

Журнал аналитической химии, 2022, T. 77, № 9, стр. 849-860
Сорбенты с ковалентно привитым полимерным слоем для гидрофильной хроматографии
А. С. Попов a, Г. С. Максимов a, О. А. Шпигун a, А. В. Чернобровкина a, *
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: chernobrovkina@analyt.chem.msu.ru
Поступила в редакцию 03.12.2021
После доработки 27.03.2022
Принята к публикации 27.03.2022
- EDN: OXHSWW
- DOI: 10.31857/S0044450222090109
Аннотация
Получены неподвижные фазы на основе силикагеля, модифицированного поливинилпиридином и различным по структуре полиэтиленимином, с использованием 1,4-бутандиолдиглицидилового эфира в качестве спейсера. Изучение и сравнение хроматографических свойств сорбентов, модифицированных полиаминами и полиэтиленгликолем, проведено с использованием теста Танака для гидрофильных неподвижных фаз и на примере разделения модельных смесей сахаров, аминокислот и водорастворимых витаминов. Показано, что природа и структура полимера в функциональном слое влияет и на селективность, и на эффективность по полярным соединениям в режиме гидрофильной хроматографии.
Гидрофильная хроматография (ГИХ) как самостоятельный метод разделения полярных соединений был предложен в 1990 г. [1]. Использование полярных неподвижных фаз в сочетании с водно-органическими смесями в качестве элюентов позволяет определять полярные нейтральные и заряженные соединения, а также обеспечивает иную селективность по сравнению с традиционно используемой обращенно-фазовой ВЭЖХ [2], возможности которой при разделении полярных аналитов ограничены. Правильный выбор сорбента является ключевым моментом в хроматографии в целом и в ГИХ в частности [3], поэтому синтез новых неподвижных фаз с улучшенной эффективностью и иной селективностью был и остается основным инструментом дальнейшего развития метода.
Формирование функционального слоя на поверхности сорбентов путем ковалентного закрепления полярных полимеров − перспективный подход к получению новых неподвижных фаз для ГИХ [4–6], поскольку обеспечивает гидрофилизацию, а также экранирование матрицы, что приводит к снижению ее влияния на удерживание аналитов и изменению селективности. Более того, использование гидрофильных полимеров может способствовать переходу в режиме ГИХ к матрицам, альтернативным силикагелю и превосходящим его по гидролитической устойчивости.
Перспективным полимером создания сорбентов для ГИХ является полиэтиленимин (ПЭИ), который может иметь как линейную, так и разветвленную структуру. Разветвленный ПЭИ нашел множество применений в синтезе новых сорбентов. Так, капилляры с модифицированными ПЭИ внутренними стенками для капиллярного электрофореза использовали для разделения белков [7, 8], органических кислот и неорганических анионов [9–11]. Кроме того, ПЭИ неоднократно применяли для создания анионообменников [12–16]. Основное преимущество использования ПЭИ для создания анионообменного функционального слоя заключается в простоте синтеза и высокой воспроизводимости его результата, так как традиционно формирование гиперразветвленного слоя посредством чередования стадий алкилирования диглицидиловыми эфирами и аминирования включает до десяти стадий и более. Одним из первых упоминаний использования ПЭИ при создании сорбентов для ГИХ является работа [17], в которой описано применение в гидрофильном режиме капиллярных колонок, модифицированных наночастицами золота с адсорбированным слоем ПЭИ. Получение более стабильных сорбентов путем ковалентного закрепления ПЭИ описано в недавних работах [6, 18]. Авторы отмечают, что модифицирование полиамином обеспечивает не только высокую гидрофильность, но и анионообменные свойства, благодаря чему становится возможным одновременное определение полярных соединений и неорганических анионов, разделение пяти органических либо пяти аминокислот [6]. Однако специфический набор модельных аналитов не позволяет в полной мере оценить достоинства таких колонок по сравнению с силикагелем, аминофазой или другими гидрофильными неподвижными фазами.
Еще одним подходящим полимером представляется поливинилпиридин (ПВП). Сорбенты, полученные с его использованием, нашли широкое применение как в ионной [19, 20], так и в сверхкритической флюидной хроматографии [21–23]. Данные о применении модифицированных ПВП неподвижных фаз в гидрофильной хроматографии отсутствуют. Стоит отметить, что меньшая гидрофильность ПВП по сравнению с ПЭИ и поэтиленгликолем (ПЭГ) может быть компенсирована вкладом гидрофобных и π–π взаимодействий при удерживании слабо полярных аналитов.
Одним из самых простых, а поэтому широко используемым способом описания свойств неподвижных фаз является изучение хроматографического поведения модельных аналитов. Такие данные позволяют классифицировать и сравнивать между собой синтезированные и уже существующие сорбенты. Однако информация об удерживании гомологов или соединений одного класса не может дать полного представления о вкладе различных вторичных взаимодействий в сложный механизм удерживания в ГИХ. Авторами работы [24] предложен тест Танака для гидрофильных неподвижных фаз, в котором набор используемых модельных аналитов позволяет количественно оценить следующие параметры: гидрофильность поверхности сорбента k(U) – определяется как фактор удерживания уридина; селективность по отношению к гомологам α(CH2); селективность по отношению к гидроксильной группе α(OH); способность к разделению пространственных изомеров α(V/A); анионо- и катионообменные свойства − α(АХ) и α(СХ) соответственно; кислотно-основную природу поверхности сорбента α(Tb/Tp). В работах [24, 25] представлены данные по различным типам селективности для множества коммерчески доступных гидрофильных колонок, которые можно использовать для сравнения и оценки свойств новых сорбентов. Так, для аминофаз характерны высокие значения параметра анионообменной селективности. Так, в работе [6] сорбент, полученный модифицированием 3-глицидилпропилсиликагеля разветвленным ПЭИ, продемонстрировал высокие значения параметров k(U) и α(AХ) по результатам теста Игрума для гидрофильных неподвижных фаз [26]. Увеличение гидрофильности поверхности и вклада анионообменных взаимодействий способствовало разделению модельной смеси пяти аминокислот менее чем за 16 мин в режиме ГИХ и семи неорганических ионов за 40 мин в режиме ионной хроматографии. Достичь таких результатов в идентичных условиях на коммерческой аминофазе Unitary NH2 авторам не удалось, что продемонстрировало преимущества модифицирования матрицы силикагеля полиэтиленимином.
Цель настоящей работы − получение новых сорбентов путем ковалентного модифицирования 3-аминопропилсиликагеля гидрофильными полимерами, такими как поливинилпиридин, линейный и разветвленный полиэтиленимин (ПЭИлин и ПЭИразв соответственно) с использованием 1,4-бутандиолдиглицидилового эфира (1,4-БДДГЭ) в качестве спейсера. Хроматографические свойства неподвижных фаз, модифицированных разными полимерами, изучали и сравнивали на примере разделения модельных смесей полярных веществ нескольких классов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приборы и материалы. В работе использовали D-(+)-рибозу, D-(+)-глюкозу, D-(+)-фруктозу, D-(+)-лактозы моногидрат, D-(+)-мальтозы моногидрат, D-(+)-сахарозу, D-(+)-ксилозу, рибофлавин, пиридоксина гидрохлорид, цианокобаламин, аскорбиновую кислоту, тиамин, аденозин, урацил, теобромин, теофиллин, уридин, видарабина моногидрат, 5-метилуридин, 2'-дезоксиуридин, N,N,N-триметилфениламмоний хлорид, п-толуолсульфонат натрия (>98.0%, “TCI”, Япония); D,L-фенилаланин, D,L-пролин, D,L-серин, L-изолейцин, L-лейцин, L-метионин, L-аспарагиновую кислоту, L-глутаминовую кислоту, L-гистидин, L-лизин, L-аргинин х. ч. (Serva, Германия); L-аспарагин, β-аланин, глицин х. ч. (Merck, Германия); L-глутамин, L-валин х. ч. (Reanal, Венгрия); никотинамид, никотиновую кислоту (>99%, Sigma-Aldrich, США); толуол х. ч. (Компонент-Реактив, Россия), ацетонитрил HPLCgradientgrade (Panreac, Германия); ледяную уксусную кислоту (99.5%), ортофосфорную кислоту (85%) х. ч., хлорид калия ч. д. а. (Panreac, Испания); этанол ч. д. а., ацетат аммония ч. (Лабтех, Россия); ацетон ч. д. а. (ХимМед, Россия); 1,4-диоксан ч.д.а. (Компонент-Реактив, Россия); разветвленный полиэтиленимин (Mw = 800, Mn = = 600, Sigma-Aldrich, США), линейный полиэтиленимин (Mn = 5000, Sigma-Aldrich, США), поли-4-винилпиридин (Mn = 6000, Sigma-Aldrich, США), диглицидиловый эфир полиэтиленгликоля (Mw = 500, Sigma-Aldrich, США).
Для синтеза сорбентов использовали силикагель с привитыми аминопропильными радикалами Диасфер-110-Амин (БиоХиммак СТ, Россия) со сферическими частицами диаметром 5 мкм, 1.52% N.
Эксперименты проводили с использованием системы ВЭЖХ, состоящей из хроматографа Dionex 3000 с двухканальным градиентным насосом, автоматической системой ввода пробы, термостатом колонки и диодно-матричным детектором. Хроматограммы регистрировали с помощью персонального компьютера и программного пакета Chromeleon 7 (Thermo Fisher Scientific, США). Для определения сахаров использовали систему ВЭЖХ, состоящую из изократического ВЭЖХ-насоса, шестиходового крана-дозатора и рефрактометрического детектора (Agilent Technologies, США). Хроматограммы регистрировали с помощью персонального компьютера и программного пакета ChemStation (Agilent Technologies, США).
Для синтеза сорбентов применяли следующее оборудование: термостат (“Memmert”, Германия), вакуумный насос серии “Laboport” (“KNF Neuberger”, Германия), механическую мешалку Eurostar (IKA-Werke, Германия). В работе использовали стальные колонки размером 100 × 3 мм, которые заполняли при помощи пневмонасоса Knauer K-1900 (Knauer, Германия).
Синтез сорбентов и заполнение хроматографических колонок. На первой стадии точную навеску аминопропилсиликагеля помещали в круглодонную колбу объемом 100 мл, добавляли 1,4-БДДГЭ (40 мМ на 1 г матрицы), растворенный в деионизованной воде из расчета 20 мл на 1 г матрицы. Суспензию непрерывно перемешивали в течение 60 мин механической мешалкой при 60°С. Продукт отфильтровывали на стеклянном фильтре и промывали большим объемом воды, затем сушили на воздухе. На второй стадии полученный продукт помещали в круглодонную колбу объемом 100 мл, добавляли поливинилпиридин/линейный полиэтиленимин/разветвленный полиэтиленимин, взятый в массовом соотношении матрица : полимер 1 : 0.1 и растворенный в деионизованной воде из расчета 20 мл на 1 г матрицы. Суспензию непрерывно перемешивали в течение 60 мин механической мешалкой при 60°С. Продукт отфильтровывали на стеклянном фильтре и промывали большим количеством воды, затем сушили на воздухе. Хроматографические колонки заполняли суспензионным способом при давлении 250 бар, используя в качестве подвижной фазы ацетонитрил. Предполагаемые структуры сорбентов и их обозначения представлены в табл. 1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтезировали три неподвижные фазы на основе 3-аминопропилсиликагеля путем ковалентного присоединения поли-4-винилпиридина, а также линейного и разветвленного полиэтиленимина, в качестве спейсера использовали бифункциональный 1,4-бутандиолдиглицидиловый эфир. Условия проведения синтеза, обеспечивающие получение неподвижных фаз с наилучшими для ГИХ свойствами, а также хроматографические характеристики сорбента С-ПЭГ [4] использовали для сравнения. Природу и структуру полимера, формирующего поверхностный слой, варьировали для установления факторов, влияющих на селективность и эффективность сорбентов, а также получения неподвижной фазы с лучшими параметрами для ГИХ. Для изучения хроматографических характеристик новых сорбентов использовали тест Танака для гидрофильных неподвижных фаз [24, 25] на примере разделения различных классов полярных соединений: сахаров, аминокислот и водорастворимых витаминов.
Результаты теста Танака представлены в табл. 2. Как видно, ковалентное присоединение полимеров приводит к увеличению гидрофильности k(U) поверхности неподвижных фаз, оцениваемой как фактор удерживания уридина, по сравнению с матрицей для всех сорбентов, кроме модифицированного поливинилпиридином С-Б-ПВП. Наибольшие значения гидрофильности демонстрируют сорбенты С-ПЭГ и С-Б-ПЭИлин, модифицированные линейными гидрофильными полимерами. При этом для С-Б-ПЭИразв, полученного в результате присоединения разветвленного ПЭИ, характерно несколько меньшее значение k(U) по сравнению с сорбентом с ПЭИлин. Это может быть следствием формирования тонкого поверхностного слоя в случае линейного полимера, в то время как разветвленный ПЭИ способствует образованию большего по толщине и более конформационно подвижного функционального слоя, массоперенос в котором затруднен. Кроме того, на поверхности сорбента с ПЭИразв больше стерически доступных гидрофобных этиленовых фрагментов, что также может вносить вклад в уменьшение гидрофильности неподвижной фазы. Таким образом, следует учитывать, что толщина функционального слоя оказывает влияние на вклады различных взаимодействий в механизм удерживания для сорбентов, полученных с использованием одинаковых по природе, но различных по пространственной структуре полимеров.
Таблица 2.
Результаты теста Танака для гидрофильных неподвижных фаз
| Селективность | Сорбент | ||||
|---|---|---|---|---|---|
| матрица | С-ПЭГ | С-Б-ПВП | С-Б-ПЭИразв | С-Б-ПЭИлин | |
| k(U) | 1.94 | 2.88 | 2.09 | 2.40 | 3.15 |
| α(CH2) | 1.27 | 1.32 | 1.18 | 1.50 | 1.25 |
| α(OH) | 1.94 | 1.87 | 1.67 | 1.90 | 1.99 |
| α(V/A) | 1.23 | 1.37 | 0.98 | 1.40 | 1.55 |
| α(AX) | 2.98 | 20.23 | 17.20 | 28.00 | 15.90 |
| α(CX) | 1.12 | 0 | 0 | 0 | 0.29 |
| α(Tb/Tp) | 0.82 | 0.64 | 0.57 | 0.45 | 0.61 |
Все четыре сорбента обладают сопоставимыми значениями параметров метиленовой α(CH2) и гидроксильной α(OH) селективности, отличными от единицы и схожими с данными для матрицы. Это позволяет предположить хорошую селективность рассматриваемых сорбентов по отношению к полярным соединениям одного гомологического ряда, а также олиго- и полиолам, таким как углеводы или многоатомные спирты. Кроме того, для всех сорбентов характерны несколько более высокие значения пространственной селективности α(V/A), кроме С-Б-ПВП с α(V/A) ≈ 1. В результате модифицирования поливинилпиридином происходит увеличение удерживания аденозина А (по сравнению с матрицей значение k’ увеличивается от 2.61 до 3.38), в то время как фактор удерживания видарабина V практически не меняется: k’(V) составляет 3.21 и 3.32 для матрицы и С-Б-ПВП соответственно. Стоит отметить, что различие в структуре полимеров, формирующих функциональный слой сорбентов С-Б-ПЭИразв и С-Б-ПЭИлин, не влияет на рассмотренные параметры.
Для всех четырех сорбентов характерен значительный вклад анионообменных взаимодействий в механизм удерживания – параметр α(AX) в 5−10 раз больше, чем для матрицы. Это ожидаемое следствие увеличения степени замещения атомов азота матрицы, в том числе формирования аммониевых групп в функциональном слое сорбентов в результате ковалентного модифицирования аминопропилсиликагеля. Однако высокое значение параметра α(AX) в случае сорбента С-Б-ПВП может являться следствием не только большого вклада электростатических взаимодействий, но и удерживания за счет дополнительных π–π взаимодействий между модифицированной поливинилпиридином поверхностью и п-толуолсульфонатом – маркером параметра α(AX). Одновременно с этим присоединение полимеров к матрице приводит к устранению вклада катионообменных взаимодействий α(СХ), что свидетельствует об экранировании диссоциировавших силанольных групп. Такой подход – модифицирование путем присоединения гидрофильного полимера – может быть использован при необходимости контроля вклада катионообменных взаимодействий или заряда матрицы при создании сорбентов для решения конкретных прикладных задач. Кроме того, малые значения параметра кислотно-основных свойств поверхности сорбентов α(Tb/Tp) < 1 и их уменьшение по сравнению с матрицей дополнительно свидетельствуют об осно́вной природе поверхности неподвижных фаз и увеличении степени замещенности атомов азота 3-аминопропилсиликагеля.
Полученные величины параметров гидрофильности k(U) и анионообменных свойств α(AX) для С-Б-ПЭИразв согласуются со значениями этих параметров для сорбента на основе силикагеля, модифицированного разветвленным ПЭИ, полученными в работе [6].
Углеводы. Сахара – моно- и олигосахариды – полярные вещества, являющиеся удобными аналитами для изучения свойств новых неподвижных фаз в режиме гидрофильной хроматографии. В ГИХ удерживание полярных нейтральных соединений, таких как углеводы, реализуется преимущественно по распределительному механизму. Отмечается также вклад образования водородных связей [27], адсорбционных взаимодействий [28, 29], замедленной мутаротации [30, 31] в удерживание сахаров.
Шкалы селективности относительно рибозы и эффективности по углеводам представлены на рис. 1. Показано, что для всех четырех сорбентов характерен порядок удерживания сахаров, ожидаемо коррелирующий с увеличением гидрофильности аналитов (табл. 3). Более того, времена удерживания дисахаридов увеличиваются с ростом гидрофильности поверхности сорбентов: С-Б-ПВП < C-Б-ПЭИразв < С-Б-ПЭИлин ≈ С-ПЭГ. Стоит отметить, что в случае С-ПЭГ наблюдали лучшее разрешение и иной порядок элюирования пары мальтоза/лактоза, чем для других сорбентов, модифицированных азотосодержащими полимерами. Однако при этом С-ПЭГ обеспечивал худшее разрешение пары рибоза/фруктоза. Из представленных данных видно, что сорбенты С-Б-ПВП и С-Б-ПЭИразв демонстрируют меньшие времена удерживания сахаров и несколько худшую селективность в отношении пар рибоза/фруктоза, фруктоза/глюкоза, мальтоза/лактоза по сравнению с С-Б-ПЭИлин и С-ПЭГ, что согласуется с уменьшением их гидрофильности по результатам теста Танака (табл. 1). Кроме того, модифицирование линейными гидрофильными полимерами, такими как ПЭГ и ПЭИлин, способствует получению сорбентов с более высокой эффективностью по сахарам – до 16 000 и 12 000 тт/м соответственно, в то время как присоединение разветвленного полиэтиленимина обеспечивает эффективность до 5000 тт/м. Это может быть следствием уменьшения сопротивления массопереносу при уменьшении толщины функционального слоя в случае использования линейных полимеров. Хроматограммы модельной смеси углеводов представлены на рис. 2. В случае сорбента С-Б-ПВП ввиду низкой селективности и эффективности добиться разделения смеси сахаров не удалось.
Рис. 1.
Шкалы селективности относительно рибозы и диаграммы эффективности по сахарам (Rib – рибоза, Fru – фруктоза, Glu – глюкоза, Suc – сахароза, Mal – мальтоза, Lac – лактоза). Подвижная фаза – деионизованная вода−ацетонитрил (15 : 85, по объему), скорость потока 1 мл/мин, рефрактометрическое детектирование.
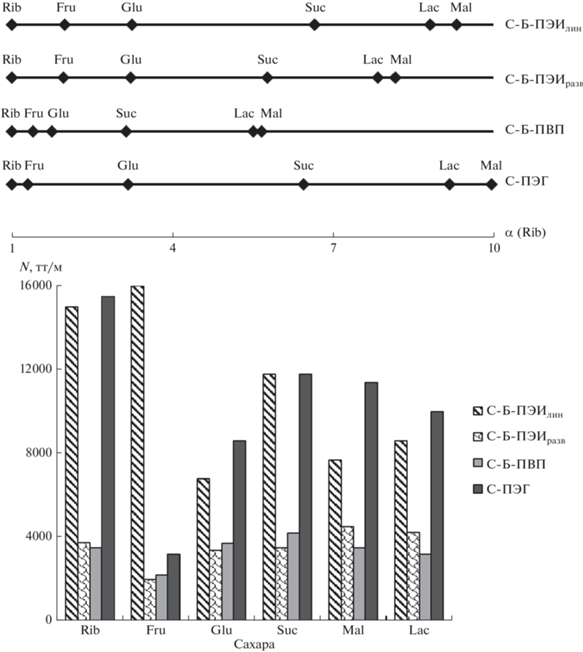
Таблица 3.
Параметры гидрофобности модельных аналитов
| Углеводы | Аминокислоты | ||||
|---|---|---|---|---|---|
| аналит | lgP* | аналит | lgP* | аналит | lgP* |
| Рибоза | –2.66 | Фенилаланин | –1.51 | Пролин | –2.38 |
| Фруктоза | –1.78 | Изолейцин | –1.82 | Аспарагин | –5.22 |
| Глюкоза | –3.12 | Метионин | –2.64 | Гистидин | –3.45 |
| Сахароза | –4.50 | Лейцин | –1.82 | Глицин | –3.64 |
| Мальтоза | –5.26 | Валин | –2.31 | Аланин | –3.22 |
| Лактоза | –5.26 | Тирозин | –1.99 | Серин | –3.69 |
Рис. 2.
Хроматограммы модельной смеси углеводов (500–1000 мг/л; Xyl – ксилоза). Объем пробы – 20 мкл. Подвижная фаза – деионизованная вода−ацетонитрил (15 : 85, по объему), скорость потока 1 мл/мин, рефрактометрическое детектирование.

Аминокислоты. Аминокислоты, представляющие собой класс полярных цвиттер-ионных соединений, также являются удобной группой модельных аналитов для изучения свойств новых сорбентов в режиме гидрофильной хроматографии. Популярность метода ГИХ при определении аминокислот обусловлена отсутствием стадии дериватизации во время пробоподготовки. В качестве подвижной фазы использовали 5 мМ фосфатный буферный раствор с pH 6.5 [4].
Из представленных шкал селективности (рис. 3) видно, что С-ПЭГ обеспечивает лучшую селективность по отношению к аминокислотам среди серии сорбентов. С-Б-ПЭИразв также продемонстрировал селективность по отношению к наиболее полярным аминокислотам – аспарагину, гистидину, глицину, аланину и серину, однако эффективность не превысила 12000 тт/м. Сочетание высокой эффективности (до 35000 тт/м) и достаточной селективности обеспечило разрешение семи аминокислот на сорбенте С-Б-ПЭИлин (рис. 4).
Рис. 3.
Шкалы селективности относительно фенилаланина и диаграммы эффективности по аминокислотам (1 – фенилаланин, 2 – изолейцин, 3 – метионин, 4 – лейцин, 5 – валин, 6 – тирозин, 7 – пролин, 8 – аспарагин, 9 – гистидин, 10 – глицин, 11 – аланин, 12 – серин). Подвижная фаза – 5 мМ фосфатный буферный раствор (pH 6.5)−ацетонитрил (15 : 85, по объему), скорость потока 1 мл/мин, УФ-детектирование, 210 нм.

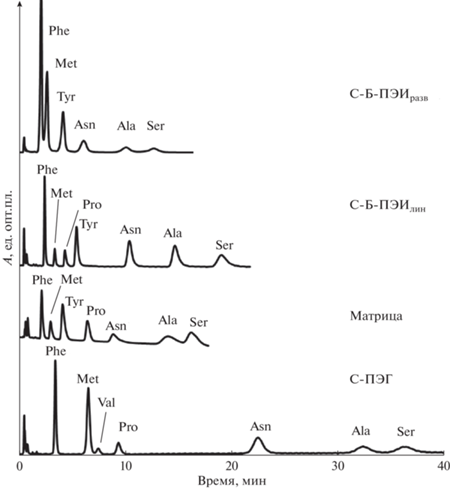
Рис. 4.
Хроматограммы модельных смесей аминокислот (фенилаланин – 10 мг/л, тирозин – 100 мг/л, остальные – 200 мг/л). Объем пробы – 20 мкл. Подвижная фаза – 5 мМ фосфатный буферный раствор (pH 6.5)−ацетонитрил (15 : 85, по объему), скорость потока 1 мл/мин, УФ-детектирование, 254 нм.
Все четыре сорбента характеризуются схожим порядком удерживания, в целом коррелирующим с уменьшением параметра гидрофобности Ханша аналитов (табл. 3). Факторы удерживания наиболее гидрофильных аминокислот – аспарагина, гистидина, глицина, аланина и серина – на сорбентах, модифицированных полиаминами, коррелируют с параметром гидрофильности по тесту Танака (kU) и увеличиваются в ряду: С-Б-ПВП < < C-Б-ПЭИразв < С-Б-ПЭИлин < С-ПЭГ, как и в случае сахаров. Таким образом, наилучшее удерживание аминокислот обеспечивает использование ПЭГ в функциональном слое неподвижной фазы. Кроме того, для сорбентов, модифицированных ПЭИ, наблюдали обратный по сравнению с двумя другими неподвижными фазами порядок удерживания пролина и тирозина, при котором менее гидрофильный тирозин имеет большие факторы удерживания, чем пролин. Такая селективность может быть вызвана вкладом водородных связей ОH-группы тирозина с аминогруппами фаз, модифицированных полиэтиленимином. Более того, модифицирование разветвленным полиэтиленимином привело к обратному порядку элюирования пары изолейцин/лейцин по сравнению с матрицей и сорбентами, модифицированными линейными полимерами (рис. 3), включая ПЭИлин. Это, как видно, не связано со структурой полимера и может быть следствием лучшего экранирования матрицы полиамином разветвленной структуры.
Сорбент С-Б-ПВП продемонстрировал слабое удерживание аминокислот и низкую эффективность до 8000 тт/м, в результате чего достигнуть разделения модельной смеси не удалось. Модифицирование разветвленным ПЭИ привело к получению сорбента с несколько лучшей селективностью в отношении аминокислот и эффективностью до 12 000 тт/м, что обеспечило разделение семи аминокислот за 18 мин с использованием смеси 1 мМ фосфатного буферного раствора и ацетонитрила в соотношении 15 : 85 ( по объему). Лучшая эффективность – до 40 000 тт/м – достигнута для колонки с сорбентом С-ПЭГ, однако время разделения модельной смеси восьми аминокислот в изократическом режиме составило 40 мин, чтобы обеспечить разделение аланина и серина. Использование сорбента С-Б-ПЭИлин позволило разделить аналогичную смесь из восьми аминокислот в тех же условиях в два раза быстрее (менее чем за 20 мин) с эффективностью до 35 000 тт/м. Стоит отметить, что С-ПЭГ обладает лучшей селективностью в отношении пары фенилаланин/метионин и трудноразделимой пары изолейцин/лейцин, однако характеризуется низким разрешением пары тирозин/пролин, в результате чего их одновременное определение невозможно. В то же время использование сорбента С-Б-ПЭИлин, демонстрирующего иной порядок удерживания аминокислот валин-пролин-тирозин, позволило достичь разделения пары тирозин/пролин, однако неразрешимой оказалась пара валин/пролин, разделяемая до базовой линии на С-ПЭГ. Таким образом, при анализе реальных объектов выбор С-ПЭГ или С-Б-ПЭИлин будет обусловлен набором разделяемых аминокислот. Хроматограммы модельной смеси аминокислот для полученных сорбентов и исходной матрицы представлены на рис. 4. Модифицирование аминопропилсиликагеля ПЭИлин позволило повысить селективность для пары аланин/серин, а также эффективность по отношению к аминокислотам.
Водорастворимые витамины. Водорастворимые витамины характеризуются большим разнообразием структур, физико-химических свойств и гидрофильностью, поэтому широко применяются для демонстрации возможностей неподвижных фаз для ГИХ. Использование витаминов в качестве модельных аналитов для тестирования сорбентов может дать полезную информацию для оценки вкладов различных типов взаимодействий, а также позволит оценить разделяющие способности изучаемых сорбентов по сравнению с коммерчески доступными, возможности которых также продемонстрированы в литературе на примере витаминов. Для сравнения рассматриваемых неподвижных фаз хроматографические параметры удерживания витаминов определяли с использованием в качестве подвижной фазы смеси 100 мМ ацетатно-аммонийного буферного раствора с pH 5.8 и ацетонитрила (10 : 90, по объему). Выбор природы буферного раствора обусловлен хорошей растворимостью ацетата аммония в растворах с высоким содержанием ацетонитрила. УФ-детектирование осуществляли при длине волны 270 нм.
Как видно из представленных на рис. 5 данных, для всех четырех сорбентов порядок элюирования витаминов в режиме ГИХ согласуется со свойствами сформированных функциональных слоев, которые подтверждены параметрами теста Танака. Так, наиболее гидрофильный тиамин (lgP = –4.8) имеет малые факторы удерживания на всех сорбентах и в зависимости от природы привитого полимера его удерживание изменяется в ряду С-Б-ПЭИ < С-Б-ПВП < С-ПЭГ, что, по-видимому, вызвано уменьшением числа пространственно доступных аммониевых групп в структуре функционального слоя сорбентов. Увеличение степени замещения аминогрупп матрицы в результате модифицирования, а также присоединения ПЭИ, в том числе образование четвертичных аммониевых групп, приводит к увеличению положительного заряда поверхности. В выбранных условиях анализа удерживание тиамина, также несущего положительный заряд, регулируется в большей степени силами электростатического отталкивания [32], тем самым обеспечивая альтернативный порядок по сравнению с немодифицированным силикагелем [33]. Следует отметить, что удерживание цианокобаламина В12 согласуется с гидрофильностью сорбента и увеличивается в ряду: С-Б-ПВП < С-Б-ПЭИразв < < С‑Б-ПЭИлин < С-ПЭГ, что может свидетельствовать о большом вкладе распределительного механизма в его удерживание. Кроме того, высокая анионообменная селективность всех четырех сорбентов, установленная по результатам теста Танака (табл. 2), обусловливает сильное удерживание аскорбиновой и никотиновой кислот, поэтому разделения модельной смеси семи витаминов во всех случаях достигали с использованием градиентного режима.
Рис. 5.
Шкалы селективности относительно никотинамида и диаграммы эффективности по витаминам. Подвижная фаза – 100 мМ ацетатно-аммонийный буферный раствор (pH 5.8)−ацетонитрил (10 : 90, по объему), скорость потока 1 мл/мин, УФ-детектирование, 270 нм.

Для экспрессного разделения слабоудерживаемых витаминов (никотинамид, В1, В2 и В6) варьировали долю водной фазы в составе элюента в диапазоне 8–12 об. %. Таким образом было достигнуто приемлемое разрешение Rs > 1.4 пары тиамин/рибофлавин на С-ПЭГ, а также пары пиридоксин/тиамин на С-Б-ПЭИразв. Во всех случаях при увеличении доли воды в подвижной фазе наблюдали ожидаемое для ГИХ уменьшение времен удерживания витаминов. Оптимальное содержание водной фазы составило: 4 об. % – для С-Б-ПЭИразв, 8 об. % – для С-ПЭГ и 12 об. % – для С-Б-ПЭИлин. Достичь полного разделения модельной смеси на С-Б-ПВП не удалось из-за низкой эффективности 3000–11 000 тт/м. На сорбенте, модифицированном разветвленным ПЭИ, время разделения витаминов в градиентном режиме составило 25 мин с эффективностью до 21 000 тт/м. Высокая эффективность сорбентов С-ПЭГ и С-Б-ПЭИлин (до 35 000 тт/м) позволила улучшить разрешение и уменьшить продолжительность анализа анализа до 16 мин. Хроматограммы витаминов представлены на рис. 6. Следует добавить, что при уменьшении доли буферного раствора на первом шаге градиентного элюирования на сорбенте С-ПЭГ стало возможным разделение тиамина и рибофлавина до базовой линии, а также изменение их порядка удерживания по сравнению с данными рис. 5 вследствие большего вклада электростатического отталкивания тиамина при уменьшении концентрации противоиона, экранирующего положительно заряженные группы в функциональном слое сорбента.
Рис. 6.
Хроматограммы модельных смесей витаминов (рибофлавин – 4 мг/л, никотинамид – 10 мг/л, остальные – 50–200 мг/л). Подвижная фаза – 100 мМ ацетатно-аммонийный буферный раствор (pH 5.8)−ацетонитрил, градиентный режим: для С-ПЭГ: 0–5 мин 8% буферного раствора, 5–6 мин 8–20% буферного раствора, 6–16 мин 20% буферного раствора; для С-Б-ПЭИлин: 0–4 мин 12% буферного раствора, 4–6 мин 12–20% буферного раствора, 6–16 мин 20% буферного раствора; для С-ПВП: 0–3 мин 10% буферного раствора, 3–4 мин 10–20% буферного раствора, 4–10 мин 20% буферного раствора, 10–11 мин 20–10% буферного раствора, 11–20 мин 10% буферного раствора. Объем пробы – 20 мкл. Скорость потока 1 мл/мин, УФ-детектирование, 270 нм.

* * *
Таким образом, сравнение хроматографических характеристик неподвижных фаз, полученных на основе 3-аминопропилсиликагеля путем ковалентного присоединения различных по природе, структуре и гидрофильности полимеров, показало, что увеличение времен удерживания полярных соединений различных классов коррелирует с ростом гидрофильности поверхности сорбентов. Использование наиболее гидрофобного поливинилпиридина привело к получению неподвижной фазы с невысокими эффективностью и селективностью в ГИХ. Модифицирование разветвленным полиэтиленимином в наибольшей степени способствует изменению селективности по сравнению с матрицей. При этом использование полимеров с линейной структурой (ПЭИлин и ПЭГ) приводит к значительному росту эффективности синтезируемых сорбентов по сахарам, аминокислотам и витаминам. Предлагаемый подход к получению неподвижных фаз обеспечивает высокую анионообменную селективность сорбентов, что является интересной альтернативой коммерчески доступным гидрофильным сорбентам на основе силикагеля благодаря иной селективности по отношению к заряженными аналитам.
Работа выполнена при поддержке Российского научного фонда (грант № 20-13-00140) с использованием оборудования, приобретенного за счет средств Программы развития Московского университета.
Список литературы
Alpert A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds // J. Chromatogr. A. 1990. V. 499. P. 177.
Huber J.F.K., Pawlowska M., Markl P. Selection of the solid support – A crucial step for the realization of solvent generated liquid-liquid chromatography // J. Chromatogr. A. 1990. V. 500. P. 280.
Чернобровкина А.В., Смоленков А.Д., Шпигун О.А. Гидрофильная хроматография – перспективный метод определения полярных веществ // Лаборатория и производство. 2018. № 4. С. 76.
Попов А.С., Максимов Г.С., Смоленков А.Д., Шпигун О.А., Чернобровкина А.В. Новые сорбенты для гидрофильной хроматографии на основе силикагеля, ковалентно модифицированного полиэтиленгликолем // Вестн. Моск. ун-та. Сер. 2: Химия. 2021. Т. 62. С. 164.
Qiu H., Loukotková L., Sun P., Tesařová E., Bosáková Z., Armstrong D. W. Cyclofructan 6 based stationary phases for hydrophilic interaction liquid chromatography // J. Chromatogr. A. 2011. V. 1218. P. 270.
Peng Ya., Hou Ya., Zhang F., Shen G., Yang B. A hyperbranched polyethylenimine functionalized stationary phase for hydrophilic interaction liquid chromatography // Anal. Bioanal. Chem. 2016. V. 408. P. 3633.
Erim F.B., Cifuentes A., Poppe H., Kraak J.C. Performance of a physically adsorbed high-molecular-mass polyethyleneimine layer as coating for the separation of basic proteins and peptides by capillary electrophoresis // J. Chromatogr. A. 1995. V. 708. P. 356.
Towns J.K., Regnier F.E. Polyethyleneimine-bonded phases in the separation of proteins by capillary electrophoresis // J. Chromatogr. A. 1990. V. 516. P. 69.
Nutku M.S., Erim F.B. Polyethyleneimine-coated capillary electrophoresis capillaries for the analysis of organic acids with an application to beverages // J. Microcolumn. Sep. 1999. V. 11. P. 541.
Nutku M.S., Erim F.B. The use of cationic polymer for the separation of inorganic anions by capillary electrophoresis // J. High. Resolut. Chromatogr. 1998. V. 21. P. 505.
Sedyohutomo A., Suzuki H., Fujimoto C. Determination of inorganic anions by capillary ion-exchange chromatography using polyethylenimine-coated octadecyl-bonded phases // Anal. Sci. 2012. V. 28. P. 625.
Лошин, А.А., Глазков Е.С., Затираха А.В., Смоленков, А.Д., Шпигун О.А. Хроматографические свойства анионообменников на основе силикагеля, поверхностно модифицированных полиэтиленимином // Сорбционные и хроматографические процессы. 2015. Т. 15. С. 390.
Alpert A.J., Regnier F.E. Preparation of a porous microparticulatee anion-exchange chromatography support for proteins // J. Chromatogr. 1979. V. 185. P. 375.
Rounds M., Kopaciewicz W., Regnier F. Factors contributing to intrinsic loading capacity in silica-based packing materials for preparative anion-exchange protein chromatography. // J. Chromatogr. A.1986. V. 362. P. 187.
Pearson J., Regnier F. High-performance anion-exchange chromatography of oligonucleotides // J. Chromatogr. A. 1983. V. 255. P. 137.
Drager R., Regnier F. High-performance anion-exchange chromatography of oligonucleotides // Anal. Biochem. 1985. V. 145. P. 47.
Lv Y.Q., Lin Z.X., Svec F. Hypercrosslinked large surface area porous polymer monoliths for hydrophilic interaction liquid chromatography of small molecules featuring zwitterionic functionalities attached to gold nanoparticles held in layered structure // Anal. Chem. 2012. V. 84. P. 8457.
Jiang L.W., Jin Y., Marcus R.K. Polyethylenimine modified poly (ethylene terephthalate) capillary channeled-polymer fibers for anion exchange chromatography of proteins // J. Chromatogr. A. 2015. V. 1410. P. 200.
Takeuchi T., Kawasaki T., Lim L.W. Separation of inorganic anions on a pyridine stationary phase in ion chromatography // Anal. Sci. 2010. V. 26. P. 511.
Auler L.M., Silva C.R., Collins K.E., Collins C.H. New stationary phase for anion-exchange chromatography // J. Chromatogr. A. 2005. V. 1073. P. 147.
West C., Lemasson E., Nagai K., Shibata T., Franco P., Bertin S., Hennig P., Lesellier E. Characterization of novel polymer-based pyridine stationary phases for supercritical fluid chromatography // Chromatographia. 2019. V. 82. P. 143.
Nagai K., Shibata T., Shinkura S., Ohnishi A. Poly (4-vinylpyridine) based novel stationary phase investigated under supercritical fluid chromatography conditions // J. Chromatogr. A. 2018. V. 1572. P. 119.
Dunkle M., West C., Pereira A., der Plas S.V., Madder A., Farrell W. Synthesis of stationary phases containing pyridine, phenol, aniline and morpholine via click chemistry and their characterization and evaluation in supercritical fluid chromatography // Scientia Chromatographica. 2014. V. 6. P. 85.
Kawachi Y., Ikegami T., Takubo H., Ikegami Y., Miyamoto M., Tanaka N. Chromatographic characterization of hydrophilic interaction liquid chromatography stationary phases: Hydrophilicity, charge effects, structur-al selectivity, and separation efficiency // J. Chromatogr. A. 2011. V. 1218. P. 5903.
Dolci M., Chromatographic Characterization of Stationary Phases for Hydrophilic Interaction Liquid Chromatography. Runcorn, Cheshire, UK: Thermo Fisher Scientific, 2013.
Dinh N. P., Jonsson T., Igrum K. Probing the interaction mode in hydrophilic interaction chromatography // J. Chromatogr. A. 2011. V. 1218. P. 5880.
Fu Q., Guo Z., Liang T., Zhang X., Xua Q., Liang X. Chemically bonded maltose via click chemistry as stationary phase for HILIC // Anal. Methods. 2010. V. 2. P. 217.
Fu Q., Liang T., Zhang X., Du Yu., Guo Z., Liang X. Carbohydrate separation by hydrophilic interaction liquid chromatography on a 'click'maltose column // Carbohydr. Res. 2010. V. 345. P. 2690.
Fu. Q., Tu L., Li Z., Xu X., Ke Ya., Jin Yu, Liang X. Separation of carbohydrates using hydrophilic interaction liquid chromatography // Carbohydr. Res. 2013. V. 379. P. 13.
Kotoni D., D`Acquarica I., Ciogli A., Villani C., Capitani D., Gasparrini F. Design and evaluation of hydrolytically stable bidentate urea-type stationary phases for hydrophilic interaction chromatography // J. Chromatogr. A. 2012. V. 1232. P. 196.
Jenkins K. HILIC separation of carbohydrates using BEH amide particle technology // J. Chromat. Today. 2015. V. 8. P. 13.
Карцова Л.А., Бессонова Е.А., Сомова В.Д. Гидрофильная хроматография // Журн. аналит. химии. 2019. Т. 74. № 5. С. 323 (Kartsova L.A., Bessonova E.A., Somova V.D. Hydrophilic interaction chromatography // J. Anal. Chem. 2019. V. 74. P. 415.)
Porter K., Lodge J. K. Determination of selected water-soluble vitamins (thiamine, riboflavin, nicotinamide and pyridoxine) from a food matrix using hydrophilic interaction liquid chromatography coupled with mass spectroscopy // J. Chromatogr. B. 2021. V. 1171. Article 122541.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии