

Журнал аналитической химии, 2022, T. 77, № 9, стр. 837-848
Способ корректировки результатов электронно-зондового энергодисперсионного элементного анализа порошковых материалов
Д. Э. Пухов a, *, А. А. Лаптева b
a Ярославский филиал Физико-технологического института имени К.А. Валиева Российской академии наук
150007 Ярославль, ул. Университетская, 21, Россия
b Ярославский государственный университет им. П.Г. Демидова
150003 Ярославль, ул. Советская, 14, Россия
* E-mail: puhov2005@yandex.ru
Поступила в редакцию 30.10.2021
После доработки 24.11.2021
Принята к публикации 01.12.2021
- EDN: WLHHNQ
- DOI: 10.31857/S0044450222090110
Аннотация
Установлены основные закономерности количественных изменений в энергодисперсионных спектрах при определении элементного состава порошкообразных материалов. Показано, что в большинстве случаев при определенной энергии зондирующих электронов расчет относительных массовых долей элементов в образцах порошков приводит к результатам, не отличающимся от полученных на эталонных полированных поверхностях образцов. При обобщении экспериментальных данных получена эмпирическая зависимость, связывающая параметры характеристических фотонов с величиной ускоряющего напряжения, необходимого для получения корректного соотношения массовых концентраций элементов при анализе порошкообразных материалов. Предложена методика корректировки результатов элементного анализа порошковых образцов.
Электронно-зондовый энергодисперсионный анализ – распространенный и достаточно эффективный экспресс-метод определения элементного состава материалов. Системы энергодисперсионного микроанализа компактны, легко интегрируемы в электронно-микроскопические системы. Детекторы рентгеновских фотонов позволяют обнаруживать примесные элементы с пределом детектирования около 0.05–0.1 мас. % и проводить анализ с относительной погрешностью в пределах 10% для элементов с концентрацией 1–10 мас. % [1–4].
Общими неизменными требованиями для получения корректных результатов расчета относительных долей элементов являются однородность состава в пределах зоны генерации рентгеновских фотонов, строго определенная ориентация плоскости поверхности образца и высокая степень ее гладкости, обычно достигаемая разными способами полирования. В большинстве случаев неидеальная гладкость поверхности образца является главной причиной смещения результатов количественного анализа. При этом качество обработки поверхности влияет в большей степени на рентгеновские диагностические линии с небольшой энергией. Так, моделирование методами Монте-Карло показывает, что борозды глубиной и шириной около 100 нм, ориентированные перпендикулярно направлению движения фотонов от поверхности образца к детектору, приводят к относительному занижению на 5% интенсивностей K-линий кислорода (0.51 кэВ) и L-линий железа (0.71 кэВ) в спектре оксида железа. Занижение интенсивностей на величину менее 1% достигается лишь при уменьшении размеров таких неровностей до 20 нм [4, 5]. При анализе в точке может наблюдаться как завышение, так и занижение интенсивности низкоэнергетической линии относительно более высоко энергетической в зависимости от положения точки анализа относительно направления на детектор. Это сопровождается отклонениями результатов вычисления массового содержания элементов от нескольких десятков до сотни процентов [1, 4]. При сканировании вдоль линии или растром геометрические эффекты вследствие усреднения ориентации локальных участков поверхности сказываются в меньшей степени.
Наиболее сильно влияние геометрии поверхности должно сказываться при количественном анализе по характеристическим линиям с энергией менее 200 эВ, включая К-линию лития. В последние годы показана возможность диагностирования фотонов с такими энергиями с использованием детекторов большой площади с низким уровнем шума [6–8]. Геометрические неоднородности должны влиять на результаты определения легких элементов в максимальной степени при локальном сканировании по участкам с размером менее 50 нм, достигаемом в низковольтной сканирующей микроскопии [9].
В целом трудности количественного электронно-зондового анализа порошковых образцов обсуждаются уже около 50 лет [10]. Выделяют следующие основные причины их возникновения. Во-первых, это так называемый массовый эффект или эффект уменьшения области генерации рентгеновских фотонов, связанный с увеличением доли отраженных электронов при падении на неровности или грани отдельных частиц, ориентация которых по отношению к электронному зонду отличается от нормальной [4, 11, 12]. Массовый эффект наблюдается и для индивидуальных микрочастиц, если их объем меньше области генерации рентгеновского излучения. В таком случае область генерации имеет обрыв, а измеряемая интенсивность характеристической линии занижена по сравнению с интенсивностью, измеренной при тех же условиях на массивном полированном образце.
Во-вторых, в зависимости от положения точки анализа на неровной поверхности по отношению к детектору пробег фотона в материале может уменьшаться или увеличиваться. Отношение регистрируемых интенсивностей низкоэнергетических линий к интенсивностям линий с высокой энергией может быть в разы больше или меньше отношения, наблюдаемого для образца с гладкой поверхностью вследствие сильно различающихся коэффициентов поглощения [4, 5, 11]. Данная закономерность носит название “эффект поглощения”.
В качестве третьего и менее значимого фактора указывается обрыв области генерации флуоресцентного излучения даже для достаточно крупных частиц с размерами порядка десятков микрометров. Данный эффект обусловливает необходимость дополнительной корректировки при анализе отдельных частиц, спектр характеристического рентгеновского излучения которых содержит близкие по энергии диагностические линии [4, 10].
Одним из возможных способов достижения удовлетворительных результатов элементного анализа в случае неровных поверхностей и индивидуальных частиц является использование метода пик/фон. В его основе лежит наблюдаемое явление, состоящее в том, что отношение интенсивностей характеристического и тормозного излучений с одинаковой энергией мало зависит от формы и размеров образца. Метод заключается во введении поправки в интенсивность характеристической линии элемента в составе исследуемого неровного образца в виде коэффициента, являющегося отношением интенсивности тормозного фона для массивного полированного образца и исследуемого материала [4]. Интенсивность тормозного излучения от полированного образца измеряется на одноэлементных стандартах или стандартах, близких по составу к исследуемому веществу. В другом случае она может быть получена моделированием на основе закономерностей возникновения непрерывного рентгеновского излучения [11, 13, 14]. Однако отмечается, что в случае малых частиц, для которых сильно выражен массовый эффект, наблюдается анизотропия генерации характеристического и непрерывного рентгеновского излучения [13]. Это требует внесения дополнительных изменений в экспериментально определяемую интенсивность фона с учетом размеров и геометрии частиц. Также к сложностям правильного подбора аппроксимирующей кривой для фона тормозного излучения приводит использование низких ускоряющих электронный зонд напряжений, снижающих проявление массового эффекта [15]. Кроме того, во всех случаях анализа частиц или массивных образцов с шероховатой поверхностью вследствие относительно низкой детектируемой интенсивности непрерывного фонового излучения неизбежно возникает существенная погрешность при определении отношения пик/фон. Это влечет необходимость значительного увеличения времени набора энергодисперсионного спектра. В целом же метод пик/фон достаточно эффективен, позволяет уменьшить относительную погрешность расчета концентрации элементов до величин в пределах от нескольких до десяти процентов в зависимости от содержания элементов в материале.
Другим подходом для учета массового эффекта и эффекта поглощения является теоретическое моделирование, учитывающее ограничения поверхностью частицы зоны генерации и поглощения рентгеновских фотонов. Подобные исследования ведутся с 70-х годов прошлого века [10, 16–19]. Теоретическая корректировка особенностей геометрии образца является трудоемкой и дает хорошие результаты с относительной погрешностью порядка 1–3% лишь при исследовании состава одиночных частиц строго определенных формы и размеров в матрице известного состава [20].
В качестве альтернативного пути предлагается корректировка экспериментальных спектров на основании постоянно пополняющихся баз экспериментальных данных и данных о моделировании генерации и поглощения рентгеновских фотонов для частиц разного состава и морфологии [21]. Относительно практического применения для анализа такой подход предполагает, что среди моделированных и/или экспериментальных данных должны обнаруживаться энергодисперсионные спектры материалов, которые при огромном разнообразии возможных форм и размеров частиц близко совпадают по геометрии с исследуемым образцом.
В случае с порошковыми материалами геометрия поверхности образца носит случайный характер. В работе [22] предложено теоретическое определение отношения интенсивностей линий элементов методом интегрирования с учетом моделируемого распределения плотности генерации характеристического излучения под наклонной плоскостью со случайным углом наклона и ориентацией относительно детектора рентгеновских фотонов. Получено совпадение с экспериментальными зависимостями относительных интенсивностей от энергии электронов. Трудоемкость и длительность расчетов может вызвать затруднения для включения описываемого теоретического аппарата в алгоритмы автоматической корректировки результатов анализа. Тем не менее одним из важных выявленных фактов является то, что при некоторых энергиях зондирующих электронов отношения интенсивностей диагностических линий некоторых или всех пар элементов в порошковых материалах совпадают с таковыми для плоскополированных образцов того же состава. Это предполагает возможность получения более точных результатов количественного анализа при определенных значениях ускоряющего напряжения.
В данной работе рассматривается способ элементного дисперсионного анализа порошкообразных материалов на основании эмпирического подбора энергии сканирующих электронов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Энергодисперсионные спектры получали на двулучевой СЭМ-ФИП системе Quanta 3D 200i (FEI, Нидерланды) с приставкой элементного анализа EDAX Apollo X (Ametek Inc., США). Диапазон ускоряющих напряжений 3–30 кВ, рабочий отрезок при наборе энергодисперсионных спектров 15 мм, угол сбора рентгеновских фотонов относительно направления движения электронов зонда 50.5°. Использовали режимы высокого вакуума для полупроводниковых образцов и низкого вакуума с заполнением рабочей камеры парами воды под давлением 70–80 Па для диэлектрических материалов. Спектры обрабатывали в программной среде EDAХ Genesis (Ametek Inc., США). Для вычисления массовых долей элементов использовали метод ZAF-коррекции. Количественный анализ в случае наличия низкоэнергетических диагностических линий (менее 0.8 кэВ) проводили с использованием эталонных образцов в виде соединений.
Объекты анализа. В качестве анализируемых материалов выбрали образцы, содержащие менее 1 мас. % примесных элементов. Использовали полупроводниковые материалы: карбид кремния (SiC) в виде шлифовальных порошков механических фракций F220, F600, F1200, F2000 и полированной поверхности муассанита; минералы касситерит (SnO2), стибнит (Sb2S3), пирит (FeS2), куприт (Cu2O), цинкит (ZnO), халькопирит (CuFeS2), арсенопирит (FeAsS), теннантит (Cu12As4S13), дискразит (Ag3Sb), Zn-замещенный минерал изоморфного ряда тетраэдрит-теннантит (Cu(3 – x)ZnxSbS(3 – y)Asy). Диэлектрические минералы: кварц (SiO2), корунд (Al2O3), кианит (Al2O(SiO4)), флюорит (CaF2), исландский шпат (CaCO3), целестин (SrSO4), виллемит (Zn2SiO4), микроклин (KAlSi3O8), сколецит (CaAl2Si3O10·3H2O), энстатит (Mg1.92Fe0.08(Si2O6)), спессартин (Ca0.17Mg0.70Mn2.31Al2(SiO4)3). В качестве контрольных использовали полированные поверхности образцов, перевод в порошкообразное состояние проводили дроблением и растиранием в агатовой ступке, для минералов с высокой твердостью применяли шлифующую резку инструментами с алмазными абразивами. Поверхность порошков в кювете выравнивали полированной пластиной из поликристаллического корунда. Визуально контролировали горизонтальность поверхности при размещении в камере микроскопа и отсутствие неровностей более 300 мкм в зоне сканирования.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Общие закономерности изменения интенсивностей диагностических линий на примере бинарных соединений. Относительные отклонения регистрируемых интенсивностей диагностических рентгеновских линий в энергодисперсионных спектрах порошкообразных образцов оценивали попарно по величине K:
где I1 и I2 – интенсивности диагностических линий элементов соответственно с меньшей и большей энергий для порошкового материала, $I_{1}^{0}$ и $I_{2}^{0}$ – интенсивности тех же линий в контрольном спектре, полученном с полированной поверхности.Такой подход позволяет избежать влияния таких факторов, как непропорциональное изменение интенсивностей линий с разными энергиями при изменении энергии зондирующих электронов, различий в чувствительности детектора к рентгеновским фотонам разных энергий. Использование величины K также допускает получение спектров контрольного и порошкообразного образца при разных значениях силы тока сканирующего электронного пучка.
Наиболее сильное искажение относительных величин диагностических пиков наблюдается для карбида кремния. Исследованы несколько размерных фракций порошка (рис. 1). Величина K, рассчитанная для линий углерода С(K-L3) и суммарной линии кремния Si(K-L3, K-M3), зависит от энергии зондирующих электронов, как показано на рис. 2а. В зависимости от энергии зондирующих электронов может происходить как относительное занижение рефлекса углерода, так и завышение. Соответственно в результатах количественного анализа доля углерода может оказаться завышенной или заниженной. Не обнаружена зависимость величины K от среднего размера частиц в порошке, за исключением самой мелкоразмерной фракции. Этот факт мы объясняем тем, что для мелких фракций материала сильно сказывается массовый эффект, когда для большого количества частиц размеры зоны генерации рентгеновского излучения начинают превосходить размеры самих частиц. Область генерации в таком случае переходит через поверхность соседней частицы, которая может оказаться ориентированной на детектор и обеспечить слабое поглощение фотонов. Частичным подтверждением этому является результат моделирования размера области генерации излучения углерода (рис. 2б) методами Монте-Карло с помощью программы CASINO v.2.48. (https://www.gel.usherbrooke.ca/casino/). Описание программного продукта приведено в работах [23–26].
Рис. 1.
СЭМ-изображения порошков SiC со средними линейными размерами частиц 62 мкм (а), 11 мкм (б), 3.4 мкм (в), 1.4 мкм (г); длина масштабного отрезка приведена в микрометрах.

Рис. 2.
Зависимость характеристики K от энергии электронного зонда для разных размерных фракций частиц SiC и их смеси одинаковых пропорций по массе (а); моделирование глубины генерации характеристического излучения углерода в составе SiC в зависимости от энергии индуцирующих электронов (б).
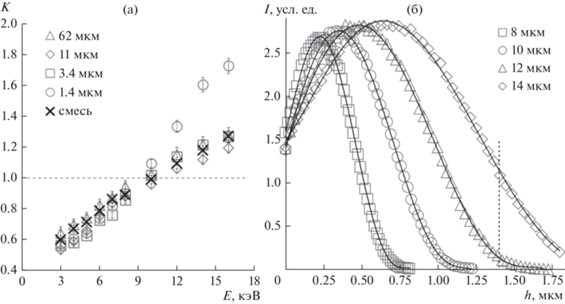
Резкие различия в поведении характеристики К для мелкоразмерной фракции проявляются в тот момент, когда глубина и сопоставимый с ней радиус области генерации характеристического излучения углерода начинают превосходить средний размер частицы. Аналогично массовый эффект присутствует и для линии кремния. Но вследствие больших различий в массовых коэффициентах поглощения С(K-L3) и Si(K-L3, K-M3), 25200 и 390 см2/г соответственно, относительное уменьшение поглощения характеристических фотонов кремния пренебрежимо мало.
По результатам прочих экспериментов какой-либо зависимости характеристики K от размера частиц порошка не обнаружено. Так, в случае порошков SnO2, полученных дроблением и дополнительным растиранием, зависимости величины К, определенной для интенсивностей линий O(K-L3) и суммарных Sn(L3-M5,4; L3-N5; L2‑M4; L1-M3), от энергии зондирующих электронов идентичны (рис. 3).
Рис. 3.
СЭМ-изображения порошков SnO2, полученных дроблением (а) и дополнительным растиранием (б); соответствующие зависимости характеристики К от энергии электронов зонда (в). Длина масштабного штриха указана в микрометрах.

Еще одной особенностью отношения интенсивностей К является то, что зависимость К от энергии электронов зонда может практически отсутствовать, если энергии диагностических линий и коэффициенты поглощения в образце соответствующих фотонов отличаются на небольшую величину. Так, при анализе порошка дискразита (Ag3Sb) определенной достоверной зависимости не установлено для линий L-серии серебра Ag(L3-M5,4; L3-N5; L2-M4; L1-M3) и L-серии сурьмы Sb(L3-M5,4; L3-N5; L2-M4; L1-M3) c энергиями максимального пика 2.98 и 3.61 кэВ (рис. 4а). При этом для основной линии серии сурьмы вследствие близости к краю поглощения серебра полный коэффициент ослабления (μ/ρ)Sb(L) = 1290 см2/г превосходит таковой для серебра (μ/ρ)Ag(L) = 580 см2/г. Если рентгеновские излучательные серии имеют более значимые различия по энергиям, наложения энергии высокоэнергетических фотонов на край поглощения более легким элементом не происходит. В таком случае зависимость относительных интенсивностей K от энергии зондирующих электронов становится более выраженной. Для примера на рис. 4а данная зависимость показана для К-серии серы S(К-L3,-M3) и L-серии сурьмы Sb(L3-M5,4; L3-N5; L2-M4; L1-M3) с энергиями основной линии 2.31 и 3.61 кэВ соответственно. Равенство K = 1 задает интервал ускоряющих напряжений, требуемых для корректного элементного анализа Sb2S3, 27–30 кВ. Действительно, анализ при энергии электронов зонда 28 кэВ приводит к результатам, соответствующим стехиометрии стибнита.
Рис. 4.
Зависимость характеристики K от энергии электронного зонда для пар диагностических линий, полученная для порошков Sb2S3, Ag3Sb, SiO2, Al2O3 (а) и Сu2O, ZnO (б).
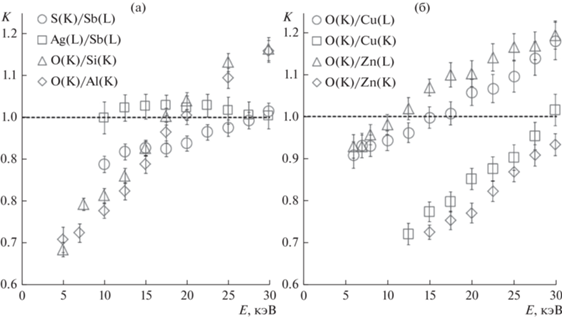
Элементы Si и Al имеют соседние порядковые номера. Это обусловливает в соответствующих оксидах SiO2 и Al2O3 близость коэффициентов ослабления для диагностической линии O(K-L3) и схожие зависимости параметра K для пар линий O(K)/Si(K) и O(K)/Al(K) от ускоряющего напряжения. Кроме того, практически неразличимы размеры и форма областей генерации характеристических фотонов элементов. В данном и подобных случаях алгоритмы корректировки результатов расчета содержания соседствующих по порядковому номеру элементов могут быть одинаковыми. В частности, правильные результаты количественного определения элементов в порошке (и SiO2, и Al2O3) достигаются при ускоряющем напряжении 17 ± 0.5 кВ.
Напротив, зависимости интенсивностей диагностических линий соседних по порядку элементов Cu и Zn в оксидах Cu2O и ZnO по отношению к линии кислорода от энергии индуцирующих излучение электронов несколько различаются (рис. 4б). Коэффициенты ослабления для диагностических линий O(K-L3), Сu, Zn(L3-M4,5; L2-M4; L3-M1), Cu, Zn(K-L2,3; K-M3) в соответствующих оксидах имеют лишь небольшие отличия. Но при этом глубина области генерации для характеристических фотонов в Cu2O и ZnO имеет существенные различия. Так, например, при моделировании области генерации рентгеновского излучения максимум плотности и максимальная глубина генерации для линии Zn(K-L2,3; K-M3) в оксиде цинка при энергии электронов 15 кэВ составляют 190 и 950 нм. В оксиде меди для линии Cu(K-L2,3; K-M3), имеющей меньшую энергию, соответствующие параметры имеют значения 140 и 760 нм. Таким образом, в материалах, резко отличающихся по поглощению и рассеиванию первичных электронов зонда, для количественного определения близких и даже совпадающих по порядковому номеру элементов оптимальные значения энергии электронов зонда могут не совпадать.
Многоэлементные соединения. В отличие от бинарных, в многоэлементных соединениях равенство K = 1 для интенсивностей диагностических линий двух элементов при определенном ускоряющем напряжении не означает, что при данном напряжении правильным окажется также отношение рассчитанных массовых долей этих элементов. Результат расчета также определяется количеством прочих элементов в составе. В качестве примера на рис. 5а приведены зависимости параметра от энергии зондирующих электронов для пар диагностических линий O(K-L3)/Ca(K-L2,3; K-M3) и С(K-L3)/O(K-L3) в порошке исландского шпата (CaCO3).
Рис. 5.
Зависимость характеристики K от энергии электронного зонда для пар диагностических линий, полученная для порошков CaCO3, Al2O(SiO4) (а) и CaAl2Si3O10·3H2O (б).
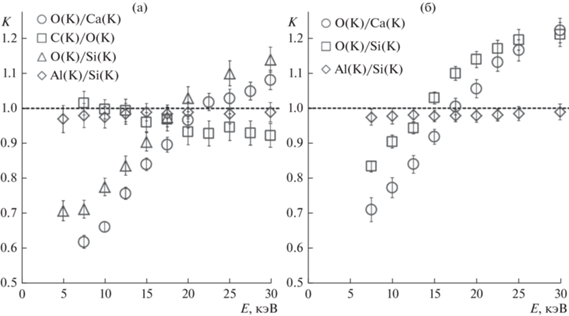
Корректные по сравнению с полированным образцом отношения интенсивностей линий O(K)/Ca(K) и С(K)/O(K) наблюдаются при ускоряющих напряжениях 22.5 и 10 кВ соответственно. Отношения рассчитанных массовых долей WtO/WtCа и WtС/WtO оказались соответствующими стехиометрии CaCO3 при близких, но несколько отличающихся значениях ускоряющего напряжения 21.5 и 9 кВ соответственно. При этом правильному отношению WtO/WtCа в результатах расчета сопутствует занижение количества углерода, правильному отношению WtС/WtO – завышение кальция. Полностью скорректированный результат получается при совместном решении уравнений
(2)
$\begin{gathered} {\text{а}}){\text{ }}W{{t}_{{\text{O}}}}{\text{/}}W{{t}_{{{\text{Cа}}}}} = {{Z}_{{{\text{O,Ca}}}}}\left( {21{\text{ кВ}}} \right); \hfill \\ {\text{б) }}W{{t}_{{\text{С}}}}{\text{/}}W{{t}_{{\text{O}}}} = {{Z}_{{{\text{C,O}}}}}\left( {9{\text{ кВ}}} \right); \hfill \\ {\text{в}}){\text{ }}W{{t}_{{\text{С}}}} + W{{t}_{{\text{O}}}} + W{{t}_{{{\text{Cа}}}}} = {\text{ }}1. \hfill \\ \end{gathered} $Таким образом, для уточнения результатов анализа трехэлементных порошковых материалов достаточно при определенных ускоряющих напряжениях определить правильные отношения массовых долей двух пар элементов.
При наличии в составе элементов, соседствующих в периодической системе, но при отсутствии в энергодисперсионном спектре краев поглощения около диагностических линий, можно считать, что отношения массовых долей этих элементов будут правильными в широком диапазоне ускоряющих напряжений (рис. 5а). Так при анализе спектров порошка кианита (Al2O(SiO4)) установлено, что для корректировки результатов элементного анализа достаточно подобрать ускоряющее напряжение, при котором в правильном соотношении находятся массовые доли пар элементов WtO и WtSi или WtO и WtAl. В случае с Al2O(SiO4) это напряжение составляет 17.5 кВ и численно совпадает с энергией электронов 17.5 кэВ, при которой выполняется равенство К = 1 для пары линий O(K)/Si(K).
Аналогичный пример представлен на рис. 5б для порошкообразного сколецита (CaAl2Si3O10·3H2O), имеющего в составе четыре диагностируемых энергодисперсионным методом элемента. В случае четырехэлементного состава достаточно определить корректные отношения массовых долей для трех независимых пар элементов. Правильное в пределах погрешности эксперимента соотношение массовых долей Al и Si получено во всем использованном диапазоне ускоряющих напряжений. Отношения массовых долей WtO/WtCа и WtO/WtSi совпадают с эталонными при энергиях зондирующих электронов 19 и 16 кэВ.
Для соединений элементов, допускающих анализ по диагностическим линиям двух серий излучения, спектр возможных комбинаций пар линий значительно расширяется. На рис. 6 представлены зависимости параметра К от энергии электронов зонда нескольких пар характеристических линий в энергодисперсионных спектрах халькопирита (CuFeS2) и арсенопирита (FeAsS). При количественной обработке спектров CuFeS2 правильный результат по составу можно получить, используя девять комбинаций отношений массовых долей элементов. Аналогичный расчет для FeAsS оказался возможен лишь по семи комбинациям. Найденный для интенсивностей диагностических линий мышьяка L-серии As(L3-M5,4; L2-M4; L1-M2; L3-M1) и железа K-серии Fe(K-L2,3; K-M3) параметр К не принимает единичного значения в доступном интервале энергий зондирующих электронов. Также и отношение массовых долей WtAs/WtFe, определенное по указанным выше линиям, оказалось заниженным по сравнению с результатами анализа на эталоне вплоть до ускоряющего напряжения 30 кВ.
Рис. 6.
Зависимость характеристики К от энергии электронного зонда для пар диагностических линий, полученная для порошков CuFeS2 (а), (б) и FeAsS (в), (г).

Обобщение экспериментальных данных. Первостепенными факторами, влияющими на генерацию характеристических рентгеновских фотонов и на регистрируемую интенсивность диагностических линий, являются объем области генерации и коэффициент ослабления µ/ρ потока фотонов в веществе. В свою очередь глубина и радиальный размер области генерации в данном материале и при заданной энергии индуцирующих электронов определяется энергией E характеристических фотонов. По совокупности проведенных измерений для каждой пары элементов в составе порошковых образцов определили энергии Е0 зондирующих электронов, при которых рассчитываемые массовые доли элементов Wti и Wtj находятся в таком же соотношении, как для полированных образцов того же состава. Эмпирически подобрана функция f(µ/ρ, E) параметров характеристических фотонов, имеющая экспериментально установленную связь с энергией Е0. Функция имеет следующий вид:
(3)
$f\left( {\frac{\mu }{\rho },E} \right) = {{\left[ {\ln \left( {{{{\left( {\frac{\mu }{\rho }} \right)}}_{i}} - {{{\left( {\frac{\mu }{\rho }} \right)}}_{j}}} \right)} \right]}^{{1.5}}}\sqrt {\frac{{{{{\left( {{{E}_{j}}{\text{/}}{{E}_{i}}} \right)}}^{{0.5}}}}}{{{{E}_{j}} - {{E}_{i}}}}} ,$Рис. 7.
Эмпирическая зависимость ускоряющего напряжения, при котором определяются корректные отношения массовых долей элементов, от функции параметров характеристических фотонов.

Фактически приведенная графическая зависимость выступает в роли градуировочного графика для подбора ускоряющего напряжения, при котором вычисляемые массовые доли пары элементов находятся в правильном соотношении между собой. При этом возникает несколько ограничений и допущений. Во-первых, для характеристических фотонов с энергиями Ei и Ej должно выполняться условие
Это означает, что фотоны с меньшей энергией должны испытывать большее ослабление. Так, не были приняты в расчет такие комбинации диагностических линий, как C(K)/O(K) в CaCO3, Al(K)/Si(K) для некоторых включающих одновременно оба элемента материалов, как это наблюдается для кианита (Al2O(SiO4)) и микроклина (KAlSi3O8). Однако в большинстве случаев, когда значение энергии диагностической линии одного элемента не находится близко к краю поглощения в другом элементе, условие (4) выполняется.
Во-вторых, отношение вычисляемых массовых долей элементов Wti/Wtj должно иметь явную зависимость от ускоряющего напряжения с достоверно фиксируемым значением Е0. Например, в рассмотрение не вошла пара элементов Al и Si при их совместном нахождении в образце и выполнении условия (4) в случае сколецита (CaAl2Si3O10·3H2O) и спессартина (Ca0.17Mg0.70Mn2.31Al2(SiO4)3). Также для спессартина к таким комбинациям линий относится Mg(K)/Al(K). В подобных ситуациях параметр относительных интенсивностей К близок к единице, а отношение Wti/Wtj в порошковом образце совпадает с эталонным в широком интервале энергий электронного пучка. Дополнительной иллюстрацией данного ограничения являются пары характеристических линий Fe(L)/Cu(L) и Fe(K)/Cu(K) на рис. 6. В целом для выраженной зависимости от ускоряющего напряжения отношения Wti/Wtj, вычисляемого по линиям с близкой энергией, коэффициенты ослабления для данных линий должны различаться между собой не менее чем на 30%.
В-третьих, в качестве максимальной величины Е0 принято значение не более 31 кВ, получаемое в ряде случаев ближней аппроксимацией зависимости Wti/Wtj от ускоряющего напряжения до достижения величиной Wti/Wtj эталонного значения.
Эмпирический метод корректировки результатов анализа. Анализ однородных порошковых материалов предлагается осуществлять по следующей схеме.
1) Энергодисперсионные спектры образца собираются при двух или нескольких значениях ускоряющего напряжения. Сканирование следует осуществлять растром по максимально доступной площади материала. В соответствии с общими принципами энергодисперсионного анализа минимальная энергия электронного пучка должна вдвое превышать энергию индуцируемых характеристических фотонов. В то же время по возможности следует избегать двадцатикратного и более превышения энергии, если в дальнейшем предполагается расчет содержания элементов без использования эталонных спектров. Спектры обрабатываются количественно, определяются концентрации элементов, производится усреднение. В зависимости от энергии электронов зонда вычисляемая доля элемента в порошке может быть завышена или занижена. Поэтому Wtср в ряде случаев может оказаться очень близкой к величине Wt0, полученной при анализе полированного образца. По той же причине установленные на этом этапе значения Wtср массовых долей характеризуются относительно широким доверительным интервалом случайной погрешности. В табл. 1 сравниваются результаты Wtср и Wt0, полученные для трех случайно выбранных ускоряющих напряжений на порошковом и полированном образцах соответственно для нескольких материалов. Отличия вычисляемых усредненных концентраций элементов Wtср и в дальнейшем их скорректированных величин Wtкорр от эталонных значений Wt0 охарактеризованы через среднее относительное отклонение
(5)
${{\varepsilon }_{{(ср, корр)}}} = \frac{1}{n}\sum\limits_{i = 1}^n {\frac{{\left| {W{{t}_{{(ср, корр) i}}} - W{{t}_{{0i}}}} \right|}}{{W{{t}_{{0i}}}}}} \times 100\% .$Таблица 1.
Результат корректировки количественного анализа для нескольких порошкообразных материалов и наборов диагностических линий
| Элемент (серия)/энергия, кэВ | Wt0, % | Wtср, % | εср, % | Wtкорр, % | εкорр, % |
|---|---|---|---|---|---|
| CaF2 | |||||
| F(K)/0.67 | 48.62 ± 0.23 | 46.61 ± 6.4 | 4.0 | 48.44* | 0.36 |
| Ca(K)/3.69 | 51.38 ± 0.26 | 53.39 ± 6.4 | 51.56* | ||
| Zn2SiO4 | |||||
| O(K)/0.52 | 28.75 ± 0.19 | 29.82 ± 1.3 | 2.2 | 28.77* | 0.53 |
| Si(K)/1.74 | 12.54 ± 0.09 | 12.35 ± 0.50 | 12.38 | ||
| Zn(L)/1.01 | 58.71 ± 0.31 | 57.83 ± 0.81 | 58.84* | ||
| Zn2SiO4 | |||||
| O(K)/0.52 | 28.64 ± 0.20 | 26.61 ± 2.1 | 8.7 | 28.36 | 1.3 |
| Si(K)/1.74 | 12.42 ± 0.09 | 10.81 ± 0.41 | 12.17 | ||
| Zn(K)/8.64 | 58.94 ± 0.35 | 62.59 ± 2.5 | 59.45 | ||
| Cu(3 – x)ZnxSbS(3 – y)Asy** | |||||
| Сu(L)/0.93 | 37.68 ± 0.22 | 36.86 ± 0.65 | 1.1 | 37.56* | 0.60 |
| Zn(L)/1.01 | 6.72 ± 0.43 | 6.56* ± 2.0 | 6.64* | ||
| As(L)/1.28 | 14.15 ± 0.20 | 14.28* ± 1.4 | 14.36 | ||
| S(K)/2.31 | 28.89 ± 0.18 | 28.76* ± 0.48 | 28.85* | ||
| Sb(L)/3.61 | 12.66 ± 0.16 | 13.13 ± 1.4 | 12.59* | ||
| Cu(3 – x)ZnxSbS(3 – y)Asy** | |||||
| Сu(K)/8.05 | 39.82 ± 0.18 | 42.28 ± 1.0 | 3.4 | 40.62 | 1.3 |
| Zn(K)/8.64 | 7.52 ± 0.33 | 8.00 ± 0.7 | 7.77* | ||
| As(L)/1.28 | 13.65 ± 0.21 | 13.07 ± 1.3 | 13.47* | ||
| S(K)/2.31 | 27.35 ± 0.16 | 24.15 ± 1.2 | 26.28 | ||
| Sb(L)/3.61 | 11.66 ± 0.16 | 12.49 ± 0.6 | 11.86 | ||
Значения Wtср являются первыми приближениями для вычисления коэффициентов ослабления характеристических линий в образце.
2) По значениям Wtср для пар диагностических линий с энергиями Еi,j на основании справочных данных определяются массовые коэффициенты ослабления (µ/ρ)i,j и в соответствии с выражением (3) вычисляется функция f(µ/ρ,E). Отвечающее функции f(µ/ρ,E) значение Е0 (рис. 7) принимается в качестве ускоряющего напряжения, при котором для данной пары диагностических линий будет получено скорректированное отношение Wti/Wtj соответствующих элементов. Для бинарных соединений достаточно провести анализ и расчет состава при найденном таким образом значении Е0. Так, единичный анализ порошка CaF2 при ускоряющем напряжении 22.5 кВ привeл к значениям Wtкорр, не отличающимся в пределах погрешности эксперимента от Wt0 для полированного образца (табл. 1).
3) Вычисление f(µ/ρ, E) невозможно, если для данных диагностических линий не выполнено условие (4). Если нет возможности уточнения результатов анализа по другой комбинации энергодисперсионных пиков, то анализ можно ограничить определением Wtср.
4) В ряде случаев f(µ/ρ, E) может принимать значения, выходящие из интервала, для которого установлена экспериментальная зависимость f(µ/ρ, E) и Е0. Примером из табл. 1 являются пары линий элементов Zn(L)/Si(K), Si(K)/Zn(K) в Zn2SiO4 и линий элементов S(K)/Cu(K), S(K)/Zn(K) в образце Cu(3–Х)ZnXSbS(3–Y)AsY. Значения функции f(µ/ρ, E) для них составили менее 12. В такой ситуации дальнейший расчет состава следует проводить по другим комбинациям диагностических линий или использовать минимальные или максимальные доступные значения ускоряющего напряжения.
5) Для состава материала, включающего n элементов, достаточной для анализа является n – 1 независимая комбинация диагностических линий. Для выбранных комбинаций линий и при соответствующих значениях Е0 проводят повторный набор энергодисперсионных спектров, из которых определяют уточненные отношения массовых долей Wti/Wtj = Zi,j. Окончательно элементный состав материала рассчитывается решением системы уравнений, аналогичной (2):
(6)
$\left\{ \begin{gathered} W{{t}_{1}}{\text{/}}W{{t}_{2}} = {{Z}_{{1,2}}} \hfill \\ ... \hfill \\ W{{t}_{{n - 1}}}{\text{/}}W{{t}_{n}} = {{Z}_{{n - 1,n}}} \hfill \\ \sum\limits_n^{i = 1} {W{{t}_{i}} = 1} \hfill \\ \end{gathered} \right..$Вычисленные величины Wti будут являться скорректированными значениями массовых долей элементов. Примеры единичных расчетов уточненных значений массовых долей приведены в табл. 1. Лучший результат достигается при использовании комбинаций характеристических линий с меньшим диапазоном энергий.
* * *
Предлагаемый на основе исследования особенностей энергодисперсионных спектров однородных порошковых материалов способ уточнения результатов анализа их элементного состава не требует трудоемких вычислений. Он может быть ингегрирован в качестве дополнительного алгоритма в программное обеспечение энергодисперсионных анализаторов для автоматического подбора оптимальных ускоряющих напряжений и расчета скорректированных концентраций элементов.
К недостаткам изложенного метода относится приверженность представленной на рис. 7 функциональной связи параметров характеристических фотонов и оптимальных для анализа энергий электронов зонда определенной аппаратной геометрии. В представленном виде эта зависимость справедлива для небольшого интервала углов между направлением сбора рентгеновских фотонов и направлением сканирующего зонда вблизи 50° при условии, что электронный пучок падает нормально к поверхности образца. Однако подобные обсуждаемым экспериментальные данные могут быть получены и обобщены для любой другой геометрии аналитической аппаратной схемы.
Использовали аналитическое оборудование Центра коллективного пользования “Диагностика микро- и наноструктур” (Facilities Sharing Centre ”Diagnostics of Micro- and Nano Structures“).
Работа выполнена в рамках государственного задания Ярославскому Филиалу Физико-технологического института им. К.А. Валиева РАН (Valiev Institute of Physics and Technology of RAS, Yaroslavl Branch) от Минобрнауки РФ по теме № FFNN-2022-0018.
Список литературы
Newbury D.E., Ritchie N.W.M. Performing elemental microanalysis with high accuracy and high precision by scanning electron microscopy/silicon drift detector energy-dispersive X-ray spectrometry (SEM/SDD-EDS) // J. Mater. Sci. 2015. V. 50. № 2. P. 493. https://doi.org/10.1007/s10853-014-8685-2
Newbury D.E., Ritchie N.W.M. Is scanning electron spectroscopy/energy dispersive X-ray spectrometry (SEM/EDS) quantitative? // Scanning. 2013. V. 35. № 3. P. 141. https://doi.org/10.1002/sca.21041
Лаврентьев Ю.Г., Карманов Н.С., Усова Л.В. Электронно-зондовое определение состава минералов: Микроанализатор или сканирующий электронный микроскоп? // Геология и геофизика. 2015. Т. 56. № 8. С. 1473. DOI: (Lavrent’ev Y.G., Karmanov N.S., Usova L.V. Electron probe microanalysis of minerals: Microanalyzer or scanning electron microscope? // Russ. Geol. Geophys. 2015. V. 56. № 8. P. 1154. https://doi.org/10.1016/j.rgg.2015.07.00610.1016/j.rgg.2015.07.006)https://doi.org/10.15372/GiG20150806
Goldstein J.I., Newbury D.E., Michael J.R., Ritchie N.W.M., Scott J.H.J., Joy D.C. Scanning Electron Microscopy and X-ray Microanalysis, 4rd Ed. New York: Springer, 2018. 550 p. https://doi.org/10.1007/978-1-4939-6676-9
Newbury D.E., Ritchie N.W.M. Quantitative SEM/EDS, step 1: What constitutes a sufficiently flat specimen? // Microsc. Microanal. 2013. V. 19. № 2. P. 1244. https://doi.org/10.1017/S1431927613008210
Hovington P., Timoshevskii V., Burgess S., Demers H., Statham P., Gauvin R., Zaghib K. Can we detect Li K X-ray in lithium compounds using energy dispersive spectroscopy? // Scanning. 2016. V. 38. № 6. P. 571. https://doi.org/10.1002/sca.21302
Béranger M. Study on the use of silicon drift detector to get information on light emitted by luminescent materials // AJPA. 2019. V. 7. № 2. P. 34. https://doi.org/10.11648/j.ajpa.20190702.11
Li X., Holland J., Burgess S., Bhadare S., Yamaguchi S., Birtwistle D., Statham P., Rowlands N. Detection of lithium X-rays by EDS // Microsc. Microanal. 2013. V. 19. № 2. P. 1136. https://doi.org/10.1017/S1431927613007678
Burgess S., Li X., Holland J. High spatial resolution energy dispersive X-ray spectrometry in the SEM and the detection of light elements including lithium // Microsc. Microanal. 2013. V. 27. № 4. P. 8.
Armstrong J.T., Buseck P.R. Quantitative chemical analysis of individual microparticles using the electron microprobe: Theoretical // Anal. Chem. 1975. V. 47. № 13. P. 2178. https://doi.org/10.1021/ac60363a033
Newbury D.E. Electron probe microanalysis of rough targets: Testing the peak-to-local background method // Scanning. 2004. V. 26. № 3. P. 103. https://doi.org/10.1002/sca.4950260302
Small J.A. The analysis of particles at low accelerating voltages (≤10 kV) with energy dispersive X-ray spectroscopy (EDS) // J. Res. Natl. Inst. Stan. 2002. V. 107. № 6. P. 555. https://doi.org/10.6028/jres.107.047
Trincavelli J., Van Grieken R.E. Peak-to-background method for standardless electron microprobe analysis of particles // X-Ray Spectrom. 1994. V. 23. P. 254. https://doi.org/10.1002/xrs.1300230605
Labar J.L., Torok S.B. A Peak-to-background method for electron-probe X-ray microanalysis applied to individual small particles // X-Ray Spectrom. 1992. V. 21. P. 183. https://doi.org/10.1002/xrs.1300210407
Hovington P., Lagace M., Rodrigue L. X-ray analysis of rough surfaces at low energy // Microsc. Microanal. 2002. V. 8. № 2. P. 1472. https://doi.org/10.1017.S1431927602103990
Armstrong J.T., Buseck P.R. A general characteristic fluorescence correction for the quantitative electron microbeam analysis of thick specimens, thin films and particles // X-Ray Spectrom. 1985. V. 14. № 4. P. 172. https://doi.org/10.1002/xrs.1300140408
Armstrong J.T. Quantitative elemental analysis of individual microparticles with electron beam instruments / Electron Probe Quantification / Eds. Heinrich K.J.F., Newbury D.E. New York: Plenum Press., 1991. P. 261. https://doi.org/10.1007/978-1-4899-2617-315
Gauvin R., Hovington P., Drouin D. Quantification of spherical inclusions in the scanning electron microscope using Monte Carlo simulations // Scanning. 1995. V. 17. № 4. P. 202. https://doi.org/10.1002/sca.4950170401
Storms H.M., Janssens K.H., Torok S.B., Van Grieken R.E. Evaluation of the Armstrong-Buseck correction for au-tomated electron probe X-ray microanalysis of particles // X-Ray Spectrom. 1989. V. 18. P. 45. https://doi.org/10.1002/xrs.1300180203
Paoletti A., Bruni B.M., Gianfagna A., Mazziotti-Tagliani S., Pacella A. Quantitative energy dispersive X-ray analysis of submicrometric particles using a scanning electron microscope // Microsc. Microanal. 2011. V. 12. № 5. P. 710. https://doi.org/10.1017/S1431927611000432
Ritchie N.W.M. Using DTSA-II to simulate and interpret energy dispersive spectra from particles // Microsc. Microanal. 2010. V. 16. № 3. P. 248. https://doi.org/10.1017 /S1431927610000243
Пухов Д.Э., Лаптева А.А. Учет неровности поверхности при электронно-зондовом энергодисперсионном анализе материалов в виде порошков // Поверхность. Рентгеновские, синхротронные и нейтронные исследования. 2020. № 9. С. 28. (Pu-khov D.E., Lapteva A.A. Taking into account the surface roughness in the electron-probe energy-dispersive analysis of powder materials // J. Surf. Investig-X-Ra. 2020. V. 14. № 5. P. 889. https://doi.org/10.1134/S102745102005014610.1134/S1027451020050146)https://doi.org/10.31857/S1028096020090149
Drouin D., Couture A.R., Joly D., Tastet X., Aimez V., Gauvin R. CASINO V2.42 – A fast and easy-to-use modeling tool for scanning electron microscopy and microanalysis users // Scanning. 2007. V. 29. № 3. P. 92. https://doi.org/10.1002/sca.20000
Drouin D., Hovington P., Gauvin R. CASINO: A new Monte Carlo code in C language for electron beam interaction – Part II: Tabulated values of Mott cross section // Scanning. 1997. V. 19. № 1. P. 20. https://doi.org/10.1002/sca.4950190103
Hovington P., Drouin D., Gauvin R. CASINO: A new Monte Carlo code in C language for electron beam interaction – Part I: Description of the program // Scanning. 1997. V. 19. № 1. P. 1. https://doi.org/10.1002/sca.4950190101
Hovington P., Drouin D., Gauvin R., Joy D.C., Evans N. CASINO: A new Monte Carlo code in C language for electron beam interaction – Part III: Stopping power at low energies // Scanning. 1997. V. 19. № 1. P. 29. https://doi.org/10.1002/sca.4950190104
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии