

Биоорганическая химия, 2019, T. 45, № 3, стр. 227-237
Никотиновые ацетилхолиновые рецепторы человека. Часть II: не-нейрональная холинергическая система
М. А. Шулепко 1, 2, М. Л. Бычков 2, Д. С. Кульбацкий 1, 2, Е. Н. Люкманова 1, 2, *
1 ФГБУН Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, д. 16/10, Россия
2 Биологический факультет МГУ им. М.В. Ломоносова
119234 Москва, Ленинские горы, д. 1, стр. 12, Россия
* E-mail: ekaterina-lyukmanova@yandex.ru
Поступила в редакцию 16.10.2018
После доработки 20.10.2018
Принята к публикации 24.10.2018
Аннотация
Широко известна роль никотиновых ацетилхолиновых рецепторов (nAChR) в функционировании центральной и периферической нервных систем. В последнее время появились данные об экспрессии некоторых подтипов nAChR в иммунных клетках и тканях эпителиального происхождения. Эти рецепторы не-нейронального происхождения вовлечены в регуляцию воспалительных процессов, а также в процессы клеточной пролиферации, дифференцировки, миграции и апоптоза. Не-нейрональные nAChR участвуют также в возникновении и развитии нескольких видов никотин-индуцированного рака. Изучение этих рецепторов в эпителиальных и раковых клетках позволило обнаружить новые способы регуляции nAChR с помощью эндогенных лигандов, а также предположить новые механизмы передачи внутриклеточного сигнала, не связанные с движением ионов через канал рецептора. В обзоре представлена современная информация о функции не-нейрональных nAChR человека.
ВВЕДЕНИЕ
Никотиновые ацетилхолиновые рецепторы (nAChR), помимо центральной и периферической нервных систем, экспрессируются в эпителиальных клетках, клетках иммунной системы, а также в митохондриях [1–3]. Такие рецепторы получили название не-нейрональных nAChR. Обнаружено, что не-нейрональные nAChR участвуют в развитии воспалительных процессов [4, 5] и поддержании гомеостаза клеток эпителия [1, 6]. Присутствующие в митохондриальной мембране nAChR регулируют через каскад сигнальных киназ начальные события апоптоза [3]. Не-нейрональные nAChR участвуют в развитии опухолей эпителиального происхождения [7]. Особенно хорошо изучена функция не-нейрональных nAChR в развитие рака легкого и опухолей желудочно-кишечного тракта при табакокурении [8].
По всей видимости, молекулярные механизмы функционирования не-нейрональных nAChR отличаются от таковых для nAChR в нервной системе. Значительную роль могут играть механизмы передачи сигнала, напрямую не связанные с открытием канала рецептора. Передача сигнала в этом случае может осуществляться посредством активации G-белков, связанных с внутриклеточным доменом рецептора, и путем активации других внутриклеточных мессенджеров [9].
Еще одним развивающимся направлением в исследовании nAChR человека в последние два десятилетия стало изучение модуляции его работы эндогенными лигандами. Эти лиганды относятся к семейству Ly6/uPAR, имеют “трехпетельную” структуру и могут быть прикрепленными к мембране с помощью GPI-якоря или секретируемыми [10]. Стоит отметить, что к семейству Ly6/uPAR в частности, относятся α-нейротоксины яда змей, классические инструменты изучения nAChR [11].
КОМПОНЕНТЫ НЕ-НЕЙРОНАЛЬНЫХ ХОЛИНЕРГИЧЕСКИХ СИГНАЛЬНЫХ СИСТЕМ
ACh не только играет роль медиатора в нервно-мышечных синапсах и ЦНС, но и выступает в качестве сигнальной молекулы в не-нейрональных тканях млекопитающих, бактериях, простейших и водорослях [4]. Рецепторы ACh присутствуют практически во всех тканях человеческого организма, и для полноценного функционирования холинергической сигнальной системы необходимы также компоненты, отвечающие за синтез ACh, его транспорт и деградацию (рис. 1, [12]).
Рис. 1.
Компоненты не-нейрональной холинергической системы, отвечающие за синтез, транспорт, деградацию и передачу сигналов. Ac – ацетат, Ac-CoA – ацетил-кофермент А, AСh – ацетилхолин, AChE – ацетилхолинэстераза, BChE – бутирилхолинэстераза, CarAT – каротин-ацетилтрансфераза, Ch – холин, ChAT – холин-ацетилтрансфераза, CHT-1 – высокоаффинный транспортер холина 1 типа, CTL – гомолог холинового транспортера, mAChR – мускариновый ацетилхолиновый рецептор, nAChR – никотиновый ацетилхолиновый рецептор, OCT – транспортер органических катионов, SLURP – секретируемые белки семейства Ly-6/uPAR, VAChT – везикулярный транспортер ацетилхолина (адаптировано из работы [12]).

Основным механизмом синтеза ACh в клетках нервной системы и вне ее является перенос холин-ацетилтрансферазой (ChAT) ацетильной группы с молекулы ацетил-кофермента А на молекулу холина, хотя в некоторых случаях (например, в скелетной мускулатуре), эта реакция может осуществляться карнитин-ацетилтрансферазой (CarAT). Скорость синтеза ACh в клетках лимитируется поступлением холина, который транспортируется внутрь клеток в нервной системе высокоаффинным транспортером холина 1 (CHT-1), а вне нервной системы – гомологами. CTL1, CTL2... CTL5 – это разные белки холинового транспортера CTL1-5 и транспортером органических катионов (OCT). В нейрональных клетках ACh запасается в везикулах с помощью специфического везикулярного транспортера ACh (VAChT), однако в не-нейрональных клетках, которые не экспрессируют VAChT, секреция ACh может происходить напрямую из цитоплазмы с помощью OCT и, предположительно, медиатофора [13]. Секретированный ACh взаимодействует с nAChR и мускариновыми ацетилхолиновыми рецепторами (mAChR), экспрессированными на поверхности клеток, а его избыток подвергается гидролизу с помощью внеклеточных ферментов ацетилхолинэстеразы (AChE) и бутирилхолинэстеразы (BChE).
ХОЛИНЕРГИЧЕСКИЕ МЕХАНИЗМЫ В ИММУННОЙ СИСТЕМЕ
В отличие от холинергических механизмов в нервной системе и в нервно-мышечных окончаниях, для клеток иммунной системы не характерно присутствие компонентов везикулярной системы транспорта ACh, что делает невозможным быструю передачу холинергического сигнала. Однако связь между холинергической передачей сигналов и иммунной системой была впервые продемонстрирована на примере электрической стимуляции блуждающего нерва, которая приводила к выделению значительных количеств ACh в селезенке [14]. Позднее было установлено, что в основных органах иммунной системы существуют нервные окончания, локализованные около мест скопления В-лимфоцитов, Т-лимфоцитов и макрофагов и идущие из ствола мозга и периферических нервных узлов, и, таким образом, автономная нервная система может принимать участие в регуляции воспалительных процессов [14]. Известно, что моноциты периферической крови содержат мРНК субъединиц α4, α5, α7, α9, α10, β1, β2 и β4 nAChR [5, 15].
Холинергическая компонента передачи сигнала обнаружена в финальной стадии воспалительного рефлекса (рис. 2), в котором афферентные нейроны активируются экзогенными и эндогенными продуктами воспаления, что, в свою очередь приводит к передаче эфферентных сигналов через блуждающий нерв в селезенку, где адренергические нейроны стимулируют Т-лимфоциты, способные секретировать ACh. Выброс ACh в селезенке стимулирует nAChR α7-типа (α7-nAChR) на поверхности макрофагов, что в свою очередь приводит к ингибированию активации ядерного фактора NF-κB и подавляет синтез и секрецию провоспалительного цитокина HMGB1 [16]. Кроме того, при стимуляции блуждающего нерва во время сепсиса, выброс ACh Т-клетками способствует подавлению синтеза провоспалительного цитокина TNFα [17]. Нарушения данного механизма приводят к развитию хронических воспалительных реакций [2]. Например, у мышей, нокаутных по гену, кодирующему субъединицу α7-nAChR (CHRNA7), при введении эндотоксинов наблюдался повышенный уровень провоспалительных цитокинов TNFα, IL-1β и IL-6, при этом стимуляция блуждающего нерва не снижала уровень провоспалительных цитокинов [18]. В связи с этим интересно отметить, что обработка никотином приводит к значительному снижению экспрессии субъединиц α3, α5, α6 и α7 nAChR в Т-лимфоцитах [19].
Рис. 2.
Воспалительный рефлекс. Пример рефлекторной цепи, регулирующей воспалительный процесс, которая состоит из афферентных и эфферентных сигналов, передаваемых через блуждающий нерв в ответ на молекулярные маркеры инфекций и повреждений. Активация адренергических нейронов в селезенке приводит к высвобождению норадреналина в непосредственной близости от Т-клеток, которые способны секретировать ACh в ответ на активацию β-адренергических рецепторов (β2-АR). ACh взаимодействует с α7-nAChR на поверхности макрофагов, что активирует рецептор и ингибирует синтез и секрецию цитокинов TNFα, IL-1, IL-18 и HMGB1 (рисунок адаптирован из работы [14]).

Стимуляция блуждающего нерва с помощью ACh приводит к ингибированию активации макрофагов селезенки по JAK2/STAT3-сигнальному пути [20]. Более того, стимуляция блуждающего нерва может влиять на иммунный ответ, останавливая миграцию В-лимфоцитов и подавляя продукцию ими антител [21]. В настоящее время холинергический контроль воспалительных процессов рассматривается в качестве перспективной стратегии для терапии некоторых патологий. Например, показано, что стимуляция блуждающего нерва с помощью ACh сокращает воспаление при овариэктомии у крыс за счет ингибирования NF-κB [22].
Холинергическая система, помимо участия в контроле секреции различных цитокинов, принимает также участие и в регуляции дифференцировки CD4+-Т-клеток. Так, снижение экспрессии β2- и β4-субъединиц nAChR и mAChR М3-типа приводит к дифференцировке CD4+-T-лимфоцитов в Т-хелперные клетки I типа [15]. Кроме того, показано, что nAChR регулируют уровень экспрессии CD40 в В-лимфоцитах, контролируя тем самым их активацию [23].
Регуляция клеточных процессов в иммунных клетках, возможно, осуществляется через nAChR-опосредованные метаботропные пути передачи сигнала. Показано, что в Т-лимфоцитах при стимуляции никотином происходит высвобождение ионов Ca2+ из внутриклеточных депо, при этом антагонисты α7-nAChR не влияют на этот процесс [24]. Возможно, именно передача сигнала через α7-nAChR по метаботропному пути способствует активации JAK2/STAT3-сигнального пути в иммунных клетках, что в итоге приводит к ингибированию NF-κB и ингибированию продукции провоспалительных цитокинов [9, 25].
ХОЛИНЕРГИЧЕСКАЯ СИГНАЛЬНАЯ СИСТЕМА ЛЕГКИХ
Ткани легкого содержат как компоненты классической нейрональной холинергической системы (в парасимпатических нервах), так и не-нейрональной холинергической системы. Существование не-нейрональной системы в легком подтверждается тем фактом, что культуры мелкоклеточного и не-мелкоклеточного рака легкого могут синтезировать и секретировать ACh, что приводит к аутокринной регуляции роста клеток через активацию nAChR и mAChR [6, 26]. Как и в случае иммунной системы, в клетках легочного эпителия секреция ACh, в основном, происходит из цитоплазмы через относительно медленные и неселективные транспортеры OCT. Скорость и направление транспорта ACh через ОСТ зависит от градиента концентрации ACh, что делает не-нейрональную холинергическую систему в легких более приспособленной для передачи длительных сигналов небольшой интенсивности. Показано, что регуляция работы α7-nAChR, например, с помощью трехпетельного эндогенного белка человека Lynx1, способствует контролю секреции муцина посредством снижения фосфорилирования киназы Src и уменьшения экспрессии мРНК GABAА-рецепторов и белка MUC5A, участвующего в формировании слизистых выделений [27]. Таким образом, α7-nAChR является перспективной мишенью для лечения заболеваний, связанных с курением и сопутствующей этому гиперсекреции муцина.
ХОЛИНЭРГИЧЕСКАЯ РЕГУЛЯЦИЯ ГОМЕОСТАЗА КЛЕТОК ЭПИТЕЛИЯ
Эпителий выполняет барьерную функцию, и передача внутри- и межклеточных сигналов посредством адренергической и холинергической систем играет важную роль в поддержании его гомеостаза. ACh участвует в регуляции многих процессов в эпителиальных клетках: пролиферации, миграции, дифференцировки и апоптоза [1]. В ходе дифференцировки кератиноцитов экспрессия субъединиц nAChR, экспонированных на поверхности цитоплазматической мембраны, меняется, что необходимо для обеспечения различных функций клеток в разных слоях эпидермиса. В клетках базального слоя обнаружены α3, α5, α7, α9, α10, β2 и β4 субъединицы nAChR, в нижних клетках шипикового слоя экспрессируется в основном α9 субъединица, а в верхних клетках шипикового слоя экспрессируются также α7, α10 и β1 субъединицы nAChR (рис. 3а). В зернистом слое эпидермиса присутствуют α7, α10 и β1 субъединицы nAChR [28]. На экспрессию nAChR в эпидермисе оказывают влияние различные факторы: возраст, курение, кожные аллергические реакции. Этим могут объясняться различия в данных о локализации nAChR, приведенные в работах разных авторов [29]. Следует отметить, что функциональные nAChR представляют собой гомо- и гетерорецепторы, состоящие из разных субъединиц. В базальном слое присутствуют α3β2-, α3β4- и α9-nAChR, в шипиковом слое содержится больше α5-содержащих гетерорецепторов α3(β2/β4)α5-nAChR и α9-nAChR. В зернистом слое особую роль играет гомопентамерный α7-nAChR [1].
Рис. 3.
Влияние nAChR на гомеостаз клеток эпителия. (а) Экспрессия различных типов субъединиц nAChR в слоях эпидермиса [1, 28]. Эндогенный регулятор SLURP-2 влияет на физиологические процессы кератиноцитов, взаимодействуя с различными подтипами nAChR и mAChR [40]. (б) Эффекты, оказываемые nAChR на разных стадиях развития кератиноцитов (преобладающий тип рецептора/биологический эффект) [1, 29, 30].

Известно, что ингибирование α7-nAChR приводит к остановке миграции кератиноцитов [30], а ингибирование экспрессии α3 и α9 субъединиц nAChR и mAChR М3-типа, нарушает формирование межклеточных контактов и адгезию кератиноцитов (рис. 3б, [31]).
α7-nAChR играет важную регуляторную роль также и в дифференцировке клеток эпидермиса (рис. 3б): ингибирование передачи сигнала через этот рецептор вызывает остановку терминальной дифференцировки кератиноцитов и способствует продолжению деления клеток [32]. Наконец, на поздних стадиях развития клеток кожи активация α7- и α9-содержащих nAChR и ингибирование mAChR приводят к апоптозу кератиноцитов, что сопровождается увеличением секреции ACh [33]. Холинергическая система играет также важную роль в проявлении реакции кератиноцитов на стресс, индуцированный механическими повреждениями или инфекцией. Так, показано, что при инфекции активация α7-nAChR подавляет экспрессию антимикробных пептидов кератиноцитами [34]. Обобщенная схема участия nAChR в физиологии кератиноцитов приведена на рис. 3б.
nAChR регулируют гомеостаз не только клеток эпидермиса, но и фибробластов дермы. Известно, что активация α3-содержащих nAChR регулирует клеточный цикл (повышая экспрессию белка р21) и процессы апоптоза (за счет активации каспазы-3 и антиапоптотического белка bcl-2) фибробластов дермы [35]. Кроме того, при активации nAChR никотином усиливается экспрессия коллагена α1, эластина и металлопротеиназы матрикса ММР-1, что указывает на вовлечение никотиновых рецепторов в контроль не только пролиферации фибробластов дермы, но и ремоделирования этими клетками своего микроокружения [35].
Предполагается, что холинергическая регуляция гомеостаза эпителия осуществляется посредством как ионотропного (в результате открытия канала и возникновения тока ионов кальция через мембрану клетки), так и метаботропного (активация внутриклеточных сигнальных каскадов без открытия канала рецептора) путей передачи сигналов через nAChR. Вторичными мессенджерами ионотропного сигналинга являются кальмодулин-зависимая киназа CaMKII, протеинкиназа С и PI3K/AKT-сигнальный путь [30], метаботропный путь опосредован RAS/RAF-1/MEK1/ERK-киназным каскадом [36].
ЭНДОГЕННЫЕ АУТО/ПАРАКРИННЫЕ РЕГУЛЯТОРЫ ГОМЕОСТАЗА КЛЕТОК ЭПИТЕЛИЯ SLURP
Основным активатором nAChR в клетках эпителия является ACh, однако в регуляции работы холинергической системы могут принимать участие также и эндогенные модуляторы nAChR. Так, секреция в межклеточное пространство позволяет белкам человека SLURP действовать в качестве ауто/паракринных регуляторов, контролирующих различные физиологические процессы как самих секретирующих клеток, так и их соседей, в том числе находящихся на значительном расстоянии (рис. 4а, [37]). На линии кератиноцитов слизистой оболочки полости рта Het1-A показано, что SLURP-1 демонстрирует антипролиферативную активность, избирательно взаимодействуя с α7-nAChR [38, 39]. В то же время SLURP-2 способен замедлять или ускорять рост кератиноцитов, взаимодействуя с nAChR и mAChR различных подтипов [40]. Возможно, SLURP-2 является универсальным регулятором клеточных процессов эпителия, взаимодействуя с ацетилхолиновыми рецепторами различных подтипов на разных стадиях клеточного развития (рис. 3а, [40]). Кроме того, показано, что белки SLURP, регулируя экспрессию различных интегринов, ускоряют заживление ран на коже и слизистых оболочках [41].
Рис. 4.
Влияние SLURP-1 на гомеостаз клеток эпителия. (а) Схема секреции SLURP-1 из внутриклеточных депо в ответ на присутствие SLURP-1 во внеклеточной среде. Предполагается, что данный механизм лежит в основе ауто/паракринной регуляции клеточного гомеостаза с помощью SLURP-1 [43]. (б) Механизм регуляции экспрессии NF-κB в кератиноцитах посредством SLURP-1. Активация α7-nAChR с помощью SLURP-1 приводит к усилению экспрессии гена NF-κB за счет активации сигнального каскада RAF-1/MEK1/ERK, которая происходит по метаботропному пути вследствие фосфорилирования JAK2 и по Ca2+-зависимому пути, вследствие активации CaMKII/PKC [42]. NF-κB – ядерный фактор “каппа-би”, JAK – Янус-киназа, RAS – малая GTP-аза, RAF-1 – cерин-терониновая протеинкиназа, MEK – митоген активируемая киназа, ERK – внеклеточные регулируемые сигналом киназы, CaMKII – Ca/кальмодулин-зависимая киназа, PKC – протеинкиназа С.

Один из предполагаемых путей передачи сигнала в кератиноцитах связан с фосфорилированием тирозинкиназы JAK2, ассоциированной с α7-nAChR. Показано, что взаимодействие SLURP-1 с α7-nAChR приводит к активации сигнального каскада JAK2/RAS/RAF-1/MEK/ERK по неионному механизму, и Ca2+-опосредованной активации сигнального каскада CaMKII/PKC [42]. В результате происходит увеличение экспрессии транскрипционного фактора NF-κB, регулятора воспалительных и пролиферативных клеточных процессов, который принимает участие также и в дифференцировке клеток (рис. 4б, [42]). Эффекты SLURP-1 сохраняются и при ингибировании тока через канал α7-nAChR с помощью специфического антагониста α-бунгаротоксина, что также указывает на наличие неионного пути холинергической регуляции гомеостаза эпителия [38].
Следует отметить, что белки SLURP вовлечены в патогенез многих заболеваний кожи: точечные мутации в гене SLURP-1 связаны с развитием кератодермы Mal de Meleda [44], гиперэкспрессия SLURP-2 обнаружена при псориазе [45], а нокаут гена SLURP-2 приводит к развитию ладонно-подошвенной кератодермы [46].
НИКОТИНОВЫЕ РЕЦЕПТОРЫ АЦЕТИЛХОЛИНА В РАЗВИТИИ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ
Известно, что табакокурение способствует развитию опухолей легкого, шеи, желудка, поджелудочной железы, желчного пузыря, печени, толстой кишки, молочной железы, шейки матки, мочевого пузыря и рака почек [7]. Один из предполагаемых молекулярных механизмов развития и прогрессии рака, вызванного табакокурением, включает прямую активацию α7-nAChR никотином и его производными–нитрозаминами (NNK и NNN), также содержащимися в табачном дыме [47]. Например, NNK и NNN индуцируют злокачественную трансформацию кератиноцитов путем активации nAChR [37]. Активация α7-nAChR приводит к экспрессии эпидермального фактора роста (EGF), васкулярного эпидермального фактора роста (VEGF) и норадреналина, которые в свою очередь активируют рецепторы EGFR, VEGFR и β-адренорецепторы, стимулируя таким образом митогенные и антиапоптотические сигнальные пути в опухолевых клетках [7]. Кроме того показано, что воздействие никотина на кератиноциты может приводить к увеличению экспрессии α5- и α7-содержащих nAChR, усиливая тем самым ответ клеток на последующую стимуляцию никотином [48].
Активация nAChR в опухолевых клетках индуцирует увеличение концентрации внутриклеточного кальция, что приводит к запуску различных сигнальных путей с участием β-аррестина, киназ Src, ERK, RAF-1 [49, 50], PKC [51], JAK2 [52], а также и транскрипционных факторов NF-κB, E2F, Sp1, GATA4, GATA6 и STAT3 [49–53]. Эти внутриклеточные сигнальные пути активируют транскрипцию генов, активирующих пролиферацию и способствующих прохождению клетки через контрольные точки клеточного цикла, и запускают синтез многих антиапоптотических белков, включая ингибитор каспаз XIAP [54]. Таким образом, при активации nAChR никотином и нитрозаминами в опухолевых клетках усиливается пролиферация и вырабатывается устойчивость к апоптозу (рис. 5).
Рис. 5.
Роль α7-nAChR в развитии и прогрессии злокачественных неоплазий. Активация α7-nAChR увеличивает чувствительность опухолевых клеток к никотину и может стимулировать передачу сигналов через рецепторы факторов роста, запускающие митогенные и антиапоптотические сигнальные пути в клетках. Активация α7-nAChR также вызывает секрецию проонкогенных нейромедиаторов и контролирует миграцию опухолевых клеток. Воздействие эндогенных ингибиторов nAChR может снизить онкогенные эффекты, возникающие при активации α7-nAChR [43]. АKT – серин/треониновая протеинкиназа Rac-α, β-AR – β-адренергические рецепторы, cAMP– циклоаденозинмонофосфат, СОХ-2 – циклооксигеназа 2, EGFR – рецептор эпидермального фактора роста, ERK – внеклеточные регулируемые сигналом киназы, Jak – Янус-киназа, NF-κB – ядерный фактор “каппа-би”, PGE – простагландин Е, PI3K – фосфоинозитид-3-киназа, РКА – протеинкиназа А, РКС – протеинкиназа С, RAF – cерин-терониновая протеинкиназа, RAS – малые GTP-азы, SLURP-1/2 – секретируемые белки семейства Ly-6/uPAR, Src – киназа семейства SFK, STAT – передатчик сигнала и активатор транскрипции, VEGFR – рецептор фактора роста эндотелия сосудов, XIAP – Х-связанный ингибитор терминальных каспаз.

nAChR могут играть важную роль не только в патогенезе солидных опухолей, но и в развитии лейкемий. ACh, секретируемый злокачественно трансформированными Т-лимфоцитами, может служить аутокринным фактором роста и регулировать пролиферацию клеток лейкемии посредством изменения кальциевой динамики в клетках. Кроме того, в Т-клетках лейкемии значительно снижена активность ацетилхолинэстеразы, что дополнительно усиливает сигналы ACh. Однако, в целом считается, что большую роль в развитии лейкемий играют не nAChR, а mAChR [55].
Кроме контроля роста опухолевых клеток, nAChR участвуют также в регуляции их миграции (метастазирования). Было показано, что активация α7-nAChR никотином вызывает эпителиально-мезенхимальный переход и миграцию опухолевых клеток различных опухолевых линий [56]. Также известно, что нокдаун гена α3-nAChR усиливает миграцию клеток опухоли пищевода [57], а нокдаун гена α5-nAChR увеличивает скорость миграции клеток карциномы легкого [58]. Интересно отметить, что нокдаун гена α7-nAChR в ассоциированных с опухолью макрофагах усиливает миграцию клеток колоректальных карцином [59]. По-видимому, nAChR не только играют важную роль в контроле миграции опухолевых клеток, но и обеспечивают регуляцию подвижности клеток опухоли макрофагами [59]. Для активации миграции под действием никотина могут быть использованы различные внутриклеточные сигнальные пути. Например, миграция клеток карциномы толстой кишки опосредуется активацией МАР-киназы р38 [60], а для регуляции метастазирования клеток рака кишечника в макрофагах запускается JAK2/STAT3-сигнальный путь [59].
Активация α7-nAChR никотином и нитрозаминами не только стимулирует пролиферацию и миграцию клеток опухолей, но и способствует росту новых сосудов в опухолевой массе (ангиогенезу). Рост новых сосудов необходим для снабжения быстроделящейся опухолевой массы кислородом и ростовыми факторами и, в итоге, приводит к выходу открепившихся опухолевых клеток в просвет сосудов и метастазированию в различные органы. Было показано, что никотин усиливает рост сосудов в опухоли желудка in vivo посредством активации ERK1/2, циклооксигеназы-2 (СОХ-2) [60] и простагландина Е [62].
Интересно отметить, что эндогенный белок человека Lynx1, также относящийся к семейству Ly6/uPAR [10], является регулятором никотин-индуцированного увеличения уровня экспрессии α7-nAChR в раковых клетках легкого [27, 63]. Более того, показано, что снижение уровня экспрессии Lynx1 приводит к стимуляции роста клеток карциномы легкого, а направленное увеличение экспрессии Lynx1 с помощью лентивирусной трансдукции приводит, напротив, к снижению пролиферации раковых клеток [63].
БЕЛКИ SLURP КОНТРОЛИРУЮТ РОСТ РАКОВЫХ КЛЕТОК ЭПИТЕЛИАЛЬНОГО ПРОИСХОЖДЕНИЯ
У пациентов с кератодермой Mal de Meleda, заболеванием, причиной которого являются мутации в гене SLURP-1, зачастую развиваются также плоскоклеточная карцинома кожи и меланома [64]. Показано, что в опухолевых клетках кожи экспрессия гена SLURP-1 снижена, и это снижение коррелирует со стадией течения опухолевого заболевания [64]. Снижение экспрессии гена SLURP-1 также наблюдается при раке кишечника [65] и легкого [66], а снижение экспрессии генов обоих белков SLURP-1 и SLURP-2 обнаружено в кератиноцитах при онкогенной трансформации, индуцированной нитрозаминами NNK и NNN [67]. При этом онкогенный эффект нитрозаминов может быть снижен в результате предварительной инкубации кератиноцитов с рекомбинантными препаратами SLURP или суперпродукции SLURP [67, 68]. Кроме того, показано, что инкубация эпителиальных клеток с рекомбинантным SLURP-1 отменяет вызванное нитрозаминами увеличение экспрессии онкогенов [69, 70]. Таким образом, белки SLURP являются протекторами при нитрозамин-индуцированной онкогенной трансформации эпителиальных клеток.
Недавно показано, что SLURP-1 способен подавлять миграцию и инвазивность клеток рака поджелудочной железы (PDAC) и что этот эффект опосредован взаимодействием SLURP-1 именно с α7-nAChR. Относительно высокий уровень белка SLURP-1 в крови у пациентов с данным типом рака коррелирует с большей продолжительностью жизни после удаления первичной опухоли [71].
Перспектива использования рекомбинантных препаратов SLURP для подавления роста раковых клеток эпителиального происхождения недавно была продемонстрирована на ряде клеточных линий карцином кожи, легкого, кишечника и груди [43, 72]. Сравнение антипролиферативной активности SLURP-1 по отношению к раковым клеткам и нормальным кератиноцитам выявило наличие “фармакологического окна” (рис. 6). На клетках аденокарциномы кожи A431 показано, что рекомбинантный SLURP-1 вызывает снижение экспрессии α7-nAChR на поверхности раковых клеток и высвобождение эндогенного SLURP-1 из внутриклеточных депо во внеклеточное пространство [43]. Это, по-видимому, приводит к стимуляции соседних клеток по паракринному механизму, вызывая секрецию SLURP-1 и усиливая, таким образом, полученный сигнал (рис. 4а).
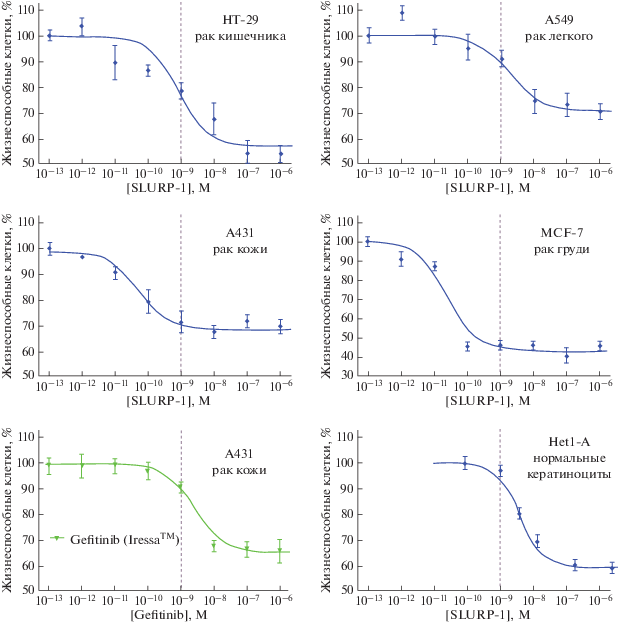
Рис. 6.
Дозозависимые кривые влияния SLURP-1 на пролиферацию линий раковых клеток HT-29, А549, A431 и MCF7. Отсутствие заметного антипролиферативного эффекта SLURP-1 на линию нормальных кератиноцитов Het-1A в наномолярной концентрации демонстрирует наличие “фармакологического окна”. Для сравнения приведена дозозависимая кривая влияния на пролиферацию клеток А431 противоракового препарата гефитиниб (Iressa) [43].
ЗАКЛЮЧЕНИЕ
Долгое время считалось, что основной функцией nAChR является участие в нервно-мышечной передаче. Затем были обнаружены никотиновые рецепторы в центральной нервной системе и была показана их ключевая роль в когнитивных процессах. В последнее время стало ясно, что развитие некоторых видов рака, причем не только рака легкого, связано с употреблением никотина, указывая на участие никотиновых рецепторов и в процессах развития опухолей. Обнаружение в самых разных тканях человеческого организма эндогенных модуляторов nAChR (Lynx, Lypd6, SLURP) [10] также подтверждает высокую распространенность этого рецептора и его вовлечение во множество жизненно-важных процессов. Получение новых знаний о структуре и функции nAChR имеет большое значение, в том числе для разработки новых лекарственных препаратов.
Список литературы
Grando S.A., Pittelkow M.R., Schallreuter K.U. // J. Invest. Dermatol. 2006. V. 126. P. 1948–1965.
Tracey K.J. // J. Clin. Invest. 2007. V. 117. P. 289–296.
Uspenska K., Lykhmus O., Obolenskaya M., Pons S., Maskos U., Komisarenko S., Skok M. // Front. Pharmacol. 2018. V. 9. P. 626.
Wessler I., Kilbinger H., Bittinger F., Unger R., Kirkpatrick C.J. // Life Sci. 2003. V. 72(18–19). P. 2055–2061.
Fujii T., Mashimo M., Moriwaki Y., Misawa H., Ono S., Horiguchi K., Kawashima K. // J. Pharmacol. Sci. 2017. V. 134. P. 1–21.
Kummer W., Krasteva-Christ G. // Curr. Opin. Pharmacol. 2014. V. 16. P. 43–49.
Grando S.A. // Nat. Rev. Cancer. 2014. V. 14. P. 419–429.
Schuller H.M. // Nat. Rev. Cancer. 2009. V. 9. P. 195–205.
Corradi J., Bouzat C. // Mol. Pharmacol. 2016. V. 90. P. 288–299.
Васильева Н.А., Локтюшов Е.В., Бычков М.Л., Шенкарев З.О., Люкманова Е.Н. // Успехи биологической химии. 2018. Т. 57. С. 303–330.
Tsetlin V.I., Hucho F. // FEBS Lett. 2004. V. 557(1–3). P. 9–13.
Beckmann J., Lips K.S. // Pharmacology. 2013. V. 92 (5–6). P. 286–302.
Israël M., Dunant Y. // J. Physiol. Paris. 1998. V. 92. P. 123–128.
Andersson U., Tracey K.J. // J. Exp. Med. 2012. V. 209. P. 1057–1068.
Qian J., Galitovskiy V., Chernyavsky A.I., Marchenko S., Grando S.A. // Genes Immun. 2011. V. 12. P. 222–230.
Wang H., Liao H., Ochani M., Justiniani M., Lin X., Yang L., Al-Abed Y., Wang H., Metz C., Miller E.J., Tracey K.J., Ulloa L. // Nat. Med. 2004. V. 10. P. 1216–1221.
Rosas-Ballina M., Olofsson P.S., Ochani M., Valdés-Ferrer S.I., Levine Y.A., Reardon C., Tusche M.W., Pavlov V.A., Andersson U., Chavan S., Mak T.W., Tracey K.J. // Science. 2011. V. 334(6052). P. 98–101.
Wang H., Yu M., Ochani M., Amella C.A., Tanovic M., Susarla S., Li J.H., Wang H., Yang H., Ulloa L., Al-Abed Y., Czura C.J., Tracey K.J. // Nature. 2003. V. 421(6921). P. 384–388.
Kimura R., Ushiyama N., Fujii T., Kawashima K. // Life Sci. 2003. V. 72(18–19). P. 2155–2158.
de Jonge W.J., van der Zanden E.P., The F.O., Bijlsma M.F., van Westerloo D.J., Bennink R.J., Berthoud H.R., Uematsu S., Akira S., van den Wijngaard R.M., Boeckxstaens G.E. // Nat. Immunol. 2005. V. 6. P. 844–851.
Mina-Osorio P., Rosas-Ballina M., Valdes-Ferrer S.I., Al-Abed Y., Tracey K.J., Diamond B. // Mol. Med. 2012. V. 18. P. 618–627.
Li P., Liu H., Sun P., Wang X., Wang C., Wang L., Wang T. // Exp. Gerontol. 2016. V. 74. P. 43–55.
Skok M.V., Grailhe R., Agenes F., Changeux J.P. // Life Sci. 2007. V. 80(24–25). P. 2334–2336.
Razani-Boroujerdi S., Boyd R.T., Dávila-García M.I., Nandi J.S., Mishra N.C., Singh S.P., Pena-Philippides J.C., Langley R., Sopori M.L. // J. Immunol. 2007. V. 179. P. 2889–2898.
de Jonge W.J., Ulloa L. // Br. J. Pharmacol. 2007. V. 151. P. 915–929.
Friedman J.R., Richbart S.D., Merritt J.C., Brown K.C., Nolan N.A., Akers A.T., Lau J.K., Robateau Z.R., Miles S.L., Dasgupta P. // Pharmacol. Ther. 2018.
Fu X.W., Rekow S.S., Spindel E.R. // Am. J. Physiol. Lung Cell Mol. Physiol. 2012. V. 303. P. 661–668.
Kurzen H., Berger H., Jäger C., Hartschuh W., Näher H., Gratchev A., Goerdt S., Deichmann M. // J. Invest. Dermatol. 2004. V. 123. P. 937–949.
Kurzen H., Schallreuter K.U. // Exp. Dermatol. 2004. V. 4. P. 27–30.
Chernyavsky A.I., Arredondo J., Marubio L.M., Grando S.A. // J. Cell Sci. 2004. V. 117. P. 5665–5679.
Nguyen V.T., Chernyavsky A.I., Arredondo J., Bercovich D., Orr-Urtreger A., Vetter D.E., Wess J., Beaudet A.L., Kitajima Y., Grando S.A. // Exp. Cell Res. 2004. V. 294. P. 534–549.
Arredondo J., Nguyen V.T., Chernyavsky A.I., Bercovich D., Orr-Urtreger A., Kummer W., Lips K., Vetter D.E., Grando S.A. // J. Cell Biol. 2002. V. 159. P. 325–336.
Nguyen V.T., Ndoye A., Hall L.L., Zia S., Arredondo J., Chernyavsky A.I., Kist D.A., Zelickson B.D., Lawry M.A., Grando S.A. // J. Cell Sci. 2001. V. 114. P. 1189–1204.
Radek K.A., Elias P.M., Taupenot L., Mahata S.K., O’Connor D.T., Gallo RL. // Cell Host Microbe. 2010. V. 7. P. 277–289.
Arredondo J., Hall L.L., Ndoye A., Nguyen V.T., Chernyavsky A.I., Bercovich D., Orr-Urtreger A., Beaudet A.L., Grando S.A. // Lab. Invest. 2003. V. 83. P. 207–225.
Chernyavsky A.I., Arredondo J., Karlsson E., Wessler I., Grando S.A. // J. Biol. Chem. 2005. V. 280. P. 39220–39228.
Grando S.A. // J. Pharmacol. Sci. 2008. V. 106. P. 174–179.
Lyukmanova E.N., Shulepko M.A., Kudryavtsev D.S., Bychkov M.L., Kulbatskii D.S., Kasheverov I.E., Astapova M.V., Feofanov A.V., Thomsen M.S., Mikkelsen J.D., Shenkarev Z.O., Tsetlin V.I., Dolgikh D.A., Kirpichnikov M.P. // PLoS One. 2016. V. 11. e0149733.
Arredondo J., Chernyavsky A.I., Webber R.J., Grando S.A. // J. Invest. Dermatol. 2005. V. 125. P. 1236–1241.
Lyukmanova E.N., Shulepko M.A., Shenkarev Z.O., Bychkov M.L., Paramonov A.S., Chugunov A.O., Kulbatskii D.S., Arvaniti M., Dolejsi E., Schaer T., Arseniev A.S., Efremov R.G., Thomsen M.S., Dolezal V., Bertrand D., Dolgikh D.A., Kirpichnikov M.P. // Sci. Rep. 2016. V. 6. 30698.
Chernyavsky A.I., Kalantari-Dehaghi M., Phillips C., Marchenko S., Grando S.A. // Wound Repair Regen. 2012. V. 20. P. 103–113.
Chernyavsky A.I., Arredondo J., Galitovskiy V., Qian J., Grando S.A. // Am. J. Physiol. Cell Physiol. 2010. V. 299. P. 903–911.
Lyukmanova E.N., Bychkov M.L., Sharonov G.V., Efremenko A.V., Shulepko M.A., Kulbatskii D.S., Shenkarev Z.O., Feofanov A.V., Dolgikh D.A., Kirpichnikov M.P. // Br. J. Pharmacol. 2018. V. 175(11). P. 1973–1986.
Perez C., Khachemoune A. // Am. J. Clin. Dermatol. 2016. V. 17. P. 63–70.
Tsuji H., Okamoto K., Matsuzaka Y., Iizuka H., Tamiya G., Inoko H. // Genomics. 2003. V. 81. P. 26–33.
Allan C.M., Procaccia S., Tran D., Tu Y., Barnes R.H. 2nd, Larsson M., Allan B.B., Young L.C., Hong C., Tontonoz P., Fong L.G., Young S.G., Beigneux A.P. // J. Invest. Dermatol. 2016. V. 136. P. 436–43.
Schaal C., Chellappan S.P. // Mol. Cancer Res. 2014. V. 12. P. 14–23.
Arredondo J., Chernyavsky A.I., Jolkovsky D.L., Pinkerton K.E., Grando S.A. // FASEB J. 2008. V. 22(5). P. 1356–1368.
Trombino S., Cesario A., Margaritora S., Granone P., Motta G., Falugi C., Russo P. // Cancer Res. 2004. V. 64. P. 135–145.
Dasgupta P., Rastogi S., Pillai S., Ordonez-Ercan D., Morris M., Haura E., Chellappan S. // J. Clin. Invest. 2006. V. 116. P. 2208–2217.
Jull B.A., Plummer H.K. 3rd., Schuller H.M. // J. Cancer. Res. Clin. Oncol. 2001. V. 127. P. 707–717.
Momi N., Ponnusamy M.P., Kaur S., Rachagani S., Kunigal S.S., Chellappan S., Ouellette M.M., Batra S.K. // Oncogene. 2013. V. 32. P. 1384–1395.
Brown K.C., Perry H.E., Lau J.K., Jones D.V., Pulliam J.F., Thornhill B.A., Crabtree C.M., Luo H., Chen Y.C., Dasgupta P. // J. Biol. Chem. 2013. V. 288. P. 33049–33059.
Dasgupta P., Kinkade R., Joshi B., Decook C., Haura E., Chellappan S. // Proc. Natl. Acad. Sci. U S A. 2006. V. 103. P. 6332–6337.
Dobrovinskaya O., Valencia-Cruz G., Castro-Sánchez L., Bonales-Alatorre E.O., Liñan-Rico L., Pottosin I. // Front. Pharmacol. 2016. V. 7. 290.
Dasgupta P., Rizwani W., Pillai S., Kinkade R., Kovacs M., Rastogi S., Banerjee S., Carless M., Kim E., Coppola D., Haura E., Chellappan S. // Int. J. Cancer. 2009. V. 124. P. 36–45.
Zhao Y., Zhou W., Xue L., Zhang W., Zhan Q. // PLoS One. 2014. V. 9. e90836.
Krais A.M., Hautefeuille A.H., Cros M.P., Krutovskikh V., Tournier J.M., Birembaut P., Thépot A., Paliwal A., Herceg Z., Boffetta P., Brennan P., Hainaut P.L. // Carcinogenesis. 2011. V. 32. P. 1388–1395.
Fei R., Zhang Y., Wang S., Xiang T., Chen W. // Oncol. Rep. 2017. V. 38. P. 2619–2628.
Xiang T., Fei R., Wang Z., Shen Z., Qian J., Chen W. // Oncol. Rep. 2016. V. 35. P. 205–210.
Shin V.Y., Wu W.K., Ye Y.N., So W.H., Koo M.W., Liu E.S., Luo J.C., Cho C.H. // Carcinogenesis. 2004. V. 25. P. 2487–2495.
Wong H.P., Yu L., Lam E.K., Tai E.K., Wu W.K., Cho C.H. // Toxicol. Sci. 2007. V. 97. P. 279–287.
Fu X.W., Song P.F., Spindel E.R. // Int. Immunopharmacol. 2015. V. 29. P. 93–98.
Bergqvist C., Kadara H., Hamie L., Nemer G., Safi R., Karouni M., Marrouche N., Abbas O., Hasbani D.J., Kibbi A.G., Nassar D., Shimomura Y., Kurban M. // Int. J. Dermatol. 2018. V. 57. P. 162–170.
Pettersson A., Nylund G., Khorram-Manesh A., Nordgren S., Delbro D.S. // Auton. Neurosci. 2009. V. 148. P. 97–100.
Russo P., Cardinale A., Margaritora S., Cesario A. // Life Sci. 2012. V. 91. P. 1087–1092.
Arredondo J., Chernyavsky A.I., Grando S.A. // Life Sci. 2007. V. 80. P. 2243–2247.
Arredondo J., Chernyavsky A.I., Grando S.A. // Biochem. Pharmacol. 2007. V. 74. P. 1315–1319.
Kalantari-Dehaghi M., Parnell E.A., Armand T., Bernard H.U., Grando S.A. // Int. Immunopharmacol. 2015. V. 29. P. 99–104.
Kalantari-Dehaghi M., Bernard H.U., Grando S.A. // Life Sci. 2012. V. 91. P. 1122–1125.
Throm V.M., Männle D., Giese T., Bauer A.S., Gaida M.M., Kopitz J., Bruckner T., Plaschke K., Grekova S.P., Felix K., Hackert T., Giese N.A., Strobel O. // Oncotarget. 2018. V. 9. P. 11734–11751.
Lyukmanova E.N., Shulepko M.A., Bychkov M.L., Shenkarev Z.O., Paramonov A.S., Chugunov A.O., Arseniev A.S., Dolgikh D.A., Kirpichnikov M.P. // Acta Naturae. 2014. V. 6. P. 60–66.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия