

Журнал аналитической химии, 2023, T. 78, № 2, стр. 159-165
Автоматизированная жидкостная микроэкстракция фторхинолонов для их последующего хроматографического определения
И. И. Тимофеева a, *, **, К. А. Барбаянов a, А. В. Булатов a
a Санкт-Петербургский государственный университет, Институт химии
198504 Санкт-Петербург, Университетский просп., 26, Россия
* E-mail: i.i.timofeeva@spbu.ru
** E-mail: timofeeva.irina.7@gmail.com
Поступила в редакцию 30.04.2022
После доработки 11.07.2022
Принята к публикации 19.07.2022
- EDN: AUSOSY
- DOI: 10.31857/S0044450223020135
Аннотация
Разработан автоматизированный способ дисперсионной жидкостной микроэкстракции антибиотиков фторхинолонового ряда на принципах циклического инжекционного анализа. Способ предполагает диспергирование экстрагента газовой фазой, которая образуется in situ в экстракционной камере проточного анализатора. В качестве экстрагента для выделения и концентрирования фторхинолонов изучен глубокий эвтектический растворитель на основе терпеноида и смеси гидрофильной и гидрофобной карбоновых кислот, и обоснована возможность его применения. Гидрофильная карбоновая кислота в составе экстрагента выступает донором протонов для образования углекислого газа-диспергатора в присутствии растворенного в водной фазе карбоната натрия. На примере определения фторхинолонов в сточных водах показана возможность сочетания разработанного способа с методом высокоэффективной жидкостной хроматографии с флуориметрическим детектированием. Пределы обнаружения (3σ) для офлоксацина, флероксацина и норфлоксацина составили 0.3 мкг/л.
Как правило, инструментальный химический анализ проб сложного состава включает процедуры разделения и концентрирования с целью устранения мешающего влияния матричных компонентов проб и снижения пределов обнаружения целевых аналитов. Однако такая пробоподготовка остается наиболее длительной и трудоемкой в общей схеме химического анализа и предполагает большой расход реагентов и проб на ее выполнение.
Новые возможности для инструментального химического анализа открыли методы жидкостной микроэкстракции, обеспечивающие возможность эффективного предконцентрирования целевых аналитов, миниатюризацию и экологическую безопасность пробоподготовки [1–3]. Миниатюризация состоит в уменьшении масштабов процедур на всех стадиях выполнения анализа с целью снижения расходов проб, реагентов и образующихся отходов и, как следствие, повышения его экологической безопасности. Повысить экспрессность жидкостной микроэкстракции возможно путем ее автоматизации на принципах проточных методов, в которых основной акцент сделан на замену ручных рутинных процедур, составляющих основу стадии пробоподготовки, простыми легко автоматизируемыми операциями объединения и смешения потоков пробы, растворов реагентов и экстрагентов [4–6].
В последнее время особое внимание уделяют поиску и разработке новых эффективных экстракционных систем для выделения различных классов аналитов, в том числе в условиях проточного анализа [7]. К “зеленым” экстрагентам последнего поколения относят глубокие эвтектические растворители (ГЭР) [8, 9]. Такие экстрагенты состоят из двух или более прекурсоров, которые способны образовывать между собой водородные связи, что приводит к существенному снижению температуры плавления ГЭР по сравнению с температурами плавления исходных компонентов. Как правило, ГЭР находятся в жидком состоянии при нормальных условиях. Комбинируя прекурсоры ГЭР можно получать “дизайнерские” гидрофобные [10] и гидрофильные [11] растворители с требуемыми экстракционными свойствами.
Фторхинолоны находят обширное применение при фармакотерапии широкого круга заболеваний человека и животных. Выведение фторхинолонов и их метаболитов из организма осуществляется почками, главным образом в неизменном виде [12], поэтому фторхинолоны попадают в сточные воды и загрязняют объекты окружающей среды. Важной задачей является разработка экспрессных и чувствительных способов определения антибиотиков в сточных водах.
Цель данной работы − разработка автоматизированного способа дисперсионной жидкостной микроэкстракции в ГЭР, реализуемого на принципах циклического инжекционного анализа (ЦИА). Способ предполагает диспергирование экстрагента газовой фазой (углекислым газом), которая образуется in situ в экстракционной камере проточного анализатора. С целью подтверждения эффективности предложенного способа дисперсионной жидкостной микроэкстракции изучали возможность выделения антибиотиков фторхинолонового ряда (офлоксацина, флероксацина и норфлоксацина) из проб сточных вод для последующего их определения методом высокоэффективной жидкостной хроматографии с флуориметрическим детектированием (ВЭЖХ-ФД).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Рабочие растворы фторхинолонов (офлоксацина, флероксацина и норфлоксацина) готовили непосредственно перед работой последовательным разбавлением деионизованной водой стандартного раствора (1.0 мг/л), полученного растворением соответствующих навесок аналитов в 0.01 М растворе NaOH. Стандартный раствор фторхинолонов устойчив при хранении в закрытом сосуде в холодильнике при 5°C в течение одного месяца.
Глубокие эвтектические растворители готовили в стеклянном стакане путем смешивания 1 мл гептановой кислоты, 22.4 г расплавленного ментола и 13 мл муравьиной кислоты. Смесь хранили в закрытом сосуде в холодильнике при 5°C.
Фосфатный буферный раствор (pH 6.4) готовили перед анализом путем смешивания 0.05 М раствора Na2HPO4 и 0.05 М раствора NaH2PO4 в соотношении 1 : 3.
Все реактивы имели квалификацию не ниже ч. д. а.
Пробы сточных вод получали из ГУП “Водоканал Санкт-Петербурга”. Перед анализом все пробы предварительно фильтровали через фильтр “синяя лента”.
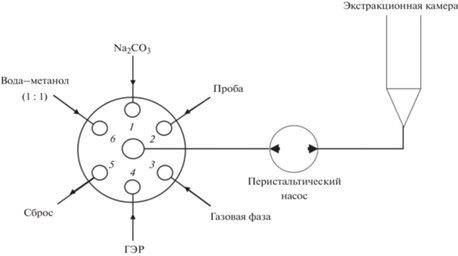
Гидравлическая схема ЦИА (рис. 1) включала: соленоидный кран-переключатель (Cole-Parmer, США); перистальтический насос (MasterFlex L/S, Cole-Parmer, США), обеспечивающий реверс направления потока (скорость потока – от 0.5 до 6.0 мл/мин); экстракционную камеру – полипропиленовую трубку конусообразной формы объемом 5 мл; трубки для коммуникаций из политетрафторэтилена (внутренний диаметр 0.5 мм). Система управлялась с помощью компьютера.
Хроматографический анализ выполняли с помощью жидкостного хроматографа с флуориметрическим детектором LC-20 (Shimadzu, Япония). Для определения фторхинолонов устанавливали длины волн возбуждения и флуоресценции 278 и 466 нм соответственно. Хроматографическое разделение осуществляли на колонке Luna C18 (250 × 4.6 мм, размер частиц 5 мкм, Phenomenex, США) в изократическом режиме при 40°C. Подвижная фаза представляла собой смесь метанола и фосфатного буферного раствора (рН 6.4) в соотношении 45 : 55. Скорость потока подвижной фазы составляла 0.7 мл/мин. Времена удерживания норфлоксацина, флероксацина и офлоксацина составляли 6.3, 9.7 и 11.3 мин соответственно.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
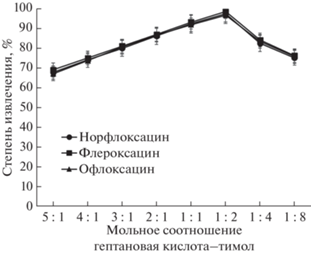
На предварительном этапе для микроэкстракционного выделения фторхинолонов в статических условиях изучали возможность применения гидрофобных ГЭР на основе тимола и гептановой кислоты с различным соотношением прекурсоров. Тимол широко применяется в качестве акцептора водородной связи для приготовления устойчивых в водной фазе ГЭР [13]. Гептановую кислоту выбрали в качестве донора водородной связи, поскольку она обеспечивает возможность получения менее вязких ГЭР по сравнению с более длинноцепочечными карбоновыми кислотами. Высокая вязкость растворителей ограничивает их применение в проточном анализе. ГЭР и водный раствор аналитов (200 мкг/л) встряхивали при соотношении фаз 1 : 30 в течение 5 мин, после чего выполняли хроматографический анализ полученных фаз. Установили, что максимальные степени извлечения наблюдаются при мольном соотношении гептановой кислоты и тимола 1 : 2 соответственно (рис. 2).
Рис. 2.
Влияние состава глубокого эвтектического растворителя на эффективность экстракции фторхинолонов (концентрация аналитов 200 мкг/л).
Для автоматизации дисперсионной жидкостной микроэкстракции фторхинолонов в ГЭР разработали гидравлическую схему (рис. 1), которая предполагает коммутацию крана-переключателя, перистальтического насоса и сообщающейся с атмосферой экстракционной камеры. Схема обеспечивает возможность диспергирования фазы ГЭР потоком углекислого газа, который образуется в результате химической реакции гидрокарбонат-ионов с прекурсором ГЭР непосредственно в экстракционной камере.
Учитывая необходимость в доноре протонов для реакции образования углекислого газа в присутствии гидрокарбонат-ионов, в состав ГЭР на основе гептановой кислоты и тимола (1 : 2) вводили муравьиную/уксусную кислоту, которая растворяется и в водной фазе, и в фазе ГЭР. Муравьиную/уксусную кислоту вводили в ГЭР в количествах, необходимых для полной нейтрализации водной фазы, чтобы обеспечить условия для извлечения молекулярных форм аналитов. Установили, что в присутствии муравьиной кислоты наблюдается более интенсивное выделение углекислого газа и перемешивание фаз (диспергирование) в экстракционной камере, так как этот прекурсор ГЭР является более сильной кислотой, чем уксусная (pKa 3.75 и 4.76 соответственно).
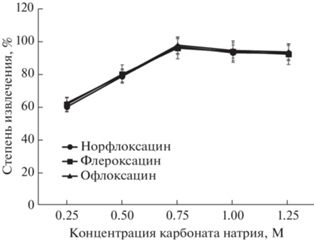
Карбонат натрия использовали как источник гидрокарбонат-ионов в растворе пробы. Для оптимизации концентрации карбоната натрия в водной фазе проводили серию экспериментов в условиях ЦИА. В экстракционной камере последовательно смешивали 2 М раствор Na2CO3 (0.6, 1.3, 1.9, 2.5, 3.1 мл) с водным раствором антибиотиков (4.4, 3.7, 3.1, 2.5, 1.9 мл). Далее в экстракционную камеру подавали ГЭР на основе тимола и гептановой кислоты (100 мкл; мольное соотношение 1 : 2) и муравьиной кислоты (от 100 до 490 мкл). Результаты, представленные на рис. 3, показывают, что оптимальная концентрация Na2CO3 составляет 0.75 М, так как, начиная с этой концентрации, степени извлечения аналитов достигают максимального значения, и дальнейшее увеличение концентрации реагента не влияет на эффективность массопереноса.
Рис. 3.
Влияние концентрации карбоната натрия на эффективность экстракции фторхинолонов (концентрация аналитов 200 мкг/л).
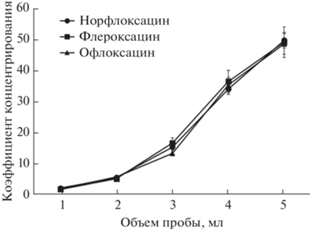
Объем пробы влияет на соотношение фаз и достигаемые коэффициенты концентрирования. С целью снижения пределов обнаружения изучили возможность увеличения соотношения объемов фаз. Для этого в экстракционную камеру последовательно подавали порции 2 М раствора Na2-CO3 и водного раствора аналитов в различных соотношениях, получая в результате от 1 до 5 мл водной фазы с постоянными концентрациями Na2CO3 (0.75 М) и аналитов (200 мкг/л). Далее к полученному раствору добавляли трехкомпонентный ГЭР, состоящий из тимола и гептановой кислоты (мольное соотношение 1 : 2) и муравьиной кислоты (от 60 до 295 мкл). Как видно из рис. 4, объем водной фазы 5 мл обеспечивает максимальные коэффициенты концентрирования. Дальнейшее увеличение объема пробы невозможно, так как при этом происходит частичное растворение фазы экстрагента в пробе, что затрудняет отбор органической фазы и приводит к невоспроизводимым результатам.
Рис. 4.
Влияние объема пробы на эффективность экстракции фторхинолонов (концентрация аналитов 200 мкг/л).
Изучали влияние времени разделения фаз в экстракционной камере на прецизионность в интервале от 30 с до 10 мин. При этом дополнительно в экстракционную камеру подавали поток атмосферного воздуха (скорость потока 3.0 мл/мин), который инициировал разрушение эмульсии. При перемешивании фаз в течение менее 5 мин значения sr составляли от 20 до 45%. Для воспроизводимого разделения фаз требовался барботаж в течение 5 мин, при этом sr ≤ 10%.
На основании полученных результатов разработали способ определения фторхинолонов в водных средах. На первом этапе с помощью перистальтического насоса через кран-переключатель в экстракционную камеру подавали 1.9 мл 2 М раствора Na2CO3 (канал 1) и 3.1 мл пробы (канал 2). Для перемешивания двух растворов в камеру в течение 20 с подавали атмосферный воздух (канал 3). Далее вводили 390 мкл ГЭР (гептановая кислота, ментол и муравьиная кислота, 1 : 2 : 48) (канал 4), после чего подавали атмосферный воздух в течение 5 мин. Скорость потоков составляла 3.0 мл/мин. На втором этапе (после разделения фаз) с помощью перистальтического насоса выполняли слив нижней (водной) фазы из камеры, а фазу экстракта перекачивали в виалу для последующего ВЭЖХ-ФД-анализа. На заключительном этапе проводили промывку всех коммуникаций системы смесью метанола и дистиллированной воды (1 : 1) (канал 6).
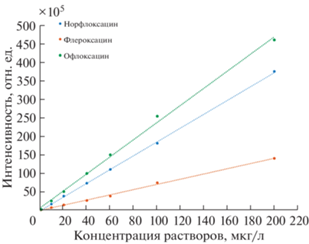
Для построения градуировочных зависимостей (рис. 5) использовали стандартные растворы аналитов, которые проводили через все стадии микроэкстракции. Способ обеспечил диапазоны определяемых концентраций офлоксацина, флероксацина и норфлоксацина от 1 до 200 мкг/л при объеме пробы 5 мл. Пределы обнаружения (3σ) для всех аналитов составили 0.3 мкг/л, пределы определения (10σ) для всех аналитов – 1 мкг/л. Для этого уровня концентраций соотношение сигнал/шум составило 8, 11 и 16 для норфлоксацина, флероксацина и офлоксацина соответственно. Относительная неисключенная систематическая погрешность для уровня концентраций аналитов 1 мкг/л не превышала ±20% (P = = 0.95). При этом значения sr внутрилабораторной прецизионности не превышали 9% (n = 6). Производительность пробоподготовки – 10 проб в час. Время хроматографического анализа – 20 мин. Способ позволил исключить ручные манипуляции и стадию центрифугирования.
Аналитические возможности способа подтверждены при анализе сточных вод. В пробах антибиотики не были обнаружены (табл. 1). Правильность полученных результатов проверяли методом введено−найдено. Хроматограммы, полученные при анализе сточной воды с различными добавками офлоксацина, флероксацина и норфлоксацина, представлены на рис. 6. Относительную степень извлечения фторхинолонов рассчитывали по формуле:
Таблица 1.
Результаты анализа сточных вод (n = 3, P = 0.95)
| Образец сточной воды | Аналит | Введено, мкг/л |
Найдено, мкг/л |
Относительная степень извлечения, % |
|---|---|---|---|---|
| № 1 | Норфлоксацин | 0 | <ПО* | |
| 15 | 12.1 ± 1.2 | 81 | ||
| 30 | 25.0 ± 1.5 | 83 | ||
| 45 | 38.6 ± 1.9 | 86 | ||
| Флероксацин | 0 | <ПО | ||
| 15 | 13.5 ± 1.4 | 90 | ||
| 30 | 24.4 ± 0.8 | 81 | ||
| 45 | 38.7 ± 2.1 | 86 | ||
| Офлоксацин | 0 | <ПО | ||
| 15 | 13.2 ± 0.9 | 88 | ||
| 30 | 25.6 ± 1.2 | 85 | ||
| 45 | 37.4 ± 0.9 | 83 | ||
| № 2 | Норфлоксацин | 0 | <ПО | |
| 15 | 14.7 ± 0.8 | 98 | ||
| 30 | 27.1 ± 1.0 | 90 | ||
| 45 | 41.7 ± 1.5 | 93 | ||
| Флероксацин | 0 | <ПО | ||
| 15 | 13.7 ± 0.7 | 91 | ||
| 30 | 28.6 ± 1.3 | 95 | ||
| 45 | 40.5 ± 1.0 | 90 | ||
| Офлоксацин | 0 | <ПО | ||
| 15 | 14.8 ± 0.9 | 99 | ||
| 30 | 27.7 ± 0.6 | 92 | ||
| 45 | 41.2 ± 1.4 | 92 | ||
| № 3 | Норфлоксацин | 0 | <ПО | |
| 15 | 12.6 ± 0.7 | 84 | ||
| 30 | 24.7 ± 1.2 | 82 | ||
| 45 | 40.0 ± 1.4 | 89 | ||
| Флероксацин | 0 | <ПО | ||
| 15 | 13.4 ± 1.1 | 89 | ||
| 30 | 26.0 ± 0.9 | 87 | ||
| 45 | 39.7 ± 1.3 | 88 | ||
| Офлоксацин | 0 | <ПО | ||
| 15 | 13.9 ± 0.6 | 93 | ||
| 30 | 26.7 ± 1.0 | 89 | ||
| 45 | 37.3 ± 0.6 | 83 |
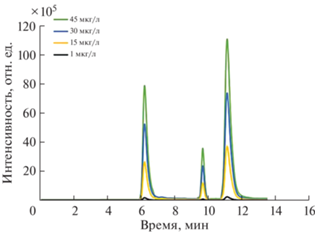
Рис. 6.
Хроматограммы, полученные при анализе сточной воды с добавками фторхинолонов (времена удерживания норфлоксацина, флероксацина и офлоксацина: 6.3, 9.7 и 11.3 мин соответственно).
Относительная степень извлечения превышала 81%, что подтверждает отсутствие существенного мешающего влияния компонентов матрицы на извлечение и определение аналитов. В соответствии с данными [14] для уровня концентраций аналита 10 мкг/л значение относительной степени извлечения в диапазоне от 60 до 115% является допустимым.
* * *
Разработан автоматизированный способ дисперсионной жидкостной микроэкстракции, основанный на диспергировании глубокого эвтектического растворителя потоком углекислого газа, который образуется в результате химической реакции. Представлена гидравлическая схема для реализации дисперсионной жидкостной микроэкстракции на принципах циклического инжекционного анализа. Для микроэкстракционного выделения офлоксацина, флероксацина и норфлоксацина изучена возможность применения глубокого эвтектического растворителя на основе ментола, гептановой и муравьиная кислот. Предложенный способ определения фторхинолонов в сточных водах обеспечивает пределы обнаружения (0.3 мкг/л), сопоставимые с приведенными в работах [15–17]. В отличие от описанных ранее способов дисперсионной микроэкстракции, разработанный способ исключает необходимость применения токсичных экстрагентов, полярных растворителей для диспергирования экстрагентов и центрифугирования для разделения фаз.
Авторы выражают благодарность РНФ (№ 21-13-00020, https://rscf.ru/project/21-13-00020/) за финансовую поддержку в проводимых исследованиях, а также ГУП “Водоканал Санкт-Петербурга” за предоставленные пробы.
Список литературы
Крылов В.А., Крылов А.В., Мосягин П.В., Маткивская Ю.О. Жидкофазное микроэкстракционное концентрирование примесей // Журн. аналит. химии. 2011. Т. 66. С. 341.
Дмитриенко С.Г., Апяри В.В., Толмачева В.В., Горбунова М.В. Жидкостная экстракция органических соединений в каплю экстрагента. Обзор обзоров // Журн. аналит. химии. 2021. Т. 76. № 8. С. 675. https://doi.org/10.31857/S0044450221080041
Дмитриенко С.Г., Апяри В.В., Толмачева В.В., Горбунова М.В. Дисперсионная жидкостно-жидкостная микроэкстракция органических соединений. Обзор обзоров // Журн. аналит. химии. 2020. Т. 75. № 10. С. 867. https://doi.org/10.31857/S0044450220100059
Золотов Ю.А. Проточный химический анализ: монография. М.: Наука, 2014. 428 с.
Цизин Г.И., Статкус М.А., Золотов Ю.А. Сорбционное и экстракционное концентрирование микрокомпонентов в проточных системах анализа // Журн. аналит. химии. 2015. Т. 70. № 11. С. 1123.
Vakh C., Falkova M., Timofeeva I., Moskvin A., Moskvin L., Bulatov A. Flow analysis: A novel approach for classification // Crit. Rev. Anal. Chem. 2016. V. 46 P. 374. https://doi.org/10.1080/10408347.2015.1087301
Вах К.С., Тимофеева И.И., Булатов А.В. Автоматизация микроэкстракционного концентрирования на принципах циклического инжекционного анализа // Журн. аналит. химии. 2019. Т. 74. № 11. С. 846. https://doi.org/10.1134/S106193481911011X
Smith E.L., Abbott A.P., Ryder K.S. Deep eutectic solvents (DESs) and their applications // Chem. Rev. 2014. V. 114. № 21. P. 11060. https://doi.org/10.1021/cr300162p
Shishov A., Bulatov A., Locatelli M., Carradori S., Andruch V. Application of deep eutectic solvents in analytical chemistry. A review // Microchem. J. 2017. V. 135. P. 33. https://doi.org/10.1016/j.microc.2017.07.015
Cao J., Su E. Hydrophobic deep eutectic solvents: The new generation of green solvents for diversified and colorful applications in green chemistry // J. Clean. Prod. 2021. V. 314. Article 127965. https://doi.org/10.1016/j.jclepro.2021.127965
Ma Y., Wang Q., Zhu T. Comparison of hydrophilic and hydrophobic deep eutectic solvents for pretreatment determination of sulfonamides from aqueous environments // Anal. Methods. 2019. V. 11. P. 5901. https://doi.org/10.1039/C9AY02244A
Turnidge J. Pharmacokinetics and pharmacodynamics of fluoroquinolones // Drugs. 1999. V. 58. P. 29. https://doi.org/10.2165/00003495-199958002-00006
Martins M.A.R., Crespo E.A., Pontes P.V.A., Silva L.P., Bülow M., Maximo G.J., Batista E.A.C., Held C., Pinho S.P., Coutinho J.A.P. Tunable hydrophobic eutectic solvents based on terpenes and monocarboxylic acid // ACS Sustain. Chem. Eng. 2018. V. 6. P. 8836. https://doi.org/10.1021/acssuschemeng.8b01203
Taverniers I., De Loose M., Van Bockstaele E. Trends in quality in the analytical laboratory. II. Analytical method validation and quality assurance // Trends Anal. Chem. 2004. V. 23. P. 535. https://doi.org/10.1016/j.trac.2004.04.001
Herrera-Herrera A.V., Hernández-Borges J., Borges-Miquel T.M., Rodríguez-Delgado M.Á. Dispersive liquid-liquid microextraction combined with ultra-high performance liquid chromatography for the simultaneous determination of 25 sulfonamide and quinolone antibiotics in water samples // J. Pharm. Biomed. Anal. 2013. V. 75. P. 130. https://doi.org/10.1016/j.jpba.2012.11.026
Selahle S.K., Nomngongo P.N. Determination of fluoroquinolones in the environmental samples using vortex assisted dispersive liquid-liquid microextraction coupled with high performance liquid chromatography // Int. J. Environ. Anal. Chem. 2020. V. 100. P. 282. https://doi.org/10.1080/03067319.2019.1636042
Herrera-Herrera A.V., Hernández-Borges J., Borges-Miquel T.M., Rodríguez-Delgado M.Á. Dispersive liquid–liquid microextraction combined with nonaqueous capillary electrophoresis for the determination of fluoroquinolone antibiotics in waters // Electrophoresis. 2010. V. 31. P. 3457. https://doi.org/10.1002/elps.201000285
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии