

Биоорганическая химия, 2019, T. 45, № 3, стр. 315-325
Синтез и биологическая активность конъюгатов 3,4,6-три-О-ацетил-N-ацетилглюкозамина и тетраацетилглюкопиранозы с алкилфосфатами
Р. Р. Шарипова 1, Б. Ф. Гарифуллин 1, А. С. Сапунова 1, А. Д. Волошина 1, М. А. Кравченко 2, В. Е. Катаев 1, *
1 Институт органической и физической химии им. А.Е. Арбузова ФИЦ Казанский научный центр РАН
420088 Казань, ул. Арбузова, 8, Россия
2 Уральский научно-исследовательский институт фтизиопульмонологии – филиал ФГБУ “НМИЦ ФПИ” Минздрава России
620039 Екатеринбург, ул. 22 Партсъезда, 50, Россия
* E-mail: kataev@iopc.ru
Поступила в редакцию 19.09.2018
После доработки 01.10.2018
Принята к публикации 11.10.2018
Аннотация
Синтезированы конъюгаты 3,4,6-три-О-ацетил-N-ацетилглюкозамина и тетраацетилированной глюкопиранозы с алкилфосфатами. Выявлена зависимость их антибактериальной и антитуберкулезной активности от длины алкильного заместителя у фосфатной группы. Так, конъюгаты с децильным заместителем проявили лучшую in vitro антитуберкулезную активность против Mycobacterium tuberculosis H37Rv (МИК 3 мкг/мл), но показали самое слабое антибактериальное действие в отношении Streptococcus aureus и Bacillus cereus (МИК 125 и более мкг/мл). И, наоборот, конъюгаты с цетильным заместителем продемонстрировали лучшую антибактериальную активность in vitro в отношении S. aureus и B. cereus (МИК 16 мкг/мл), но проявили самую низкую в изученных рядах производных антитуберкулезную активность in vitro (МИК 12 мкг/мл).
ВВЕДЕНИЕ
Пептидогликан (PG) является важной макромолекулой, которая окружает бактерии и микобактерии, придает им форму и защищает их от разрушающего действия собственного высокого осмотического давления. Ингибирование синтеза PG приводит к лизису бактериальных клеток, поэтому все ферменты, участвующие в его биосинтезе, являются привлекательными мишенями для дизайна новых антибактериальных и антимикобактериальных (антитуберкулезных) агентов. Из всего разнообразия ферментов внимание исследователей сосредоточено на N-ацетилглюкозамин-1-фосфат-уридилтрансферазе (GlmU-T) и PG-гликозилтрансферазах (PG-Ts), которые катализируют последние стадии биосинтеза PG [1–3].
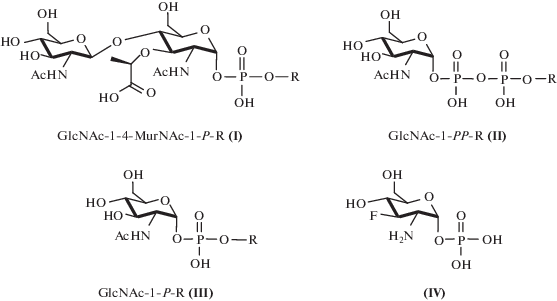
В последнее десятилетие была синтезирована большая серия ингибиторов GlmU- и PG-трансфераз с модификацией углеводного остатка, фосфатной группы и алкильного заместителя (рис. 1) [3–9]. Наиболее известные ингибиторы имеют дисахаридный фрагмент, состоящий из N-ацетилглюкозамина (GlcNAc) и фрагментов N-ацетилмурамовой кислоты (MurNAc) (соединения (I) [4]) или имеют только фрагмент GlcNAc (соединения (II), (III) [5–8]). Кроме того, известные ингибиторы подразделяются на дифосфаты (соединения (II) [5–8]) и монофосфаты (соединения (I), (III) [4, 7]). Было показано, что ингибиторы (II), (III) являются альтернативными субстратами для бактериальных глюкозил- и галактозилтрансфераз и способны в некоторой степени ингибировать активность различных бактериальных ферментов [4–8]. Очень интересно, что простейшее соединение (IV) (рис. 1) ингибирует GlmU-трансферазу M. tuberculosis в миллимолярных концентрациях [9]. Казалось бы, если какое-либо соединение ингибирует любой фермент на пути к пептидогликану бактерии, оно также должно ингибировать рост самой бактерии. Удивительно, но, насколько нам известно, в литературе нет сообщений о том, как синтезированные ингибиторы бактериальных глюкозил- или галактозилтрансфераз угнетают рост самих бактерий. В литературе можно найти только одно сравнение антиферментативной и антибактериальной активности ингибиторов гликозилтрансфераз [4]. Причем полученные результаты [4] удивительны. Так, лучшие ингибиторы (III) PG-гликозилтрансферазы PBP1b E. coli продемонстрировали отсутствие антибактериальной активности [4].
Принимая во внимание эти данные, мы решили проверить, появится ли антибактериальная активность у соединений типа (III), если ацетилировать гидроксильные группы углеводного остатка.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В настоящей работе сообщается о синтезе, антибактериальной и антитуберкулезной активности конъюгатов 3,4,6-три-O-ацетил-N-ацетил-D-глюкозаминa и тетраацетилированной D-глюкопиранозы с алкилфосфатами.
В качестве исходного соединения для синтеза целевых конъюгатов (далее гликофосфатов) на основе 3,4,6-три-О-ацетил-N-ацетил-D-глюкозамина (XII) и (XIV) использовали коммерческий гидрохлорид глюкозамина (V) (схема 1 ). Он был ацилирован аналогично [10], и полученный 1,3,4,6-тетра-О-ацетил-N-ацетил-D-глюкозамин (VI) региоселективным удалением защиты с аномерной гидроксильной группы метиламином в метаноле аналогично [11] был превращен в 3,4,6-три-О-ацетил-N-ацетил-D-глюкозамин (VII). Затем аномерную гидроксильную группу производного (VII) подвергли фосфонилированию салицилхлорфосфитом (2-хлор-2H,4H-1,3,2-бензодиоксафосфорин-4-он) (IX) [12], который был получен из салициловой кислоты (VIII) аналогично [13]. H-Фосфонат (X) выделили с выходом 34% после хроматографирования. В спектре 1H-ЯМР H-фосфоната (X) наряду с характеристичными сигналами 3,4,6-три-О-ацетил-N-ацетилглюкозамина (VII) появились сигналы в виде триплета при 1.36 м.д. (JH-H 7.3 Гц) и квартета при 3.08 м.д. (JH-H 7.3 Гц), что соответствовало резонансу протонов триэтиламмониевой группы, а также дублет при 6.9 м.д. (JP-H 668 Гц), соответствующий резонансу протона Р–Н. Аномерный протон резонировал в виде мультиплета в области 5.61−5.69 м.д., что указывало на образование смеси аномеров. В спектре 31P-ЯМР соединения (X) присутствовал синглет при 1.93 м.д., соответствующий резонансу атома фосфора в H-фосфонатах.
На следующей стадии провели реакцию H-фосфоната (X), активированного пивалоилхлоридом, с деканолом. Полученный таким образом гликофосфонат (XI) затем окисляли водным раствором йода и с выходом 23% (после хроматографирования) получили целевой гликофосфат (XII). Его образование было подтверждено, в первую очередь, исчезновением в спектре 1Н-ЯМР сигнала протона P−H при 6.90 м.д. и появлением в спектре 31P-ЯМР синглета при –2.27 м.д., соответствующего фосфатной группе в моноанионной форме (${\text{PO}}_{4}^{ - }$). Масс-спектральные данные также указывали на образование гликофосфата (XII). В спектре MALDI гликофосфата (XII) наблюдался пик при m/z 566.4 [M]– (C24H41NO12P). Аномерный протон гликофосфата (XII) резонировал в виде дублета дублетов при 5.51 м.д. (3JH1, P 7.34 Гц, 3J H1, H2 3.22 Гц), что указывало на образование индивидуального α-аномера.

Схема 1 . Синтез гликофосфатов (XII) и (XIV). Условия и реагенты: (i) Ac2O, Py, 0 → 20°С, 24 ч; (ii) CH3NH2, CH3OH, THF, rt, 0.5 ч; (iii) PCl3, толуол, 110°С, 12 ч; (iv) Et3N, H2O, THF, rt, 12 ч; (v) CH3(CH2)9OH, PivCl, Py, –15°С, 2 ч; (vi) 1. I2, H2O, 20°С, 1 ч; 2. Na2S2O3; (vii) CH3(CH2)15OH, PivCl, Py, –15°С, 2 ч, rt.
Для определения влияния длины алкильного заместителя на биологическую активность, кроме гликофосфата (XII), взаимодействием Н-фосфоната (X) с цетиловым спиртом был синтезирован гликофосфат (XIV), выделенный после хроматографии с выходом 10%. Его образование было подтверждено отсутствием в спектре 1H-ЯМР сигнала протона P−H и появлением в спектре 31P-ЯМР синглета при –2.43 м.д., соответствующего фосфатной группе в моноанионной форме (${\text{PO}}_{4}^{ - }$). Спектр MALDI гликофосфата (XIV) содержал пик при m/z 650.6 [M]– (C30H53NO12P). Аномерный протон гликофосфата (XIV) наблюдался в спектре 1H-ЯМР в виде дублета дублетов при 5.51 м.д. (3JH1, P 7.18 Гц, 3JH1, H2 3.53 Гц) что указывало на α-ориентацию гликозидной связи.
Для оценки влияния на биологическую активность гликофосфатов (XII) и (XIV) остатка глюкозамина были синтезированы гликофосфаты (XX) и (XXII), в которых вместо 3,4,6-три-О-ацетил-N-ацетилглюкозамина содержалась тетраацетилированная D-глюкопираноза (схема 2 ). Исходное соединение, D-глюкопираноза (XV), была ацетилирована аналогично [14], затем аномерную защитную группу региоселективно удалили раствором ацетата гидразина аналогично [15], и полученная таким образом 2,3,4,6-тетра-O-ацетил-α/β-D-глюкопираноза (XVII) была вовлечена в реакцию с салицилхлорфосфитом (IX), которая привела к H-фосфонату (XVIII), выделенному колоночной хроматографией с выходом 44%. В его спектре 1H-ЯМР, наряду с характерными сигналами тетраацетилированной глюкопиранозы (XVII), наблюдался триплет при 1.28 м.д. (JH-H 7.3 Гц) и квартет при 3.0 м.д. (JH-H 7.3 Гц), соответствующие триэтиламмониевой группе, а также дублет при 6.91 м.д. (JP-H 638.9 Гц), соответствующий протону P−H. В спектре 31P‑ЯМР соединения (XVIII) присутствовал синглет при 0.77 м.д., соответствующий резонансу атома фосфора в H-фосфонатах. В спектре MALDI H-фосфоната (XVIII) наблюдался пик при m/z 513.2 [M] (C20H36NO12P). Аномерный протон, резонирующий в виде дублета дублетов при 5.75 м.д. (JH1, P 8.9 Гц, JH1, H2 3.4 Гц), указывал на образование α-аномера.

Схема 2 . Синтез гликофосфатов (XX) и (XXII). Условия и реагенты: (i) Ac2O, Py; 0 → 20°С, 24 ч; (ii) H3N+${\text{NH}}_{3}^{ + }$2AcO–, DMF, 0 → 20°С, 3 ч; (iii) THF, Et3N, 20°С, 12 ч; (iv) CH3(CH2)9OH, PivCl, Py, –15°С, 2 ч; (v) 1. H2O, I2, 20°С, 1 ч, 2. Na2S2O3; (vi) CH3(CH2)15OH, PivCl, Py, –15°С, 2 ч.
Гликофосфаты (XX) и (XXII) были получены из H-фосфоната (XVIII) в полном соответствии с процедурой синтеза гликофосфатов (XII) и (XIV) (схема 1 ), описанной выше. Гликофосфат (XX) был получен с выходом 53% после хроматографии. В его спектре MALDI присутствовал пик при m/z 567.1 [M]– (C24H40O13P–). Атом фосфора резонировал в спектре 31P-ЯМР в виде синглета при –2.69 м.д., что указывало на наличие фосфатной группы в моноанионной форме (${\text{PO}}_{4}^{ - }$). Аномерный протон в спектре 1H-ЯМР резонировал в виде дублета дублетов при 5.72 м.д. (JH1, P 7.8, JH1, H2 3.3 Гц), что указывало на образование индивидуального α-аномера. Гликофосфат (XXII) был получен с выходом 35% после хроматографии. В его спектре MALDI наблюдался пик при m/z 675.2 [M + Na]+ (C30H53NaO13P). В спектре 31P-ЯМР гликофосфата (XXII) присутствовал только один синглет при –2.31 м.д., а аномерный протон в спектре 1H-ЯМР резонировал в виде дублета дублетов при 5.57 м.д. (JH1, P 7.9, JH1, H2 3.5 Гц), что указывало на образование гликофосфата (XXII) в виде индивидуального α-аномера.
Отметим различие между нашим методом получения гликофосфатов (XII), (XIV) и методами получения конъюгатов глюкозамина с фосфатными или дифосфатными фрагментами, описанными в литературе [4–8, 16]. Во-первых, это способ получения ключевого производного глюкозамина (VII) с защищенными гидроксильными группами в положениях С3, С4, С6 и свободной аномерной гидроксильной группой, которая далее подвергается фосфонилированию. 3,4,6-Три-О-ацетил-N-ацетилглюкозамин (VII) легко получали региоселективным удалением ацетильной защиты аномерной гидроксильной группы моносахарида (VI) раствором метиламина в метаноле. Во-вторых, мы использовали другую процедуру [12] для получения конъюгатов с фосфатной группой из производного глюкозамина (VII). Обычно (см., например, [4]) моносахарид (VII) фосфорилируется реакцией с дибензил-N,N-диизопропилфосфорамидатом и 1Н-тетразолом с последующим окислением перекисью водорода, дебензилированием и обработкой гидроксидом трет-бутиламмония. Полученная соль далее вовлекается в реакции с алифатическими бромидами в присутствии молекулярных сит. В своем исследовании мы алкилировали не фосфат глюкозамина, а H-фосфонат (Х), который был легко получен реакцией 3,4,6-три-О-ацетил-N-ацетилглюкозамина (VII) с салицилхлорфосфитом (IХ) [13]. Получение целевых гликофосфатов (XII) и (XIV) из H-фосфоната (X) проводили в одну стадию окислением H-фосфонатов (XI) и (XIII) до фосфатов (XII) и (XIV) йодом на заключительном этапе процесса. Аналогичным образом синтезировали гликофосфаты (XX) и (XXII) на основе 2,3,4,6-три-O-ацетил-α-D-глюкопиранозы. Выходы гликофосфатов (XII) и (XIV) были небольшими, но условия реакции будут доработаны.
Для определения влияния на биологическую активность гликофосфатов (XII) и (XIV) наличия алкильного заместителя был синтезирован гликофосфат (XXV) (схема 3 ). Оксазолин (XXIII) получали реакцией 1,3,4,6-тетра-О-ацетил-N-ацетилглюкозамина (VI) с триметилсилилтрифторметансульфонатом (TMS-OTf) аналогично [17]. Гликофосфат (XXV) синтезировали из оксазолина (XXIII) по известной методике [18], но без выделения и очистки дибензилфосфата (XXIV). Гликофосфат (XXV) в виде монотриметиламмониевой соли получили с выходом 27%. В его спектре 31P-ЯМР присутствовал синглет при 1.94 м.д., соответствующий фосфатной группе в моноанионной форме (${\text{PO}}_{4}^{ - }$). В спектре MALDI фосфата (XXV) наблюдался пик при m/z 426.3 [M]– (C14H21NO12P). В отличие от гликофосфатов (XII), (XIV), (XX), (XXII), аномерный протон гликофосфата (XXV) присутствовал в спектре 1H-ЯМР в виде триплета при 5.18 м.д. (3JH1, -2 = 3JH1, P = 10 Гц), что указывало на образование β-аномера.
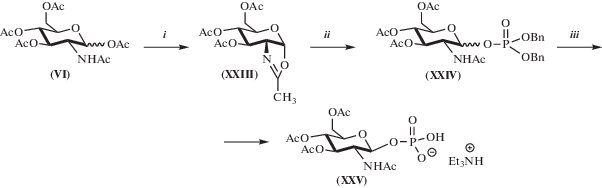
Схема 3 . Синтез фосфата (XXV). Условия и реагенты: (i) TMS-OTf, C2H4Cl2, 50°C, 30 ч; (ii) (PhCH2O)2P(O)OH, Ag2CO3, CH2Cl2/CH3CN/Et2O, 0–20°C, 18 ч; (iii) H2, Pd/C, MeOH, Et3N, 20°C, 4–8 ч.
Соединения (VI), (X), (XII), (XIV), (XX), (XXII), (XXV) и (XXVI) тестировали на антитуберкулезную активность в отношении Mycobacterium tuberculosis H37Rv (МБТ), а также на антимикробную активность в отношении грамположительных бактерий (Staphylococcus aureus ATCC 209p и Bacillus cereus ATCC 8035), грамотрицательных бактерий (Escherichia coli CDCF-50 и Pseudomonas aeruginosa ATCC 9027) и грибов (Aspergillus niger BKMF-1119, Trichophyton mentagrophytes var. gypseum 1773 и Candida albicans 855-653). Оказалось, что все изученные соединения ингибируют in vitro рост МБТ (табл. 1). Хотя их антитуберкулезная активность в 30–125 раз ниже, чем активность противотуберкулезного препарата Изониазид, тем не менее антитуберкулезная активность соединений (X), (XIV), (XXII) и (XXV) была равна активности противотуберкулезного препарата Пиразинамид. Более того, антитуберкулезная активность соединений (VI) и (XXVI) превышала активность Пиразинамида в 2 раза, а активность соединений (XII) и (XX) превышала активность Пиразинамида в 4 раза (табл. 1). Соединения (VI), (XX), (XXV), (XXVI) оказались неактивными против всех бактерий и грибов, используемых при антимикробном тестировании. Только гликофосфаты (XII), (XIV), (XXII) проявили антибактериальную активность против S. aureus и B. cereus (табл. 1). В отношении других бактерий и грибов, использованных при скрининге, эти соединения оказались неактивными. Анализируя значения МИК, представленные в табл. 1, можно сделать следующие важные выводы. Во-первых, именно алкильный заместитель обуславливает появление антибактериальной активности у тетраацетилированной глюкопиранозы и 3,4,6-три-О-ацетил-N-ацетилглюкозамина. Что касается антитуберкулезной активности, то она примерно одинакова для производных глюкозамина (VI), (XXVI) и гликофосфатов (XII) и (XX). Во-вторых, гликофосфаты (XIV) и (XXII) с гексадецильным заместителем показали лучшую антибактериальную активность (МИК 16 мкг/мл), чем гликофосфаты (XII) и (XX) с децильным заместителем. Действительно, гликофосфаты (XIV) и (XXII) с гексадецильным заместителем ингибировали in vitro рост S. aureus в четыре раза лучше, чем антибиотик Хлорамфеникол, в то время как гликолипид (XII) с децильным спейсером был равен по активности Хлорамфениколу, а гликофосфат (XX) вообще был не активен в отношении бактерий и грибов, используемых при тестировании. В-третьих, среди изученных соединений природа углеводного остатка гликофосфата не влияла на его антибактериальную активность, более важной оказалась длина алкильного заместителя. Так, можно видеть, что антибактериальная in vitro активность в отношении S. аureus и B. cereus гликофосфата (XIV), содержащего остаток глюкозамина, и гликофосфата (XXII), содержащего глюкопиранозильный остаток, одинакова. В-четвертых, длина алкильного заместителя значительно влияет на антитуберкулезную активность исследуемых соединений. Гликофосфаты (XII) и (XX) с децильным заместителем в четыре раза лучше ингибировали in vitro рост МБТ (МИК 3 мкг/мл), чем гликофосфаты (XIV) и (XXII) с гексадецильным фрагментом (МИК 12 мкг/мл). В-пятых, длина алкильного заместителя в изученных гликофосфатах влияет на антитуберкулезную и антибактериальную активность по-разному. Так, гликофосфаты (XII) и (XX) с децильной цепочкой продемонстрировали лучшую антитуберкулезную активность in vitro против МБТ (МИК 3 мкг/мл), но одновременно показали самую слабую антибактериальную активность против S. aureus (МИК 62 и более 500 мкг/мл соответственно) и B. cereus (МИК 125 и более 500 мкг/мл соответственно). И наоборот, гликофосфаты (XIV) и (XXII) с цетильной цепочкой показали лучшую антибактериальную активность in vitro против S. aureus (МИК 16 мкг/мл) и против B. cereus (МИК 62.5 мкг/мл), но проявили самую низкую антитуберкулезную активность in vitro (MИК 12 мкг/мл) против МБТ.
Таблица 1.
Антитуберкулезная и антимикробная активность синтезированных соединений
| Минимальная ингибирующая концентрация (МИК), мкг/мл | |||
|---|---|---|---|
| M. tuberculosis H37Rv |
S. aureus ATCC 209p |
B. cereus ATCC 8035 |
|
 |
6 | >500 | >500 |
 |
6 | >500 | >500 |
 |
12 | – | – |
 |
12 | >500 | >500 |
 |
3 | 62 | 125 |
 |
12 | 16 | 62 |
 |
3 | >500 | >500 |
 |
12 | 16 | 62 |
| Изониазид | 0.1 | − | − |
| Пиразинамид | 12 | − | − |
| Хлорамфеникол | − | 62 | 62 |
Таким образом, в этом исследовании мы показали, что ацетилирование гидроксильных групп углеводного остатка в гликофосфатах типа (III) [4] приводит к появлению антибактериальной и антитуберкулезной активности.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры 1Н, 13С, 31P ЯМР (δ, м.д.; КССВ J, Гц) регистрировали на спектрометре Avance-400 (Bruker, Германия) с частотой 400 МГц (1Н) и 100.6 МГц (13С, 31P) в CDCl3. Масс-спектры МАLDI получены на времяпролетном масс-спектрометре UltraFlex III TOF/TOF (Bruker Daltonik GmbH, Бремен, Германия) в линейном режиме. Лазер Nd:YAG, λ 355 нм. Данные обрабатывали с помощью программы FlexAnalysis 3.0 (Bruker Daltonik GmbH, Бремен, Германия). Измерения проводили в диапазоне m/z 200–6000, фиксировали отрицательно заряженные ионы. Использовали металлическую мишень, в качестве матрицы использовали п-нитроанилин. Образцы растворяли в метаноле (10–3 мг/мл). Оптическое вращение измеряли на поляриметре Perkin Elmer-341 (Perkin Elmer, США) при λ = 589 нм, 20°C. Полноту протекания реакций и чистоту веществ контролировали методом тонкослойной хроматографии на пластинах Sorbfil (ООО “Имид” Краснодар, Россия), вещества обнаруживали обработкой пластин 5% раствором серной кислоты с последующим нагреванием до 120°C.
Все реакции, чувствительные к воздуху и/или влаге, проводились в атмосфере аргона в безводных растворителях, которые предварительно очищали и сушили (при необходимости) в соответствии со стандартными процедурами.
Соединения (VI) [10], (VII) [11], (IX) [13], (XVI) [14], (XVII) [15], (XXIII) [17] и (XXVI) [19, 20] были синтезированы по известным методикам. Спектральные данные соединений (VI), (VII), (IX) согласуются с опубликованными в литературе [10, 11, 13]. Спектральные данные соединений (XVI), (XVII) и (XXIII) согласуются с данными, опубликованными в литературе [21–23]. Характеристики соединения (XXVI) соответствовали литературным [19, 20]. Использовали коммерческие гидрохлорид D-глюкозамина (V) и D-глюкопиранозу (XV) (Acros, Бельгия).
3,4,6-Три-О-ацетил-2-дезокси-2-ацетамидо-α/β-D-глюкопиранозил-Н-фосфонат триэтиламмония (X). К раствору 0.67 г (2 ммоль) 3,4,6-три-О-ацетил-N-ацетилглюкозамина (VII) [11] и 1.87 мл триэтиламина (18.5 ммоль) в 10 мл сухого THF добавляли при перемешивании 0.42 г (2 ммоль) салицилхлорфосфита (IX) [13], перемешивали 3 ч, добавляли 1 мл воды и перемешивали еще 1 ч. Реакционную смесь упаривали досуха, полученный остаток хроматографировали на силикагеле (элюент − CH2Cl2−CH3OH, 40 : 1 → 5 : 1 с добавлением 1% Et3N по объему). Получили 0.33 г (34%) соединения (X) в виде бесцветного масла, $[\alpha ]_{D}^{{20}}$ +56.7 (c 0.8, CHCl3). Найдено, %: C 47.05; H 7.49; N 5.53; P 5.98. C20H37N2O11P. Вычислено, %: C 46.87; H 7.28; N 5.47; P 6.04. Спектр 1Н-ЯMР: 1.36 [т, 9H, J 7.30, N+(CH2CH3)3], 1.97, 2.01, 2.07 (все с, 12Н, 4CH3CO), 3.08 [кв, 6Н, J 7.3, N+(CH2CH3)3], 4.09−4.27 (м, 3H, H2, H6a, H6b), 4.36−4.46 (м, 1H, H5), 5.17 (т, 1H, J 9.62, H4), 5.27–5.35 (м, 1H, H3), 5.61−5.69 (м, 1H, H1), 6.90 (д, 1H, J1,P 668.04, P–H), 6.90−7.05 [м, 1H, NHC(O)CH3], 11.73 (уш. с, 1H, HN+). Спектр 13C-ЯMР: 8.7 (CH3CH2N+), 20.7, 20.8 [3 C(O)CH3], 23.0 (CH3C(O)NH), 45.9 (CH2N+), 52.1 (C2), 61.9 (C6), 68.4 (C4), 69.0 (C3), 71.2 (C5), 93.1 (C1, J 3.9), 169.6, 170.9, 171.1, 171.2 (4 C=O). Спектр 31P-ЯMР: +1.93.
Общая методика синтеза гликофосфатов (XII) и (XIV). К охлажденному до –20°С раствору Н-фосфоната (X) (1 экв.) в 10 мл пиридина добавляли при перемешивании сначала 2 экв. декан-1-ола или гексадекан-1-ола, затем раствор 4.3 экв. пивалоилхлорида (Piv-Cl) в 5 мл пиридина. Перемешивали 1 ч, затем добавляли 1 мл воды и 1 экв. йода. Продолжали перемешивание 2 ч, затем по каплям добавляли 1 М раствор Na2S2O3 до исчезновения окраски йода. Реакционную смесь соломенно-желтого цвета упаривали досуха, полученный остаток хроматографировали на силикагеле (элюент − CH2Cl2−CH3OH, 40 : 1 → 5 : 1 с добавлением 1% Et3N по объему), после чего выделяли децилгликофосфат (XII) (23%, бесцветное масло) и гексадецилгликофосфат (XIV) (10%, бесцветное масло).
2-Ацетамидо-2-дезокси-3,4,6-три-О-ацетил-α-D-глюкопиранозилдецилфосфат триэтиламмония (XII). $[\alpha ]_{D}^{{20}}$ +26.6 (c 0.642, CH3OH). Найдено, %: C 54.09; H 8.78; N 4.21; P 4.55. C30H57N2O12P. Вычислено, %: C 53.88; H 8.59; N 4.19; P, 4.63. Масс-спектр MALDI-TOF MS: m/z 566.4 [M]–. Рассчитана М– 566.2 (C24H41NO12P–). Спектр 1Н-ЯМР: 0.86 (т, 3H, J 7.0, H10'), 1.18 (уш.с, 4H, H-9', H-8'), 1.21–1.28 (м, 10H, H3'−H-7'), 1.32 [т, 9H, J 7.30, N+(CH2CH3)3], 1.56–1.64 (м, 2H, H2'), 1.94, 1.98, 1.99, 2.06 (все с, 12Н, 4 CH3CO), 3.06 [кв, 6Н, J 7.31, N+(CH2CH3)3], 3.83−3.94 (м, 2H, H1'), 4.05−4.10 (м, 1H, H2), 4.18−4.25 (м, 2H, H6a, H6b), 4.30−4.40 (м, 1H, H5), 5.16 (т, 1H, J 9.80, H4), 5.26-5.32 (м, 1H, H3), 5.51 (дд, 1H, J1,P 7.34, J1,2 3.22, H1), 7.06 [д, 1H, J2,NH 9.35, NHC(O)CH3], 11.98 (уш.с, 1H, HN+). Спектр 13C-ЯМР: 8.7 (CH3CH2N+), 14.1 (C10'), 20.8, 22.7, 23.0 [3 C(O)CH3, CH3C(O)NH], 25.9, 27.4, 29.4, 29.5, 29.6, 29.7, 31.9 (C2'−C9'), 45.7 (CH2N+), 52.2 (C2), 61.9 (C6), 66.2 (C1'), 68.4 (C4), 68.7 (C3), 71.5 (C5), 94.1 (d, C1, JC,P 5.4), 169.5, 170.8, 171.0, 182.2 (4 C=O). Спектр 31P-ЯМР: –2.27.
2-Ацетамидо-2-дезокси-3,4,6-три-О-ацетил-α-D-глюкопиранозилцетилфосфат триэтиламмония (XIV). $[\alpha ]_{D}^{{20}}$ +10.5 (c 0.846, CH2Cl2). Найдено, %: C 57.35; H 9.20; N 3.63; P 4.08. C36H69N2O12P. Вычислено, %: C 57.43; H 9.24; N 3.72; P 4.11. Масс-спектр MALDI-TOF MS: m/z 650.6 [M]–. Рассчитана М– 650.3 (C30H53NO12P–). Спектр 1Н-ЯМР: 0.86 (т, 3H, J 6.8, H16'), 1.19 (уш. с, 4H, H15', H14'), 1.22–1.27 (м, 22H, H3'−H13'), 1.36 [т, 9H, J 7.33, N+(CH2CH3)3], 1.55–1.65 (м, 2H, H2'), 1.94, 1.98, 1.99, 2.06 (все с, 12Н, 4 CH3CO), 3.07 [кв, 6Н, J 7.33, N+(CH2–CH3)3], 3.85−3.94 (м, 2H, H1'), 4.05−4.11 (м, 1H, H2), 4.17−4.27 (м, 2H, H6a, H6b), 4.32−4.40 (м, 1H, H5), 5.16 (т, 1H, J 9.68, H4), 5.29 (т, 1H, J 9.96, H3), 5.51 (дд, 1H, J1,P 7.18, J1,2 3.53, H1), 7.17 (д, 1H, J2,NH 9.64, NHC(O)CH3). Спектр 13C-ЯМР: 8.8 (CH3CH2N+), 14.2 (C16'), 20.8, 20.9 [3 C(O)CH3, NHC(O)CH3], 22.8, 25.9, 27.4, 29.5, 29.8, 32.0 (C2'−C15'), 45.8 (CH2N+), 52.3 (C2), 62.0 (C6), 63.6 (C1'), 68.7 (С3, C4, С5), 94.2 (C1), 169.5, 170.9, 182.6 (4 C=O). Спектр 31P-ЯМР: –2.43.
2,3,4,6-Тетра-О-ацетил-α-D-глюкопиранозил-Н-фосфонат триэтиламмония (XVIII). К раствору 6.2 г (17.8 ммоль) тетраацетата глюкопиранозы (XVII) [15] и 17.3 мл триэтиламина (124.6 ммоль) в 35 мл THF добавляли при перемешивании 3.23 г (16 ммоль) салицилхлорфосфита (IX) [13], перемешивали 9 ч, добавляли 15.4 мл воды и перемешивали еще 1 ч. Реакционную смесь упаривали досуха, полученный остаток хроматографировали на силикагеле (элюент − CH2Cl2−CH3OH, 50 : 1 с добавлением 1% Et3N по объему). Получили 4 г соединения (XVIII) (44%) в виде гигроскопичного белого порошка, $[\alpha ]_{D}^{{20}}$ +48.4 (c 0.9, CHCl3). Найдено, %: C 46.82; H 7.11; N 2.81; P 6.00. C20H36NO12P. Вычислено, %: C 46.78; H 7.07; N 2.73; P 6.03. Масс-спектр MALDI-TOF MS: m/z 513.2 [M]. Рассчитана М 513.2 (C20H36NO12P). Спектр 1Н-ЯМР: 1.28 [т, 9H, J 7.30, N+(CH2CH3)3], 1.96, 1.98, 2.01, 2.04 (все с, 12Н, 4CH3CO), 3.0 [кв, 6Н, J 7.30, N+(CH2CH3)3], 4.07 (дд, 1H, J6a,6b 12.4, J6a,5 2.3, H6a), 4.19 (дд, 1H, J6a,6b 12.4, J6b,5 3.39, H6b), 4.26−4.30 (м, 1H, H5), 4.90−4.94 (м, 1H, H2), 5.07 (т, 1H, J 9.8, H4), 5.50 (т, 1H, J 9.8, H3), 5.75 (дд, 1H, J1,P 8.9, J1,2 3.4, H1), 6.91 (д, 1H, J1,P 638.9, P–H). Спектр 13C-ЯМР: 8.5 (CH3CH2N+), 20.49, 20.57, 20.59, 20.61 [4 C(O)CH3], 45.7 (CH2N+), 61.6 (C6), 68.2 (C3), 68.3 (C4), 70.1 (C5), 70.5 (C2), 91.1 (C1), 169.5, 169.8, 169.9, 170.6 (4 C=O). Спектр 31P-ЯМР: +0.77.
Общая методика синтеза гликофосфатов (XX), (XXII). К охлажденному до –20°С раствору Н-фосфоната (XVIII) (1 экв.) в 10 мл пиридина добавляли при перемешивании сначала 2 экв. декан-1-ола или гексадекан-1-ола, затем раствор 4.3 экв. пивалоилхлорида в 5 мл пиридина. Перемешивали 1 ч, затем добавляли 1 мл воды и 1 экв. йода. Продолжали перемешивание 2 ч, затем по каплям добавляли 1 М раствор Na2S2O3 до исчезновения окраски йода. Реакционную смесь соломенно-желтого цвета отгоняли досуха, полученный остаток хроматографировали на силикагеле), после чего выделяли гликофосфаты (XX) (53%, бесцветное масло) и (XXII) (35%, бесцветное масло).
2,3,4,6-Тетра-О-ацетил-α-D-глюкопиранозилдецилфосфат триэтиламмония (XX). Фосфат (XX) выделяли флеш-хроматографией на силикагеле (элюент CH2Cl2/CH3OH, 100 : 1 → 100 : 2, 1 об. % Et3N). Выход 53%, бесцветное масло; $[\alpha ]_{D}^{{20}}$ +18.2° (с 1.00, CH2Cl2). Найдено,%: C 53.79; H 8.49; N 2.05; P 4.68. C30H56NO13P. Вычислено, %: C 53.80; H 8.43; N 2.09; P 4.62. Масс-спектр MALDI-TOF MS: m/z 567.1 [M]–. Рассчитана М– 567.2 (C24H40O13P–). Спектр 1Н-ЯМР: 0.86 (т, 3Н, J 6.9, H10'), 1.18−1.23 (м, 14Н, H3'−H-7'), 1.31 [т, 9H, J 7.3, N+(CH2CH3)3], 1.39–1.44 (м, 2Н, H-8'), 1.56–1.65 (м, 2Н, H2'), 1.98, 1.99, 2.02, 2.05 (все с, 12Н, 4CH3CO), 3.07 [кв, 6Н, J 7.3, N+(CH2CH3)3], 3.84–3.95 (м, 2H, H1'), 4.05–4.34 (м, 3H, H5, H6a, H6b), 4.89–4.96 (м, 1H, H2), 5.11 (т, 1H, J 9.8, H4), 5.52 (т, 1H, J 9.8, H3), 5.72 (дд, 1H, J1,P 7.8, J1,2 3.3, H1). Спектр 13C-ЯМР: 8.5 (CH3CH2N+), 14.1 (C10'), 20.6, 20.7 [4 C(O)CH3], 22.6, 25.7, 27.1, 29.3, 29.4, 29.6, 30.8, 31.9 (C2'–C9'), 45.6 (CH2N+), 61.6 (C6), 67.9 (C1'), 68.3 (C4), 68.3 (C3), 70.2 (C5), 70.3 (C2), 91.9 (C1), 169.6, 170.1, 170.7 (4 C=O). Спектр 31P-ЯМР: –2.69.
2,3,4,6-Тетра-О-ацетил-α-D-глюкопиранозилцетилфосфат триэтиламмония (XXII). Фосфат (XXII) выделяли флеш-хроматографией на силикагеле (элюент CH2Cl2/CH3OH, 100 : 1 → 100 : 1.5, 1 об. % Et3N). Выход 35%, бесцветное масло; $[\alpha ]_{D}^{{20}}$ +11.2° (с 1.00, CH2Cl2). Найдено, %: C 57.41; H 9.01; N 1.78; P 4.15. C36H68NO13P. Вычислено, %: C 57.35; H 9.09; N 1.86; P 4.11. Масс-спектр MALDI-TOF MS: m/z 675.2 [M + Na]+. Рассчитана [M + + Na]+ 675.3 (C30H53NaO13P+). Спектр 1Н-ЯМР: 0.75 (т, 3Н, J 7.0, H16’), 1.03−1.23 (м, 26Н, H3'−H15'), 1.28 [т, 9H, J 7.3, N+(CH2–CH3)3], 1.45–1.53 (м, 2Н, H2'), 1.88, 1.90, 1.92, 1.95 (все с, 12Н, 4CH3CO), 3.07 [кв, 6Н, J 7.3, N+(CH2CH3)3], 3.52–3.76 (м, 2H, H1'), 3.96–4.16 (м, 3H, H5, H6a, H6b), 4.76–4.80 (м, 1H, H2), 4.98 (т, 1H, J 9.7, H4), 5.38 (т, 1H, J 9.6, H3), 5.57 (дд, 1H, J1,P 7.9, J1,2 3.5, H1). Спектр 13C-ЯМР: 8.7 (CH3CH2N+), 14.2 (C10'), 20.8 [C(O)CH3], 22.8, 25.9, 27.4, 29.5, 29.6, 29.8, 30.1, 30.8, 32.0 (C2'–C15'), 45.9 (CH2N+), 61.8 (C6), 66.2 (C1'), 68.5 (C4), 68.9 (C3), 70.3 (C5), 70.8 (C2), 92.0 (C1), 169.8, 170.3, 170.9 (4 C=O). Спектр 31P-ЯМР: –2.31.
3,4,6-Три-О-ацетил-2-дезокси-2-ацетамидо-β-D-глюкопиранозил-О-фосфат триэтиламмония (XXV). К раствору 1.69 г (5.13 ммоль) оксазолина (XXIII) [17] в 37 мл 1,2-дихлорэтане добавляли 1.71 г (6.16 ммоль) дибензилфосфата, реакционную смесь перемешивали при комнатной температуре в течение 19 ч. Реакционную смесь концентрировали и сушили в вакууме. Полученный продукт (XXIV) использовали далее без дальнейшей очистки. В 50 мл метанола растворяли 1.7 г (2.8 ммоль) фосфата (XXIV) и добавляли 1.6 г 10% Pd/C в атмосфере аргона. Раствор гидрировали при энергичном перемешивании 3 ч при комнатной температуре, фильтровали через целит. Целит промывали горячим метанолом (10 мл) и триэтиламином (0.4 мл). Растворители удаляли в вакууме, остаток промывали смесью CH2Cl2/диэтиловый эфир. Осадок отфильтровывали, фильтрат упаривали в вакууме. Получили 0.4 г (27%) соединения (XXV) в виде бесцветного сиропа, $[\alpha ]_{D}^{{20}}$ + 34.4 (c 0.9, CH3OH). Найдено, %: C 45.51; H 7.01; N 5.23; P 5.89. C20H37N2O12P. Вычислено, %: C 45.45; H 7.06; N 5.30; P 5.86 . Масс-спектр MALDI-TOF MS: m/z 426.3 [M]– . Рассчитана М– 426.1 (C14H21NO12P–). Спектр 1Н-ЯМР: 1.17 [т, 9H, J 7.3, N+(CH2CH3)3], 1.86, 1.87, 1.89, 1.95 (все с, 12Н, CH3CO), 2.95 (кв, 6Н, J 7.3, N+(CH2CH3)3), 3.91−3.99 (м, 1Н, H6a), 4.05–4.17 (м, 3H, H2, H5, H6b), 4.98 (т, 1H, J 9.5, H4), 5.08 (д, 1H, J 3.2, H3), 5.18 (т, 1H, J 10, H1), 6.44 (д, 1H, J2,NH 9.5, NH), 9.37 (уш. с, 1H, N+H). Спектр 13C-ЯМР: 8.7 (CH3CH2N+), 20.7, 20.8, 22.8 [3 C(O)CH3 ], 23.2 (CH3C(O)NH), 45.5 (CH2N+), 52.6 (C2), 62.4 (C6), 67.3 (C4), 68.7 (C3), 71.6 (С5), 91.6 (d, J 6.7, C1), 169.6 (NHC=O), 170.7, 170.9, 171.4 (3 C=O). Спектр 31P- ЯМР: +1.94.
Биологическая активность. Антимикробную активность соединений (VI), (XII), (XIV), (XX), (XXII), (XXV), (XXVI) изучали методом серийных разведений в жидких питательных средах по методикам [24, 25], определяя минимальную ингибирующую концентрацию (МИК), вызывающую задержку роста и размножения тест-микроорганизмов. Использовали культуры грамположительных бактерий: Staphylococcus aureus АТСС 209p, Bacillus cereus АТСС 8035; грамотрицательных бактерий Escherichia coli CDC F-50, Pseudomonas aeruginosa ATCC 9027 и грибов Aspergillus niger BKMF-1119, Trichophyton mentagrophytes var. gypseum 1773, Candida albiсans 855-653. В качестве контроля использовали антибиотик Хлорамфеникол.
Рис. 1.
Некоторые представители ингибиторов ферментов, участвующих в биосинтезе пептидогликанов бактерий и микобактерий. R = пренил (–C5H9); фарнезил (–C15H25); феноксигексил (–C6H12OPh); децил (–C10H21); феноксиундецил (–C11H22OPh); цетил (–C16H33); феноксицетил (–C16H32OPh).
Изучение антитуберкулезной активности соединений (VI), (X), (XII), (XIV), (XX), (XXII), (XXV), (XXVI) проводили методом вертикальной диффузии [26] на плотной питательной среде “Новая” с использованием лабораторного штамма Mycobacterium tuberculosis H37Rv (МБТ). Питательную среду разливали в пробирки по 5 мл, засевали по 0.1 мл суспензией микобактерий разведенной по стандарту мутности 10 ед. ГКИ, и помещали в термостат на 24 ч для выращивания МБТ. Через сутки пробирки ставили в вертикальное положение и по свободному краю пробирки закапывали по 0.3 мл субстанции соединений (VI), (X), (XII), (XIV), (XX), (XXII), (XXV), (XXVI) в водном растворе DMSO в концентрациях 12.5, 6.2, 3.1, 1.5, 0.7, 0.35, 0.1 мкг/мл. Затем пробирки помещали в термостат и выдерживали в стерильных условиях при температуре 37°С в течение 10 суток. Оценку роста МБТ проводили по стандартной методике, согласно которой появление зон задержки роста МБТ более 10 мм свидетельствует о наличии туберкулостатических свойств. В качестве контроля использовали противотуберкулезные препараты Изониазид и Пиразинамид, ингибировавшие рост МБТ при МИК 0.1 мкг/мл и 12 мкг/мл соответственно.
Список литературы
van Heijenoort J. // Nat. Prod. Rep. 2001. V. 18. P. 503–519.
Rani C., Khan I.A. // Eur. J. Pharm. Sci. 2016. V. 83. P. 62–70.
Derouaux A., Sauvage E., Terrak M. // Frontier Immun. 2013. V. 4. P. 1–6.
Dumbre S., Derouaux A., Lescrinier E., Piette A., Joris B., Terrak M., Herdewijn P. // J. Amer. Chem. Soc. 2012. V. 134. P. 9343–9351.
Montoya-Peleaz P.J., Riley J.G., Szarek W.A., Valvano M.A., Schutzbach J.S., Brockhausen I. // Bioorg. Med. Chem. Lett. 2005. V. 15. P. 1205–1211.
Brockhausen I., Larsson E.A., Hindsgaul O. // Bioorg. Med. Chem. Lett. 2008. V. 18. P. 804–807.
Riley J.G., Xu C., Brockhausen I. // Carbohydr. Res. 2010. V. 345. P. 586–597.
Винникова А.Н., Торгов В.И., Уткина Н.С., Веселовский В.В., Дружинина Т.Н., Ванг С., Брокхаузен И., Данилов Л.Л. // Биоорган. химия. 2015. Т. 41. № 1. С. 121–123. [Vinnikova A.N., Torgov V.I., Utkina N.S., Veselovsky V.V., Druzhinina T.N., Wang S., Brockhausen I., Danilov L.L. // Russ. J. Bioorg. Chem. 2015. V. 41. P. 105–107.]
Li Y., Zhou Y., Mac Y., Li X. // Carbohydr. Res. 2011. V. 346. P. 1714–1720.
Cao Z., Qu Y., Zhou J., Liu W., Yao G. // J. Carbohydr. Chem. 2015. V. 34. P. 28–40.
Gorityala B.K., Lu Z., Leow M.L., Ma J., Liu X.-W. // J. Amer. Chem. Soc. 2012. V. 134. P. 15229–15232.
Изместьев Е.С., Андреева О.В., Шарипова Р.Р., Кравченко М.А., Гарифуллин Б.Ф., Стробыкина И.Ю., Катаев В.Е., Миронов В.Ф. // Ж. орг. хим. 2017. Т. 53. № 1. С. 56–61. [Izmest’ev E.S., Andreeva O.V., Sharipova R.R., Kravchenko M.A., Garifullin B.F., Strobykina I.Yu., Kataev V.E., Mironov V.F. // Russ. J. Org. Chem. 2017. V. 53. P. 51–56.]
Young R.W. // J. Amer. Chem. Soc. 1952. V. 74. P. 1672–1673.
Filice M., Guisan J.M., Terreni M., Palomo J.M. // Nat. protocols. 2012. V. 7. P. 1783–1796.
Fusari M., Fallarini S., Lombardi G., Lay L. // Bioorg. Med. Chem. 2015. V. 23. P. 7439–7447.
Chen C., Liu B., Xu Y., Utkina N., Zhou D., Danilov L., Torgov V., Veselovsky V., Feng L. // Carbohydr. Res. 2016. V. 430. P. 36–43.
Saneyoshi H., Yamamoto Y., Kondo K., Hiyoshi Y., Ono A. // J. Org. Chem. 2017. V. 82. P. 1796–1802.
Wang S., Czuchry D., Liu B., Vinnikova A.N., Gao Y., Vlahakis J.Z., Szarek W.A., Wang L., Feng L., Brockhausen I. // J. Bacteriol. 2014. V.196. P. 3122–3133.
Chauviere G., Bouteille B., Enanga B., de Albuquerque C., Croft S.L., Dumas M., Perie J. // J. Med. Chem. 2003. V. 46. P. 427–440.
Kyas A., Feigel M. // Helv. Chim. Acta. 2005. V. 88. P. 2375–2396.
Débieux J.-L., Cosandey A., Helgen C., Bochet C.G. // Eur. J. Org. Chem. 2007. P. 2073–2077.
Bock K., Guzman J.F.-B., Refn S. // Carbohydr. Res. 1992. V. 232. P. 353–357.
Chambers D.J., Evans G.R., Fairbanks A.J. // Tetrahedron. 2004. V. 60. P. 8411–8419.
National Committee for Clinical Laboratory Standards, Methods for dilution antimicrobial susceptibility. Tests for bacteria that grow aerobically. Sixth edition: Approved standard. M7–A5, NCCLS, Wayne, Pa., USA. 2000.
National Committee for Clinical Laboratory Standards, Reference method for broth dilution antifungal susceptibility testing of conidium–forming filamentous fungi: Proposed standard. M38–P, NCCLS, Wayne, Pa., USA. 1998.
Kudoh S., Kudoh T. // Kekkaku. 1973. V. 48. P. 501–512.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия