

Биоорганическая химия, 2019, T. 45, № 1, стр. 27-39
Липид II как мишень действия антибиотиков нового поколения: структурно-динамические исследования
И. С. Панина *, А. О. Чугунов *, **, Р. Г. Ефремов *, **
* Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН, Россия, Москва
** Национальный исследовательский университет “Высшая школа экономики”, Россия, Москва
Поступила в редакцию 25.05.2018
После доработки 27.05.2018
Принята к публикации 28.05.2018
Аннотация
Ввиду нарастающей проблемы антибиотикорезистентности в медицине, большое внимание уделяется антимикробным веществам, действующим на небелковые молекулы, устроенные более консервативно, чем белки. Одной из наиболее перспективных и изученных таких мишеней является компонент биосинтетического пути формирования бактериальной клеточной стенки – липид II, который присутствует только в мембране бактерий и имеет консервативную химическую структуру. Существуют несколько классов природных антибиотиков, действующих на липид II, часть из которых блокируют синтез пептидогликанового слоя клеточной стенки, связывая липид II в прочный комплекс, а другие имеют дополнительный бактерицидный механизм, заключающийся в нарушении целостности мембраны. В обзоре рассмотрены перспективы использования таких антибактериальных веществ в качестве новых лекарств для борьбы с устойчивыми к антибиотикам патогенами. Основной акцент сделан на исследованиях структуры липида II и механизмов его распознавания антибиотиками, а также на компьютерном моделировании их взаимодействия.
ВВЕДЕНИЕ
После того, как в середине XX века антимикробные препараты прочно вошли в арсенал медицины, человечество столкнулось с новой проблемой – способностью бактерий вырабатывать устойчивость к действию антибиотиков [1]. Проблема развития антибиотикорезистентности усиливается за счет повсеместного (и зачастую неправильного) применения антимикробных препаратов в медицине и ветеринарии, а также за счет использования этих соединений при кормлении скота [2]. В этой связи поиск и разработка новых антибактериальных веществ становятся критически важными задачами, привлекающими внимание исследователей и врачей по всему миру.
Вероятность развития резистентности бактерий к разным соединениям различна [3]. Среди соединений с пониженной способностью вызывать резистентность большой интерес представляют, например, вещества, молекулярные мишени которых имеют консервативное химическое строение. Установление молекулярного механизма действия уже известных подобных антибиотиков является фундаментальной задачей, и ее решение, наряду с пониманием молекулярных принципов, лежащих в основе появления резистентности, поможет в разработке новых “устойчивых к резистентности” антибиотиков.
Одной из наиболее перспективных небелковых мишеней действия антибиотиков является компонент биосинтетического пути формирования бактериальной клеточной стенки – липид II, который присутствует в мембране всех бактерий, но при этом отсутствует в эукариотических клетках [3–6]. Будучи “узким местом” в этом жизненно важном для микроорганизмов процессе [7], липид II получил нарицательное имя “ахиллесова пята” бактерий. Консервативное химическое строение этой молекулы, в которой гидрофобный полиизопреновый хвост, погруженный в мембрану, соединяется с мономером пептидогликана клеточной стенки через пирофосфатный линкер (см. рис. 1), позволяет рассчитывать на то, что к некоторым действующим на него антибиотикам не будет развиваться резистентность.
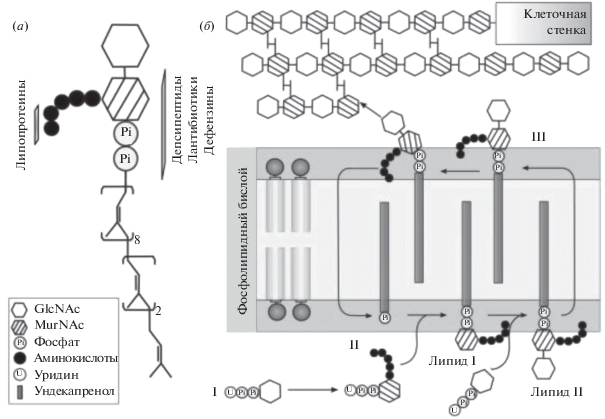
Рис. 1.
Схематичное изображение структуры липида II (а) и схема синтеза бактериальной клеточной стенки (б). (а) Липид II состоит из остатков сахаров (MurNAc и GlcNAс), пентапептида (L-Ala-D-γ-Glu-L-Lys-D-Ala-D-Ala), пирофосфатного линкера (показан как PiPi) и ундекапренольного “хвоста”. Отмечены участки взаимодействия с антибиотиками разных классов. (б) – на стадии I под действием ферментов формируются основные единицы (UDP-GlcNAс и UDP-MurNAc-пентапептид) из промежуточных метаболитов [8, 9]. Конденсация основных единиц осуществляется путем ферментативного превращения локализующегося в мембране ундекапренилфосфата (фосфорилированного изопреноидного спирта, содержащего 11 звеньев (55 атомов углерода)) в липид I [10], а затем в липид II (стадия II) [11]. На завершающей стадии (III) молекула-предшественник клеточной стенки – липид II – переправляется с помощью различных флиппаз [5] на периплазматическую сторону бактериальной мембраны, где мономерное звено пептидогликана встраивается в растущую цепь [12]. Вновь образовавшийся ундекапренилфосфат возвращается на внутриклеточную сторону бислоя, и цикл повторяется [13].
Для рационального дизайна новых антибиотиков необходимо понимать пространственную организацию фармакофора, позволяющего не только эффективно взаимодействовать с мишенью, но и изначально распознавать ее среди других молекул. Поскольку липид II, как и любой другой компонент клеточной мембраны, является сложным объектом для экспериментальных методов структурной биологии, он представляет интерес для изучения методами компьютерного моделирования. В предлагаемом обзоре мы рассмотрим известные классы антибактериальных веществ, мишенью которых является молекула липида II, а также перспективы их использования в качестве новых лекарств для борьбы с резистентными к антибиотикам бактериями. Основной акцент будет сделан на исследованиях структуры липида II и механизмов его распознавания антибиотиками, а также на компьютерном моделировании процессов межмолекулярного взаимодействия.
ЛИПИД II. СТРОЕНИЕ, ФУНКЦИЯ В КЛЕТКЕ И БИОСИНТЕЗ
Клеточная стенка является одним из жизненно важных компонентов бактериальной клетки, прочная ячеистая структура которой противодействует осмотическому давлению, возникающему между внутренней и внешней средой клетки. Клеточная стенка бактерий представляет собой полимер пептидогликана, структурные блоки которого (дисахаридные группы, соединенные поперечными пептидными цепочками) синтезируются внутри клетки как часть специфической молекулы-предшественника – липида II [5], располагающегося в мембране бактерий (рис. 1а). Головка липида II представляет собой мономер клеточной стенки и состоит из двух остатков сахаров (N-ацетилмурамовой кислоты (MurNAc) и N-ацетилглюкозамина (GlcNAс)) и присоединенного к MurNAc пентапептида. К гидрофильной головке присоединяется через пирофосфатный линкер длинный гидрофобный бактериопренольный хвост, располагающийся в неполярной части мембраны. Биосинтез пептидогликана начинается в цитоплазме клетки и протекает в три стадии (рис. 1б). Молекула липида II выполняет роль трансмембранного переносчика мономерных звеньев пептидогликана к постоянно обновляющейся клеточной стенке бактерий.
Несмотря на жизненно важную функцию, выполняемую липидом II, его количество в клетке невелико. Клеточная стенка в грамположительных бактериях обычно составляет около 20 слоев [14], причем каждый слой содержит огромное число субъединиц пептидогликана. Для ее строительства и обновления в клетках содержатся десятки тысяч молекул липида II, например, в бактериях рода Bacillus megaterium было обнаружено 34 000 единиц [15, 16]. Для грамотрицательных бактерий, пептидогликановая оболочка которых значительно тоньше (в среднем около 1.5 слоя [17]), число молекул липида II в любой момент времени не превышает 2000 на клетку [18]. В сравнении с фосфолипидами (основным липидным компонентом мембраны) количество липида II действительно крайне мало.
Липид II также важен для бактерий, у которых нет клеточной стенки, например, для представителей рода Wolbachia. Несмотря на отсутствие ферментов, необходимых для синтеза пептидогликана, у этих микроорганизмов сохранилась способность продуцировать липид II, который вовлечен в процесс деления клетки [19].
Учитывая принципиальную роль липида II в жизнедеятельности бактерий, а также относительно небольшое количество доступного липида II, нетрудно понять, что эта молекула является мишенью действия множества природных антибиотиков. Кроме того, целенаправленное блокирование этой молекулы – многообещающий путь для разработки новых антибактериальных веществ [20]. Далее будут описаны известные антибактериальные агенты, мишенями которых являются различные части молекулы липида II, а также представлены возможные механизмы адаптивной резистентности бактерий.
ЛИПИД II КАК МИШЕНЬ ДЕЙСТВИЯ ПРИРОДНЫХ АНТИБИОТИКОВ
Катионные антимикробные пептиды (АМП), продуцируемые всеми живыми организмами, включая сами бактерии, естественным образом взаимодействуют с отрицательно заряженными компонентами мембраны клетки (в первую очередь – с фосфолипидами, несущими у бактерий, в отличие от эукариот, отрицательный заряд). Адсорбировавшись на поверхности мембраны под влиянием электростатических сил, АМП, благодаря своей амфифильной природе, нарушают целостность липидного бислоя [21–24]. Долгое время считалось, что все АМП убивают бактерии неспецифическим путем: встраиваясь в мембрану клетки и формируя в ней поры или разрушая ее за счет детергент-подобного действия [25–27]. Однако позднее выяснилось, что некоторые АМП проявляют антибактериальную активность за счет селективного связывания со своей мишенью, приводящего к ингибированию метаболических процессов [28]. Одним из важнейших таких процессов является синтез клеточной стенки, а одна из ключевых вовлеченных в него молекул – это вышеупомянутый липид II. Молекула липида II является мишенью нескольких классов АМП, действие которых многократно усиливается [21, 29] благодаря их “заякориванию” на липиде II:
Гликопептиды (например, ванкомицин) блокируют кросс-сшивание пептидогликановых звеньев между собой. Ванкомицин связывается с С-концевым дипептидом D-Ala-D-Ala-пептидогликановой головки липида II посредством пяти водородных связей и дополнительных гидрофобных взаимодействий [30, 31], что препятствует росту клеточной стенки. Ванкомицин был внедрен в клиническую практику в 1958 г. [32–34], однако за счет горизонтального переноса генов появились нечувствительные к ванкомицину штаммы Staphylococcus aureus (VRSA) [32, 33]. Изучение механизмов резистентности к ванкомицину показало, что причиной является мутация диаланинового пептида в головке липида II, естественным образом закрепляющаяся под действием эволюционного отбора [35–38]. Возможным выходом является поиск и применение антибиотиков, действующих на менее вариабельные и более химически консервативные (непептидные) фрагменты молекулы липида II [39].
Немодифицированные пептиды, такие как дефензины, обладают широким спектром действия против грамположительных и грамотрицательных бактерий и убивают клетки несколькими способами [40, 41]. Дефензины являются частью врожденной иммунной системы большинства многоклеточных организмов, включая млекопитающих, беспозвоночных и растения. Известно несколько дефензинов, выделенных из различных организмов, которые специфично связывают липид II. Первым из таких дефензиновых антибиотиков был плектазин, обнаруженный в 2005 г. [42]. Исследование комплекса плектазин/липид II в мицеллах додецилфосфохолина методом ЯМР показало, что плектазин связывает липид II в области пирофосфата [43]. Взаимодействие липида II было продемонстрировано in vitro для грибковых дефензинов оризизина (из Aspergillus oryzea) и евроцина (из Eurotium amstelodami) [40, 44], а также для дефензинов беспозвоночных люцифензина (из личинок бабочки Lucilia sericata) и галлицина (из мидии Mytilus galloprovinciali) [45].
Лантибиотики представляют собой посттрансляционно модифицированные природные пептиды, содержащие такие неканонические аминокислотные остатки, как лантионин и/или метиллантионин [46–48]). В зависимости от структуры, мотива связывания с мишенью и механизма действия, лантибиотики подразделяют на две основные группы [49]. Удлиненные гибкие пептиды, несущие положительный заряд, входят в группу низина – первого открытого лантибиотика, применяемого как пищевой консервант E234 (рис. 2). Они не только блокируют встраивание новых звеньев в растущую цепь пептидогликана за счет связывания липида II, но и образуют поры в бактериальной мембране [50, 51]. Лантибиотики группы мерсацидина (рис. 2), напротив, имеют жесткую глобулярную структуру и либо являются электронейтральными, либо несут суммарный отрицательный заряд (актагардин, плантирицин C). Эти молекулы практически не нарушают целостность мембран, а для проявления их антибактериальной активности необходимы ионы Ca2+ [52]. Низин и родственные ему лантибиотики взаимодействуют с пирофосфатной группой липида II, замена которой в эволюционном процессе гораздо менее вероятна, чем мутация одной аминокислоты, а двойной механизм действия еще больше усложняет возможность защиты для клетки-мишени.
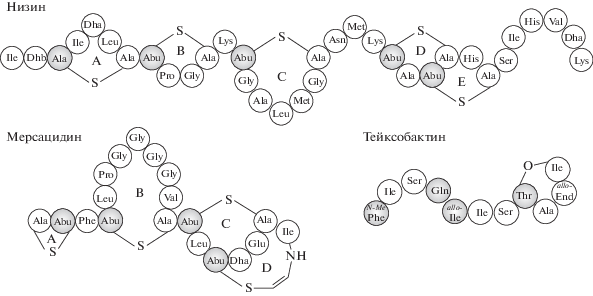
Рис. 2.
Схематичное изображение структур низина, мерсацидина и тейксобактина. Ala-S-Ala — лантионин, Abu-S-Ala — метиллантионин, allo-End — аллоэндурацидидин, allo-Ile — аллоизолейцин, Dha — дегидроаланин, Dhb — дегидроаминобутерин, MePhe — N-метилфенилаланин. Серым цветом показаны D-аминокислоты. Латинскими буквами А–Е обозначены кольцевые фрагменты в составе пептидов.
Депсипептиды – нерибосомальные пептиды, в которых остатки (гидрокси)аминокислот связаны между собой не только амидными, но и сложноэфирными связями [53]. Рамопланин, активный против широкого спектра грамположительных бактерий, был одобрен управлением по контролю за пищевыми продуктами и лекарственными препаратами США (FDA) для лечения инфекционных заболеваний желудочно-кишечного тракта, вызванных резистентными ко многим препаратам бактериями Clostridium difficile [54–56]. Ранние исследования показали, что рамопланин блокирует синтез пептидогликана путем ингибирования превращения липида I в липид II, катализируемого мембранно-связанной гликозилтрафсферазой MurG [57]. Позже было установлено, что рамопланин связывает с высокой аффинностью пирофосфатную группу, а также остаток MurNAc липида II и препятствует встраиванию головки молекулы в растущую пептидогликановую сеть [58–60]. Стехиометрия комплекса составляет 2 : 1 [61].
Открытый в 2015 г. тейксобактин (рис. 2) дополняет существующий список антибиотиков, связывающих липид II. Тейксобактин убивает бактерии путем ингибирования синтеза пептидогликана за счет связывания с консервативными фрагментами молекулы липида II – пирофосфатом и MurNAc. Специальные эксперименты по получению резистентных к тейксобактину мутантов не привели к положительным результатам: устойчивость к нему у бактерий не вырабатывается. Было показано, что даже при четырехкратном превышении минимальной ингибирующей концентрации (МИК) не обнаруживается устойчивых штаммов Staphylococcus aureus или Mycobacterium tuberculosis. Кроме того, резистентных штаммов S. aureus не было выделено и после того, как клетки были высеяны в течение 27 дней на средах, содержащих тейксобактин в концентрациях ниже МИК [62]. Это позволило предположить, что развитие резистентности к тейксобактину – трудновыполнимая задача для патогенных микроорганизмов.
Кроме агентов, действующих непосредственно на молекулу липида II, известны еще и различные соединения, ингибирующие его синтез (D-циклосерин, фосфомицин, туникамицин) или рециклизацию (возможность повторного использования ундекапренольного хвоста молекулы) (бацитрацин) [4].
Таким образом, существует целый ряд соединений, нацеленных на липид II, представляющий собой привлекательную мишень для высокоизбирательного антибактериального воздействия. Сложная “модульная” структура липида II позволяет различным антибиотикам использовать в качестве сайтов связывания разные группы молекулы (рис. 1а). Одни антибактериальные агенты взаимодействуют с легкомодифицируемой пептидной частью молекулы, что приводит к ожидаемому развитию устойчивости к их действию. Другие специфически распознают более консервативные части, такие как пирофосфат. Именно они считаются перспективными прототипами антибиотиков нового поколения, развитие резистентности к которым будет, по крайней мере, сильно отсрочено во времени.
ЛИПИД II В ЛИПИДНОМ БИСЛОЕ: МОЛЕКУЛЯРНЫЕ МОДЕЛИ
Липид II – молекула, сильно отличающаяся по структуре и свойствам от фосфолипидов мембраны, в которую она погружена. В то же время она очень слабо представлена в мембране, и поэтому экспериментальное изучение ее структуры и динамики методами структурной биологии затруднено. Перспективной альтернативой является компьютерное моделирование, в частности, метод молекулярной динамики (МД), хорошо зарекомендовавший себя как инструмент исследования конформационной динамики липидов и их смесей в бислойных мембранах in silico [63]. За последние пять лет опубликовано много теоретических исследований липида II в мембранном окружении, а также его комплексов с различными лигандами.
Исследование структуры и динамики липида II в фосфолипидных бислоях различного состава с помощью моделирования методом МД показало, что молекула расположена в мембране таким образом, что ее головка “плавает” на поверхности бислоя, пирофосфатная группа находится на том же уровне, что и фосфатные группы липидов, а длинный “хвост” все время остается внутри гидрофобной зоны бислоя, принимая разнообразные конформации (рис. 3 ) [64]. Первая половина хвоста молекулы (5 звеньев, отсчитывая от пирофосфата) проникает внутрь по направлению к центру мембраны, тогда как оставшиеся 6 звеньев (конец хвоста) более подвижны и преимущественно расположены между двух монослоев, придавая цепи “L-образную” конформацию (70% состояний в МД). Однако конец хвоста может также находиться около противоположной поверхности мембраны, пронизывая ее насквозь (“I-образная” конформация; 18%), или даже на той же, что и головка молекулы (“V-образная” конформация; 12%) [64].
Пирофосфатная группа определяет взаимную ориентацию “хвоста” и “головки” липида II. Анализ результатов МД в бислое позволяет выделить три основных конформации, описываемые значением псевдодвугранного угла (Головка)-О–Р–Р–О-(Хвост): около –60°, +60° и 180° [65].
Поскольку молекулы липида II локализуются преимущественно в неупорядоченных жидкокристаллических доменах бактериальных мембран [66], преимущественно вблизи полюсов деления клетки [67], эффективная локальная концентрация липида II может быть не такой уж и низкой, и его молекулы могут взаимодействовать друг с другом. Помимо этого, известно, что в поровом комплексе, формируемом низином, присутствуют четыре молекулы липида II [68]. В этой связи интересно было бы узнать, способен ли липид II образовывать олигомеры в нативной среде. Опубликованные на этот счет результаты противоречивы. Моделирование конформационной динамики липида II в бислое, имитирующем мембрану бактерий S. aureus (54% фосфатидилглицерин, лизилфосфатидилглицерин и 10% кардиолипин), показало катион-опосредованную димеризацию липида II [69], чего не наблюдалось в другом исследовании – в мембране, соответствующей грамположительным бактериям (пальмитоилолеилфосфатидилглицерин (POPG) и пальмитоилолеилфосфатидилэтаноламин (POPE) в соотношении 3 : 1 [70]) [64].
Присутствие липида II оказывает влияние на локальные свойства моделируемого бислоя, проявляющееся в перегруппировках конкретных типов липидов вокруг исследуемой молекулы [64, 69]. Кроме того, липид II изменяет характеристики поверхности бактериальной мембраны. Подвижный хвост молекулы возмущает окружающие липиды, что приводит к формированию долгоживущего (не менее 300–400 нс) гидрофобного “атолла” вокруг головки молекулы (рис. 4). Такое долговременное возмущение свойств поверхности бислоя может играть важную роль в распознавании липида II как мишени внеклеточными агентами. Интересно, что данный эффект не обнаруживается при моделировании липида II в цвиттерионной “эукариотической” мембране (чистый POPC). Мозаичный характер чередования гидрофильных и гидрофобных свойств, присущий чистому бислою, не изменяется при наличии в системе липида II. Это указывает на существенную роль липидного окружения, а также на взаимную адаптацию свойств нативной мембраны и липида II [64].
Рис. 3.
Конформации “хвоста” липида II в мембране, наблюдаемые в ходе МД (данные не опубликованы). Липид II изображен в CPK-представлении: “L-образная” конформация “хвоста” показана белым цветом; “V-образная” — серым; “I-образная” — черным; головка молекулы — белым; пирофосфатная группа — серым со штриховкой. Линиями показаны липиды мембраны; малыми сферами — атомы фосфора липидов. Молекулы воды с целью упрощения не представлены.

Рис. 4.
Двумерная карта распределения гидрофобных свойств на поверхности мембраны POPG/POPE [64] (вид вдоль нормали к плоскости мембраны). Степень гидрофобности в точках доступной растворителю поверхности мембраны рассчитана методом молекулярного гидрофобного потенциала (МГП) и усреднена по времени траектории МД (200 нс). (а) – карта распределения МГП на поверхности липидного бислоя. (б) — карта распределения МГП на поверхности бислоя, содержащего липид II. Расположение головки липида II показано черным контуром. Усредненное положение центра масс пирофосфатной группы – черным квадратом (PPI). Гидрофильные (отрицательные значения МГП) и гидрофобные (положительные значения МГП) области поверхности показаны черным и белым цветом, соответственно (шкала справа). В присутствии липида II на поверхности бактериальной мембраны образуется амфифильный паттерн, охватывающий молекулу липида II полукольцом (черный пунктир).

Компьютерный эксперимент позволяет изучать сложные мембранные системы на молекулярном уровне, выявляя тонкие эффекты, которые пока что невозможно обнаружить экспериментальными методами. Однако без практического подтверждения он всегда будет приводить лишь к предположениям. Недавние исследования влияния липида II на степень упорядоченности ацильных цепей липидов бислоя с помощью спектроскопии комбинационного рассеяния (КР) полностью подтвердили эффекты, обнаруженные в компьютерном эксперименте, включая локальные изменения гидрофобных свойств мембраны [71].
МЕХАНИЗМ РАСПОЗНАВАНИЯ ЛИПИДА II РАЗЛИЧНЫМИ АНТИБИОТИКАМИ: ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
Понимание процессов распознавания мишени ее лигандами, а также знание пространственной структуры комплекса лиганд–мишень имеют принципиальное значение для рационального дизайна новых молекул, эффективно идентифицирующих свою мишень. Именно поэтому большинство компьютерных исследований липида II нацелены на предсказание модели его комплекса с различными антибактериальными агентами в мембране. Результаты таких работ могут стать начальным этапом на пути создания новых бактерицидных средств. Отправной точкой для проведения компьютерного моделирования зачастую становятся экспериментальные структуры изучаемых молекул или их комплексов.
Взаимодействие низин – липид II было подробно описано на молекулярном уровне в 2004 г., когда с помощью ЯМР-спектроскопии была получена структура их комплекса (PDB ID: 1WCO) в соотношении 1 : 1 в растворе диметилсульфоксида (DMSO), слабо имитирующего мембранное окружение [72]. В результате анализа этой структуры был выявлен основной мотив связывания – кольца А и В низина образуют “ловушку” вокруг MurNAc и пирофосфата молекулы липида II, формируя сеть водородных связей, донорами протона в которых являются амидные группы основной цепи низина (рис. 5а). Все лантибиотики низинового типа имеют схожую систему A/B-колец, участвующих в связывании липида II, что указывает на то, что пирофосфатная “ловушка” является общей детерминантой связывания в этом классе лантибиотиков [67]. Биохимические исследования также показали, что водородные связи, образуемые основной цепью низина, являются ключевым взаимодействием при связывании липида II, и различные аминокислоты могут быть замещены без потери активности; только остатки в позициях 3 и 7–11 являются консервативными (рис. 2). Также важен гидрофобный характер остатка в позиции 6, который, как считается, взаимодействует с изопреноидным хвостом липида II и/или бислоем [73]. Предполагается, что после первичного связывания молекул происходит кластеризация четырех таких комплексов и четырех дополнительных молекул низина, в результате чего образуется поровый комплекс низин/липид II в стехиометрическом соотношении 8 : 4 [68].
Рис. 5.
Схематичное изображение структур комплексов антибиотиков с фрагментами их потенциальных мишеней (по данным экспериментальных методов). (а) – структура комплекса низина с пирофосфатной группой липида II в DMSO, установленная методом ЯМР-спектроскопии [72]. Кольца А и В низина формируют вокруг пирофосфатной группы (изображена в СРК-представлении) “ловушку”, связывая ее пятью водородными связями (показаны пунктирными линиями). (б) – структура гидрохлорида [Ac-Δ1-5-Arg10]тейксобактина (по данным рентгеноструктурного анализа) [76]. Хлорид-анион (изображен сферой) координирован четырьмя амидными группами основной цепи пептида, а также гуанидиновой группировкой остатка Arg10. В обоих комплексах участвуют кольцевые фрагменты молекул антибиотиков, в которых амидные протоны (доноры водородных связей) ориентированы в направлении единого центра. Abu – аминобутерин; Dha – дегидроаланин.

Лантибиотики группы мерсацидина не обладают порообразующей активностью, однако ингибируют биосинтез клеточной стенки, эффективно связывая липид II в комплекс. ЯМР-исследование подобного комплекса в мицеллах DPC показало, что структурные изменения, вызванные образованием комплекса, приводят к экспонированию заряженных групп на поверхности пептида, указывая на то, что, несмотря на довольно гидрофобную природу мерсацидина, ключевую роль в механизме связывания играют электростатические взаимодействия [74]. Замена Glu17 (рис. 2) на аланин привела к потере антибактериальной активности [75], что говорит о необходимости наличия заряда в боковой цепи для связывания мерсацидином липида II.
Рентгеноструктурный анализ молекулы актагардина выявил наличие сайта связывания ионов Ca2+, во взаимодействии с которым участвует остаток Glu11. Это позволило предположить, что ион Ca2+ участвует в формировании мостика между боковой цепью остатка Glu и отрицательно заряженной пирофосфатной группой липида II [49]. Экспериментальные данные также указывают на участие остатка GlcNAc в межмолекулярном взаимодействии, поскольку мерсацидин может отличать липид I от липида II [39].
Совсем недавно была получена кристаллическая структура гидрохлорида укороченного N-ацетилированного аналога тейксобактина ([Ac-Δ1-5-Arg10]тейксобактин) [76], в которой ион Cl− связан в его особой полости, обогащенной амидными протонами (рис. 5б). Авторы предполагают, что обнаруженная полость тейксобактина и его аналогов может быть приспособлена для размещения и более крупных анионов, включая пирофосфатную группу липида II, однако эту гипотезу еще предстоит доказать. В полученной структуре анион хлора координирован водородными связями с гуанидиновой группировкой Arg10 и четырьмя амидными NH-группами, две из которых входят в состав циклического депсипептидного кольца. Такой способ взаимодействия аналогичен связыванию низина с пирофосфатной группой липида II (PDB ID: 1WCO).
Кроме того, было показано, что D-конфигурация четырех аминокислотных остатков тейксобактина (MePhe1, Gln4, allo-Ile5 и Thr8) имеет решающее значение для его биологической активности, поскольку замена их на L-аналоги в трех позициях из четырех (кроме D-Thr8) приводит к значительной потере антибактериальной активности (рис. 2) [77]. Любая модификация конфигурации остатков в кольцевой структуре тейксобактина также приводит к значительному ослаблению биологической активности. Однако интересно, что полный энантиомер аналога тейксобактина сохраняет исходную антибактериальную активность (за исключением активности в отношении Staphylococcus epidermis, где она уменьшается), что может означать, что важны не абсолютные конфигурации аминокислот, а порядок чередования оптических изомеров в молекуле [78]. Стехиометрический анализ показал, что тейксобактин, как и большинство других антибиотиков, связывается с липидом II в отношении 2 : 1 [62].
Таким образом, прямое определение структур комплексов различных агентов с липидом II в бактериальной мембране – сложновыполнимая задача. Известные в настоящее время пространственные модели имеют ряд недостатков, главный из которых – отсутствие учета естественного окружения молекул, т.е. мембраны в случае липида II и воды в случае низина. Дело в том, что в условиях стерических ограничений, накладываемых липидным бислоем, структуры и самой мишени, т.е. липида II, и его комплексов с различными антибиотиками могут существенно отличаться от экспериментальных. Тем не менее, структуры, полученные в экспериментах, демонстрируют наличие в молекулах различной природы схожих областей, обогащенных сонаправленными аминогруппами. Это свидетельствует о том, что так называемый “пирофосфатный фармакофор” должен обеспечивать жесткую пространственную ориентацию доноров водородных связей, что реализуется в природных соединениях за счет кольцевых фрагментов.
МЕХАНИЗМ РАСПОЗНАВАНИЯ ЛИПИДА II РАЗЛИЧНЫМИ АНТИБИОТИКАМИ: ДАННЫЕ КОМПЬЮТЕРНОГО МОДЕЛИРОВАНИЯ
На данный момент было проведено несколько исследований комплекса низина c липидом II в мембране in silico. Д. Коч (D. Koch) и соавт. представили результаты МД-моделирования изолированного липида II, встроенного в мембрану, состоящую из POPC, а также в составе комплекса с низином 1 : 1, где в качестве стартовой структуры использовали ЯМР-модель 1WCO из Брукхэйвенского банка данных [72]. Установлено, что связывание низина влияет на поведение липида II в бислое, уменьшая его конформационную свободу. Липид II изменяет относительное расположение своих составных частей в бислое: пентапептид сильнее экспонируется в растворитель, тогда как дисахарид, напротив, погружается внутрь мембраны. Однако конформации обеих молекул в целом остаются аналогичными экспериментальной структуре. Как было показано ранее, липид II и мембрана оказывают взаимное влияние друг на друга [64], поэтому выбранная авторами работы модельная мембрана эукариотических клеток позволяет оценить стерические ограничения, накладываемые бислоем, но не роль конкретного липидного окружения в рассматриваемом межмолекулярном взаимодействии.
Приближаясь к реальным условиям, С. Малхолланд (S. Mulholland) и др. рассмотрели этот комплекс в “нативной” стехиометрии низин : липид II = 2 : 1 на поверхности бислоя состава POPG : POPE в соотношении 3 : 1, имитирующего мембраны грамположительных бактерий [70, 79]. Для построения тройного комплекса на поверхности мембраны был проведен молекулярный докинг укороченного аналога низина (остатки 1–12) с тремя различными моделями липида II. Конформации первых двух колец А и В первой молекулы низина получились схожими с экспериментальной структурой, однако была обнаружена дополнительная водородная связь между N-концом низина и остатком D-Glu липида II. В С-концевой области пентапептида липида II был выявлен сайт связывания второй молекулы низина. Также было показано, что для липида II в составе тройного комплекса появляется новое предпочтительное состояние хвоста молекулы – так называемая “скрученная” конформация [79].
Кроме того, в работе [80] было продемонстрировано действие низина как “обычного” линейного антимикробного пептида в мембранах бактериальных клеток, не содержащих липида II. Показано, что в высоких концентрациях низин может деформировать мембраны даже в отсутствие липида II, взаимодействуя с фосфолипидами. Степень деформации мембраны зависит от ее толщины и степени олигомеризации низина.
Возможный механизм связывания молекулы липида II дефензином плектазин был предложен в результате моделирования их комплекса с помощью молекулярного докинга [43]. В этой модели амидные протоны остатков Phe2, Gly3, Cys4 и Cys37 образуют водородные связи с пирофосфатной группой, а D-Glu липида II формирует солевой мостик с N-концом и боковой цепью His18 плектазина. Стабильность и конформационную динамику этого комплекса изучали в модельной мембране бактерий S. aureus [69]. Исходный комплекс оказался неустойчивым в мембранной среде, что может быть следствием отсутствия учета влияния липидов мембраны при докинге. Конформационная лабильность липида II, а также частичная заглубленность в мембрану его групп, участвующих во взаимодействии, привели к сильному изменению стартовой структуры комплекса. В результате было получено несколько моделей слабосвязанных комплексов липида II с плектазином [69].
Исследование in silico взаимодействия дефензина копсин и его мутанта с бактериальной мембраной как в присутствии, так и в отсутствие липида II, не выявило существенных различий (<10%) в общем числе контактов пептида с липидами мембраны [81]. Учитывая, что присутствие липида II стерически затрудняет доступ пептида к части поверхности бислоя, авторы предполагают, что копсин может более эффективно встраиваться в бактериальную мембрану при связывании с липидом II. Кроме того, как и в предыдущей работе, была обнаружена высокая вариабельность способов взаимодействия с липидом II. В обеих пептидных молекулах были выявлены множественные сайты связывания остатков сахаров и боковых цепей пентапептида липида II с предпочтительным взаимодействием с остатком мурамовой кислоты MurNAc. Как и в предыдущем случае, разнообразие наблюдаемых способов связывания может быть следствием выбора начальных условий моделирования – в данном случае авторы помещали пептид на расстоянии около 2 нм от поверхности POPG/POPE-мембраны в присутствии или без липида II, с различным углом наклона в каждом из четырех стартов МД. Поскольку время МД-расчета было сильно ограничено (300 нс), моделируемая система сложная (~50 000 атомов), а стартовые условия различны для каждого вычислительного эксперимента, в данном случае возникает вопрос о воспроизводимости и сходимости результатов в независимых стартах МД, а также о влиянии стартовой конфигурации на полученные решения.
Изучение комплекса липида II с тейксобактином методом МД в водном растворе также показало возможность образования нескольких мотивов связывания [82]. Четыре предложенных модели комплекса принципиально различаются по числу образуемых межмолекулярных связей (3–5), а также группами обеих молекул, вовлеченными во взаимодействие. В трех комплексах из четырех тейксобактин связывается с разными остатками пентапептида липида II. И только в одном случае пирофосфатная группа и связанный с ней остаток MurNAc участвуют во взаимодействии с молекулой антибиотика, что соответствует экспериментальным данным [62].
Относительно более высокая эффективность взаимодействия была продемонстрирована для моделей, где в связывании участвуют фрагменты кольца тейксобактина. Это указывает на важность кольцевого мотива в распознавании липида II. В данной работе не учитывается мембранная среда, что авторы статьи рассматривают как разумное упрощение. В качестве аргумента приводится ссылка на более ранние эксперименты in vitro, в которых была показана способность изолированного липида II образовывать комплекс с тейксобактином [62]. Это, по мнению авторов, указывает на то, что мембрана не участвует в образовании комплекса. Однако естественное окружение может играть важную опосредованную роль в процессе межмолекулярного взаимодействия, формируя и/или стабилизируя подходящие конформации обоих партнеров, ограничивая таким образом разнообразие их возможных мод связывания.
Оценка эффективности взаимодействия нескольких антибиотиков с их мишенью – липидом II – с помощью метода молекулярного докинга и последующего статистического анализа конформаций комплексов показала уменьшение эффективности связывания в ряду тейксобактин > оритаванцин > далбаванцин > телаванцин > > тейкопланин > ванкомицин [83]. Эти результаты подтверждаются экспериментальными данными, приведенными в литературе.
Результаты проводимых авторами в настоящее время компьютерных экспериментов проливают свет на возможный молекулярный механизм распознавания лантибиотиками липида II в области его пирофосфатного фрагмента. Этот механизм основан на индуцированном соответствии ряда доноров водородных связей в системе связанных колец A/B низина и распознаваемого ими фрагмента липида II – его пирофосфатной группы [84]. Подробнее эти результаты будут изложены в отдельной публикации.
ЗАКЛЮЧЕНИЕ
Компьютерное атомистическое моделирование в настоящее время является одним из мощных инструментов структурных исследований мембранных процессов, в том числе начальных этапов распознавания пептидными антибиотиками их мишеней в липидных бислоях клеточных мембран. Эксперименты in silico показали, что целевая для многих антибиотиков пирофосфатная группа липида II расположена на границе раздела двух фаз (мембрана–вода), а, значит, частично недоступна для внеклеточных агентов. Это накладывает ограничения на применимость молекулярных моделей комплексов липида II с различными антибактериальными веществами, структуры которых были получены без учета мембранного окружения. Иными словами, при изучении процессов первичного молекулярного распознавания в таких сложных системах эффекты среды обязательно должны быть приняты во внимание. Как было сказано выше, в данном случае важнейшая роль принадлежит эффектам взаимной тонкой подстройки взаимодействующих партнеров, структура и динамика которых могут критическим образом зависеть от их окружения.
В случае надлежащей параметризации эмпирических силовых полей, используемых в таких расчетах, а также при выборе адекватных моделей исследуемых систем и вычислительных протоколов, компьютерное моделирование позволяет не только воспроизвести “в среднем” наблюдаемые в экспериментах детали связывания пептид/липид II, но и получить детальную динамическую картину подобных взаимодействий. В частности, можно оценить роль эффектов индуцированного соответствия молекул-партнеров, особенности их структуры и динамики в гетерогенном водно-липидном окружении. Последнее, к слову, может играть ключевую роль, как это было недавно показано для белок-белковых взаимодействий в мембранах [85, 86].
Ввиду ограниченности вычислительных ресурсов, моделируемые методом МД биологические системы приходится сильно упрощать, а расчетное время моделирования эволюции системы в среднем не превышает микросекунд. Тем не менее, с помощью метода МД, позволяющего наблюдать за процессами на атомарном уровне, можно получить ответы на следующие вопросы: каким образом происходит распознавание мишени антибиотиками в столь химически схожем окружении, которое формируют соседние липиды бислоя? Какова роль мембраны в процессе межмолекулярного распознавания и взаимодействия. Существует ли консервативный среди разных классов антибиотиков так называемый “пирофосфатный фармакофор”, являющийся ключевой детерминантой связывания антимикробных пептидов. Ответы на эти вопросы, как ожидается, станут отправной точкой для рационального дизайна новых антибиотиков, лишенных (по крайней мере, частично) фармакокинетических недостатков их природных прототипов, более селективных, активных и удобных в производстве и, главное, с отсроченным во времени развитием резистентности у патогенных микроорганизмов.
БЛАГОДАРНОСТИ
Работа выполнена в рамках Программы Президиума РАН “Молекулярная и клеточная биология”, Российского Фонда фундаментальных исследований (грант 16-04-00578), Программы фундаментальных исследований Национального исследовательского университета “Высшая школа экономики” (НИУ ВШЭ) и с использованием средств субсидии в рамках государственной поддержки ведущих университетов Российской Федерации “5-100”. Авторы благодарны СКЦ “Политехнический” СПбПУ и МСЦ РАН за предоставленные суперкомпьютерные ресурсы; а также профессору Д.С. Новику (J.S. Nowick) (Калифорнийский университет, г. Ирвайн, США) за любезно предоставленные координаты модели пространственной структуры комплекса гидрохлорида аналога тейксобактина.
Список литературы
Ventola C.L. // P T. 2015. V. 40. P. 277–283.
Villa T.G., Viñas M. New Weapons to Control Bacterial Growth. Springer Int. Pub., 2016. P. 1–13.
Arias C.A., Murray B.E. // N. Engl. J. Med. 2015. V. 372. P. 1168–1170.
Schneider T., Sahl H.-G. // Int. J. Med. Microbiol. 2010. V. 300. P. 161–169.
Scheffers D.-J., Tol M.B. // PLoS Pathog. 2015. V. 11:e1005213
Ng V., Chan W.C. // Chemistry. 2016. V. 22. P. 12606–12616.
Kramer N.E., Smid E.J., Kok J., de Kruijff B., Kuipers O.P., Breukink E. // FEMS Microbiol. Lett. 2004. V. 239. P. 157–161.
Brown E.D., Vivas E.I., Walsh C.T., Kolter R. // J. Bacteriol. 1995. V. 177. P. 4194–4197.
Mengin-Lecreulx D., Flouret B., van Heijenoort J. // J. Bacteriol. 1982. V. 151. P. 1109–1117.
Bouhss A., Trunkfield A.E., Bugg T.D.H., Mengin-Lecreulx D. // FEMS Microbiol. Rev. 2008. V. 32. P. 208–233.
Mengin-Lecreulx D., Texier L., Rousseau M., van Heijenoort J. // J. Bacteriol. 1991. V. 173. P. 4625–4636.
Typas A., Banzhaf M., Gross C.A., Vollmer W. // Nat. Rev. Microbiol. 2011. V. 10. P. 123–136.
Tatar L.D., Marolda C.L., Polischuk A.N., van Leeuwen D., Valvano M.A. // Microbiology. 2007. V. 153. P. 2518–2529.
Esko J.D., Doering T.L., Raetz C.R.H. Essentials of Glycobiology. New York: Cold Spring Harbor Laboratory Press, 2009. Chapter 20.
van Heijenoort J. // Microbiol. Mol. Biol. Rev. 2007. V. 71. P. 620–635.
Fuchs-Cleveland E., Gilvarg C. // Proc. Natl. Acad. Sci. U.S.A. 1976. V. 73. P. 4200–4204.
Labischinski H., Goodell E.W., Goodell A., Hoch-berg M.L.D. // J. Bacteriol. 1991. V. 173. P. 751–756.
van Heijenoort Y., Gómez M., Derrien M., Ayala J., van Heijenoort J. // J. Bacteriol. 1992. V. 174. P. 3549–3557.
Vollmer J., Schiefer A., Schneider T., Julicher K., Johnston K.L., Taylor M.J. Sahl H.G., Hoerauf A., Pfarr K.M. // Int. J. Med. Microbiol. 2013. V. 303. P. 140–149.
Schneider T., Sahl H.-G. // Curr. Opin. Investig. Drugs. 20105. V. 11. P. 157–164.
Zasloff M. // Nature. 2002. V. 415. P. 389–395.
Polyansky A.A., Chugunov A.O., Vassilevski A.A., Grishin E.V., Efremov R.G. // Curr. Protein Pept. Sci. 2012. V. 13. P. 644–657.
Andersson D.I., Hughes D., Kubicek-Sutherland J.Z. // Drug Resist. Updat. 2016. V. 26. P. 43–57.
Lee T.-H., Hall K.N., Aguilar M.-I. // Curr. Top. Med. Chem. 2016. V. 16. P. 25–39.
Matsuzaki K. // Biochim. Biophys. Acta. 1999. V. 1462. P. 1–10.
Yang L., Weiss T.M., Lehrer R.I., Huang H.W. // Biophys. J. 2000. V. 79. P. 2002–2009.
Shai Y. // Biochim. Biophys. Acta. 1999. V. 1462. P. 55–70.
Le C.-F., Fang C.-M., Sekaran S.D. // Antimicrob. Agents Chemother. 2017. V. 61:e02340-16.
Arnusch C.J., Pieters R.J., Breukink E. // PLoS One. 2012. V. 7:e39768.
Barna J.C., Williams D.H. // Annu. Rev. Microbiol. 1984. V. 38. P. 339–357.
Reynolds P.E. // Eur. J. Clin. Microbiol. Infect. Dis. 1989. V. 8. P. 943–950.
Wright G. // Nature. 2015. V. 517. P. 442–444.
Marshall C.G., Lessard I.A., Park I., Wright G.D. // Antimicrob. Agents Chemother. 1998. V. 42. P. 2215–2220.
Levine D.P. // Clin. Infect. Dis. 2006. V. 42. Suppl. 1. S5–12.
Reynolds P.E., Courvalin P. // Antimicrob. Agents Chemother. 2005. V. 49. P. 21–25.
Faron M.L., Ledeboer N.A., Buchan B.W. // J. Clin. Microbiol. 2016. V. 54. P. 2436–2447.
Reynolds P.E., Depardieu F., Dutka-Malen S., Arthur M., Courvalin P. // Mol. Microbiol. 1994. V. 13. P. 1065–1070.
Grohs P., Gutmann L., Legrand R., Schoot B., Mainardi J.L. // J. Bacteriol. 2000. V. 182. P. 6228–6232.
Hsu S.-T., Breukink E., Bierbaum G., Sahl H.G., De Kruijff B., Kaptein R., Van Nuland N., Bonvin A. // J. Biol. Chem. 2003. V. 278. P. 13110–13117.
Münch D., Sahl H.-G. // Biochim. Biophys. Acta. 2015. V. 1848. P. 3062–3071.
Antcheva N., Guida F., Tossi A. Handbook of Biologically Active Peptides. Elsevier, 2013. P. 101–118.
Mygind P.H., Fischer R.L., Schnorr K.M., Hansen M.T., Sonksen C.P., Ludvigsen S., Raventos D., Buskov S., Christensen B., De Maria L., Taboureau O., Yaver D., Elvig-Jorgensen S.G., Sorensen M.V., Christensen B.E., Kjaerulff S., Frimodt-Moller N., Lehrer R.I., Zasloff M., Kristensen H.H. // Nature. 2005. V. 437. P. 975–980.
Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A.K., Mygind P.H., Raventós D.S., Neve S., Ravn B., Bonvin A.M., De Maria L., Andersen A.S., Gammelgaard L.K., Sahl H.G., Kristensen H.H. // Science. 2010. V. 328. P. 1168–1172.
Sass V., Schneider T., Wilmes M., Körner C., Tossi A., Novikova N., Shamova O., Sahl H.G. // Infect. Immun. 2010. V. 78. P. 2793–2800.
Schmitt P., Wilmes M., Pugnière M., Aumelas A., Bachère E., Sahl H.G., Schneider T., Destoumieux-Garzón D. // J. Biol. Chem. 2010. V. 285. P. 29208–29216.
Willey J.M., van der Donk W.A. // Annu. Rev. Microbiol. 2007. V. 61. P. 477–501.
Al-Mahrous M.M., Upton M. // Expert Opin. Drug Discov. 2011. V. 6. P. 155–170.
Islam M.R., Nagao J.-I., Zendo T., Sonomoto K. // Biochem. Soc. Trans. 2012. V. 40. P. 1528–1533.
Müller A., Klöckner A., Schneider T. // Nat. Prod. Rep. 2017. V. 34. P. 909–932.
Breukink E., van Kraaij C., Demel R.A., Siezen R.J., Kuipers O.P., de Kruijff B. // Biochemistry. 1997. V. 36. P. 6968–6976.
van Kraaij C., Breukink E., Noordermeer M.A., Demel R.A., Siezen R.J., Kuipers O.P., de Kruijff B. // Biochemistry. 1998. V. 37. P. 16033–16040.
Böttiger T., Schneider T., Martínez B., Sahl H.-G., Wiedemann I. // Appl. Environ. Microbiol. 2009. V. 75. P. 4427–4434.
Bionda N., Pitteloud J.-P., Cudic P. // Future Med. Chem. 2013. V. 5. P. 1311–1330.
Bassères E., Endres B.T., Dotson K.M., Alam M.J., Garey K.W. // Curr. Opin. Gastroenterol. 2017. V. 33. P. 1–7.
Han J., Chen J., Shao L., Zhang J., Dong X., Liu P., Chen D. // PLoS One. 2016. V. 11. P. e0154121.
Schneider T., Müller A., Miess H., Gross H. // Int. J. Med. Microbiol. 2014. V. 304. P. 37–43.
Reynolds P.E., Somner E.A. // Drugs Exp. Clin. Res. 1990. V. 16. P. 385–389.
Fang X., Tiyanont K., Zhang Y., Wanner J., Boger D., Walker S. // Mol. Biosyst. 2006. V. 2. P. 69–76.
Lo M.-C., Men H., Branstrom A., Helm J., Yao N., Goldman R., Walke S. // J. Am. Chem. Soc. 2000. V. 122. P. 3540–3541.
Cheng M., Huang J.X., Ramu S., Butler M.S., Cooper M.A. // Antimicrob. Agents Chemother. V. 58. P. 6819–6827.
Hu Y., Helm J.S., Chen L., Ye X.-Y., Walker S. // J. Am. Chem. Soc. 2003. V. 125. P. 8736–8737.
Ling L.L., Schneider T., Peoples A.J., Spoering A.L., Engels I., Conlon B.P., Mueller A., Schäberle T.F., Hughes D.E., Epstein S., Jones M., Lazarides L., Steadman V.A., Cohen D.R., Felix C.R., Fetterman K.A., Millett W.P., Nitti A.G., Zullo A.M., Chen C., Lewis K. // Nature. 2015. V. 517. P. 455–459.
Piggot T.J., Holdbrook D.A., Khalid S. // J. Phys. Chem. B. 2011. V. 115. P. 13381–13388.
Chugunov A., Pyrkova D., Nolde D., Polyansky A., Pentkovsky V., Efremov R. // Sci. Rep. 2013. V. 3. P. 1678.
Панина И.С., Нольде Д.Е., Чугунов А.О., Ефремов Р.Г. // Акт. вопр. биол. физ. и хим. 2016. Т. 1. С. 263–267.
de Kruijff B., van Dam V., Breukink E. // Prostaglandins Leukot. Essent. Fatty Acids. 2008. V. 79. P. 117–121.
Hasper H.E., Kramer N.E., Smith J.L., Hillman J.D., Zachariah C., Kuipers O.P., de Kruijff B., Breukink E. // Science. 2016. V. 313. P. 1636–1637.
Hasper H.E., de Kruijff B., Breukink E. // Biochemistry. 2004. V. 43. P. 11567–11575.
Witzke S., Petersen M., Carpenter T.S., Khalid S. // Biochemistry. 2016. V. 55. P. 3303–3314.
Prossnigg F., Hickel A., Pabst G., Lohner K. // Biophys. Chem. 2010. V. 150. P. 129–135.
Sosa Morales M.C., Álvarez R.M.S. // J. Raman Spectrosc. 2017. V. 48. P. 170–179.
Hsu S.-T.D., Breukink E., Tischenko E., Lutters M.A., de Kruijff B., Kaptein R., Bonvin A.M., van Nuland N.A. // Nat. Struct. Mol. Biol. 2004. V. 11. P. 963–967.
Oppedijk S.F., Martin N.I., Breukink E. // Biochim. Biophys. Acta. 2016. V. 1858. P. 947–957.
Szekat C., Jack R.W., Skutlarek D., Farber H., Bierbaum G. // Appl. Environ. Microbiol. 2003. V. 69. P. 3777–3783.
Brötz H., Bierbaum G., Leopold K., Reynolds P.E., Sahl H.G. // Antimicrob. Agents Chemother. 1998. V. 42. P. 154–160.
Yang H., Du Bois D.R., Ziller J.W., Nowick J.S. // Chem. Commun. 2017. V. 53. P. 2772–2775.
Parmar A., Iyer A., Vincent C.S., Van Lysebetten D., Prior S.H., Madder A., Taylor E.J., Singh I. // Chem. Commun. 2016. V. 52. P. 6060–6063.
Yang H., Chen K.H., Nowick J.S. // ACS Chem. Biol. 2016. V. 11. P. 1823–1826.
Mulholland S., Turpin E.R., Bonev B.B., Hirst J.D. // Sci. Rep. 2016. V. 6. P. 21185.
Prince A., Sandhu P., Kumar P., Dash E., Sharma S., Arakha M., Jha S., Akhter Y., Saleem M. // Sci. Rep. 2016. V. 6. P. 37908.
Franzoi M., van Heuvel Y., Thomann S., Schürch N., Kallio P.T., Venier P., Essig A. // Biochemistry. 2017. V. 56. P.4992–5001.
Liu Y., Liu Y., Chan-Park M.B., Mu Y. // Sci. Rep. 2017. V. 7. P. 17197.
Lungu C.N., Diudea M.V. // Comput. Aided Drug Des. 2018. V. 14. P. 29–34.
Панина И.С., Нольде Д.Е., Крылов Н.А., Ефремов Р.Г. // Акт. вопр. биол. физ. и хим. 2017. Т. 2. С. 528–532.
Bocharov E.V., Lesovoy D.M., Bocharova O.V., Urban A.S., Pavlov K.V., Volynsky P.E., Efremov R.G., Arseniev A.S. // Biochim. Biophys. Acta. 2018. V. 1862. P. 1410–1420.
Kuznetsov A.S., Polyansky A.A., Fleck M., Volynsky P.E., Efremov R.G. // J. Chem. Theory Comput. 2015. V. 11. P. 4415–4426.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия