

Биоорганическая химия, 2019, T. 45, № 3, стр. 303-314
Синтез фрагментов ДНК, содержащих β-дикетогруппу, для аффинной модификации белков
М. В. Монахова 1, Е. А. Кубарева 1, Е. А. Романова 1, А. С. Семкина 2, Д. С. Набережнов 3, Д. Н. Рао 4, Т. С. Зацепин 5, Т. С. Орецкая 1, *
1 Московский государственный университет имени М.В. Ломоносова, химический факультет и НИИ физико-химической биологии имени А.Н. Белозерского
119991 г. Москва, Ленинские горы, 1, Россия
2 Российский национальный исследовательский медицинский университет имени Н.И. Пирогова
117997 г. Москва, ул. Островитянова, 1, Россия
3 Национальный медицинский исследовательский центр онкологии имени Н.Н. Блохина
115478 г. Москва, Каширское шоссе, 24, Россия
4 Department of Biochemistry, Indian Institute of Science
560012 Bangalore, C.V. Raman Road, India
5 Сколковский институт науки и технологий
121205 г. Москва, ул. Нобеля, 3, Россия
* E-mail: oretskaya@belozersky.msu.ru
Поступила в редакцию 10.07.2018
После доработки 27.07.2018
Принята к публикации 01.08.2018
Аннотация
Для модификации гуанидиновой группы остатков аргинина в белках, взаимодействующих с ДНК, предложено использовать дикарбонильные производные олигонуклеотидов. В ДНК-фрагменты с включением 2'-амино-2'-дезоксиуридина, полученные с помощью химического автоматического синтеза, вводили β-дикетогруппу путем ацилирования 2'-аминогруппы 4,6-диоксогептановой кислотой в присутствии водорастворимого N-[3-(диметиламино)пропил]-N′-этилкарбодиимида (EDC). Продемонстрирована способность олигодезоксирибонуклеотидов, содержащих β-дикетогруппу, вступать в реакции с гуанидином, Nα-Boc-L-аргинином и Nα-Dns-L-аргинином. Введение такой модификации в одну из цепей 15-звенного ДНК-дуплекса приводит к его дестабилизации. Впервые показано образование конъюгатов белков MutS и MutL из системы репарации неканонических пар нуклеотидов E. coli с 17-звенными ДНК-дуплексами, содержащими остаток 2'-дезокси-2'-(4,6-диоксогептиламидо)уридина. Для повышения селективности модификации остатков аргинина в белках ДНК-лигандами с β-дикетогруппой реакционную смесь обрабатывали раствором гидроксиламина, что приводило к разрушению оснований Шиффа, образованных с участием остатков лизина.
ВВЕДЕНИЕ
Сиквенс-специфические ДНК-белковые контакты являются определяющими во многих биологических процессах, таких как транскрипция и ее регуляция, репарация, метилирование и гидролиз ДНК и другие. Для исследования белково-нуклеиновых комплексов сегодня применяют ряд методов, которые можно разделить на инструментальные, молекулярно-биологические и химические. Инструментальные методы, например, криоэлектронная микроскопия, рентгеноструктурный анализ и метод ядерно-магнитного резонанса, применяются для установления структуры комплексов белков с нуклеиновыми кислотами, а молекулярно-биологические и химические – в основном для характеристики отдельных стадий формирования таких комплексов в процессе функционирования.
Особое место среди химических методов занимает метод аффинной модификации белков реакционноспособными аналогами ДНК, который позволяет получить дополнительную информацию о структуре специфического комплекса и фиксировать короткоживущие макромолекулярные ассоциаты, образующиеся на разных стадиях ДНК-белкового узнавания. Существует множество способов ковалентного присоединения молекул ДНК к белку, наиболее распространенные из которых основаны на использовании ДНК-реагентов, нацеленных на специфическое взаимодействие с остатками цистеина и лизина [1, 2]. Расширение набора эффективных и специфических реакций между боковыми радикалами аминокислот в составе белка и химически модифицированными реакционноспособными звеньями нуклеиновых кислот позволит более системно изучать особенности белково-нуклеиновых взаимодействий, оценивать возможность специфического ингибирования этих белков и получать стабильные ковалентно связанные комплексы белок-ДНК.
Положительно заряженные остатки аргинина часто располагаются в активных центрах ДНК-связывающих белков [3]. Для модификации гуанидиновой группы аргинина используются α- и β-дикарбонильные соединения [4–7]. Так как α-дикетоны и диальдегиды взаимодействуют также с остатками гуанина [8], то этот тип реагентов не подходит для изучения НК-белковых комплексов. Реакция производных гуанидина с β-дикетонами проходит в щелочной среде (рН 9–11), приводит к образованию замещенных пиримидинов с высоким выходом и широко используется в синтетической практике [9, 10]. Однако как метод модификации белков эта реакция описана только для низкомолекулярных соединений, таких как 2,4-пентандион [4], пропандиаль [5], натриевая соль нитропропандиаля [11], 4,6-диоксогептановая кислота [12]. Известны примеры синтеза β-дикето- и β-диальдегидных производных нуклеиновых кислот [13–16], однако их взаимодействие с производными гуанидина не описано.
ДНК-реагент для аффинной модификации белков должен селективно взаимодействовать с остатками аминокислот белка в составе специфического комплекса. В данной работе в качестве таких реагентов предлагается использовать фрагменты ДНК, содержащие β-дикетогруппу при С2'-атоме углеводного фрагмента. Объектами исследования были выбраны белки системы репарации неканонических пар нуклеотидов (“мисматчей”) в ДНК (MMR) E. coli MutS и MutL.
Для белка MutS имеются данные РСА, демонстрирующие, что остатки аргинина непосредственно вовлечены во взаимодействие с ДНК [17], поэтому MutS является удобной моделью для валидации нового реагента для аффинной модификации. ДНК-связывающая область MutL из Е. coli детально не охарактеризована. Это обусловлено тем, что комплекс MutL с ДНК динамически подвижен, однако предполагается, что остатки Arg162, Arg266 и Arg316 АТФазного и α‑β-“сэндвичевого” субдоменов N-концевого домена участвуют во взаимодействии с ДНК [18]. Это предположение подтверждают недавние работы, выполненные для гомологичных белков MutL из дрожжей (Pms1) и бактерии Aquifex aeolicus, согласно которым остатки аргинина и лизина этих субдоменов действительно могут быть вовлечены во взаимодействие с ДНК [19, 20]. В ходе нашей работы предполагалось выяснить, возможно ли взаимодействие коротких 17-звенных ДНК-дуплексов с белком MutL из Е. coli, и ответить на вопрос, сближены ли остатки аргинина белка MutL с ДНК в отсутствие белка MutS.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез фрагментов ДНК, содержащих β-дикетогруппу
Впервые были синтезированы фрагменты ДНК с β-дикетогруппой при С2'-атоме углеводного фрагмента путем постсинтетической модификации олигонуклеотидов с включением 2'‑амино-2'-дезоксиуридина. 2'-Аминосодержащие олигонуклеотиды хорошо зарекомендовали себя в наших предшествующих исследованиях, продемонстрировав высокую реакционную способность в реакциях ацилирования [21, 22]. В работе [12] β-дикетопроизводное антибиотика цефалексина синтезировали ацилированием лактоном, полученным обработкой 4,6-диоксогептановой кислоты карбодиимидом. Первоначально для синтеза олигонуклеотидов с β-дикетогруппой мы использовали именно эту методику, однако ацилирование 2'-аминосодержащего олигонуклеотида лактоном в смеси DMSO-вода при pH 8.5 не позволяло достичь степени превращения выше 60%. Ацилирование 2'-аминосодержащего олигонуклеотида 4,6-диоксогептановой кислотой в смеси DMSO-вода в присутствии EDC при рН 9.0 (см. схему 1 ) дало возможность увеличить выход продукта реакции до 75–95%.
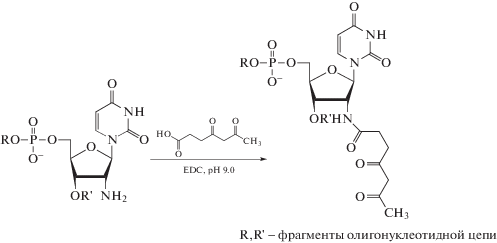
Схема 1 .
В качестве примера на рис. 1 приведена хроматограмма разделения реакционной смеси, содержащей олигонуклеотид 5'-TCGGAAAGUnCCCCTC (1), где Un – остаток 2'-амино-2'-дезоксиуридина, и продукт его модификации (2). Их строение подтверждено методом времяпролетной масс-спектрометрии. Так, рассчитанная масса олигонуклеотида 5'-TCGGAAAGUdioxCCCCTC (2), где Udiox – остаток 2'-дезокси-2'-(4,6-диоксогептиламидо)уридина (4655.0) совпадает с величиной, определенной масс-спектрометрически (4655.7). В контрольных экспериментах было показано отсутствие в условиях реакции продуктов ацилирования 4,6-диоксогептановой кислотой и ее лактоном экзоциклических аминогрупп гетероциклических оснований природных олигонуклеотидов.

Реакционная способность фрагментов ДНК, содержащих β-дикетогруппу
Для проверки способности синтезированных олигонуклеотидов с β-дикетогруппой взаимодействовать с остатками аргинина использовали олигонуклеотиды (2) и 5'-TCGGAUdioxAGTCCCCTC (3).

Схема 2 .
Реакцию олигонуклеотида (2) с гуанидином (схема 2 ) проводили в натрий-карбонатном буфере с концентрацией 0.5 М при pH 8.8 и 0.1 М при pH 9.2, однако ВЭЖХ и гель-электрофорез в ПААГ не позволили отделить продукт от исходного производного олигонуклеотида. Реакционную смесь анализировали методом MALDI-TOF. Был зафиксирован продукт присоединения гуанидина к олигонуклеотиду (2).
Для упрощения анализа реакционной смеси нами были использованы гидрофобные производные аргинина. При проведении реакции β-дикетосодержащего олигонуклеотида (2) с Nα-Вoc- и Nα-Dns-производными L-аргинина наилучшие результаты по данным обращенно-фазовой ВЭЖХ в ион-парном варианте (степень превращения 18 и 15% соответственно) были получены в том же буфере (рН 9.2).
Интересно отметить, что олигонуклеотиды с β-дикетогруппой, очищенные методом электрофореза в ПААГ, не вступали в реакцию с модельным соединением – Nα-Boc-L-аргинином. Анализ олигонуклеотида (3) после очистки в ПААГ методом масс-спектрометрии показал полное отсутствие искомого конъюгата (m/z [M]+ рассчитано: 4883.4), а масса основного продукта в анализируемом образце (m/z [M]+ найдено: 4718.0) соответствовала массе продукта присоединения акриламида к дикетопроизводному олигонуклеотида. Таким образом, зафиксировано образование аддукта с акриламидом в результате C/O-алкилирования β-дикетогруппы. Поэтому ПААГ не применялся для очистки синтезированных β-дикетопроизводных.
Также мы показали, что дикетогруппа не взаимодействует с гуанином в составе дуплекса. Для этого были сформированы ДНК-дуплексы, содержащие гуанозин “напротив” модифицированного нуклеозида в противоположной цепи. Это позволяет использовать предложенные ДНК-реагенты для аффинной модификации белков, не опасаясь их побочной реакции с самой ДНК.
Термическая устойчивость дуплексов с остатком 2'-дезокси-2'-(4,6-диоксогептиламидо)уридина
Нами впервые методом УФ-спектроскопии исследовано влияние остатка 4,6-диоксогептановой кислоты, введенного по 2'-аминогруппе 2'-дезоксиуридина одной из цепей, на термическую стабильность образованного ею и комплементарным олигодезоксирибонуклеотидом ДНК-дуплекса (табл. 1). Температура плавления дуплексов (I)–(III) понижается на 6°C при повышении рН раствора от 6.9 до 9.2. Введение остатка 2'-амино-2'-дезоксиуридина в одну из цепей дуплекса (I) приводит к его дестабилизации (ΔTпл 4°C) по сравнению с немодифицированным дуплексом (III), что согласуется с данными работ [23, 24] (рис. 2). 2'-Дезокси-2'-(4,6-диоксогептиламидо)уридин понижает Tпл на 11°C. При модификации 2'-положения углеводного фрагмента наблюдается изменение гипохромного эффекта, что свидетельствует о локальном нарушении межплоскостных взаимодействий гетероциклических оснований.


Взаимодействие ДНК-лигандов, содержащих β-дикетогруппу при С2'-атоме углеводного фрагмента, с белком MutS
Белок MutS играет ключевую роль в репарации “мисматчей”. В случае E. coli он представляет собой гомодимер [17]. MutS действует как сенсор, сканирующий ДНК в поисках неканонических пар нуклеотидов и небольших инсерционно-делеционных петель [25]. В специфическом комплексе аминокислотные остатки из обеих субъединиц MutS взаимодействуют с ДНК, однако связывание происходит асимметрично – каждая субъединица образует множество контактов, но они различны. Общая площадь поверхности ДНК-белковых контактов составляет ~1850 Å2 [26]. Большинство контактов MutS с ДНК являются гидрофильными (аминокислотные остатки взаимодействуют с углеводофосфатным остовом ДНК) и не зависят от нуклеотидной последовательности. Благодаря этому MutS способен функционировать в различных нуклеотидных контекстах [27].
Для аффинной модификации белка MutS были сконструированы 17-звенные ДНК-дуплексы (табл. 2), одна из цепей которых представляла собой олигонуклеотид с единичным включением β-дикетогруппы: 5'-ACTGGTGCTTGGCUdioxGCT (4), 5'‑AGCUdioxGCCAGGCACCAGT (5), 5'-ACTUdioxGTGCTTGGCAGCT (6), 5'-TCAGCACCCAGGGUdioxGCC (7). Нуклеотидная последовательность дуплекса (IV) соответствует последовательности дуплекса, использовавшегося при кристаллизации комплекса MutS с ДНК [17]. Дуплекс (IV) состоит из олигонуклеотида (4) с β‑дикетогруппой в модифицированном звене Udiox и ему комплементарного, и содержит G•T-пару (нуклеотид Т в модифицированной цепи), которую узнает белок MutS. Согласно анализу структуры комплекса MutS с G•T-содержащей ДНК (PDB код 1E3M) в программе PyMOL Viewer, 2'-дезокси-2'-(4,6-диоксогептиламидо)уридиновое звено в дуплексе (IV) удалено от остатка Arg500 не более чем на 7 Å, что достаточно для взаимодействия β-дикетогруппы с остатком аргинина (схема 3 ). Для контроля за ходом реакции на 5'-конец ДНК-дуплекса (IV) вводилась радиоактивная 32Р-метка или флуорофор – производное флуоресцеина (FAM).


Схема 3 .
На первом этапе проводился подбор условий для аффинной модификации MutS реакционноспособной ДНК. Варьировали соотношение концентраций ДНК-реагента (IV) и белка. Реакционную смесь инкубировали при 37°С в течение 30 мин, затем ее анализировали методом гель-электрофореза в неденатурирующих условиях (рис. 3). Наибольшая степень комплексообразования зафиксирована при 5–20-кратном избытке MutS по отношению к лиганду. Для дальнейших исследований использовали соотношение ДНК–белок 1 : 10.
Рис. 3.
Анализ в 6% ПААГ в неденатурирующих условиях продуктов связывания белка MutS с ДНК-дуплексом (IV) (1 мкМ), содержащим 5'-32Р-метку. Условия реакции: 30 мин, 37°С. Соотношение концентраций MutS - ДНК 0.5 : 1 (дорожка 1), 1 : 1 (дорожка 2), 2 : 1 (дорожка 3), 5 : 1 (дорожка 4), 10 : 1 (дорожка 5), 20 : 1 (дорожка 6). Дорожка 7 – исходный ДНК-дуплекс (IV). Положение ДНК-белковых комплексов указано стрелками.

На следующем этапе была изучена кинетика образования конъюгата белка MutS с 32P-меченным лигандом (IV). Время реакции составляло от 0.5 до 5 ч. Пробы из реакционной смеси, отобранные через определенные промежутки времени, анализировали методом гель-электрофореза в денатурирующих условиях (рис. 4). Чтобы повысить эффективность анализа, то есть зафиксировать продукт реакции и вместе с тем “задержать” в геле исходную ДНК, использовали комбинированный ПААГ, два верхних слоя которого аналогичны используемым в классическом гель-электрофорезе по Лэммли, а третий слой представлял собой 20% ПААГ с 0.1% SDS. В ходе такого анализа ДНК оставалась в 20% ПААГ, в то время как ДНК-белковый конъюгат – в 8% ПААГ. В геле наблюдалось появление дополнительной зоны, содержащей радиоактивность и имеющей существенно меньшую подвижность, чем исходный ДНК-реагент (рис. 4). Исходя из полученных данных было решено проводить аффинную модификацию белка MutS ДНК-лигандами с β-дикетогруппой в течение 3 ч. Инкубирование реакционной смеси более длительное время не увеличило значительно выход ДНК-белкового конъюгата.
Рис. 4.
Зависимость выхода конъюгата белка MutS (10 мкМ) с ДНК-лигандом (IV) (1 мкМ), содержащим 5'-32Р-метку, от времени при 37°С: (а) анализ продуктов реакции в денатурирующем ПААГ (электрофореграмма); дорожка К – исходная ДНК; над дорожками геля указано время реакции; положение ДНК-белкового конъюгата указано стрелкой; (б) график зависимости выхода продукта от времени протекания реакции; на графике приведено стандартное отклонение от среднего значения.

Рис. 5.
Анализ в денатурирующем ПААГ, содержащем SDS, продуктов ковалентного связывания белка MutS (1 мкМ) с 5'-FAM-меченными ДНК-дуплексами (IV)–(VII) (5 мкМ). Условия реакции: 3 ч, 37°С. Электрофореграммы: (а) детектирование зон по флуоресценции ДНК; (б) окрашивание геля раствором солей серебра. Дорожка М – маркер молекулярной массы белков, дорожки 1 и 2 – белок MutS и ДНК-фрагмент (V) соответственно. Остальные дорожки – реакционные смеси, содержащие MutS и ДНК-дуплексы (IV)–(VII) (указаны над дорожками геля). Положение ДНК-белковых конъюгатов указано стрелками.

Образование ковалентно связанного ДНК-белкового комплекса детектировали по появлению в геле дополнительной зоны белка с меньшей подвижностью. Для этого реакционную смесь, содержащую MutS и пятикратный избыток 5'-FAM-меченного ДНК-дуплекса (IV), инкубировали 3 ч при 37°С и анализировали методом гель-электрофореза в денатурирующих условиях. Свободную ДНК и ДНК в составе конъюгата детектировали в геле по наличию флуоресценции при 473 нм. Затем гель окрашивали раствором солей серебра для визуализации белка. Наличие белка и ДНК в зоне с меньшей подвижностью доказывало образование ДНК-белкового конъюгата (рис. 5).
Для исследования специфичности предложенных нами ДНК-реагентов использовали 5'-FAM-меченные лиганды (V)–(VI), различающиеся между собой положением реакционноспособной группы относительно “мисматча”, что приводит к изменению расстояния между модифицированным нуклеотидом в составе ДНК-дуплекса и Arg500 белка MutS (табл. 2). Дуплексы (V) и (VI) содержат некомплементарную пару G•T, но в дуплексе (V), в отличие от (IV), модифицированный нуклеотид с β-дикетогруппой располагается в противоположной цепи, содержащей остаток G “мисматча” на расстоянии 4 нт от 3'-конца. ДНК-лиганд (VI) аналогичен (IV), но модифицированный нуклеотид располагается с другой стороны от “мисматча” G•T. Учитывая индифферентность MutS к первичной структуре ДНК вокруг “мисматча” [28, 29], в качестве отрицательного контроля в реакции “кросслинкинга” использовали ДНК-дуплекс (VII) произвольной последовательности. В этом дуплексе отсутствует некомплементарная пара, то есть он является каноническим. Вместе с тем, он содержит нуклеотид с β-дикетогруппой в 4-ом положении с 3'-конца “нижней” цепи как и в случае ДНК-лиганда (IV).
Установлено, что выход ДНК-белкового конъюгата зависит от положения модифицированного нуклеотида в составе ДНК-дуплекса (рис. 5). Наибольший выход конъюгата наблюдался при взаимодействии MutS с ДНК-лигандом (IV), в комплексе с которым расстояние от β-дикетогруппы до остатка Arg минимально. C увеличением расстояния от β-дикетогруппы в составе ДНК-лигандов (V) и (VI) до остатка Arg500 в MutS до 10 Å выход ДНК-белкового конъюгата уменьшался. На основании полученных данных можно предположить, что реакция между белком и лигандом в данном случае протекает селективно. Появление на геле в ряде случаев нескольких зон, соответствующих ДНК-белковым конъюгатам, может быть обусловлено взаимодействием двух молекул 5'-FAM-меченного ДНК-фрагмента с белком. Известно, что белок MutS предпочтительнее образует комплексы с двуцепочечными ДНК-фрагментами, содержащими “мисматч”, чем с каноническими дуплексами. Меньшая эффективность образования продуктов реакции ковалентного связывания MutS с дуплексом (VII) произвольной последовательности без пары G•T по сравнению с дуплексом (IV) подтвердила эти данные.

Схема 4 .
В работе [4] показано, что соединения, содержащие β-дикетогруппу, способны, помимо остатка аргинина, модифицировать в белках остаток лизина. Особенность реакции с аминогруппой лизина состоит в том, что ее продукт может быть разрушен при воздействии гидроксиламина (схема 4 ). Для проверки селективности реакции по отношению к остаткам Arg 32Р-меченный дуплекс (IV) инкубировали 3 ч с MutS, затем в реакционную смесь добавляли раствор гидроксиламина и выдерживали 15 мин при 37°С. Анализ в ПААГ в денатурирующих условиях показал наличие белково-нуклеинового конъюгата, однако его выход уменьшился (рис. 6). Следовательно, предложенный нами ДНК-лиганд с β-дикетогруппой действительно взаимодействует с остатком аргинина. Вероятно, модифицированный ДНК-лиганд (IV) также способен взаимодействовать с одним из остатков лизина MutS. Например, остаток Lys496 сближен с тем же участком углеводофосфатного остова ДНК, что и Arg500 [17].
Рис. 6.
Анализ в денатурирующем ПААГ продуктов ковалентного связывания белка MutS (10 мкМ) с 5'-32Р-меченным ДНК-дуплексом (IV) (1 мкМ). Условия реакции: 3 ч, 37°С. Дорожка 1 – исходная ДНК, дорожка 2 – реакционная смесь, содержащая MutS и ДНК-дуплекс (IV), дорожка 3 – реакционная смесь, содержащая MutS и ДНК-дуплекс (IV), после добавления раствора гидроксиламина.

Взаимодействие ДНК-лигандов с β-дикетогруппой с белком MutL
После образования специфических контактов с “мисматч”-содержащей ДНК белок MutS связывает белок MutL, который в свою очередь выполняет функцию молекулярного координатора остальных участников системы репарации, таких как β-“зажим” (β-субъединица ДНК-полимеразы III), хеликаза UvrD и, в случае E. coli, никующая эндонуклеаза MutH [30–32]. Показано, что гомологи белка MutL способны связывать как одноцепочечную, так и двуцепочечную ДНК вне зависимости от наличия “мисматча” [33–35]. Согласно современным представлениям, ДНК-связывающий центр MutL находится в N-концевом домене белка [36]. Считается, что основные аминокислотные остатки двух субдоменов MutL – АТФазного и α-β-“сэндвичевого”, образуют положительно заряженную полость, в которой происходит связывание ДНК. Эта гипотеза была предложена для дрожжевого аналога MutL – Pms1 и подтверждена с помощью ограниченного протеолиза ДНК-белкового комплекса и методом “футпринтинга” с помощью гидроксил-радикалов [19]. Недавно такая положительно заряженная полость была обнаружена в кристаллической структуре N-концевого домена белка MutL из бактерии Aquifex aeolicus [20]. Установлено, что входящие в ее состав остатки Arg156, Lys158, Arg259 и Lys265 необходимы для эффективного связывания ДНК. Кроме того, показано, что мутантная форма MutL из E. coli с заменами на Glu остатков Arg162, Arg266 (АТP-азный субдомен) и Arg316 (α-β-“сэндвичевый” субдомен) не способна взаимодействовать с ДНК [18, 37, 38].
Эффективность комплексообразования MutL, как и других ДНК-связывающих белков, определяется длиной ДНК: чем протяженнее ДНК, тем стабильнее ДНК-белковый комплекс [20, 39]. Используя стандартный метод “торможения” в геле в неденатурирующих условиях, нам не удалось, в отличие от MutS, зафиксировать комплекс MutL E. coli с 17-звенными ДНК-дуплексами. Ранее по изменению сигнала поляризации флуоресценции было показано взаимодействие MutL E. coli только с 41-звенным дуплексом [18]. Мы предприняли попытку ковалентной фиксации MutL с помощью 5'-32Р-меченных дуплексов (IV)–(VI) в условиях, подобранных для MutS: 10‑кратный избыток ДНК-реагента по отношению к MutL, 37°С, 3 ч. Буфер, согласно рекомендациям [37], не содержал в своем составе KCl. В ходе аффинной модификации белка ДНК-реагентами с β-дикетогруппой нам удалось получить конъюгаты МutL с ДНК, то есть впервые показать взаимодействие MutL из Е. coli с короткими 17‑звенными ДНК-дуплексами и продемонстрировать взаимодействие ДНК с остатками аргинина в ДНК-связывающем центре этого белка.
Несмотря на низкую эффективность процесса, образование продуктов аффинной модификации MutL наблюдалось для всех ДНК-дуплексов, независимо от местоположения химически активной группы по отношению к G•T -паре, а также для дуплекса (VII) без “мисматча” (рис. 7). Таким образом, в отсутствие MutS взаимодействие MutL с ДНК носит неспецифический характер, что согласуется с данными работ [33–35].
Рис. 7.
Анализ в денатурирующем ПААГ продуктов ковалентного связывания белка MutL (10 мкМ) с 5'-32Р-меченными ДНК-дуплексами (1 мкМ): (V) (дорожка 2), (IV) (дорожка 3), (VI) (дорожка 4) и (VII) (дорожка 5). Дорожка 1 – исходный ДНК-дуплекс (V). Условия реакции: 3 ч, 37°С.

Как и в случае MutS, была исследована cелективность предложенных 17-звенных ДНК-реагентов по отношению к остаткам аргинина в MutL. При обработке раствором гидроксиламина реакционной смеси, содержащей ДНК-белковый конъюгат, после 3 ч инкубации его выход снижался примерно в 2 раза (данные не приведены). Этот результат свидетельствует о возможном взаимодействии модифицированного ДНК-лиганда также с остатками лизина в MutL.
В заключение следует отметить, что нами впервые предложено использовать дикарбонильные производные олигонуклеотидов для модификации гуанидиновой группы остатков аргинина в белках, взаимодействующих с ДНК. Была продемонстрирована способность фрагментов ДНК, содержащих β-дикетогруппу, вступать в реакции с гуанидином, Nα-Boc-L-аргинином и Nα-Dns-L-аргинином. Впервые показано образование конъюгатов белков MutS и MutL из системы репарации неканонических пар нуклеотидов E. coli с 17-звенными ДНК-дуплексами, содержащими остаток 2'-дезокси-2'-(4,6-диоксогептиламидо)уридина. Несмотря на невысокий выход конъюгатов ДНК с белками, дуплексы с β-дикетогруппой – это первый пример ДНК-реагентов, которые могут взаимодействовать с остатками аргинина. Для повышения селективности реакции аффинной модификации белка можно использовать обработку реакционной смеси гидроксиламином, что позволит исключить побочную реакцию ДНК-лигандов, содержащих β-дикетогруппу, с аминогруппой остатка лизина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы: 4,6-диоксогептановая кислота, Nα- Dns-L-аргинин, DMSO, N-[3-(диметиламино)пропил]-N'-этилкарбодиимид (EDC), ADP, ампициллин, рифампицин (Sigma-Aldrich, США); трео-2,3-дигидрокси-1,4-димеркаптобутан (дитиотреит, DTT), Nα-Boc-L-аргинин (Fluka, Швейцария); SDS, Трис, N-2-гидроксиэтилпиперазин-N′-2-этансульфоновая кислота (HEPES), EDTA, глицин (Хеликон, Россия); наборы белков–маркеров молекулярной массы PageRuler™ (10–200 кДа), набор ДНК–маркеров молекулярной массы GeneRuler™ (1000 п.о.) (ThermoFisher Scientific, США); бромфеноловый синий (Reanal, Венгрия); Ni-NTA-агароза (Novagen, Германия). Мини-колонки и микро-колонки для гель-фильтрации: NAP-5, NAP-10, Ultra MicroSpin G-50 (GE HealthCare, США).
Ферменты. Т4-полинуклеотидкиназа – коммерческий препарат производства “ThermoFisher Scientific”, США. Рекомбинантные белки MutS-(Δ801-853) и MutL дикого типа выделены из культуры клеток E. coli (штамм HMS174(λDE3)) методом аффинной хроматографии на Ni-NTA-агарозе по стандартной методике [40] с дальнейшим диализом в буфере, содержащем 10 мМ HEPES-KOH (pH 7.9), 1 мМ EDTA, 200 мМ KCl, 10% глицерина. Плазмидные конструкции, несущие гены белков, были любезно предоставлены проф. П. Фридхоффом,Университет имени Ю. Либиха, Германия (P. Friedhoff, Justus Liebig University, Germany).
Модифицированные олигодезоксирибонуклеотиды и олигодезоксирибонуклеотиды природного строения синтезировали на автоматическом синтезаторе ДНК/РНК ASM-2000 (БИОССЕТ, Россия) по стандартному регламенту амидофосфитного синтеза с применением коммерческих реагентов (Glen Research, США). Для получения олигонуклеотидов с включениями 2'-дезокси-2'-аминоуридина использовали 5'-O-диметокситритил-2'-трифторацетамидо-2'-дезокси-3'-(N,N-диизопропиламидо)цианэтилфосфит уридина (ChemGenes, США). Масс-спектры олигонуклеотидов регистрировали на масс-спектрометре Bruker Microflex MALDI-TOF (Германия). В качестве матрицы использовали смесь 3-гидроксипиколиновой кислоты и гидроцитрата аммония.
ВЭЖХ-анализ реакционных смесей, полученных при синтезе олигонуклеотидов, контроль чистоты и анализ продуктов их взаимодействия с низкомолекулярными соединениями проводили методом обращенно-фазовой ВЭЖХ в ион-парном варианте на хроматографе фирмы “Waters” (США). Использовали колонку 4.6 × 250 мм с сорбентом Luna С-18(2) (размер частиц 5 мкм). Условия аналитического разделения: температура колонки 45°С; элюент 48 мМ калий-фосфатный буфер (pH 7.0), содержащий 2 мМ дигидрофосфат тетрабутиламмония, специально рассчитанный градиент ацетонитрила: 5–19.6% за 1 мин, 19.6–22.2% за 4 мин, 22.2–23.4% за 5 мин, 23.4–24.5% за 10 мин, 24.5–25.7% за 20 мин (шаг элюции 1 звено в мин) и 5–21.2% за 1 мин, 21.2–23.8% за 4 мин, 23.8–25.0% за 5 мин, 25.0–26.1% за 10 мин, 26.1–27.3% за 20 мин (шаг элюции 2 звена в мин); скорость потока 1 мл/мин.
Получение модифицированных олигонуклеотидов (2)–(7), содержащих остаток 2'-дезокси-2'-(4,6-диоксогептиламидо)уридина. К раствору 4,6-диоксогептановой кислоты (1 мг, 6.3 мкмоль) и циклогексилкарбодиимида (1 мг, 6.0 мкмоль) в 50 мкл DMSO добавляли 4 нмоль олигонуклеотида, содержащего 2'-дезокси-2'-аминоуридин, в 100 мкл натрий-карбонатного буфера (0.1 М, pH 9.2), выдерживали в течение 16–18 ч. Затем проводили гель-фильтрацию раствора на колонке NAP-5. Реакционную смесь анализировали методом ВЭЖХ, строение подтверждали методом MALDI-TOF.
Изучение возможности образования ковалентной связи между тяжами ДНК-дуплекса, содержащего β-дикетогруппу. Смешивали 1.6 нмоль олигонуклеотида (2) и 2.2 нмоль комплементарного олигонуклеотида 5'-TTGAGGGGACTTTCCGAAT, 5 мкл 2 М раствора NaCl и добавляли воду до объема 150 мкл. Смесь нагревали до 80°С и оставляли охлаждаться до 20°С. Затем добавляли 100 мкл натрий-карбонатного буфера (0.1 М, pH 9.2), инкубировали 2 сут, проводили гель-фильтрацию на колонке NAP-5. Продукты реакции анализировали методом ВЭЖХ.
Реакция олигонуклеотидов, содержащих β-дикетогруппу, с гуанидином и производными аминокислот. К 4 нмоль олигонуклеотида (2), (3) добавляли раствор 1 мг гидрохлорида гуанидина (Nα-Boc-L-аргинина, Nα-Dns-L-аргинина) в 30 мкл буфера. При проведении реакции с гуанидином использовали натрий-карбонатный буфер с концентрацией 0.5 М при pH 8.8 и 0.1 М при pH 9.2, а с Dns-аргинином только последний (0.1 М, pH 9.2). С Boc-аргинином кроме карбонатного буфера (0.5 М, pH 8.8 и 0.1 М, pH 9.2) использовали также 0.1 М калий-фосфатный (pH 9.0) и 0.1 М Трис-боратный (pH 8.2). Реакционные смеси инкубировали 16–18 ч, проводили гель-фильтрацию на колонке NAP-5 и анализировали ВЭЖХ. Времена удерживания продукта присоединения Boc-аргинина и Dns-аргинина к олигонуклеотиду (2) составляют 17.8 и 18.9 мин соответственно, исходного олигонуклеотида (2) – 15.9 мин. Наличие продукта реакции подтверждали методом спектрометрии MALDI TOF. Для продукта присоединения к олигонуклеотиду (2) гуанидина m/z [M]+ найдено: 4669.6, рассчитано: 4669.0; Nα-Boc-L-аргинина – m/z [M]+ найдено: 4884.9, рассчитано: 4684.4. В случае Dns-аргинина продукт присоединения выделить не удалось.
Зависимость устойчивости ДНК-дуплексов от температуры изучали на спектрофотометре Hitachi U-2900 (Япония). Использовали термостатируемые кварцевые кюветы Hellma (Германия) c длиной оптического пути 10 мм. УФ-поглощение фиксировали при длине волны 260 нм, соблюдая следующий температурный режим: термостатирование при 25°С – 16 мин, нагрев от 20 до 80°С – 120 мин. Измерение проводили в двух растворах: 0.01 М калий-фосфатный буфер pH 6.9 c 0.1 М NaCl и 0.1 М натрий-карбонатный буфер pH 9.2 c 0.1 М NaCl. Концентрация дуплексов составляла 1 мкМ.
Для введения 5'-32P-метки 30–40 пмоль ДНК-фрагмента инкубировали в 10 мкл 50 мМ буфера Трис-HCl (pH 7.6), содержащего 10 мМ MgCl2, 5 мМ DTT, 100 мкМ спермидин и 0.3 мкМ [γ‑32P]ATP, с Т4-полинуклеотидкиназой (1 ед. акт.) в течение 1 ч при 37°С. После проведения реакции смесь очищали с помощью гель-фильтрации на колонке Ultra MicroSpin G-50. 32P-Меченные и немеченые ДНК-дуплексы формировали путем последовательной денатурации-ренатурации комплементарных цепей в соотношении 1 : 1.
Комплексообразование белка MutS с ДНК-дуплексом (IV). Смеси 32P-меченного ДНК-дуплекса (IV) (5 пмоль) с белком MutS (5–100 пмоль) инкубировали в 20 мМ буфере HEPES-KOH (pH 8.0), содержащем 125 мМ KCl, 1 мМ ADP, 5 мМ MgCl2, 1 мг/мл BSA, 1 мМ ДТТ, при 37°С в течение 20 мин. Затем пробы анализировали методом электрофореза в 6% ПААГ (18 × 22 × 0.15 см), содержащем 5.81% акриламида и 0.19% N,N′-метиленбисакриламида в буфере, содержащем 40 мМ Трис-CH3COOH (pH 7.5), 1 мМ EDTA, в течение 3 ч при температуре 4°С при силе тока 15 мА и напряжении 100 В (напряженность поля 4.5 В/см).
Визуализацию в геле зон, содержащих 32P- и FAM-метку, и обсчет данных проводили на приборах FLA-3000 (Fujifilm, Япония) и Typhoon FLA 9500 (GE HealthCare, США), используя компьютерные программы AIDA (Automatic Imaging Data Analysis Program) и ImageQuant TL (GE HealthCare, США).
Аффинная модификация белков MutS и MutL реакционноспособными ДНК-дуплексами. Белок MutS или MutL (количество варьировали от 10 до 100 пмоль) инкубировали в 10 мкл буфера 20 мМ HEPES-KOH (pH 8.0), содержащего 125 мМ KCl, 1 мМ ADP, 5 мМ MgCl2, 1 мг/мл BSA, 1 мМ DTT (в случае белка MutL буфер не содержал KCl), с меченным ДНК-дуплексом (количество варьировали от 5 до 50 пмоль) при 37°С в течение 0.5–5 ч. Продукты реакции анализировали в денатурирующем ПААГ (толщиной 1 мм) в 25 мМ буфере Трис-HCl, содержащем 250 мМ глицин (pH 8.3), 0.1% SDS при напряженности поля 14 В/см. Концентрирующий гель имел в своем составе 0.125 М Трис-НСl (рН 6.8), 4% акриламида, 0.1% N,N '-метиленбисакриламида, 0.1% SDS. Разделяющий гель состоял из двух слоев. Верхний содержал 0.375 М Трис-НСl (рН 8.8), 8% акриламида, 0.5% N,N′-метиленбисакриламида, 0.1% SDS; нижний – 0.375 М Трис-НСl (рН 8.8), 20% акриламида, 1% N,N′-метиленбисакриламида, 0.1% SDS. Пробы наносили на гель в 20 мкл 50 мМ буфера Трис-HCl (pH 6.8), 2.5% SDS, 10% глицерина, 0.5% β-меркаптоэтанола, 0.01% бромфенолового синего. Перед нанесением на гель пробы прогревали в течение 5 мин при 95°С. ПААГ с SDS окрашивали раствором нитрата серебра с помощью набора PageSilver Silver Staining Kit (ThermoFisher Scientific, США) по протоколу фирмы-производителя. Эксперименты проводили не менее трех раз.
Обработка конъюгатов MutS-ДНК и MutL-ДНК гидроксиламином. После проведения аффинной модификации белков MutS и MutL к реакционным смесям добавляли 1 мкл 2 М водного раствора гидрохлорида гидроксиламина, инкубировали при 37°С в течение 15 мин и анализировали методом электрофореза в денатурирующем ПААГ по методике, описанной выше.
Список литературы
Dolinnaya N.G., Zubin E.M., Kubareva E.A., Zatsepin T.S., Oretskaya T.S. // Cur. Org. Chem. 2009. V. 13. P. 1029–1049.
Verdine G.L., Norman D.P. // Annu. Rev. Biochem. 2003. V. 72. P. 337–366.
Luscombe N.M., Austin S.E., Berman H.M., Thornton J.M. // Genome Biol. 2000. V. 1. P. 1–10.
Gilbert H., O’Leary M. // Biochemistry. 1975. V. 14. P. 5194–5199.
King T. // Biochemistry. 1966. V. 5. P. 3454–3459.
Grossberg A., Pressman D. // Biochemistry. 1968. V. 4. P. 272–279.
Takahashi K. // J. Biol. Chem. 1968. V. 243. P. 6171–6179.
Basu A.K., O’Hara S.M., Valladier P., Stone K., Mols O., Marnett L.J. // Chem. Res. Toxicol. 1988. V. 1. P. 53–59.
Selig Ph. (Ed.) Guanidines as Reagents and Catalysts I. Ser. Topics in Heterocyclic Chemistry. Springer International Publishing, 2017. V. 50. P. 181.
Selig Ph. (Ed.) Guanidines as Reagents and Catalysts II. Ser. Topics in Heterocyclic Chemistry. Springer International Publishing, 2017. V. 51. P. 207.
Signor A., Bonora G., Biondi L., Nisato D., Marzotto A., Scoffone E. // Biochemistry. 1971. V. 10. P. 2748–2752.
Kinzel O., Budzikiewicz H. // J. Peptide Res. 1999. V. 53. P. 618–625.
Gao K., Orgel L.E. // Proc. Natl. Acad. Sci. USA. 1999. V. 96. P. 14837–14842.
Onizuka K., Taniguchi Y., Sasaki S. // Bioconjugate Chem. 2009. V. 20. P. 799–803.
Onizuka K., Shibata A., Taniguchi Y., Sasaki S. // Chem. Commun. 2011. V. 47. P. 5004–5006.
Onizuka K., Nishioka T., Li Z., Jitsuzaki D., Taniguchi Y., Sasaki S. // Chem. Commun. 2012. V. 48. P. 3969–3971.
Lamers M.H., Perrakis A., Enzlin J.H., Winterwerp H.H.K., de Wind N., Sixma T.K. // Nature. 2000. V. 407. P. 711–717.
Groothuizen F.S., Winkler I., Cristovao M., Fish A., Winterwerp H.H., Reumer A., Marx A.D., Hermans N., Nicholls R.A., Murshudov G.N., Lebbink G.H., Friedhoff P., Sixma T.K. // eLife. 2015. V. 4. P. e06744.
Schorzman A.N., Perera L., Cutalo-Patterson J.M., Pedersen L.C., Pedersen L.G., Kunkel T.A., Tomer K.B. // DNA Repair (Amst.). 2011. V. 10. P. 454–465.
Fukui K., Iino H., Baba S., Kumasaka T., Kuramitsu S., Yano T. // Biochim. Biophys. Acta. 2017. V. 1865. P. 1178–1187.
Кузнецова Л.Г., Волков Е.М., Романова Е.А., Ташлицкий В.Н., Орецкая Т.С., Шабарова З.А. // Биоорган. химия. 1991. Т. 17. С. 1289–1291 [Kuznetsova L.G., Volkov E.M., Romanova E.A., Tashlitsky V.N., Oretskaya T.S., Shabarova Z.A. // Bioorg. Khim. (Moscow). 1991. V. 17. P. 1289–1291].
Зацепин Т.С., Долинная Н.Г., Кубарева Е.A., Ивановская М.Г., Метелев В.Г., Орецкая Т.С. // Успехи химии. 2005. Т. 74. С. 84–103.
Кузнецова Л.Г., Романова Е.А., Волков Е.М., Ташлицкий В.Н., Орецкая Т.С., Крынецкая Н.Ф., Шабарова З.А. // Биоорган. химия. 1993. Т. 19. С. 455–466. [Kuznetsova L.G., Romanova E.A., Volkov E.M., Tashlitsky V.N., Oretskaya T.S., Krynetskaya N.F., Shabarova Z.A. // Bioorg. Khim. (Moscow). 1993. V. 19. P. 455–466].
Зубин Е.М., Анцыпович С.И., Орецкая Т.С., Романова Е.А., Волков Е.М., Ташлицкий В.Н., Шабарова З.А. // Биоорган. химия. 1997. Т. 23. С. 809–816. [Zubin E.M., Antsypovich S.I., Oretskaya T.S., Romanova E.A., Volkov E.M., Tashlitsky V.N., Shabarova Z.A. // Russian Journal of Bioorg. Chem. 1997. V. 23. Р. 730–736].
Iyer R.R., Pluciennik A., Burdett V., Modrich P.L. // Chem. Rev. 2006. V. 106. P. 302–323.
Natrajan G., Lamers M.H., Enzlin J.H., Winterwerp H.H.K., Perrakis A., Sixma T.K. // Nucl. Acids Res. 2003. V. 31. P. 4814–4821.
Sharma A., Doucette C., Biro F.N., Hingorani M.M. // J. Mol. Biol. 2013. V. 425. P. 4192–4205.
Yang Y., Sass L.E., Du C., Hsieh P., Erie D.A. // Nucl. Acids Res. 2005. V. 33. P. 4322–4334.
Tessmer I., Yang Y., Zhai J., Du C., Hsieh P., Hingorani M.M., Erie D.A. // J. Biol. Chem. 2008. V. 283. P. 36646–36654.
Modrich P. // J. Biol. Chem. 1989. V. 264. P. 6597–6600.
Au K.G., Welsh K., Modrich P. // J. Biol. Chem. 1992. V. 267. P. 12142–12148.
Friedhoff P., Li P., Gotthardt J. // DNA Repair (Amst.). 2016. V. 38. P. 50–57.
Mechanic L.E., Frankel B.A., Matson S.W. // J. Biol. Chem. 2000. V. 275. P. 38337–38346.
Bende S.M., Grafström R.H. // Nucl. Acids Res. 1991. V. 19. P. 1549–1555.
Drotschmann K., Hall M.C., Shcherbakova P.V., Wang H., Erie D.A., Brownewell F.R., Kool E.T., Kunkel T.A. // Biol. Chem. 2002. V. 383. P. 969–975.
Guarne A., Ramon-Maiques S., Wolff E.M., Ghirlando R., Hu X., Miller J.H., Yang W. // EMBO J. 2004. V. 23. P. 4134–4145.
Ban C., Junop M., Yang W. // Cell. 1999. V. 97. P. 85–97.
Robertson A., Pattishall S.R., Matson S.W. // J. Biol. Chem. 2006. V. 281. P. 8399–8408.
Hall M.C., Wang H., Erie D.A., Kunkel T.A. // J. Mol. Biol. 2001. V. 312. P. 637–647.
Feng G., Winkler M.E. // Biotechniques. 1995. V. 19. P. 956–965.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия