную группу, стабилизирующую свободный радикал и создающую стерическое препятствие для димеризации, наблюдалось восстановление образовавшегося свободного радикала [70]. Было показано, что все нуклеиновые основания и некоторые другие производные пурина и пиримидина дают анодные сигналы, благодаря формированию малорастворимых соединений с ртутью электрода, что в дальнейшем использовали для их катодного инверсионного определения в наномолярных концентрациях [71–73]. В середине 1980-х гг. группой Палечека был предложен адсорбционный инверсионный анализ нуклеиновых кислот, который позволил на несколько порядков повысить чувствительность определения концентрации ДНК. Было обнаружено, что ДНК и РНК легко иммобилизуются на поверхности ртутных электродов путем простого погружения электрода в каплю раствора нуклеиновой кислоты (3–10 мкл) на короткое время (30–180 с) [74, 75]. Благодаря сильной адсорбции, ДНК и РНК формируют стабильный слой на поверхности электрода; далее электрод промывают и проводят вольтамперометрические измерения в новом растворе, уже не содержащем аналита. Такая процедура позволила снизить объем анализируемой пробы на 2–3 порядка по сравнению с классической вольтамперометрией. Данную технику назвали “адсорбционная инверсионная вольтамперометрия с переносом электрода (АИВАПЭ)”. Комбинирование АИВАПЭ с постояннотоковой хронопотенциометрией оказалось особенно успешным в определении нуклеиновых кислот с помощью ртутных электродов [76].
В течение первых двух десятилетий для определения содержания нуклеиновых кислот использовали только ртутные электроды. Однако, несмотря на уникальные преимущества, электроды из ртути имеют ряд ограничений, включая механическую нестабильность и узкую анодную область рабочих потенциалов, только в редких случаях подходящую для окисления органических соединений. В 1980-е гг. Брабек – аспирант Палечека [77, 78], после защиты диссертации ставший сотрудником Драйхерста, вместе с последним ввел в электрохимию нуклеиновых кислот электроды из углеродных материалов [79–83]. Учитывая вклад Брабека в электрохимию белков, его смело можно назвать основателем прямого электрохимического анализа биополимеров на твердых электродах. Удивительно, что первые публикации об электрохимическом окислении высокомолекулярных нуклеиновых кислот на электродах из углеродных материалов появились только в 1978 г. [79–81]. Уже в начале 1960-х гг. Элвинг и Смит [84, 85], изучая поведение мономерных компонентов нуклеиновых кислот на электроде из графита, показали, что Гуа и Аде способны окисляться на данном типе электродов. Позже Драйхерст с соавт. [86, 87] более подробно исследовали окисление Аде и Гуа на электроде из пиролитического графита и предложили возможные механизмы электродных реакций. Важным результатом исследования электроокисления низкомолекулярных компонентов нуклеиновых кислот было открытие об окислении нуклеозидов Аде и Гуа при значительно более высоких положительных потенциалах, чем потенциалы, характерные для свободных оснований [88, 89]. Эти исследования также показали существенную разницу между потенциалами пиков окисления гуанозина и аденозина. В 1980 г. Брабек опубликовал обнаруженные им с помощью дифференциальной импульсной вольтамперометрии количественные закономерности окисления молекул ДНК из разных природных объектов на электроде из пиролитического графита [82]. Изученные молекулы ДНК отличались суммарным содержанием оснований Гуа и Цит (Гуа + Цит). Все образцы ДНК давали на вольтамперограмме “пик Г” (при потенциале около 0.9 В отн. НКЭ, рН 6.4), соответствующий окислению остатка Гуа, и “пик А” (около 1.2 В), соответствующий окислению Аде. Отмечается, что потенциалы пиков Г и А отличались на 0.28 В и не отражали содержание (Гуа + Цит) в образце ДНК. Однако было найдено, что отношение высот пиков А и Г, обозначенное K, равно отношению (Аде + Тим)/(Гуа + Цит) образца ДНК. Этот факт использовали для разработки метода определения содержания (Гуа + Цит) в ДНК согласно формуле:
где K – отношение высот пиков А и Г, соответствующих окислению остатков Аде и Гуа ДНК. Интересно, что отношение высот пиков А и Г было одним и тем же для нативной и термически денатурированной ДНК. В случае денатурированной ДНК величины сигналов заметно возрастали по сравнению с неденатурированной [82].Интерес электрохимиков к нуклеиновым кислотам был в немалой степени поддержан вручением в 1962 г. Нобелевской премии по медицине Вилкинсу, Крику и Уотсону “за открытия, касающиеся молекулярной структуры нуклеиновых кислот и их значения для передачи информации в живых системах”. К моменту первых публикаций об электрохимической активности нуклеиновых кислот была известна структура ДНК в форме двойной спирали, показанная Уотсоном и Криком [90] одновременно с Франклин и Гослингом [91] в 1953 г. Это произошло спустя почти сто лет после открытия молекулы ДНК Мишером в 1869 г. [92]. Уотсон и Крик предложили систему на основе спаренных оснований [90]. Структура ДНК включала две водородные связи для пары Аде и Тим и три водородные связи для пары Гуа и Цит. Такая молекула нуклеиновой кислоты теперь известна как B-форма ДНК, стабилизируемая связанной водой, которая идеально вписывается в минорную бороздку. Альтман в 1889 г. дал имя ДНК, а ее роль в передаче генетической информации была открыта Гриффитом в 1928 г. [92]. В 1950 г. Чаргафф с соавт. [93] обнаружили, что в ДНК количество Аде всегда равно количеству Тим, а количество Гуа всегда равно количеству Цит. Эта закономерность была крайне важна при разработке модели двойной спирали. Количество пуринов (Аде + Гуа) всегда равно количеству пиримидинов (Цит + Тим), но отношение (Гуа + + Цит)/(Аде + Тим) варьируется от вида к виду.
Таким образом, можно заключить, что к концу 1980-х годов был заложен фундамент электрохимии белков и нуклеиновых кислот и опубликованы основные виды аналитических сигналов окисления и восстановления данных биомолекул на ртутном электроде и электродах из твердых материалов. Однако, несмотря на знания о сложной пространственной организации молекул биополимеров, в электрохимии господствовало представление об одномерной структуре белков и нуклеиновых кислот при интерпретации результатов. Хотя многие процессы и явления, такие как прямой перенос электрона или биоэлектрокатализ, к этому времени были открыты, природа аналитических сигналов биополимеров не была до конца объяснена.
СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ ОБ ЭЛЕКТРОХИМИЧЕСКИХ СВОЙСТВАХ БЕЛКОВ И НУКЛЕИНОВЫХ КИСЛОТ
Сегодня ученые-электрохимики приходят к пониманию необходимости расширения представлений о редокс-процессах, протекающих на поверхности электродов с участием биополимеров, таких как белки и нуклеиновые кислоты, прежде всего, принимая во внимание их пространственную (3D) структуру [94]. Долгое время электрохимическое поведение биополимеров рассматривалось с точки зрения общего числа тех или иных электроактивных групп в первичной последовательности молекул, опираясь на число мономерных звеньев. Однако последние исследования свидетельствуют, что далеко не все потенциально электроактивные группы в полипептидной или полинуклеотидной цепи могут одновременно участвовать в электрохимических реакциях [95]. C увеличением молекулярной массы биополимера (площади его поверхности), приходящейся на одну электроактивную группу, электрохимический сигнал падает [95, 96]. С развитием компьютерных технологий и методов биоинформатики, позволяющих моделировать пространственную структуру макромолекул, становится очевидным, что электрохимический сигнал биополимера не может быть равен сумме сигналов входящих в него мономеров. Это, с одной стороны, усложняет интерпретацию аналитического сигнала, а с другой, – открывает широкие перспективы для применения электрохимии с целью решения биохимических и медицинских задач. Опираясь на структуру молекулы, с помощью электрохимического анализа ученый может зарегистрировать такие изменения, как образование биполимерных комплексов [97], агрегация [98] или деградация биополимеров [99], а также их посттрансляционные (ПТМ) [100] или пострепликативные (ПРМ) модификации [101]. Такие изменения в структуре биополимеров происходят в живых системах как в норме, так и при различных патологиях [98–105]. К тому же, если раньше господствовало четкое разделение на “электроактивные” и “неэлектроактивные” вещества, то сегодня более корректно будет сказать, что биомолекула неэлектроактивна в данных экспериментальных условиях. Возникает уверенность, что поиск подходящих условий в будущем позволит открыть новые свойства веществ, в частности, биополимеров, сахаров и липидов. Углубление понимания взаимосвязи между пространственной организацией белков и нуклеиновых кислот и их электрохимическим поведением способствует появлению принципиально новых (био)сенсорных систем как для повседневного применения, так и для биомедицинских исследований.
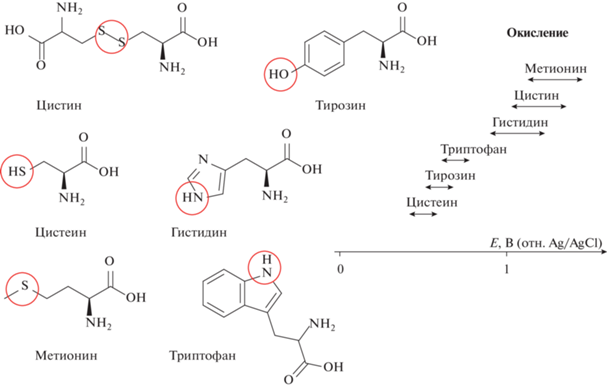
Белки. На сегодняшний день известно, что на твердых электродах белки способны давать электрохимический сигнал: (1) за счет окисления аминокислотных остатков (в диапазоне потенциалов от 0.5 до 1.5 В); (2) и/или за счет восстановления или окисления своих простетических групп (например, гема или флавина в диапазоне от –0.5 до 0.5 В отн. Ag/AgCl) [47, 51, 106–112] (рис. 3). Подробный анализ электрохимического поведения биомолекул, в частности, биополимеров, на электродах из углеродных материалов (графит, графен, углеродные нанотрубки, допированный бором алмаз) с учетом влияния структуры поверхности электрода на адсорбцию молекул дан в обзоре Фойты с соавт. [112]. Необходимо отметить, что, в отличие от ставших рутинными графитовых и стеклоуглеродных электродов, допированный бором алмаз обладает более широким рабочим диапазоном положительных потенциалов. Однако, насколько известно, до сих пор не обнаружены какие-либо принципиально новые свойства белковых молекул и нуклеиновых кислот на электродах из допированного бором алмаза [113], впрочем, как и на электродах, модифицированных графеном или углеродными нанотрубками. При этом недавно показано [114], что практически все протеиногенные аминокислоты способны окисляться на поверхности печатных графитовых электродов при потенциале 0.95 В в амперометрическом проточно-инжекционном анализе, а в некоторых случаях может быть зарегистрирован и сигнал восстановления продуктов окисления аминокислот, в частности, Трп на электроде из карандашного графита [115]. Тем не менее четких аналитически значимых сигналов восстановления протеиногенных аминокислот и их остатков пока получить не удалось. Наибольший вклад в сигнал окисления молекулы белка при потенциале 0.6–0.8 В вносят остатки Тир, Трп и Цис [109, 116–124], тогда как для окисления остатков Гис, Мет и Цис-Цис требуется потенциал около или выше 1 В (отн. Ag/AgCl, нейтральная среда) [118–124] (рис. 4). Так, показано, что пептид Aβ(1–42) на квадратно-волновой вольтамперограмме дает два четких пика при потенциалах 0.6 и 1.0 В и волну в области потенциалов 1.2–1.5 В, отнесенных соответственно к остаткам Тир (Тир-10), Гис (Гис-6, -13, -14) и Мет (Мет-35) пептида [122]. На электродах из углеродных материалов и золота окисление белков за счет аминокислотных остатков протекает как необратимый рН-зависимый процесс [95, 117–120]. Необходимо отметить, что механизм окисления остатков аминокислот в составе белка остается до конца не изученным, также как и реакции окисления большинства свободных аминокислот [109, 110, 125–127]. Требуется установление продуктов электрохимических реакций с помощью различных физико-химических методов (в частности, ЯМР и ИК-спектроскопии). Возникает закономерный вопрос о связи структуры белка с его электрохимическими свойствами. Одна из первых попыток ответить на данный вопрос была предпринята Брабеком и Морнштейном, которые писали [55]: “Бычий сыворочный альбумин, также содержащий, как и лизоцим, остатки триптофана, дает, однако, только один пик при потенциалах окисления остатков тирозина. Это поведение может быть объяснено тем фактом, что бычий сывороточный альбумин содержит примерно в 10 раз больше остатков тирозина, чем триптофана”. Таким образом, видно, что в данной работе, опубликованной в 1980 г., авторы опирались лишь на первичную структуру белка, а именно на его аминокислотную последовательность, и предполагали, что количество электроактивных остатков, таких как Тир и Трп, определяет наблюдаемое электрохимическое поведение белков. Потребовалось три десятилетия, чтобы понять, что только остатки аминокислот, расположенные на поверхности молекулы белка, с электроактивными группами, ориентированными наружу, доступны для электрохимического окисления на поверхности электрода и, следовательно, определяют сигнал электроокисления [95]. Если белки имеют достаточно жесткую пространственную структуру, то конформация пептидов часто характеризуется как “случайный клубок”. Электрохимию пептидов можно выделить в отдельную область [128]: из-за небольшой молекулярной массы сигналы их аминокислотных остатков выражены ярче и легче интерпретируются. Веским аргументом в пользу тесной связи интенсивности сигнала электроокисления со структурой белка является то, что разворачивание (денатурация) белка приводит к значительному увеличению величины тока [55, 120, 129]. Напротив, агрегация белков или пептидов приводит к уменьшению тока электроокисления [130–134], что также позволяет предположить, что не общее число электроактивных остатков, а их расположение на поверхности белковой молекулы определяет наблюдаемое электрохимическое поведение. Интересно, что крупные пептидные агрегаты, такие как агрегаты β‑амилоида, участвующего в патогенезе болезни Альцгеймера, неспособны производить какой-либо значимый сигнал окисления: во время агрегации пептидов основной вклад в общий сигнал образца вносят мономеры и мелкие олигомеры [130]. Фактически, это делает электрохимию уникальным методом мониторинга агрегации пептидов путем регистрации уменьшения тока окисления, вызванного исчерпанием пула пептидных мономеров/олигомеров при их включении в агрегаты [131–134]. Показано также, что сигнал электроокисления чувствителен к заменам отдельных аминокислот в полипептидной цепи, как это происходит в случае мутантов β-амилоида [122, 123, 135] и генетически модифицированных вариантов ацетилхолинэстеразы [117]. В последнем случае некоторые замены электроактивных остатков на условно “неэлектроактивные” приводили к увеличению сигнала электроокисления, указывая на значимость конформационных изменений, связанных с заменами, для доступности электроактивных остатков для окисления на поверхности электрода [95]. Таким образом, изменения конформации также могут влиять на интенсивность сигнала электроокисления наряду с концентрацией белка или пептида [55, 120, 122, 136].
Рис. 3.
Потенциалы сигналов окисления и восстановления белков и нуклеиновых кислот на твердых электродах.

Рис. 4.
Потенциалы и сайты окисления аминокислот тирозина, триптофана, цистеина, гистидина, метионина и цистина на твердых электродах.
Возвращаясь к активным центрам, необходимо отметить, что регистрация редокс-активности белков, несущих простетические группы, требует специальной модификации поверхности электрода, позволяющей “соединить” активный центр и электрод [46, 47, 108]. Фактически, это также подтверждает гипотезу о сохранении пространственной структуры редокс-активного белка в условиях электрохимического эксперимента на поверхности электрода [46]. Как правило, электродные реакции с вовлечением активных центров белков протекают в тонком слое на поверхности электрода и характеризуются линейной зависимостью тока пика от скорости развертки потенциала [46, 47, 106–108]. Белок Цит с является, возможно, единственным исключением из этого правила и дает обратимый сигнал иона Fe(III)/Fe(II) гема из капли раствора даже на немодифицированном электроде [39, 43]. В случае редокс-активных белков их “ориентированная” иммобилизация на поверхности электрода приобретает определяющее значение для достижения эффективного переноса электронов и биоэлектрокатализа [47, 137]. Добиться упорядоченного расположения молекул фермента на поверхности электрода, т.е. сориентировать их для наблюдения наиболее эффективного биоэлектрокатализа, возможно, с помощью проводящих полимеров или проводящих наноматериалов. Помимо этого широко используют мутантные формы ферментов со специально введенными аминокислотными заменами. Прямой перенос электрона с электрода на активный центр фермента (белка) позволяет определить концентрацию редокс-активных белков путем регистрации величины тока пика восстановления или окисления его активных центров [138–140]; а ферментативная активность белка может быть инициирована наложением соответствующего потенциала [137, 141]. Однако поиск подходящей матрицы для иммобилизации редокс-активных белков является непростой задачей, и часто оказывается невозможным добиться эффективного соединения биохимической ферментативной реакции с электродной. В некоторых случаях прямой перенос электронов между активным центром фермента (белка) и электродом недостижим по определению из-за гликозилирования или большого размера белковой глобулы. Изучая механизмы прямого биоэлектрокатализа [47], можно выявить взаимосвязь между структурой и функциями белка-фермента, что важно для понимания биохимических процессов, а также для разработки биосенсорных систем. Направленное введение аминокислотных остатков, таких как Гис и Цис, пептидов или фрагментов биотина, с использованием технологий генной инженерии позволило разработать редокс-активные ферменты с заданными электрохимическими свойствами, включая возможность их “ориентированной” иммобилизации [137, 142]. Известно, что некоторые аминокислотные замены (мутации) могут оказывать заметное влияние на ферментативную активность белков. Так, изучение электрохимической и электрокаталитической активности иммобилизованных на поверхности стеклоуглеродного электрода гемопротеинов семейства цитохромов Р450 (CYP), а именно CYP 2C9 человека и двух его полиморфных вариантов, присутствующих примерно у 35% кавказской популяции, показало более низкую каталитическую активность для форм CYP 2C9*3 (ks = = 3 ± 1 мин–1) и CYP 2C9*2 (ks = 12 ± 2 мин–1) по сравнению с диким типом CYP 2C9 (ks = 18 ± 1 мин–1) в присутствии S-варфарина – маркерного субстрата CYP 2C9 [143]. Разработанный биоэлектрохимический способ позволяет определять терапевтически значимые различия в метаболизме лекарственных средств, связанные с полиморфизмом, ответственным за неблагоприятные лекарственные реакции, в частности, у значительной части населения Кавказа. С помощью точечных аминокислотных замен в последовательности фермента можно решить и задачу увеличения скорости прямого переноса электрона в электрохимических биосенсорах, как показано на примере пероксидазы из табачной мозаики [144]. Авторы наблюдали значительное увеличение скорости прямого переноса электронов между графитовым электродом и пероксидазой из табачной мозаики после замены остатка лейцина, расположенного вблизи кармана гема на поверхности фермента, на Трп [144]. Увеличение скорости прямого переноса электронов между редокс-активным центром белка и электродом и повышение чувствительности определения субстрата фермента могут быть достигнуты с помощью изменения пространственной структуры белка, т.е. направленного разворачивания (денатурации) полипептидной цепи, как показано на примерах гемоглобина [145] и Цит с [146] по отношению к пероксиду водорода и нитрит-иону. Стоит отметить, что для гемопротеинов обнаружено множество ПТМ [102]. На вопрос, влияют ли эти ПТМ на способность иона Fe(III)/(II) гема белка отдавать или принимать электроны, можно ответить с помощью электрохимии. Например, показано [147], что миоглобин скелетных мышц лошади с электрохимически пронитрованным Тир-103 дает более низкий и менее выраженный пик востановления, чем у нативного миоглобина на циклических вольтамперограммах. К сожалению, несмотря на важность влияния ПТМ на структуру и функции редокс-активных гемопротеинов, таких как Цит с [148] или цитохром Р450 2B1 [149], изменения в интенсивности прямого переноса электронов и биоэлектрокатализе этих белков в результате ПТМ в настоящее время изучены мало.
Современные технологии позволяют получить трехмерную структуру молекулы белка с локализацией всех аминокислотных остатков. В настоящее время в Банке данных о белках (PDB, protein data base, https://www.rcsb.org) доступно множество 3D моделей структур, включая сложные белковые комплексы. Конечно, результаты компьютерного моделирования не обязательно предсказывают истинную локализацию электроактивных групп аминокислотных остатков в условиях электрохимического эксперимента, и следует иметь в виду, что на их расположение могут в некоторой степени влиять: (1) состав электролита (растворителя), (2) электрическое поле электрода и (3) степень жесткости молекулы белка как таковой. Другим важным аспектом электроокисления белка является то, что некоторые аминокислотные остатки в последовательности белка могут быть модифицированы различными функциональными группами, благодаря явлению посттрансляционной модификации [150]. Эти новые функциональные группы могут генерировать свои собственные электрохимические сигналы или влиять (подавлять или усиливать) на исходный сигнал аминокислотных остатков [100, 151]. В настоящее время как аминокислотные последовательности, так и ожидаемые ПТМ могут быть взяты для подавляющего числа белков во всеобъемлющей базе данных, известной как Универсальный белковый ресурс (UniProt, https://www.uniprot.org). Таким образом, используя Банк данных о белках и Универсальный белковый ресурс, становится возможным предсказать электрохимическое поведение на электроде практически любой белковой молекулы, что делает интерпретацию этого поведения исключительно на основе аминокислотной последовательности белка (как, например, в работах [118, 152]) довольно упрощенной. Учет пространственной структуры белка для интерпретации его электрохимических сигналов позволяет глубже понять реальные процессы, происходящие на поверхности электрода [153].
Нуклеиновые кислоты. Известно, что в процессах восстановления и окисления нуклеиновых кислот на твердых электродах участвуют остатки азотистых оснований (Гуа, Аде, Тим, Цит и Ура) [79, 82, 94, 96, 97, 112, 154–161]. Однако среди различных типов электродов только электрод из пиролитического графита позволил зарегистрировать как восстановление, так и окисление остатков азотистых оснований в нуклеозидах [162] и олигонуклеотидах [159] из-за широкого рабочего окна потенциалов от –2 до 2 В (отн. Ag/AgCl; ацетатный буферный раствор, рН 5). Кроме того и сигналы окисления продуктов восстановления, и сигналы восстановления продуктов окисления азотистых оснований в нуклеозидах и олигонуклеотидах также зарегистрированы на пиролитическом графите [159, 162] (рис. 3). Реакции восстановления или окисления нуклеиновых оснований или их остатков требуют достаточно высоких отрицательных (примерно от –1.5 до ‒2.0 В) или положительных (примерно от 1.0 до 1.5 В) потенциалов соответственно [79, 82, 86, 87, 154, 155, 158, 161–173]. Если сигналы окисления или восстановления оснований Гуа и Аде и их остатков в молекулах нуклеиновых кислот достаточно легко получить в различных экспериментальных условиях и на различных типах твердых электродов, то Тим, Цит и Ура требуют более высоких положительных или отрицательных потенциалов и более жестких условий эксперимента, и, как следствие, атрибуция их сигналов часто довольно сомнительна (рис. 5). Так, окисление мононуклеотидов на углеродных электродах проявлялось в виде анодных пиков при потенциалах 0.9–1.0 В для гуанозинмонофосфата, 1.2–1.3 В для аденозинмонофрсфата, 1.4–1.5 В для тимидинмонофосфата и 1.5–1.6 В для цитидинмонофосфата (отн. Ag/AgCl, фосфатный буферный раствор, рН 7.4) [163, 174]. Интересно, что добавление сахара и фосфатных групп к азотистому основанию (с образованием нуклеозида или нуклеотида) сдвигает максимум потенциала окисления молекул к более положительным значениям и наоборот [86, 87, 164, 165]. Однако процессы окисления и восстановления нуклеиновых оснований в молекулах ДНК происходят при потенциалах, отличных от потенциалов, характерных для соответствующих свободных нуклеозидов или нуклеотидов, что указывает на то, что процессы окисления остатков оснований в мономерных и полимерных молекулах протекают по разным механизмам и дают разные продукты [159, 162]. В то время как механизмы и продукты электрохимического окисления или восстановления свободных азотистых оснований, их нуклеозидов и нуклеотидов достаточно хорошо изучены [55, 86, 87, 159, 162, 164–168] (рис. 5), аналогичные подробные исследования для остатков оснований в полимерных молекулах ДНК или РНК отсутствуют, поэтому какие-либо выводы на основе знаний о мономерах оказываются довольно умозрительными [162]. Как окисление, так и восстановление нуклеиновых оснований могут включать образование промежуточных продуктов в виде радикалов и их химические реакции, которые по своей природе чувствительны к расположению активных фрагментов [162]. Многие факторы, такие как последовательность и структура нуклеиновых кислот, концентрация аналита, свойства поверхности электрода, рН и состав фонового электролита, могут влиять на механизмы электродных реакций нуклеиновых оснований [162]. Надо сказать, что на сегодняшний день на твердых электродах не зарегистрировано сигналов восстановления остатков оснований высокомолекулярных ДНК или РНК из природных источников. В то же время сигналы окисления и восстановления получены для всех остатков оснований в коротких синтетических олигонуклеотидах определенной последовательности с преобладанием сигналов окисления остатков Гуа и Аде [159, 161, 173–176]. Однако необходимо отметить, что в большинстве работ, к сожалению, не приводится никакой информации о чистоте олигонуклеотидов или о дополнительной очистке полученных синтетических препаратов, так же как и о конформации молекул однонитевой ДНК (онДНК).
Рис. 5.
Схемы реакций первичного окисления гуанина и аденина на электродах из углеродных материалов [83].

Что касается высокомолекулярной двунитевой ДНК (днДНК) из природных источников, то ее окисление на твердых электродах за счет остатков оснований, впервые продемонстрированное в пионерских работах Брабека [82, 154, 155], до сих пор остается спорным [112, 158, 169, 175]. На вольтамперограммах, снятых на электродах из углеродных материалов, молекулы днДНК дают только два пика окисления: около 0.7–0.9 В и 1.0–1.2 В (отн. Ag/AgCl, нейтральная среда), которые приписывают остаткам Гуа и Аде соответственно [79, 82, 83, 155, 172]. Выдвинута гипотеза, что электроактивные группы этих азотистых оснований, не участвующие в образовании водородных связей в двойной спирали ДНК, остаются свободными для электроокисления на поверхности электрода [82, 154, 155]. Также обнаружено, что токи пиков окисления для нативной днДНК заметно ниже по сравнению с сигналами денатурированной ДНК [82, 95, 154, 155] и закономерно уменьшаются с увеличением логарифма молекулярной массы ДНК как для нативных, так и для денатурированных биополимеров [96]. Для объяснения наблюдаемых различий в электрохимическом поведении нативной и денатурированной ДНК высказано предположение, что ДНК адсорбируется на поверхности электрода, и гибкая полимерная цепь денатурированной ДНК может лучше прилегать или повторять неровную поверхность электрода, чем более жесткая молекула нативной днДНК [154, 155, 95 ]. В результате количество сегментов ДНК, находящихся в контакте с поверхностью электрода или в непосредственной близости от нее так, чтобы их можно было электроокислить, выше для денатурированной ДНК, чем для нативной днДНК. На самом деле, это яркий пример рассмотрения молекулы ДНК как одномерной структуры. Действительно, данная гипотеза частично подтверждена выводами о том, что предварительная поляризация электрода при положительных потенциалах (от 0.2 до 1.3 В) в течение нескольких минут требовалась для надежной регистрации сигналов окисления как от нативной, так и от денатурированной ДНК и даже от небольших олигонуклеотидов на углеродных и золотых электродах [96, 154, 155, 169–171, 174]. Это объяснялось более эффективной адсорбцией отрицательно заряженных биомолекул на положительно заряженных электродах. Например, обнаружено, что после предполяризации электрода из высокоупорядоченного пиролитического графита при 0.4 В (отн. Ag/AgCl) в течение 15 мин днДНК из тимуса теленка дает выраженные пики окисления остатков Гуа и Аде, которые не могут быть получены без предполяризации [171]. Однако в другой работе сигнал окисления остатков Гуа нативной днДНК из тимуса теленка не удалось получить даже после предполяризации [79]. Более того, Ванг с соавт. [177], используя амперометрический проточно-инжекционый анализ при потенциале 1 В, продемонстрировали, что нативная днДНК из тимуса теленка дает на угольно-пастовых электродах более высокие токи окисления, чем денатурированная ДНК. В настоящее время все чаще признается, что азотистые основания, скрытые внутри двойной спирали ДНК, могут быть труднодоступны или недоступны совсем для электродных реакций [157, 178]. Таким образом, результаты экспериментов по электрохимическому окислению днДНК пока вызывают больше вопросов, чем ответов.
Одним из объяснений наблюдаемых расхождений в электрохимическом поведении ДНК может быть деградация, особенно с учетом известной зависимости тока электроокисления от размера ДНК [96]. Деградация может привести к образованию низкомолекулярных фрагментов ДНК и/или, возможно, свободных мономеров, которые могут существенно повлиять на результат электрохимического эксперимента. Действительно, деградация ДНК продемонстрирована для днДНК из тимуса теленка, иммобилизованной на поверхности золотого электрода при потенциале 0.5 В (отн. Ag/AgCl) [179]. Процедура предполяризации электрода потенциально может способствовать такой деградации. Присутствие в образце ионов металлов (или их комплексов) в комбинации с наложением на электрод отрицательного потенциала может разрушать молекулы ДНК, как показано для ДНК и комплексов Cu(II)-бипиридил при –0.6 В [180]. Тем не менее, деградирует ли ДНК на электродах из углеродных материалов во время электрохимического эксперимента, в настоящее время до конца не известно [155].
Хотя использование в исследованиях днДНК из природных источников представляется крайне привлекательным из-за доступности и низкой стоимости коммерческих препаратов, такие препараты страдают неоднородностью размеров молекул и потенциального присутствия как онДНК, так и днДНК, как справедливо указывали Брабек и Куделка в 1980 г. [96]. К сожалению, во многих электрохимических работах (например, [171, 172, 177, 181]) отсутствует биохимическая характеристика используемых образцов ДНК, что затрудняет сравнение результатов, полученных разными авторами, и их интерпретацию. Очевидно, что ампликоны, полученные с помощью полимеразной цепной реакции (ПЦР), являющиеся идеальными фрагментами днДНК заданной длины, которая может варьироваться от десятков до сотен пар оснований, – более подходящая модель для изучения электроокисления днДНК. Их применение позволило бы лучше понять механизмы, лежащие в основе электроокисления днДНК в электрохимическом эксперименте. Недавно показано [175], что электроокисление ДНК природного происхождения на печатных графитовых электродах протекает преимущественно путем окисления остатков Гуа и Аде молекул онДНК, что выражается двумя сигналами при потенциалах около 0.75 и 1.05 В (фосфатный буферный раствор, рН 7.4) соответственно. Для онДНК обнаружено, что реакции окисления обоих остатков контролируются диффузией. В молекулах днДНК, даже таких коротких, как молекулы длиной 24 нуклеотида, остатки этих оснований, по-видимому, недоступны для окисления на углеродных электродах. Можно сделать вывод, что сигналы электроокисления для конкретного гетерогенного препарата ДНК природного происхождения продуцируются преимущественно низкомолекулярными фрагментами онДНК [175]. Такое электрохимическое поведение ДНК хорошо согласуется с результатами, полученными для белковых молекул, в которых электроактивные аминокислотные остатки, скрытые внутри белковой глобулы [95] или пептидного агрегата [128, 130, 132], недоступны для электродных реакций.
Хотя прямая электрохимия нуклеиновых кислот характеризуется высокими потенциалами окисления (от 1 до 2 В) и восстановления (от –1 до –2 В) и относительно низкими значениями регистрируемых токов, некоторые системы электрохимических (био)сенсоров разработаны на основе снижения сигналов электроокисления при образовании двойной спирали [99, 178, 182]. С целью улучшения аналитических характеристик широко используют электроды, модифицированные различными материалами, позволяющие достичь электрокатализа в отношении нуклеиновых кислот и их мономеров и, как следствие, повысить чувствительность анализа [156]. Альтернативная стратегия, предложенная группой Хочека и Фойты [183, 184], основана на прямом электрохимическом зондировании ДНК с модифицированными основаниями. Разработана палитра электрохимически активных групп, которые могут быть введены в последовательности ДНК путем полимеразного включения химически модифицированных нуклеотидов [183, 184]. Такой подход позволил значительно повысить чувствительность и селективность электрохимического детектирования нуклеиновых кислот на различных электродах. Более того, стало возможным мультипотенциальное редокс-кодирование нуклеиновых кислот. Данный подход может иметь широкий спектр аналитических применений, включая регистрацию мутаций, повреждения, гибридизации, амплификации ДНК, а также анализ взаимодействия ДНК с белками. Электрохимические свойства нуклеиновых кислот потенциально могут быть использованы для мониторинга различных ПРМ ДНК и РНК [185, 101 ]. Подобно аминокислотным остаткам в белках, большое количество модифицированных нуклеозидов идентифицировано в ДНК и РНК живых организмов, вирусов, митохондрий и хлоропластов в результате нормальных и патогенных ферментативных или неферментативных процессов [185]. Разработаны различные электрохимические стратегии для обнаружения ДНК или РНК как потенциальных онкологических биомаркеров, например, метилированой ДНК промоторов генов, циркулирующей опухолевой ДНК, вирусных нуклеиновых кислот или коротких некодирующих молекул РНК, в частности, микроРНК [101]. В завершение необходимо отметить, что селективность электрохимического определения веществ, прежде всего, задается методами пробоподготовки, выделения и концентрирования. Aнализу биообразцов на наличие ДНК вирусов или бактерий предшествует этап направленной амплификации, реализуемый за счет ПЦР или изотермической амплификации [186] и позволяющий выявить вплоть до одной молекулы нуклеиновой кислоты заданной последовательности в образце.
Нанопоровое секвенирование биополимеров. В современной биоэлектрохимии можно выделить еще одно перспективное направление, основанное на изменение проводимости при прохождении биомолекулы через канал наноразмеров, так называемую “нанопору”, – “нанопоровое секвенирование”. Считываемый кондуктометрический сигнал обусловлен особенностями строения биополимеров. Нанопоровое секвенирование развивается с начала 1990-х гг. и не так давно позволила успешно секвенировать ДНК [187]. Данный способ регистрации электрохимического сигнала биомолекулы открывает возможности для изучения популяционных неоднородностей и конформационной динамики систем, начиная от отдельных ДНК и заканчивая отдельными белками [188]. Нанопоры, обеспечивающие размещение отдельных объектов анализа в ограниченном пространстве, преобразуют поведение одной молекулы в регистрируемый электрохимический сигнал с высоким соотношением сигнал/шум. Большой объем исследований посвящен электрохимическому обнаружению различных объектов, начиная с нуклеиновых кислот, пептидов, белков и биомолекулярных комплексов и заканчивая органическими низкомолекулярными и высокомолекулярными молекулами с помощью нанопор. Благодаря последовательному удержанию части молекулы в нанопоре, новые механизмы считывания сигнала проливают свет на связь структуры одной молекулы с ее кондуктометрической активностью. Так, с помощью нанопоры, изготовленной из мутантного белка аэролизина T232K/K238Q с усиленным электростатическим взаимодействием на участке T232K и высоким отталкивающим барьером на участке K238Q, было изучено фосфорилирование 9-членного пептида Тау-белка, участвующего в патогенезе болезни Альцгеймера [189]. Сигнал, производимый датчиком на основе аэролизина T232K/K238Q, позволил почти со 100%-ной точностью идентифицировать характерное распределение нефосфорилированного Тау-пептида, пептида pS262-Тау, pT263-Тау и пептида pS262/pT263-Тау (где “p” указывает на фосфорилированную аминокислоту). Превосходная чувствительность данной белковой нанопоры обусловлена чрезвычайно низкой скоростью транслокации, которая увеличивает продолжительность считывания сигнала до десятков или сотен миллисекунд для пептида из девяти аминокислотных остатков [189].
В настоящее время компания Oxford Nanopore Technologies выпускает серию устройств для секвенирования ДНК под брендом Oxford Nanopore (https://nanoporetech.com). В России эксклюзивным дистрибьютором продукции Oxford Nanopore Technologies является компания SkyGen (https://www.skygen.com). Компания SkyGen регулярно проводит конференции и семинары и оказывает научную поддержку пользователям секвенаторов Oxford Nanopore. В устройствах Oxford Nanopore регистрируется изменение ионного тока в момент прохождения биомолекулы через нанопору. Информация об изменении тока используется для идентификации анализируемой биомолекулы. В приборах, изготовленых по технологии Oxford Nanopore, для создания пор в мембранах используются специально разработанные и запатентованные порообразующие белки. Порообразующие белки распространены в природе. Например, белок α-гемолизин и аналогичные белковые поры естественным образом существуют в клеточных мембранах, где они действуют в качестве каналов для переноса ионов или молекул в клетки и из клеток. Белок α-гемолизин представляет собой гептамер с порой внутренним диаметром 1 нм. В том же диапазоне лежат размеры многих биомолекул, включая ДНК. Поры достаточно стабильны. Необходимо отметить, что технология нанопорового секвенирования была бы неосуществима на практике без биоинформатической обработки регистрируемого сигнала. Компания также разработала и запатентовала электронику, которая позволяет параллельно проводить регистрацию сигналов нескольких нанопор, собирать и анализировать данные в режиме реального времени. Самый простой вариант прибора, основанного на данной технологии, – это MinION Oxford Nanopore Technologies, который позволяет считывать всю последовательность ДНК, соответствующею десяткам килобаз и ограниченную только собственной длиной исследуемых молекул [190]. Компания Oxford Nanopore ведет поиск новых решений для создания нанопор, обладающих свойствами, которые могут улучшить производительность приборов. Белковые нанопоры достаточно прочны, легко воспроизводимы при низких затратах и легко модифицируются. Однако будущие поколения сенсорных устройств на основе нанопор, вероятно, будут использовать нанопоры, изготовленные из синтетических материалов. Хотя в настоящее время твердотельные нанопоры не обладают химической специфичностью белковых.
Таким образом, электрохимия биополимеров основана на измерении сигналов восстановления или окисления редокс-активных центров белков или/и окисления их аминокислотных остатков, а также окисления или восстановления азотистых оснований в составе ДНК или РНК (рис. 3). Показано, что практически все протеиногенные аминокислоты подвержены специфическому электрохимическому окислению (при потенциале от 0.5 до 1.5 В); в то же время все нуклеотиды, входящие в состав ДНК и РНК, способны как необратимо окисляться (при потенциале от 1 до 2 В), так и восстанавливаться (при потенциале от –1 до –2 В) за счет остатков азотистых оснований на электродах из углеродных материалов (графит, стеклоуглерод). Электрохимические реакции свободных аминокислот и нуклеиновых оснований, а также их остатков в белках и нуклеиновых кислотах требуют систематического исследования с целью идентификации продуктов реакций и установления механизмов. Кроме того, электрохимия биополимеров в настоящее время нуждается в характеристике рядов из белковых молекул и нуклеиновых кислот с известными пространственной структурой, последовательностью мономеров и молекулярной массой для выявления общих закономерностей “структура–свойства”. Использование в электрохимических исследованиях препаратов без дополнительной биохимической характеристики и, при необходимости, дополнительной очистки затрудняет интерпретацию и сравнение экспериментальных результатов разных научных групп. Требуется рассмотрение электрохимического поведения белков и нуклеиновых кислот с точки зрения их пространственной структуры. Современный уровень технологий позволяет моделировать пространственную структуру одной биомолекулы и регистрировать сверхнизкие токи с высоким разрешением сигнал/шум. С помощью электрохимических подходов сегодня стало возможным определение нуклеотидной последовательности молекул ДНК, а расшифровка первичной структуры белков уже не за горами.
* * *
В заключении сопоставим две рассмотренные области электрохимии биополимеров: белков и нуклеиновых кислот. Первые работы по изучению электроактивности нуклеиновых кислот появились примерно на 30 лет позже пионерских исследований в области полярографии белков и к настоящему времени, переживая бурный рост, догоняют и даже опережают их. Интересно, что одни и те же ученые внесли заметный вклад в развитие этих двух параллельных направлений – электрохимии ДНК и белков: Палечек, Брабек, Фойта, Ванг, Оливейра-Бретт и др. Те, кто начинал с изучения электрохимии ДНК, затем перешел к белкам, и наоборот. Можно только восхищаться стойкостью Палечека, который до конца своих дней оставался верен ртутному электроду, изучая поведение различных классов биополимеров. Временем основания электрохимического анализа биополимеров на твердых электродах можно считать конец 1970-х начало 1980-х годов, когда Брабек опубликовал результаты своих исследований по электроокислению молекул ДНК и белков на электродах из углеродных материалов. В данном обзоре обобщены данные об известных на сегодняшний день собственных электрохимических свойствах биополимеров, проявляющихся на немодифицированных электродах. Зная фундаментальные свойства, исследователи в зависимости от конкретной аналитической задачи всегда смогут подобрать систему, где эти свойства проявятся желаемым образом. Развитие электрохимии белков и нуклеиновых кислот расширяет области применения электроанализа в целом – не только для количественного определения, но и для качественного анализа изменений структуры молекул. Моделируя сложные биологические системы и процессы in vitro, ученые могут исследовать влияние различных внешних и внутренних факторов, таких как рН среды или мутаций. Таким образом, электрохимия дает нам особую информацию о свойствах биомолекул, которая в комбинации с другими физико-химическими методами позволяет глубже заглянуть в молекулярные механизмы процессов в живых организмах.
Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021–2030 гг.).
Список литературы
Кудрявцев П.С. Курс истории физики. М.: Просвещение, 1982. 448 с.
Химическая энциклопедия / Под ред. Кнунянца И.Л. М.: Советская энциклопедия, 1988.
Березовчук А.В. Физическая химия: конспект лекций. М.: Эксмо, 2015. 150 с.
Чизмаджев Ю.А. Биоэлектрохимия: из прошлого в будущее // Соросовский образовательный журн. 2000. Т. 6. № 3. С. 23.
Clark L.C. and Lyons C. Electrode systems for continuous monitoring in cardiovascular surgery // Ann. N.Y. Acad. Sci. 1962. V. 102. № 1. P. 29.
Theavenot D.R., Toth K., Durst R.A., Wilson G.S. Electrochemical biosensors: Recommended definitions and classification // Pure Appl. Chem. 1999. V. 71. № 12. P. 2333.
Turner A.P.F. Biosensors: Sense and sensibility // Chem. Soc. Rev. 2013. V. 42. № 8. P. 3184.
Labuda J., Bowater R.P., Fojta M., Gauglitz G., Glatz Z., Hapala I., Havliš J., Kilar F., Kilar A., Malinovská L., Sirén H.M.M., Skládal P., Torta F., Valachovič M., Wimmerová M., Zdráhal Z., Hibbert D.B. Terminology of bioanalytical methods (IUPAC Recommendations 2018) // Pure Appl. Chem. 2018. V. 90. № 7. P. 1121.
Lubert K.H., Kalcher K. History of electroanalytical methods // Electroanalysis. 2010. V. 22. № 17–18. P. 1937.
Shikata M. The electrolysis of nitrobenzene with the mercury dropping cathode // Trans. Faraday Soc. 1925. V. 21. № August. P. 42.
Китаев Ю.П., Троепольская Т.В., Будников Г.К. Промежуточные продукты в электрохимических реакциях. М.: Наука, 1982. 216 с.
Зиятдинова Г.К., Будников Г.К. Возможности электроаналитической химии в решении проблем фармацевтического анализа / Фармацевтический анализ (Серия “Проблемы аналитической химии”. Т. 16) / Под ред. Будникова Г.К., Гармонова С.Ю. М.: АРГАМАК-МЕДИА, 2013. С. 230.
Zavolskova M.D., Nikitina V.N., Maksimova E.D., Karyakina E.E., Karyakin A.A. Constant potential amperometric flow-injection analysis of ions and neutral molecules transduced by electroactive (conductive) polymers // Anal. Chem. 2019. V. 91. № 12. P. 7495.
Komkova M.A., Zarochintsev A.A., Karyakina E.E., Karyakin A.A. Electrochemical and sensing properties of Prussian Blue based nanozymes “artificial peroxidase” // J. Electroanal. Chem. 2020. V. 872. Article 114048.
Супрун Е.В. Электрохимические биосенсорные системы для анализа клинически значимых белков и пептидов. Дис. … докт. биол. наук. Москва: Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 2017. 290 с.
Heyrovsky J., Babicka J. Polarographic studies with the dropping mercury cathode. Part XIII – The effect of albumins // Collect. Czech. Chem. Commun. 1930. V. 2. P. 370.
Herles F., Vancura A. Research on the cause of the characteristic ‘‘wave’’ on the polarographic curve of human serum // Bull. Int. Acad. Sci. Boheme. 1932. V. 33. P. 119.
Bobrowski A., Zarebski J. Catalytic systems in adsorptive stripping voltammetry // Electroanalysis. 2000. V. 12. № 15. P. 1177.
Tomschik M., Havran L., Fojta M., Palecek E. Constant current chronopotentiometric stripping analysis of bioactive peptides at mercury and carbon electrodes // Electroanalysis. 1998. V. 10. № 6. P. 403.
Palecek E., Ostatna V., Masarik M., Bertoncini C.W., Jovin T.M. Changes in interfacial properties of a-synuclein preceding its aggregation // Analyst. 2008. V. 133. № 1. P. 76.
Zivanovic M., Aleksic M., Ostatna V., Doneux T., Palecek E. Polylysine-catalyzed hydrogen evolution at mercury electrodes // Electroanalysis. 2010. V. 22. № 17–18. P. 2064.
Palecek E., Ostatna V. Electroactivity of nonconjugated proteins and peptides. Towards electroanalysis of all proteins // Electroanalysis. 2007. V. 19. № 23. P. 2383.
Brdicka R. Polarographic studies with the dropping mercury kathode. Part XXXI. A new test for proteins in the presence of cobalt salts in ammoniacal solutions of ammonium chloride // Coll. Czech. Chem. Commun. 1933. V. 5. P. 112.
Brdicka R. Polarographic studies with the dropping mercury kathode. Part XXXII. Activation of hydrogen in sulphydryl group of some thio-acids in cobalt salts solutions // Coll. Czech. Chem. Commun. 1933. V. 5. P. 148.
Brdicka R. Polarographic investigations in serological cancer diagnosis // Nature. 1937. V. 139. № 3528. P. 1020.
Walker I.C., Reimann S.P. Polarographic studies of human blood sera // Am. J. Cancer. 1939. V. 37. № 4. P. 585.
Millar G.J. Studies on the polarography of proteins 1. The relation of wave heights to protein concentration and origin of wave III // Biochem. J. 1953. V. 53. № 3. P. 385.
Millar G.J. Studies on the polarography of proteins 2. The influence of pH and concentration of ammonia and ammonium ion upon the polarographic waves // Biochem. J. 1953. V. 53. № 3. P. 393.
Kalous V., Pavlicek Z. Polarographic characterization of proteins // Biochim. Biophys. Acta 1962. V. 57. № 1. P. 44.
Kizek R., Trnkova L., Palecek E. Determination of metallothionein at the femtomole level by constant current stripping chronopotentiometry // Anal. Chem. 2001. V. 73. № 20. P. 4801.
Raspor B., Paic M., Erk M. Analysis of metallothioneins by the modified Brdička procedure // Talanta. 2001. V. 55. № 1. P. 109.
Fojta M., Fojtova M., Havran L., Pivonkova H., Dorcak V., Sestakova I. Electrochemical monitoring of phytochelatin accumulation in Nicotiana tabacum cells exposed to sub-cytotoxic and cytotoxic levels of cadmium // Anal. Chim. Acta. 2006. V. 558. № 1–2. P. 171.
Cecil R., Weitzman P.D.J. The electroreduction of the disulphide bonds of insulin and other proteins // Biochem. J. 1964. V. 93. № 1. P. 1.
Zikan J., Kalous V. Polarographic reaction of proteins and peptides. II. Catalytic and reduction waves of insulin S-sulphopeptides and of S-sulphocysteine // Coll. Czech. Chem. Commun. 1967. V. 32. № 1. P. 246.
Kolthoff I.M., Stricks W., Tanaka N. The polarographic prewaves of cystine (RSSR) and dithiodiglycolic acid (TSST) and the oxidation potentials of the systems RSSR-RSH and TSST-TSH // J. Am. Chem. Soc. 1955. V. 77. № 18. P. 4739.
Stricks W., Kolthoff I.M. Polarography of glutathione // J. Am. Chem. Soc. 1952. V. 74. № 18. P. 4646.
Stankovich M.T., Bard A.J. The electrochemistry of proteins and related substances: I. Cystine and cysteine at the mercury electrode // J. Electroanal. Chem. 1977. V. 75. № 2. P. 487.
Ralph T.R. The electrochemistry of l-cystine and l-cysteine: Part 1: Thermodynamic and kinetic studies // J. Electroanal. Chem. 1994. V. 375. № 1. P. 1.
Betso S.R., Klapper M.H., Anderson L.B. Electrochemical studies of heme proteins. Coulometric, polarographic and combined spectroelectrochemical methods for reduction of the heme prosthetic group in cytochrome c // J. Am. Chem. Soc. 1972. V. 94. № 23. P. 8197.
Scheller F., Janchen M., Lampe J., Prümke H.J., Blanck J., Palecek E. Studies on electron transfer between mercury electrode and hemoprotein // Biochim. Biophys. Acta. 1975. V. 412. № 1. P. 157.
Scheller F. Functional properties of adsorbed hemoproteins // Bioelectrochem. Bioenerg. 1977. V. 4. № 4. P. 490.
Ludwig R. Direct electron transfer to enzymes / Encyclopedia of Applied Electrochemistry / Eds. Kreysa G., Ota K., Savinell R.F. N.Y.: Springer, 2014. P. 330.
Yeh P., Kuwana T. Reversible electrode reaction of cytochrome c // Chem. Lett. 1977. V. 6. № 10. P. 1145.
Eddowes M.J., Hill H.A.O. A novel method for investigation of the electrochemistry of metalloproteins: Cytochrome c // J. Chem. Soc. Chem. Commun. 1977. № 21. P. 771b.
Eddowes M.J., Hill H.A.O., Uosaki K. The electrochemistry of cytochrome c. Investigation of the mechanism of the 4,4'-bipyridyl surface modified gold electrode // J. Electroanal. Chem. 1980. V. 116. P. 527.
Rusling J.F. Liquid crystal surfactant films for electrochemical catalysis // Microporous Mater. 1994. V. 3. № 1–2. P. 1.
Karyakin A.A. Principles of direct (mediator free) bioelectrocatalysis // Bioelectrochemistry. 2012. V. 88. P. 70.
Березин И.В., Богдановская В.А., Варфоломеев С.Д., Тарасевич М.Р., Ярополов А.И. Биокатализ. Равновесный кислородный потенциал в присутствии лакказы // Доклады Академии наук. 1978. Т. 240. С. 615.
Ярополов А.И., Маловик В., Варфоломеев С.Д., Березин И.В. Электровосстановление перекиси водорода на электроде с иммобилизованной пероксидазой // Доклады Академии наук. 1979. Т. 249. № 6. С. 1399.
Yaropolov A.I., Karyakin A.A., Varfolomeyev S.D., Berezin I.V. Mechanism of H2-electrooxidation with immobilized hydrogenase // Bioelectrochem. Bioenerg. 1984. V. 12. № 3–4. P. 267.
Bioelectrochemistry: Fundamentals, Experimental Techniques, and applications / Ed. Bartlett P.N. Chichester: John Wiley & Sons, 2008.
Koryta J., Pradac J. Electrode processes of the sulfhydryl-disulfide system: I. Cystine at a platinum electrode // J. Electroanal. Chem. Interfacial Electrochem. 1968. V. 17. № 1–2. P. 167.
Koryta J., Pradac J. Electrode processes of the sulfhydryl-disulfide system: II. Cystine at a gold electrode // J. Electroanal. Chem. Interfacial Electrochem. 1968. V. 17. № 1–2. P. 177.
Koryta J., Pradac J. Electrode processes of the sulfhydryl-disulfide system: Electrode processes of the sulfhydryl-disulfide system: III. Cysteine at platinum and gold electrodes // J. Electroanal. Chem. Interfacial Electrochem. 1968. V. 17. № 1–2. P. 185.
Brabec V., Mornstein V. Electrochemical behaviour of proteins at graphite electrodes: I. Electrooxidation of proteins as a new probe of protein structure and reactions // Biochim. Biophys. Acta: Protein Structure. 1980. V. 625. № 1. P. 43.
Reynaud J.A., Malfoy B., Bere A. The electrochemical oxidation of three proteins: RNAase A, bovine serum albumin and concanavalin A at solid electrodes // J. Electroanal. Chem.: Interfacial Electrochem. 1980. V. 116. P. 595.
Paleček E. Past, present and future of nucleic acids electrochemistry // Talanta. 2002. V. 56. № 5. P. 809.
Palecek E., Fojta M., Jelen F., Vetterl V. Electrochemical analysis of nucleic acids, Bioelectrochemistry / The Encyclopedia of Electrochemistry. V. 9. Ch. 12 / Eds. Bard A.J., Stratmann M., Weinheim, Germany: Wiley-VCH Verlag, 2002. P. 365.
Paleček E. Oszillographische Polarographie der Nucleinsäuren und ihrer Bestandteile // Naturwissenschaften. 1958. V. 45. № 8. P. 186.
Palecek E. Oscillographic polarography of highly polymerized deoxyribonucleic acid // Nature. 1960. V. 188. P. 656.
Paleček E. Oszillographische Polarographie der Nucleinsäurekomponenten // Coll. Czech. Chem. Commun. 1960. V. 25. № 9. P. 2283.
Bendich A. The Nucleic Acids / Eds. Chargaff E., Davidson J.N. N.Y.: Academic Press, 1955. V. 1. P. 119.
Berg H. Polarographic studies on nucleic acids and nucleases. I. Polarographic determination of proteins in the presence of nucleic acids // Biochemische Zeitschrift. 1957. V. 329. № 3. P. 274.
Miller I.R. The structure of DNA and RNA in the water-mercury interface // J. Mol. Biol. 1961. V. 3. № 3. P. 229.
Miller I.R. Temperature dependence of the adsorption of native DNA in a polarized water-mercury interface // J. Mol. Biol. 1961. V. 3. P. 357.
Marmur J., Lane D. Strand separation and specific recombination in deoxyribonucleic acids: Biological studies // Proc. Natl. Acad. Sci. U.S.A. 1960. V. 46. № 4. P. 453.
Marmur J., Rownd R., Schildkraut C.L. Progress in Nucleic Acid Research / Eds. Davidson J.N., Cohn W.E. London: Academic Press, 1963. V. 1. P. 231.
Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms // J. Mol. Biol. 1961. V. 3. № 2. P. 208.
Cummings T.E., Elving P.J. Electrochemical reduction of thymine in dimethylsulfoxide auto-protonation of the radical-anion and reduced free-radical // J. Electroanal. Chem. 1979. V. 102. P. 237.
Cummings T.E., Elving P.J. Electrochemical reduction of uracil in dimethyl-sulfoxide – autoprotonation of anion radical // J. Electroanal. Chem. 1978. V. 94. P. 123.
Paleček E. Reaction of nucleic acid bases with the mercury electrode: Determination of purine derivatives at submicromolar concentrations by means of cathodic stripping voltammetry // Anal. Biochem. 1980. V. 108. № 1. P. 129.
Paleček E., Jelen F., Hung M.A., Lasovsky J. 445 – Reaction of the purine and pyrimidine derivatives with the electrode mercury // Bioelectrochem. Bioenerg. 1981. V. 8. № 6. P. 621.
Paleček E. Determination of pseudouridine at submicromolar concentrations by cathodic stripping voltammetry at a mercury electrode // Anal. Chim. Acta. 1985. V. 174. P. 103.
Paleček E. Adsorptive transfer stripping voltammetry: Determination of nanogram quantities of DNA immobilized at the electrode surface // Anal. Biochem. 1988. V. 170. № 2. P. 421.
Teijeiro C., Nejedly K., Palecek E. Cyclic voltammetry of submicrogram quantities of supercoiled, linear and denatured DNAs with DNA-modified mercury electrode // J. Biomol. Struct. Dyn. 1993. V. 11. № 2. P. 313.
Paleček E., Tomschik M., Stankova V., Havran L. Chronopotentiometric stripping of DNA at mercury electrodes // Electroanalysis. 1997. V. 9. № 13. P. 990.
Brabec V., Paleček E. The reducibility of polynucleotides and their a.c. polaro-graphic behavior // J. Electroanal. Chem. 1970. V. 27. № 1. P. 145.
Brabec V. Interaction of nucleic acids with electrically charged surfaces: VII. The effect of ionic strength of neutral medium on the conformation of DNA adsorbed on the mercury electrode1 // Biophys. Chem. 1980. V. 11. № 1. P. 1.
Brabec V., Dryhurst G. Electrochemical behavior of natural and biosynthetic polynucleotides at the pyrolytic graphite electrode A new probe for studies of polynucleotide structure and reactions // J. Electroanal. Chem. 1978. V. 89. № 1. P. 161.
Brabec V., Dryhurst G. Electrochemical oxidation of polynucleotides at graphite electrodes – New probe of nucleic-acids structure // Studia Biophysica. 1978. V. 67. P. 23.
Brabec V., Dryhurst G. Electrochemical oxidation of polyadenylic acid at graphite electrodes // J. Electroanal. Chem. 1978. V. 91. № 2. P. 219.
Brabec V. Electrochemical oxidation of nucleic acids and proteins at graphite electrode. Qualitative aspects // J. Electroanal. Chem. 1980. V. 116. P. 69.
Brabec V. Nucleic acid analysis by voltammetry at carbon electrodes // J. Electroanal. Chem. 1981. V. 128. P. 437.
Smith D.L., Elving P.J. Analytical utilization of the polarographic and voltammetric behavior of purines and pyrimidines // Anal. Chem. 1962. V. 34. № 8. P. 930.
Elving P.J., Smith D.L. The graphite electrode. An improved technique for voltammetry and chronopotentiometry // Anal. Chem. 1960. V. 32. № 13. P. 1849.
Dryhurst G., Elving P.J. Electrochemical oxidation of adenine: reaction products and mechanisms // J. Electrochem. Soc. 1968. V. 115. № 10. P. 1014.
Dryhurst G., Pace G.F. Electrochemical oxidation of guanine at the pyrolytic graphite electrode // J. Electrochem. Soc. 1970. V. 117. № 10. P. 1259.
Dryhurst G. Adsorption of guanine and guanosine at the pyrolytic graphite electrode: Implications for the determination of guanine in the presence of guanosine // Anal. Chim. Acta. 1971. V. 57. № 1. P. 137.
Dryhurst G. Electrochemical determination of adenine and adenosine: Adsorption of adenine and adenosine at the pyrolytic graphite electrode // Talanta. 1972. V. 19. № 6. P. 769.
Watson J.D., Crick F.H.C. Molecular structure of nucleic acids // Nature. 1953. V. 171. P. 737.
Franklin R.E., Gosling R.G. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate // Nature. 1953. V. 17. P. 156.
Wilson W.D. Nucleic Acids in Chemistry and Biology / Eds. Blackburn G.M., Gait M.J., New York: Oxford University Press, 1996, P. 329.
Zamenhof S., Brawerman G., Chargaff E. On the desoxypentose nucleic acid from several microorganisms // Biochim. Biophys. Acta. 1952. V. 9. P. 402.
Suprun E.V. Direct electrochemistry of proteins and nucleic acids: the focus on 3D structure // Electrochem. Commun. 2021. V. 125. Article 106983.
Suprun E.V., Zharkova M.S., Morozevich G.E., Veselovsky A.V., Shumyantseva V.V., Archakov A.I. Analysis of redox activity of proteins on the carbon screen printed electrodes // Electroanalysis. 2013. V. 25. P. 2109.
Brabec V., Koudelka J. Oxidation of deoxyribonucleic acid at carbon electrodes. The effect of the quality of the deoxyribonucleic acid sample // J. Electroanal. Chem.: Interfacial Electrochem. 1980. V. 116. P. 793.
Chiorcea-Paquim A.M., Oliveira-Brett A.M. Redox behaviour of G-quadruplexes // Electrochim. Acta. 2014. V. 126. P. 162.
Veloso A., Kerman K. Advances in electrochemical detection for study of neurodegenerative disorders // Anal. Bioanal. Chem. 2013. V. 405. P. 5725.
Fojta M., Daňhel A., Havran L., Vyskočil V. Recent progress in electrochemical sensors and assays for DNA damage and repair // Trends Anal. Chem. 2016. V. 79. P. 160.
Suprun E.V. Protein post-translational modifications – A challenge for bioelectrochemistry // Trends Anal. Chem. 2019. V. 116. P. 44.
Bartosik M., Jirakova L. Electrochemical analysis of nucleic acids as potential cancer biomarkers // Curr. Opin. Electrochem. 2019. V. 14. P. 96.
Lin Y.W. Structure and function of heme proteins regulated by diverse post-translational modifications // Arch. Biochem. Biophys. 2018. V. 641. P. 1.
Soskic V., Groebe K., Schrattenholz A. Nonenzymatic posttranslational protein modifications in ageing // Exp. Gerontol. 2008. V. 43. P. 247.
Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease // Physiol. Rev. 2007. V. 87. P. 315.
Humphrey S.J., James D.E., Mann M. Protein phosphorylation: A major switch mechanism for metabolic regulation // Trends Endocrinol. Metab. 2015. V. 26. P. 676.
Scheller F.W., Wollenberger U., Lei C., Jin W., Ge B., Lehmann C., Lisdat F., Fridman V. Bioelectrocatalysis by redox enzymes at modified electrodes // Rev. Mol. Biotechnol. 2002. V. 82. P. 411.
Ferapontova E.E., Shleev S., Ruzgas T., Stoica L., Christenson A., Tkac J., Yaropolov A.I., Gorton L. Direct electrochemistry of proteins and enzymes // Perspect. Bioanal. 2005. V. 1. P. 517.
Scheller F.W., Bistolas N., Liu S., Janchen M., Katterle M., Wollenberger U. Thirty years of haemoglobin electrochemistry // Adv. Coll. Interface Sci. 2005. V. 116. P. 111.
Paleček E., Tkáč J., Bartosik M., Bertók T., Ostatná V., Paleček J. Electrochemistry of nonconjugated proteins and glycoproteins. Toward sensors for biomedicine and glycomics // Chem. Rev. 2015. V. 115. P. 2045.
Suprun E.V., Shumyantseva V.V., Archakov A.I. Protein electrochemistry: Application in medicine. A review // Electrochim. Acta. 2014. V. 140. P. 72.
Vacek J., Zatloukalova M., Novak D. Electrochemistry of membrane proteins and protein–lipid assemblies // Curr. Opinion Electrochem. 2018. V. 12. P. 73.
Hasoň S., Daňhel A., Schwarzová-Pecková K., Fojta M., Carbon electrodes in electrochemical analysis of biomolecules and bioactive substances: Roles of surface structures and chemical groups / Nanotechnology and Biosensors / Eds. Nikolelis D.P., Nikoleli G.P. Elsevier. 2018. P. 51.
Baluchová S., Daňhel A., Dejmková H., Ostatná V., Fojta M., Schwarzová-Pecková K. Recent progress in the applications of boron doped diamond electrodes in electroanalysis of organic compounds and biomolecules – A review // Anal. Chim. Acta. 2019. V. 1077. P. 30.
Suprun E.V., Karpova E.V., Radko S.P., Karyakin A.A. Advanced electrochemical detection of amino acids and proteins through flow injection analysis and catalytic oxidation on Prussian Blue // Electrochim. Acta. 2020. V. 331. Article 135289.
Özcan A., Şahin Y. A novel approach for the selective determination of tryptophan in blood serum in the presence of tyrosine based on the electrochemical reduction of oxidation product of tryptophan formed in situ on graphite electrode // Biosens. Bioelectron. 2012. V. 31. P. 26.
Vestergaard M., Kerman K., Saito M., Nagatani N., Takamura Y., Tamiya E. A Rapid label-free electrochemical detection and kinetic study of Alzheimer’s amyloid beta aggregation // J. Am. Chem. Soc. 2005. V. 127. P. 11892.
Somji M., Dounin V., Muench S.B., Schulze H., Bachmann T.T., Kerman K. Electroanalysis of amino acid substitutions in bioengineered acetylcholinesterase // Bioelectrochemistry. 2012. V. 88. P. 110.
Enache T.A., Oliveira-Brett A.M. Peptide methionine sulfoxide reductase A (MsrA): Direct electrochemical oxidation on carbon electrodes // Bioelectrochemistry. 2013. V. 89. P. 11.
Topal B.D., Özkan S.A., Uslu B. Direct electrochemistry of native and denatured alpha-2-macroglobulin by solid electrodes // J. Electroanal. Chem. 2014. V. 719. P. 14.
Chiku M., Nakamura J., Fujishima A., Einaga Y. Conformational change detection in nonmetal proteins by direct electrochemical oxidation using diamond electrodes // Anal. Chem. 2008. V. 80. P. 5783.
Dorcak V., Ostatna V., Palecek E. Electrochemical reduction and oxidation signals of angiotensin peptides. Role of individual amino acid residues // Electrochem. Commun. 2013. V. 31. P. 80.
Suprun E.V., Khmeleva S.A., Radko S.P., Kozin S.A., Archakov A.I., Shumyantseva V.V. Direct electrochemical oxidation of amyloid-β peptides via tyrosine, histidine, and methionine residues // Electrochem. Commun. 2016. V. 65. P. 53.
Suprun E.V., Radko S.P., Khmeleva S.A., Mitkevich V.A., Archakov A.I., Makarov A.A., Shumyantseva V.V. Electrochemical oxidation of amyloid-beta peptide isoforms on carbon screen printed electrodes // Electrochem. Commun. 2017. V. 75. P. 33.
Enache T.A., Oliveira-Brett A.M. Alzheimer’s disease amyloid beta peptides in vitro electrochemical oxidation // Bioelectrochemistry. 2017. V. 114. P 13.
Nguyen N.T., Wrona M.Z., Dryhurst G. Electrochemical oxidation of tryptophan // J. Electroanal. Chem.: Interfacial Electrochem. 1986. V. 199. P. 101.
Enache T.A., Oliveira-Brett A.M. Phenol and para-substituted phenols electrochemical oxidation pathways // J. Electroanal. Chem. 2011. V. 655. P. 9.
Enache T.A., Oliveira-Brett A.M. Pathways of electrochemical oxidation of indolic compounds // Electroanalysis. 2011. V. 23. P. 1337.
Sęk S., Vacek J., Dorčák V. Electrochemistry of peptides // Curr. Opinion Electrochem. 2019. V. 14. P. 166.
Ostatna V., Cernocka H., Kurzatkowska K., Palecek E. Native and denatured forms of proteins can be discriminated at edge plane carbon electrodes // Anal. Chim. Acta. 2012. V. 735 P. 31.
Suprun E.V., Khmeleva S.A., Kiseleva Y.Y., Radko S.P., Archakov A.I., Shumyantseva V.V. Quantitative aspects of electrochemical detection of amyloid-β aggregation // Electroanalysis. 2016. V. 28. P. 1977.
Suprun E.V., Radko S.P., Andreev E.A., Khmeleva S.A., Kozin S.A., Makarov A.A., Archakov A.I., Shumyantse-va V.V. Electrochemical detection of Zn(II)- and Cu(II)-induced amyloid-β aggregation: Quantitative aspects and application to amyloid-β isoforms // J. Electroanal. Chem. 2017. V. 791. P. 152.
Suprun E.V., Radko S.P., Farafonova T.E., Mitkevich V.A., Makarov A.A., Archakov A.I., Shumyantseva V.V. Application of an electrochemical method to evaluation of amyloid-β aggregation inhibitors: Testing the RGKLVFFGR-NH2 peptide antiaggregant // Electroanalysis. 2017. V. 29. P. 2906.
Suprun E.V., Radko S.P., Kozin S.A., Mitkevich V.A., Makarov A.A. Electrochemical detection of Zn(II)-induced amyloid-β aggregation: Insights into aggregation mechanisms // J. Electroanal. Chem. 2018. V. 830–831. P. 34.
Suprun E.V., Radko S.P., Kozin S.A., Mitkevich V.A., Makarov A.A. Application of electrochemical method to a comparative study of spontaneous aggregation of amyloid-β isoforms // J. Electroanal. Chem. 2020. V. 861. Article 113938.
Suprun E.V., Khmeleva S.A., Radko S.P., Archa-kov A.I., Shumyantseva V.V. Electrochemical analysis of amyloid-β domain 1-16 isoforms and their complexes with Zn(II) ions // Electrochim. Acta. 2016. V. 187. P. 677.
Chikae M., Fukuda T., Kerman K., Idegami K., Miura Y., Tamiya E. Amyloid-β detection with saccharide immobilized gold nanoparticle on carbon electrode // Bioelectrochemistry. 2008. V. 74. P. 118.
Ferapontova E.E., Grigorenko V.G., Egorov A.M., Borchers T., Ruzgas T., Gorton L. Direct electron transfer in the system gold electrode–recombinant horseradish peroxidases // J. Electroanal. Chem. 2001. V. 509. P. 19.
Ashe D., Alleyne T., Iwuoha E. Serum cytochrome c detection using a cytochrome c oxidase biosensor // Biotechnol. Appl. Biochem. 2007. V. 46. P. 185.
Suprun E.V., Shilovskaya A.L., Lisitsa A.V., Bulko T.V., Shumyantseva V.V., Archakov A.I. Electrochemical immunosensor based on metal nanoparticles for cardiac myoglobin detection in human blood plasma // Electroanalysis. 2011. V. 23. P. 1051.
Pandiaraj M., Sethy N.K., Bhargava K., Kameswara-rao V., Karunakaran C. Designing label-free electrochemical immunosensors for cytochrome c using nanocomposites functionalized screen printed electrodes // Biosens. Bioelectron. 2014. V. 54. P. 115.
Shumyantseva V.V., Kuzikov A.V., Masamrekh R.A., Bulko T.V., Archakov A.I. From electrochemistry to enzyme kinetics of cytochrome P450 // Biosens. Bioelectron. 2018. V. 121. P. 192.
Gilardi G., Fantuzzi A., Sadeghi S.J. Engineering and design in the bioelectrochemistry of metalloproteins // Curr. Opin. Struct. Biol. 2001. V. 11. P. 491.
Panicco P., Castrignanò S., Sadeghi S.J., Di Nardo G., Gilardi G. Engineered human CYP2C9 and its main polymorphic variants for bioelectrochemical measurements of catalytic response // Bioelectrochemistry. 2021. V. 138. Article 107729.
Olloqui-Sariego J.L., Zakharova G.S., Poloznikov A.A., Calvente J.J., Hushpulian D.M., Gorton L., Andreu R. Influence of tryptophan mutation on the direct electron transfer of immobilized tobacco peroxidase // Electrochim. Acta. 2020. V. 351. Article 136465.
Wu H., Wang X., Qiao M., Zhang H., Jin X., Fan S. Enhancing sensitivity of hemoglobin-based electrochemical biosensor by using protein conformational intermediate // Sens. Actuators B: Chem. 2015. V. 221. P. 694.
Lancellotti L., Borsari M., Bellei M., Bonifacio A., Bortolotti C.A., Di Rocco G., Ranieri A., Sola M., Battistuz-zi G. Urea-induced denaturation of immobilized yeast iso-1 cytochrome c: Role of Met80 and Tyr67 in the thermodynamics of unfolding and promotion of pseudoperoxidase and nitrite reductase activities // Electrochim. Acta. 2020. V. 363. Article 137237.
Gómez-Mingot M., Alcaraz L.A., Heptinstall J., Donaire A., Piccioli M., Montiel V., Iniesta J. Electrochemical nitration of myoglobin at tyrosine 103: Structure and stability // Arch. Biochem. Biophys. 2013. V. 529. P. 26.
Díaz-Moreno I., García-Heredia J.M., Díaz-Quinta-na A., Teixeira M., Miguel A. Nitration of tyrosines 46 and 48 induces the specific degradation of cytochrome c upon change of the heme iron state to high-spin // Biochim. Biophys. Acta. 2011. V. 1807. P. 1616.
Roberts E.S., Lin H.L., Crowley J.R., Vuletich J.L., Osawa Y., Hollenberg P.F. Peroxynitrite-mediated nitration of tyrosine and inactivation of the catalytic activity of cytochrome P450 2B1 // Chem. Res. Toxicol. 1998. V. 11. P. 1067.
Walsh C.T., Garneau-Tsodikova S., Gatto G.J., Jr. Protein posttranslational Modifications: The chemistry of proteome diversifications // Angew. Chem. Int. Ed. 2005. V. 44. P. 7342.
Suprun E.V., Radko S.P., Farafonova T.E., Kozin S.A., Makarov A.A., Archakov A.I., Shumyantseva V.V. Electrochemical detection of protein post-translational modifications: Phosphorylation and nitration of amyloid-beta (1–16) // Electrochim. Acta. 2017. V. 258. P. 1182.
Fernandes I.P., Oliveira-Brett A.M. Caveolin proteins electrochemical oxidation and interaction with cholesterol // Bioelectrochemistry. 2020. V. 133. Article 107451.
Супрун Е.В., Жаркова М.С., Веселовский А.В., Арчаков А.И., Шумянцева В.В. Электрохимическое окисление тромбина на печатных графитовых электродах // Электрохимия. 2017. Т. 53. С. 108.
Brabec V. Study of thermal and acid denaturation of DNA by means of voltammetry at graphite electrodes // Biopolymers. 1979. V. 18. P. 2397.
Brabec V. Interaction of nucleic acids with electrically charged surfaces: VI. A comparative study on the electrochemical behaviour of native and denatured dnas at graphite electrodes // Biophys. Chem. 1979. V. 9. P. 289.
de-los-Santos-Álvarez P., Lobo-Castañón M.J., Miranda-Ordieres A.J., Tunon-Blanco P. Electrochemistry of nucleic acids at solid electrodes and its applications // Electroanalysis. 2004. V. 16. P. 1193.
Oliveira-Brett A.M. Electrochemical DNA Assays, Bioelectrochemistry: Fundamentals, Experimental Techniques, and Applications / Ed. Bartlett P.N. Chichester: John Wiley & Sons, 2008. P. 411.
Ferapontova E.E. DNA electrochemistry and electrochemical sensors for nucleic acids // Annu. Rev. Anal. Chem. 2018. V. 11. P. 197.
Špaček J., Daňhel A., Hasoň S., Fojta M. Label-free detection of canonical DNA bases, uracil and 5‑methylcytosine in DNA oligonucleotides using linear sweep voltammetry at a pyrolytic graphite electrode // Electrochem. Commun. 2017. V. 82. P. 34.
Rodrigues Pontinha A.D., Chiorcea-Paquim A.M., Eritja R., Oliveira-Brett A.M. Quadruplex nanostructures of d (TGGGGT): Influence of sodium and potassium ions // Anal. Chem. 2014. V. 86. P. 5851.
Vidláková P., Pivoňková H., Kejnovská I., Trnková L., Vorlíčková M., Fojta M., Havran L. G-quadruplex-based structural transitions in 15-mer DNA oligonucleotides varying in lengths of internal oligo(dG) stretches detected by voltammetric techniques // Anal. Bioanal. Chem. 2015. V. 407. P. 5817.
Špaček J., Fojta M., Wang J. Electrochemical reduction and oxidation of six natural 2'-deoxynucleosides at a pyrolytic graphite electrode in the presence or absence of ambient oxygen // Electroanalysis. 2019. V. 3. P. 2057.
Oliveira-Brett A.M., Piedade J.A.P., Silva L.D., Diculescu V.C. Voltammetric determination of all DNA nucleotides // Anal. Biochem. 2004. V. 332. P. 321.
Goncalves L.M., Batchelor-McAuley C., Barros A.A., Compton R.G. Electrochemical oxidation of adenine: a mixed adsorption and diffusion response on an edge-plane pyrolytic graphite electrode // J. Phys. Chem. C. 2010. V. 114. P. 14213.
Li Q., Batchelor-McAuley C., Compton R.G. Electrochemical oxidation of guanine: Electrode reaction mechanism and tailoring carbon electrode surfaces to switch between adsorptive and diffusional responses // J. Phys. Chem. B. 2010. V. 114. P. 7423.
Oliveira Brett A.M., Matysik F.M. Voltammetric and sonovoltammetric studies on the oxidation of thymine and cytosine at a glassy carbon electrode // J. Electroanal. Chem. 1997. V. 429. P. 95.
Ferapontova E.E. Electrochemistry of guanine and 8-oxoguanine at gold electrodes // Electrochim. Acta. 2004. V. 49. P. 1751.
Goyal R.N., Singh U.P., Abdullah A.A. Electrochemical oxidation of uracil and 5-halouracils at pyrolytic graphite electrode // Indian J. Chem. 2003. V. 42A. P. 42.
Ferapontova E.E., Domínguez E. Direct electrochemical oxidation of DNA on polycrystalline gold electrodes // Electroanalysis. 2003. V. 15. P. 629.
Brett C.M., Oliveira Brett A.M., Serrano S.H. On the adsorption and electrochemical oxidation of DNA at glassy carbon electrodes // J. Electroanal. Chem. 1994. V. 366. P. 225.
Wu L., Zhou J., Luo J., Lin Z. Oxidation and adsorption of deoxyribonucleic acid at highly ordered pyrolytic graphite electrode // Electrochim. Acta. 2000. V. 45. P. 2923.
Yao T., Wasa T., Musha S. The anodic voltammetry of deoxyribonucleic acid at a glassy carbon electrode // Bull. Chem. Soc. Jpn. 1978. V. 51. P. 1235.
Brotons A., Mas L.A., Metters J.P., Banks C.E., Iniesta J. Voltammetric behaviour of free DNA bases, methylcytosine and oligonucleotides at disposable screen printed graphite electrode platforms // Analyst. 2013. V. 138. P. 5239.
Stempkowska I., Ligaj M., Jasnowska J., Langer J., Filipiak M. Electrochemical response of oligonucleotides on carbon paste electrode // Bioelectrochemistry. 2007. V. 70. P. 488.
Suprun E.V., Kutdusova G.R., Khmeleva S.A., Radko S.P. Towards deeper understanding of DNA electrochemical oxidation on carbon electrodes // Electrochem. Commun. 2021. V. 124. Article 106947.
Shumyantseva V.V., Bulko T.V., Tikhonova E.G., Sanzhakov M.A., Kuzikov A.V., Masamrekh R.A., Pergushov D.V., Schacher F.H., Sigolaeva L.V. Electrochemical studies of the interaction of rifampicin and nanosome/rifampicin with dsDNA // Bioelectro-chemistry. 2021. V. 140. Article 107736.
Wang J., Chen L., Chicharro M. Trace measurements of nucleic acids using flow injection amperometry // Anal. Chim. Acta. 1996. V. 319. P. 347.
Diculescu V.C., Chiorcea–Paquim A.M., Oliveira-Brett A.M. Applications of a DNA-electrochemical biosensor // Trends Anal. Chem. 2016. V. 79. P. 23.
Ферапонтова Е.Э., Шиповсков С.В. Электрохимически инициированное окислительное повреждение двухцепочечной ДНК из тимуса теленка, адсорбированной на золотых электродах // Биохимия. 2003. Т. 68. № 1. С. 118. (Ferapontova E.E., Shipovskov S.V. Electrochemically induced oxidative damage of double-stranded calf thymus DNA adsorbed on gold electrodes // Biochemistry (Moscow). 2003. V. 68. P. 99.)
Yang Z.S., Wang Y.L., Zhang Y.Z. Electrochemically induced DNA cleavage by copper-bipyridyl complex // Electrochem. Commun. 2004. V. 6. P. 158.
Honeychurch K.C., O’Donovan M.R., Hart J.P. Voltammetric behaviour of DNA bases at a screen-printed carbon electrode and its application to a simple and rapid voltammetric method for the determination of oxidative damage in double stranded DNA // Biosens. Bioelectron. 2007. V. 22. P. 2057.
Ferapontova E.E. Basic concepts and recent advances in electrochemical analysis of nucleic acids // Curr. Opin. Electrochem. 2017. V. 5. P. 218.
Hocek M., Fojta M. Nucleobase modification as redox DNA labelling for electrochemical detection // Chem. Soc. Rev. 2011. V. 40. P. 5802.
Fojta M. Redox labeling of nucleic acids for electrochemical analysis of nucleotide sequences and DNA damage / Biosensors for Security and Bioterrorism Applications / Eds. Nikolelis D.P., Nikoleli G.-P. N.Y.: Springer, 2016. P. 309.
Oberacher H., Erb R., Plattner S., Chervet J.P. Mechanistic aspects of nucleic-acid oxidation studied with electrochemistry-mass spectrometry // Trends Anal. Chem. 2015. V. 70. P. 100.
Zhao Y., Chen F., Li Q., Wang L., Fan C. Isothermal amplification of nucleic acids // Chem. Rev. 2015. V. 115. P. 12491.
Long Y.-T. Nanopore confinement for single-molecule measurement of proteins // Small Methods. 2020. V. 4. Article 2000695.
Lin Y., Ying Y.-L., Long Y.-T. Nanopore confinement for electrochemical sensing at the single-molecule level // Curr. Opinion Electrochem. 2018. V. 7. P. 172.
Li S., Wu X.Y., Li M.Y., Liu S.C., Ying Y.L., Long Y.T. T232K/K238Q aerolysin nanopore for mapping adjacent phosphorylation sites of a single Tau peptide // Small Methods. 2020. V. 4. № 11. Article 2000014.
Laver T., Harrison J., O’Neill P.A., Moore K., Farbos A., Paszkiewicz K., Studholme D.J. Assessing the performance of the Oxford nanopore technologies MinION // Biomol. Detect. Quantif. 2015. V. 3. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии