

Журнал аналитической химии, 2023, T. 78, № 2, стр. 99-107
Автоматизация пробоподготовки на принципах хроматомембранных методов разделения при анализе водных и воздушных сред
Л. Н. Москвин a, *, А. Л. Москвин b, О. В. Родинков a, Н. М. Якимова a
a Санкт-Петербургский государственный университет, Институт химии
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
b Санкт-Петербургский национальный исследовательский
университет информационных технологий, механики и оптики
197101 Санкт-Петербург, Кронверкский просп., 49, Россия
* E-mail: moskvinln@yandex.ru
Поступила в редакцию 05.07.2022
После доработки 18.07.2022
Принята к публикации 18.07.2022
- EDN: CDLRBW
- DOI: 10.31857/S004445022302007X
Аннотация
В статье обобщен накопленный опыт применения хроматомембранных методов (ХММ) для автоматизации химического анализа водных и воздушных сред. Особый акцент сделан на проточных методах непрерывного анализа, где ХММ часто не имеют альтернативы.
Пробоподготовка до сих пор остается наиболее сложной для автоматизации стадией химического анализа, особенно в тех случаях, когда она включает концентрирование аналитов, а анализ выполняется в непрерывном режиме. Пробоподготовка в анализе жидких и газовых сред, как правило, решает две задачи. Во-первых, концентрирования аналитов, а во-вторых, их перевод в другое фазовое состояние для их определения наиболее простыми и доступными методами. Например, для одновременного определения “кислых” газов (HCl, HF, SO2, NO2 и т.д.) в воздухе возможно использование ионного хроматографа после их предварительного выделения из газовой фазы в водный раствор. Или, наоборот, для определения летучих органических веществ (ЛОВ) в воде обычно прибегают к газовой экстракции, чтобы перенести ЛОВ в газовую фазу для их последующего определения методом газовой хроматографии. Для решения тех и других задач пробоподготовки чаще всего находят применение методы жидкостно-жидкостной экстракции, жидкостной абсорбции из газовой фазы в водные растворы и газовой экстракции. Общий принцип разделения веществ, лежащий в основе этих методов, – различия в их межфазном распределении. В течение длительного времени оставались неизменными принципиальные схемы реализации процессов межфазного распределения: смешение и расслаивание фаз в статических условиях или создание потока одной фазы относительно другой, находящейся в стационарном состоянии. За редкими исключениями любая из этих схем, независимо от используемой системы фаз, позволяет осуществить разделение при периодическом вводе пробы исходной смеси веществ в зону ее контакта с фазой, извлекающей аналиты, и при периодическом выводе аналитов из извлекающей фазы или при использовании в качестве пробы самой извлекающей фазы с выделенными в нее аналитами. А в тех случаях, когда существует возможность непрерывного разделения, например, в противоточной жидкостно-жидкостной экстракции, как правило, возникают серьезные проблемы с расслаиванием фаз в потоке. Отсюда – объективные трудности, с которыми приходится сталкиваться при автоматизации стадии пробоподготовки с использованием любого из упомянутых выше методов.
Хроматомембранные процессы и основанные на их принципе методы разделения. Новые возможности для решения задач пробоподготовки открыли хроматомембранные методы (ХММ) – новые методы разделения [1], основанные на проявлении капиллярных эффектов в гидрофобных пористых средах [2, 3]. Хроматомембранные массообменные процессы (ХММП), лежащие в основе ХММ, принципиально осуществимы в системах жидкость–газ или жидкость–жидкость. Для осуществления ХММП необходимы бипористые матрицы – пористые структуры с двумя типами однородных по размерам пор, различающихся размерами, изготовленные из гидрофобного материала. При этом единственная жидкая фаза или одна из двух жидких фаз должна быть настолько полярной, чтобы исключалась смачиваемость ею материала, из которого изготовлены гидрофобные пористые матрицы. Дополнительным обязательным требованием является определенный размер открытых пор в этих пористых матрицах. Одни из них относятся к категории микро- или мезопор (в дальнейшем по отношению к ним используется одна обобщающая характеристика – микропоры). Вторые относятся к категории макропор, существенно превосходя первые по размерам. Критерием выбора размеров пор каждого типа является возникающее в них капиллярное давление Рк по отношению к полярной жидкой фазе (ПЖФ).
В применяемых для осуществления ХММП бипористых матрицах размер микропор должен быть таким, чтобы в условиях проведения эксперимента давление ПЖФ во всем объеме бипористой матрицы (РПЖФ) не превышало величину Рк, возникающего в микропорах:
В этом случае последние оказываются недоступными для нее. В то же время микропоры в матрице могут служить каналами для прохождения второй фазы, участвующей в массообменном процессе: неполярной жидкости или газа. В свою очередь, размеры макропор не должны быть меньше некоторой пороговой величины, при которой возникающее в них капиллярное давление сделает их непроницаемыми для ПЖФ. В этом случае они образуют каналы для создания потока этой фазы. В результате в бипористых гидрофобных матрицах можно создать независимые, пересекающиеся под произвольными углами потоки двух фаз, массообмен между которыми будет осуществляться в местах пересечения каналов, образованных порами каждого типа, т.е. процесс массообмена будет аналогичен осуществляемому в хроматографической колонке, функционирующей в режимах обращенно-фазовой жидкостно-жидкостной хроматографии (случай двух жидких фаз) или в режиме жидкостно-газовой хроматографии (система жидкость–газ). Последнее гарантирует высокую эффективность массообмена, достигаемую в хроматографических методах.
Для того чтобы обеспечить ввод в бипористую матрицу и вывод из нее неполярной жидкой и (или) газовой фазы с соответствующих сторон массобменных матриц устанавливают плотно прижатые к ней однородно микропристые гидрофобные мембраны, требования к размерам пор в которых аналогичны требованиям к размерам микропор в бипористых матрицах.
Схема осуществления ХММП (рис. 1) соответствует прохождению потоков двух фаз, пересекающихся под прямым углом: полярная жидкость протекает через бипористую матрицу, образующую массообменное пространство от входа в массообменную камеру 5 до выхода из нее 6 (P1 и P2 – давления ПЖФ на входе в массообменную камеру и на выходе из нее соответственно), а в перпендикулярном направлении создается поток неполярной жидкости или газа от входа 3 к выходу 4 (P3 и P4 – давление на входе и выходе соответственно). Смешение потоков двух фаз в массообменной камере исключается за счет поддержания определенных соотношений давлений полярной и неполярной жидкой или газовой фаз на входе и на выходе из матрицы, в результате чего давление в пределах всего объема, занимаемого в массообменной камере неполярной фазой, поддерживается на более низком уровне, чем давление полярной фазы, т.е. минимальное давление P2 полярной фазы, создаваемое на ее выходе из массообменной камеры, должно превышать максимальное давление P3 неполярной жидкой фазы или газа на входе в нее:
Рис. 1.
Схема осуществления хроматомембранных массообменных процессов в случае потоков фаз, пересекающихся под прямым углом. 1 – бипористая гидрофобная матрица; 2 – однородномикропористые гидрофобные мембраны; 3 – вход неполярной жидкой или газовой фазы; 4 – выход неполярной жидкой или газовой фазы; 5 – вход полярной жидкой фазы; 6 – выход полярной жидкой фазы. P1, P2, P3, P4 – давления обеих фаз соответственно на входе в матрицу и на выходе из нее.
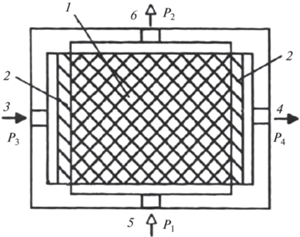
В результате газ или неполярная жидкая фаза не могут перейти из “микропор” в макрпоры. В свою очередь, их вытеснение из “микропор” полярной фазой исключается благодаря тому, что максимальная величина давления ПЖФ Р1, которое реализуется на ее входе в матрицу, поддерживается меньшей совокупной величины давления Р4 и Рк, возникающего в микропорах:
где |Pк| – абсолютная величина капиллярного давления, имеющего в данном случае отрицательное значение.Поток неполярной жидкой или газовой фазы вводится в массообменное пространство и выходит из него через гидрофобные пористые мембраны, размеры пор в которых в случае идентичных материалов совпадают с размерами микропор в бипористой матрице, образующей массообменное пространство. Граничным условием выбора материала мембран и размера пор в них является соотношение:
В рамках приведенной физико-химической модели ХММП достаточно очевидны подходы к выбору материала пористой матрицы. По своим физико-химическим свойствам материал матрицы должен быть, во-первых, химически инертным по отношению к обеим фазам, во-вторых, должен обеспечивать максимальный краевой угол смачивания полярной фазой. Подход к выбору пористой структуры материала бипористой матрицы с точки зрения эффективности процесса межфазного обмена аналогичен применяемому в хроматографических методах подходу к выбору размеров частиц носителей неподвижной фазы. В обоих случаях приходится искать компромисс между улучшением проницаемости бипористой матрицы или хроматографической колонки соответственно и ухудшением разрешения зон разделяемых веществ при увеличении размеров частиц сорбентов или носителей, а в случае ХММП – радиусов макропор.
Не столь однозначны критерии выбора радиусов “микропор”. При уменьшении их радиуса увеличивается величина возникающего в них капиллярного давления, что оправдано с точки зрения предотвращения проникновения в них полярной жидкой фазы. Но при этом снижается проницаемость матрицы для потока газа или жидкости, смачивающей поверхность пор, пропорционально квадрату их радиуса. Таким образом, возникает проблема подбора интервала радиусов “микропор”, при котором ХММП может быть осуществлен наиболее эффективно. Теоретически и экспериментально доказано [4, 5], что в идеальном случае при использовании в качестве материала матриц из политетрафторэтилена радиус микропор должен находиться в интервале от 1 до 10 мкм. При этом, как уже отмечалось выше, микропоры, также как и макропоры должны быть максимально близки друг к другу по размерам.
Пробоподготовка на принциапх хроматомембранных методов при выполнении анализов офлайн и онлайн. Еще одно достоинство хроматомембранных методов для использования их в пробоподготовке проявляется в открываемой ими возможности разделения веществ как по традиционной дискретной схеме, так и в непрерывном режиме соответственно при выполнении анализов офлайн и онлайн. Если в последнем случае для исключения смешения фаз требуется строгое выполнение условия (2), то при дискретном разделении смешение фаз исключается схемой подачи обеих фаз в хроматомембранную ячейку (ХМЯ) с перекрытием на входе и выходе из нее каналов подачи и сбора той фазы, которая в условиях эксперимента является неподвижной. Соответственно условие осуществления ХММП упрощается до
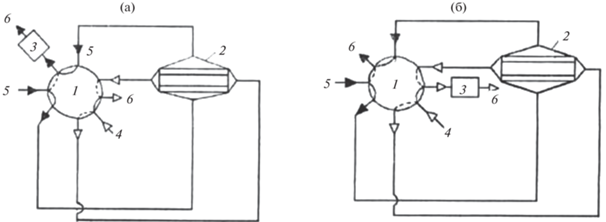
где ΔP – градиент давления, при котором в ХМЯ подается та фаза, которая выступает в роли подвижной.Принципиальные схемы осуществления ХММП в дискретном режиме приведены на рис. 2.
Рис. 2.
Принципиальные схемы проведения хроматомембранного предварительного концентрирования в дискретном режиме из водной (а) и газовой (б) фазы: 1 – кран-переключатель потоков, 2 – хроматомембранная ячейка, 3 – детектор, 4 – анализируемый (а) или абсорбирующий (б) водный раствор, 5 – органический экстрагент (а) или анализируемая газовая смесь (б), 6 – сброс.
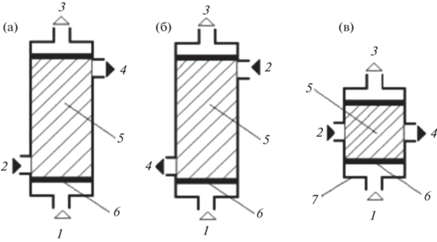
Другими важными параметрами оптимизации условий хроматомембранного разделения веществ являются направления относительного перемещения фаз в ХМЯ. В зависимости от направлений относительного перемещения фаз возможны три варианта ХММ (рис. 3). В случае параллельных потоков двух фаз, движущихся в одном направлении, реализуется прямоточный вариант метода. При встречном движении потоков – противоточный. Наконец, если потоки пересекаются под прямым углом, то осуществляется двухмерная схема ХМ-разделения. Относительные достоинства этих схем подробно обсуждаются в работах [6, 7], где рассмотрены закономерности движения зон разделяемых веществ в ХМЯ. Указанные закономерности описываются теми же уравнениями, что и в соответствующих вариантах хроматографических методов. Там же можно найти подтверждение применимости тарелочной теории хроматографии для оценки эффективности ХМ-методов и описание примеров их применения в вариантах хроматомембранной жидкостной экстракции (ХМЖЭ), хроматомембранной газовой экстракции (ХМГЭ) и хроматомембранной жидкостной абсорбции (ХМЖА).
Рис. 3.
Прямоточная (а), противоточная (б) и двухмерная (в) схемы осуществления хроматомембранных процессов. 1 и 3 – вход и выход потока неполярной жидкой или газовой фазы газа; 2 и 4 – вход и выход потока полярной жидкой фазы; 5 – массообменный слой; 6 – мембраны.
Первыми иллюстрациями возможностей применения ХМЖЭ в проточных методах анализа явились проточно-инжекционное экстракционно-фотометрическое определение меди [6] и анионных поверхностно-активных веществ [8]. В обоих случаях при минимальных временных затратах и полной автоматизации процесса предварительного концентрирования удалось достичь резкого снижения пределов обнаружения аналитов. При использовании в качестве экстрагента хлороформного раствора диэтилдитиокарбамата свинца нижняя граница диапазона определяемых концентраций Cu(II) составила 0.1 мкг/л. При хроматомембранном экстракционном предварительном концентрировании анионных поверхностно-активных веществ в форме ионных ассоциатов с метиленовым голубым при продолжительности концентрирования 5 мин нижняя граница диапазона определяемых концентраций составила 8 мкг/л.
С увеличением продолжительности цикла предварительного концентрирования нижняя граница диапазона определяемых концентраций может быть пропорционально снижена. В этом проявляются принципиальные преимущества хроматомембранного экстракционного предварительного концентрирования по сравнению с принятыми в настоящее время сегментными схемами экстракции в проточно-инжекционном анализе (ПИА) [9], которые не позволяют достичь существенного концентрирования аналитов из-за ограничений возможности варьирования соотношения объемов пробы и экстрагента.
Гибридная схема экстракционно-хроматографической и хроматомембранной пробоподготовки. Еще одной нетрадиционной возможностью ХМ-методов является разделение двухфазных смесей водных сред и органических растворителей в виде эмульсий, что позволило осуществить гибридные схемы разделения, включающие на первом этапе экстракционно-хроматографическое концентрирование аналитов с элюированием концентрата органической фазой, удерживаемой в хроматографической колонке в качестве неподвижной. На заключительном этапе проводится разделение в ХМЯ двухфазного потока, выходящего из экстракционно-хроматографической колонки, на индивидуальные компоненты с детектированием аналитов в фазе экстрагента, выходящего из ХМЯ. Подобная схема анализа позволила преодолеть основные ограничения возможностей использования ХМ-методов в схемах пробоподготовки при анализе природных и сбросных вод, существенно загрязненных взвесями неорганической и органической природы.
Присутствие взвесей приводит к засорению ими бипористых гидрофобных матриц, что требует их частой замены в ХМЯ. В этих случаях из-за возможных потерь определяемых примесей недопустима предварительная фильтрация пробы. Типичный пример – определение в воде примесей нефтепродуктов и фенолов [10].
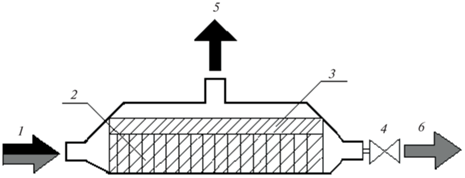
Конструкция ХМЯ для разделения двухфазных потоков приведена на рис. 4.
Рис. 4.
Схема ячейки для хроматомембранного разделения двухфазных водно-органических потоков: 1 – вход смешанного потока водной и органической фаз; 2 – массообменная бипористая матрица; 3 – мембрана; 4 – дроссель; 5 – выход органической фазы; 6 – выход водной фазы.
В предлагаемой схеме пробоподготовки элюирование из концентрирующей колонки может осуществляться как при остановке потока водной фазы, так и при непрерывном пропускании через ХМЯ потоков водной и органической фаз. Первый вариант соответствует схеме ПИА с инжекцией пробы в аналитическую систему, второй – схеме непрерывного проточного анализа (НПА).
Независимо от используемого варианта схемы взвеси, содержащиеся в пробе воды, будут засорять экстракционно-хроматографическую колонку, периодическая замена которой, учитывая простоту ее конструкции, более оправданна, чем замена ХМЯ. Важное преимущество предложенной схемы гибридной подготовки в ПИА, кроме уже отмеченного, проявляется в устранении ограничений по величине давления, под которым проба воды может подаваться в предконцентратор. Это давление в случае выделения аналитов непосредственно в ХМЯ ограничено величиной капиллярного давления, возникающего в микропорах бипористой матрицы и гидрофобных мембран, обеспечивающих выведение потока органической фазы из ячейки. В случае выделения аналитов в экстракционно-хроматографической колонке подобных ограничений нет. Соответственно, появляется возможность предварительного концентрирования при больших скоростях потока пробы с соответствующим сокращением продолжительности анализа. Результаты экспериментальной проверки предложенной схемы экстракционного предварительного концентрирования в непрерывном режиме представлены на примере определения в воде нефтепродуктов флуориметрическим методом (рис. 5).
Рис. 5.
Динамика изменения аналитического сигнала при осуществлении непрерывного проточного анализа после изменения содержания нефтепродуктов в потоке водной пробы. Концентрации нефтепродуктов в порядке их изменения в пробах, подаваемых в анализатор, приведены в форме их номеров по порядку последовательной подачи в анализатор: 1 – 0.1 мкг/л; 2 – 5 мкг/л; 3 – 50 мкг/л; 4 – 100 мкг/л; 5 – 250 мкг/л. Объемная скорость водной пробы – 7 мл/мин; органического экстрагента – 0.7 мл/мин.
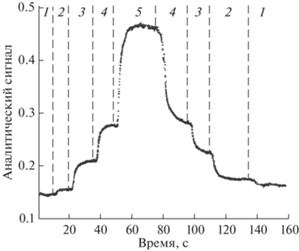
При люминесцентном детектировании в случае НПА для определения нефтепродуктов на уровне 1 мкг/л достаточен объем пробы 20 мл. Суммарная продолжительность анализа – 5 мин. Предел обнаружения дополнительно может быть снижен пропорциональным увеличением объема пробы. Возможности варьирования коэффициента концентрирования в режиме НПА определяются соотношением скоростей потоков водной и органической фаз. При соотношении скоростей потоков водной и органической фаз, равном 9, диапазон определяемых концентрацией нефтепродуктов в воде составил 5–250 мкг/л. При контроле онлайн схема НПА, в отличие от ПИА, исключает потерю информации о концентрации нефтепродуктов в анализируемой воде в период времени, соответствующий стадии элюирования концентрата из экстракционно-хроматографической колонки.
Хроматомембранная жидкостная абсорбция. С учетом значительно меньшей вязкости газообразных сред по сравнению с жидкими ХММП более эффективен для разделения веществ в системе жидкость−газ. Принципиальные схемы проточного анализа газообразных сред с хроматомембранным жидкостно-абсорбционным выделением определяемых веществ в водные растворы аналогичны применяемым для жидкостно-экстракционного выделения [11]. Иллюстрацией аналитических возможностей хроматомембранного предварительного концентрирования из газовой фазы может служить определение наиболее типичных загрязнителей воздуха: диоксидов азота [12–15], серы [16, 17], aммиака [18–20], формальдегида [21], гидразина [22], паров ацетона [23] и одновременное ионохроматографическое определение неорганических соединений, образующих в поглощающих водных растворах ионные формы [24–26].
В проточных методах анализа, когда они используются для контроля онлайн, целесообразно включение ХМЯ в качестве постоянного элемента аналитических приборов [24]. В случае ПИА периодически отбираемых проб в различных точках пробоотбора ХМЯ могут включаться в выносимые блоки для автономного пробоотбора [25]. Причем число ячеек должно соответствовать числу отбираемых проб. Выносной блок подключается к анализатору в лаборатории таким образом, чтобы обеспечить последовательное элюирование концентратов из всех ячеек. При этом одновременно регенерируется ХМЯ.
Последняя схема автономного пробоотбора в ХМЯ наиболее адекватна хроматомембранной подготовке в дискретных методах химического анализа. В частности, она использована для ионохроматографического определения в воздухе микропримесей, образующих в водных растворах диссоциирующие соединения: HF, HCl и NO2 [26].
Хроматомембранная газовая экстракция. Отдельным направлением аналитического применения хроматомембранных методов является парофазный анализ (ПФА), основанный на газоэкстракционном выделении аналитов [27]. Непрерывное ХМГЭ-выделение аналитов открыло, в частности, возможность непрерывной пробоподготовки при определении летучих органических веществ в водных средах [28–30].
ХМГЭ легко сочетается с газоадсорбционным концентрированием аналитов из потока газа-экстрагента с их последующей термодесорбцией. В англоязычной литературе подобная гибридная схема анализа называется purge and trap [31, 32]. Помимо подходов к выбору адсорбентов, рекомендуемых в работе [31], найдено оригинальное решение – композиционные поверхностно-слойные сорбенты [33], обеспечившие большую эффективность сорбции, благодаря чему описанная в работе [34] хроматомембранная версия этой схемы, помимо универсальности в плане применения в вариантах онлайн и офлайн анализа при решении задач пробоподготовки для определения летучих органических веществ, по своим возможностям превосходит все известные аналоги. В частности, она позволяет проводить отбор проб на месте, используя установки без насоса и источника питания к нему. Взаимное перемещение фаз обеспечивается установкой сосуда с пробой и ХМЯ на различной высоте, обеспечивающей протекание пробы через ячейку и всасывание в нее атмосферного воздуха в качестве газа-экстрагента.
Независимо от реализуемого варианта ПФА – дискретного или непрерывного − в обоих случаях достигается высокая воспроизводимость результатов. Относительное стандартное отклонение не превышает 0.02–0.03. Выбор оптимальных скоростей потоков фаз зависит от режима хроматомембранного процесса и осуществляется, исходя из компромисса между требованиями к пределу обнаружения и к инерционности системы контроля. Время, необходимое для достижения стационарного состояния хроматомембранного процесса, составляет от нескольких секунд до нескольких минут в зависимости от величин коэффициентов распределения, соотношений скоростей потоков фаз, выбранного режима и параметров массообменного слоя [1].
Дополнительные преимущества проявляются при определении микропримесей. Осуществление хроматомембранного процесса в непрерывном режиме позволяет проводить продувку соединительных коммуникаций и дозирующей петли газового хроматографа газом-экстрагентом, содержащим выделяемые вещества, что минимизирует адсорбцию определяемых веществ на поверхности коммуникаций и так называемый “эффект памяти” [34–36].
* * *
Расширение арсенала методов разделения за счет появления группы хроматомембранных методов позволило найти целый ряд новых методических решений в пробоподготовке, расширяющих ее возможности в трех аспектах:
– повышение коэффициентов концентрирования аналитов;
– перевод аналитов в другое агрегатное состояние, более адекватное возможностям методов их определения;
– автоматизация стадии пробоподготовки при выполнении анализов как по традиционной схеме офлайн, так и в варианте систем непрерывного аналитического контроля онлайн.
В плане уже доказанных и потенциально возможных областей применения ХММП основанные на его принципах методы разделения являются универсальными. Основными объектами, при анализе которых они уже подтвердили свою эффективность, являются объекты окружающей среды и биологические среды. Хроматомембранные методы пробоподготовки достаточно универсальны и с точки зрения аналитических методов, к которым они могут быть адаптированы. Это, в первую очередь, проточные и хроматографические методы: газовая и ионная хроматография.
Статья посвящена 300-летию Санкт-Петербургского государственного университета.
Список литературы
Москвин Л.Н., Родинков О.В. Хроматомембранные методы разделения веществ. СПб: Изд-во С.-Петербургского университета, 2014. 216 с.
Москвин Л.Н. Хроматомембранный метод разделения веществ // Доклады РАН. 1994. Т. 334. № 5. С. 599.
Moskvin L.N. Chromatomembrane method for the continuous separation of substances // J. Chromatogr. A. 1994. V. 669. P. 81.
Москвин Л.Н., Родинков О.В. Хроматомембранные методы: физико-химические принципы, аналитические и технологические возможности // Изв. Акад. Наук. Сер. хим. 2012. Т. 61. № 4. С. 719. (Moskvin L.N., Rodinkov O.V. Chromatomembrane methods: Physicochemical principles, analytical and technological possibilities // Russ. Chem. Bull. 2012. V. 61. № 4. P. 723).
Родинков О.В., Москвин Л.Н., Васькова Е.А. Оптимизация пористой структуры гидрофобной матрицы для осуществления хроматомембранных массообменных процессов // Журн. физ. химии. 2005. Т. 79. № 3. С. 539. (Rodinkov O.V., Moskvin L.N., Vaskova E.A. Optimization of the porous structure of a hydrophobic matrix for chromatomembrane mass-exchange processes // Russ. J. Phys. Chem. 2005. V. 79. № 3. P. 453.)
Moskvin L.N., Simon U., Rodinkov O.V., Grigoriev G.L. Flow-injection analysis and chromatography – A new device for extraction in gas-liquid and liquid-liquid systems // Talanta. 1994. V. 41 (10). P. 1765.
Родинков О.В., Бугайченко А.С., Москвин Л.Н. Сравнение аналитических возможностей различных схем хроматомембранной газовой экстракции // Журн. аналит. химии. 2021. Т. 76. № 9. С. 797. (Rodinkov O.V., Bugaichenko A.S., Moskvin L.N. Comparison of the analytical capabilities of different chromatomembrane gas extraction techniques// J. Anal. Chem. 2021. V. 76. № 9. P. 1051.) https://doi.org/10.31857/S0044450221090097
Москвин Л.Н., Михайлова Н.В., Николаева Д.Н. Экстракционно-фотометрическое определение анионных поверхностно-активных веществ с хроматомембранным концентрированием // Журн. аналит. химии. 1996. Т. 51. № 8. С. 845. (Moskvin L.N., Nikolaeva D.N., Mikhailova N.V. Determination of anionic surfactants in water with adsorption preconcentration // J. Anal. Chem. 1996. V. 51. № 3. P. 282.)
Ganeto F., Rios Q., Luque de Castro. Liquid—liquid extraction in continuous flow systems without phase separation // Anal. Chem. 1988. V. 60. P. 2354.
Moskvin A.L., Moskvin L.N., Moszhuchin A.V., Fomin V.V. Txtraction-cromatographic preconcentration with chromatomambrane separation of extract from aqueous phase for luminexcence determination of oil and phenols in natural water by flow analysis // Talanta. 1999. V. 50. P. 113.
Родинков О.В., Москвин Л.Н., Синицына Т.В., Григорьев Г.Л. Хроматомембранная абсорбция микропримесей полярных органических веществ из воздуха водными растворами // Журн. аналит. химии. 1998. Т. 53. № 4. С. 373. (Rodinkov O.V., Moskvin L.N., Sinitsina T.V., Grigor’ev G.L. Chromatomembrane absorption of trace impurities of polar organic substances from water to aqueous // J. Anal. Chem. 1998. V. 53. № 4. P. 326.)
Никоноров В.В., Москвин Л.Н. Новый реагент для определения нитрит-ионов // Журн. аналит. химии. 1996. Т. 51. № 8. С. 737. (Nikonorov V.V., Moskvin L.N. New reagent for determining nitrites // J. Anal. Chem. 1996. V. 51. № 7. P. 679.)
Wei Y., Oshima M., Simon J., Motomizu S. Absorption, concentration and determination of trace amounts of air pollutants by flow injection method coupled with a chromatomembrane cell system: Application to nitrogen dioxide determination // Talanta. 2002. V. 58. № 6. P. 1343.
Erxleben H., Moskvin L., Nikitina T.G., Simon J. Determination of small quantities of nitrogen oxides in air by ion chromatography using a chromatomembrane cell for preconcentration // Fresenius J. Anal. Chem. 1998. V. 361. № 2. P. 325.
Wei. Y., Oshima M., Simon J., Motomizu S. The application of the chromatomembrane cell for the absorptive sampling of nitrogen dioxide followed by continuous determination of nitrite using a micro-flow injection system // Talanta. 2002. V. 57. № 2. P. 355.
Москвин Л.Н., Никоноров В.В. Проточно-инжекционное определение диоксида серы в воздухе с предварительным хроматомембранным концентрированием // Журн. аналит. химии. 1996. Т. 51. № 8. С. 891. (Moskvin L.N., Nikonorov V.V. Flow-injection determination of sulfur dioxide in air with preliminary chromatomembrane concentration // J. Anal. Chem. 1996. V. 51. № 8. P. 891.)
Sritharathikhun P., Oshima M., Wei Y., Simon J., Motomizu S. On-line cellection/concentration and detection of sulfur dioxide in air by flow-injection spectrophotometry coupled with a chromatomambrane cell // Anal. Sci. 2004. V. 20. № 1. P. 113.
Erxleben H., Simon J., Moskvin L.N., Vladimirova L.O., Nikitina T.G. Automized procedures for the determination of ozone and ammonia contents in air by using the chromatomembrane method for gas-liquid extraction // Fresenius J. Anal. Chem. 2000. V. 366. № 4. P. 332.
Москвин А.Л., Мельниченко А.Н., Диченко О.Ю. Фотометрическое определение аммиака в воздухе рабочей зоны с хроматомембранным концентрированием // Вестн. С.-Петерб. ун-та. Сер. 4: Физика. Химия. 2011. Вып. 4. С. 55.
Москвин А.Л., Мельниченко А.Н., Диченко О.Ю. Фотометрическое определение аммиака в воздухе рабочей зоны с хроматомембранной жидкостной абсорбцией // Аналитика и контроль. 2013. Т. 17. № 4. С. 485.
Sritharathikhun P., Oshima M., Wei Y., Simon J., Motomizu S. On-line collection/concentration of trace amounts of formaldehyde in air with chromatomembrane cell and its sensitive determination by flow injection technique coupled with spectrophotometric and fluorometric // Talanta. 2005. V. 67. № 5. P. 1014.
Москвин Л.Н., Родинков О.В., Синицына Т.В. Фотометрическое определение гидразина в воздухе с хроматомембранным концентрированием // Журн. аналит. химии. 1999. Т. 54. № 1. С. 61. (Moskvin L.N., Rodinkov O.V., Sinitsyna T.V. Determination of hydrazine in air by photometry with chromatomembrane preconcentration // J. Anal. Chem. 1999. V. 54. № 1. P. 53.)
Родинков О.В., Бугайченко А.С., Москвин Л.Н. Динамическая хроматомембранная жидкостная хемосорбция микропримесей из газовой фазы в политетрафторэтиленовых матрицах, модифицированных сорбционно-активным материалом // Журн. физ. химии. 2010. Т. 84. № 8. С. 1568. (Rodinkov O.V., Bugaichenko A.S., Moskvin L.N. Dynamic chromatomembrane liquid chemisorption of trace impurities from a gas phase in polytetrafluoroethylene matrices modified with sorptionactive material // Russ. J. Phys. Chem. A. 2010. V. 84. № 8. P. 1428.) https://doi.org/10.1134/S0036024410080261
Moskvin L.N., Moskvin A.L. Chromatomembrane methods – Novel automatization possibilities of substances separation processes // Laboratory Robotics and Automatization. 1998. V. 10. P. 3.
Москвин А.Л., Москвин Л.Н., Родинков О.В. Хроматомембранный метод – новый принцип функционирования устройств для пробоподготовки в аналитических приборах // Научное приборостроение. 1999. Т. 9. № 4. С. 62.
Москвин Л.Н., Родинков О.В., Катрузов А.Н., Томилова Е.С. Ионохроматографическое определение полярных неорганических примесей в воздухе с хроматомембранным предконцентрированием // Журн. аналит. химии. 1996 Т. 51. № 11. С. 1214. (Moskvin L.N., Rodinkov O.V., Katruzov A.N., Tomilova E.S. Ion-chromatographic determination of polar inorganic impurities in air with chromatomembrane preconcentration // J. Anal. Chem. 1996. V. 51. № 11. P. 1109.)
Moskvin L.N., Rodinkov O.V. Continuous chromatomembrane headspace analysis // J. Chromatogr. A. 1996. V. 725. P. 351.
Родинков О.В., Москвин Л.Н., Зыкин И.А. Газохроматографическое определение газообразных углеводородов в водных растворах с хроматомембранной газовой экстракцией // Журн. аналит. химии. 2003. Т. 58. № 1. С. 82. (Rodinkov O.V., Moskvin L.N., Zykin I.A. Gas-chromatographic determination of gaseous hydrocarbons in aqueous solutions using chromatomembrane gas extraction // J. Anal. Chem. 2003. V. 58. № 1. P. 71.)
Майорова Н.А., Родинков О.В., Москвин Л.Н. Газохроматографическое определение метилакрилата и метилметакрилата в водных растворах с хроматомембранной газовой экстракцией и газоадсорбционным концентрированием // Вестн. С.-Петерб. ун-та. Сер. 4: Физика. Химия. 2005. Вып. 4. С. 84.
Родинков О.В., Москвин Л.Н. Хроматомембранное концентрирование микропримесей органических загрязнителей природных вод и атмосферного воздуха // Журн. аналит. химии. 2002. Т. 57. № 10. С. 1057. (Moskvin L.N., Rodinkov O.V., Chromatomembrane preconcentration of trace impurities of organic pollutants from natural waters and atmospheric air // J. Anal. Chem. 2002. V. 57. № 10. P. 894.)
Dettmer K., Engewald W. Adsorbent materials commonly used in air analysis for adsorptive enrichment and thermal desorption of volatile organic compounds // Anal. Bioanal. Chem. 2002. V. 373. P. 490.
Mayer H., Spiekermann M., Bergmann M. Determination of volatile organic compounds in water by purge&trap – Gas chromatography – Mass spectrometry // J. Mol. Struct. 1995. V. 348. P. 389.
Родинков О.В., Москвин Л.Н. Поверхностно-слойные композиционные сорбенты для экспрессного концентрирования летучих органических веществ из водных и газовых сред // Журн. аналит. химии. 2012. Т. 67. № 10. С. 908. (Rodinkov O.V., Moskvin L.N. Surface layer composite sorbents for the rapid preconcentration of volatile organic substances from aqueous solutions and gas atmospheres // J. Anal. Chem. 2012. V. 67. № 10. P. 814.) https://doi.org/10.1134/S1061934812100073
Москвин Л.Н., Родинков О.В. От жидкостно-газовой хроматографии к хроматомембранному массообменному процессу // Журн. аналит. химии. 2019. Т. 74. № 10. С. 729. (Moskvin L.N., Rodinkov O.V. From liquid–gas chromatography to a chromatomembrane mass-exchange process //J. Anal. Chem. 2019. V. 74. № 10. P. 955.) https://doi.org/10.1134/S0044450219100098
Родинков О.В., Москвин Л.Н., Майорова Н.А. Быстродействие различных схем непрерывной хроматомембранной газовой экстракции // Журн. аналит. химии. 2005. Т. 60. № 8. С. 727. (Rodinkov O.V., Moskvin L.N, Maiorova N.A. Operation rates of different schemes of continuous chromatomembrane gas extraction // J. Anal. Chem. 2005. V. 60. № 8. P. 820.) https://doi.org/10.1134/S1061934819100083
Rodinkov O.V., Moskvin L.N., Viktorova M.I., Dyakin A.A., Yakimova N.M. Chromatomembrane headspace analysis of aqueous solutions at elevated temperatures // Chromatographia. 2015. V. 78. P. 1211.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии