

Биоорганическая химия, 2019, T. 45, № 2, стр. 218-221
Оптимизация синтеза 3-гидроксигиспидина – люциферина биолюминесцентной системы высших грибов
А. И. Бубырев 1, А. С. Царькова 1, 2, З. М. Каськова 1, 2
1 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 Российский национальный исследовательский медицинский университет имени Н.И. Пирогова
117997 Москва, ул. Островитянова, д. 1, Россия
Поступила в редакцию 30.10.2018
После доработки 31.10.2018
Принята к публикации 31.10.2018
Аннотация
Активное изучение биолюминесцентной системы высших грибов, в том числе поиск и очистка люциферазы, требует использование больших количеств синтетического субстрата – люциферина. Существующая на данный момент методика его синтеза не является оптимальной и проводится с невысоким выходом целевого продукта. В данной работе синтез люциферина грибов масштабирован до граммовых количеств, найден эффективный способ его очистки на финальной стадии. Общий выход в сравнении с предыдущей методикой увеличен в 3.5 раза, стоимость синтеза упала в 3 раза.
ВВЕДЕНИЕ
Свойство живых организмов испускать свет, или биолюминесценция, успешно используется для разработки аналитических методов и визуализации биологических процессов [1]. Одной из недавно открытых является биолюминесцентная система высших грибов [2] – удачный кандидат для создания новых методов биоимиджинга [3]. В настоящей работе проведена масштабная оптимизация синтеза ключевого биолюминесцентного субстрата грибов – молекулы люциферина.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Люциферин (I), или 3-гидроксигиспидин, можно отнести к классу стирилпиронов, его биосинтетическим предшественником является распространенный вторичный метаболит грибов и растений – гиспидин (II) [2]. Логичным подходом к синтезу люциферина было бы введение гидроксильной группы в третье положение молекулы гиспидина или его защищенного производного, однако данные попытки успехом не увенчались, и синтез люциферина был реализован через конденсацию 3,4-диметоксибензальдегида с пираноном (IV), уже содержащим две гидроксильные группы [4].
Активное изучение биолюминесцентной системы высших грибов в нашей лаборатории и проведение исследований с люциферазой потребовали получения большого количества люциферина, вследствие чего появилась необходимость проведения значительной оптимизации существующей методики синтеза. Первые стадии получения производного (IV) из коммерчески доступной дегидроацетовой кислоты (III) остались без существенных изменений. Однако ключевая стадия конденсации пиранона (IV) с 3,4-диметоксибензальдегидом (V) была значительно переработана. Ранее реакцию проводили в условиях катализа метилатом магния [4]. Стадия проходила с низким выходом продукта (VII), а ее масштабирование в разы увеличивало время реакции. Оптимальное решение было найдено путем введения двухступенчатого превращения: альдольная конденсация с диизопропиламидом лития (LDA) и дегидратация получившегося гидроксипроизводного (VI) с образованием тетраметилпроизводного люциферина (VII). Выход на две стадии составил 72%, против 32% в синтезе [4].
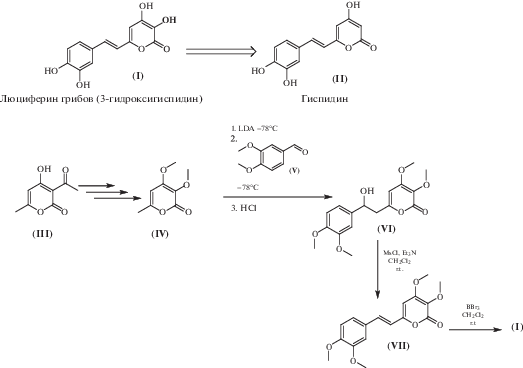
Схема 1. Усовершенствованная методика синтеза люциферина грибов.
Для финальной стадии превращения метоксильных групп в гидроксильные использовался большой избыток трибромида бора (35 экв.). После водной обработки реакционной массы и экстракции целевой продукт (I) очищали методом обращенно-фазовой ВЭЖХ. Такая схема выделения и очистки приводила к значительным потерям люциферина (I). Увеличение концентрации исходного тетраметоксипродукта (VII) с 0.15 до 0.26 М и времени реакции с 16 до 60 ч привело к снижению необходимого количества трибромида бора с 35 до 16 экв. Возможность отгонки такого избытка реагента позволила избежать водной обработки реакционной массы и потери высоко полярного продукта (I) в этой операции. Окончательно, аналитически чистый люциферин (I) с суммарным выходом 16% на 7 стадий, без потери активности был выделен осаждением из диметилформамида при добавлении воды.
Таким образом, методика синтеза люциферина грибов была существенно оптимизирована: проведено масштабирование до граммовых количеств, выход продукта увеличен в 3.5 раза, выход в ключевой стадии повышен с 32 до 72%, найден способ эффективной очистки конечного продукта без привлечения ВЭЖХ. Стоимость синтеза по сравнению с описанной ранее методикой упала в 3 раза.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры ЯМР (δ, м.д.; J, Гц) регистрировали на приборе Bruker Fourier 300 в CDCl3, ацетоне-d6, внутренний стандарт – тетраметилсилан. Масс-спектры высокого разрешения (HRMS) регистрировали на приборе Agilent 6224 TOF LC/MS System методом электрораспылительной ионизации (ESI).
Соединение (IV) получено из (III) согласно ранее описанной методике [4]. Нумерация атомов в описанных соединениях выполнена по правилам номенклатуры ИЮПАК и приводится в соответствии с [2].
6-(2-(3,4-Диметоксифенил)-2-гидроксиэтил)-3,4-диметокси-2H-пиранон (VI). К 12.5 мл раствора диизопропиламина (88.2 ммоль) в безводном THF (100 мл) при –78°C при перемешивании в атмосфере аргона прикапывали 33 мл 2.5 М раствора н-бутиллития в н-гексане (82.3 ммоль). Реакционную смесь перемешивали 45 мин. Затем в течении 45 мин прикапывали раствор пиранона (IV) (10 г, 58.8 ммоль) в безводном THF (70 мл) и оставляли на 2 ч, до выпадения серого осадка литиевого енолята. После этого добавляли по каплям 70 мл раствора 3,4-диметоксибензальдегида (V) (9.76 г, 58.8 ммоль) в безводном THF и перемешивали 30 мин при –78°C, затем приблизительно 1.5 ч при –40°C. По окончании реакции (контроль по ТСХ, элюент – диэтиловый эфир) добавляли 100 мл 3 М HCl и экстрагировали этилацетатом (3 × 75 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (2 × 100 мл), водой (2 × 100 мл), 100 мл насыщенного раствора NaCl и высушивали над Na2SO4. Реакционную смесь концентрировали в вакууме. Продукт (VI) получали в виде желтоватого масла (19.2 г), которое вводили в следующую стадию без дополнительной очистки.
(E)-6-(3,4-Диметоксистирил)-3,4-диметокси-2H-пиран-2-он (VII). К раствору соединения (VI) (19.2 г) в 150 мл сухого дихлорметана при перемешивании добавляли 20.9 мл триэтиламина (150 ммоль), затем раствор охлаждали на бане со льдом и по каплям добавляли 6.6 мл мезилхлорида (85 ммоль). Реакционную смесь перемешивали при комнатной температуре 6 ч. По окончании реакции (контроль по ТСХ, элюент – диэтиловый эфир) к раствору добавляли 2 М HCl (150 мл) и перемешивали 20 мин, водную фракцию промывали дихлорметаном (3 × 75 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (2 × 100 мл), водой (2 × 100 мл) и насыщенным раствором NaCl (1 × 100 мл), высушивали над Na2SO4, концентрировали. Из сухого остатка методом флэш-хроматографии (Rf 0.5, диэтиловый эфир) выделяли продукт (VII) в виде желтого порошка (13.5 г, 72% на две стадии). 1H-ЯМР (300 МГц, CDCl3) 7.37 (д, J 15, 1H, H8), 7.06 (дд, J 8.3, 2.0, 1H, H14), 7.00 (д, J 2.0, 1H, H10), 6.86 (д, J 8.3, 1H, H13), 6.46 (д, J 15.9, 1H, H7), 6.08 (с, 1H, H5), 3.98 (с, 3H, OCH3), 3.92 (с, 3H, OCH3), 3.90 (с, 3H, OCH3), 3.86 (с, 3H, OCH3). 13C-ЯМР (75 МГц, CDCl3) 161.2 (С2), 159.3 (С4), 155.4 (С6), 150.5 (С12), 149.3 (С11), 135.2 (С8), 128.4 (С3), 127.8 (С9), 121.6 (С14), 116.4 (С7), 111.3 (С13), 109.2 (С10), 97.0 (С5), 60.4 (OCH3), 57.3 (OCH3), 56.1 (OCH3), 55.9 (OCH3). HRMS: найдено, m/z 319.1187 [M + H]+. C17H19O$_{6}^{ + }$. Вычислено: M + H =319.1176.
(E)-6-(3,4-Дигидроксистирил)-3,4-дигидрокси-2H-пиран-2-он (I). К раствору производного (VII) (5.2 г, 16.4 ммоль) в сухом дихлорметане (60 мл) при –30°C в атмосфере аргона при перемешивании быстро прикапывали трибромид бора (24.7 мл, 260 ммоль, 65.1 г), затем смесь оставляли при перемешивании на 60 ч при комнатной температуре. Дихлорметан и избыток трибромида бора отгоняли в вакууме. Остаток растворяли в CH2Cl2 (40 мл), при охлаждении добавляли MeOH (20 мл), реакционную смесь концентрировали в вакууме, красно-коричневый остаток растворяли в DMF (6 мл), добавляли ледяную воду (150 мл). Образовавшийся осадок отфильтровывали, промывали холодной водой до нейтральной реакции, сушили в вакууме и промывали смесью диэтиловый эфир–гексан, 1 : 1 (3 × 35 мл), диэтиловым эфиром (2 × 15 мл), повторно сушили в вакууме. Продукт (I) получали в виде желтого порошка (3.86 г, 90%). 1H-ЯМР (300 МГц, ацетон-d6) 9.23 (с, 1H, OH), 8.28 (с, 1H, OH), 8.07 (с, 1H, OH), 7.58 (с, 1H, OH), 7.12 (д, J 2.0, 1H, H10), 7.10 (д, J 16.0, 1H, H8), 6.99 (дд, J 8.2, 2.0, 1H, H14), 6.86 (д, J 8.2, 1H, H13), 6.64 (д, J 16.0, 1H, H7), 6.21 (с, 1H, H5). 13C-ЯМР (75 МГц, ацетон-d6) 160.8 (C2), 150.8 (C4, С6), 146.7 (C12), 145.6 (C11), 131.4 (C8), 127.3 (C9), 123.5 (C3), 119.7 (C14), 116.3 (C7), 115.8 (C13), 113.7 (C10), 102.2 (C5). HRMS: найдено, m/z 263.0553 [M + H]+. C13H11O$_{6}^{ + }$. Вычислено: M + H =263.0550.
Список литературы
Yao Z., Zhang B.S., Prescher J.A. // Curr. Opin. Chem. Biol. 2018. V. 45. P. 148–156.
Purtov K.V., Petushkov V.N., Baranov M.S., Mineev K.S., Rodionova N.S., Kaskova Z.M., Tsarkova A.S., Petunin A.I., Bondar V.S., Rodicheva E.K., Medvedeva S.E., Oba Y., Oba Yu., Arseniev A.S., Lukyanov S., Gitelson J.I., Yampolsky I.V. // Angew. Chem. 2015. V. 54. P. 8124–8128.
Осипова З.М., Щеглов А.С., Ямпольский И.В. // Вестник РГМУ. 2018. Т. 1. С. 80–83.
Kaskova Z.M., Dörr F.A., Petushkov V.N., Purtov K.V., Tsarkova A.S., Rodionova N.S., Mineev K.S., Guglya E.B., Kotlobay A., Baleeva N.S., Baranov M.S., Arseniev A.S., Gitelson J.I., Lukyanov S., Suzuki Y., Kanie S., Pinto E., Mascio P.D., Waldenmaier H.E., Pereira T.A., Carvalho R.P., Oliveira A.G., Oba Y., Bastos E.L., Stevani C.V., Yampolsky I.V. // Sci. Adv. 2017. V. 3. P. e1602847.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия