

Биоорганическая химия, 2019, T. 45, № 3, стр. 333-336
Получение нового аналога хромофора зеленого флуоресцентного белка из производного коричного альдегида
С. О. Зайцева 1, *, М. С. Баранов 1
1 ФГБУН Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
* E-mail: snezhana_zaitseva@mail.ru
Поступила в редакцию 30.11.2018
После доработки 04.12.2018
Принята к публикации 19.12.2018
Аннотация
Новое производное хромофора зеленого флуоресцентного белка (Z)-5-((E)-4-(4-гидроксифенил)аллилиден)-2,3-диметил-3,5-дигидро-4H-имидазол-4-он, было получено с использованием карбоксиимидата на ключевой стадии синтеза. Сравнение оптических свойств полученного соединения и классического аналога хромофора GFP – 5-(4-гидроксибензилиден)-3,5-дигидро-4Н-имидазол-4-она – показало, что максимумы поглощения и эмиссии созданного нами производного смещаются в более длинноволновую область с ростом размера сопряженной π-системы бензилиденового фрагмента.
Синтетические аналоги хромофоров флуоресцентных белков – различные бензилиденимидазолоны – являются предметом пристального внимания исследователей вот уже почти четыре десятилетия. Интенсивная окраска, небольшой размер, высокая растворимость и легко варьируемые с помощью заместителей спектральные характеристики позволяют успешно использовать данные соединения в химических и биологических исследованиях [1]. В качестве флуоресцентных [2] и флуорогенных [3] красителей такие молекулы могут применяться при визуализации разнообразных клеточных структур, мечения различных объектов, разработке ионных или рН-чувствительных сенсоров [4, 5].
Основной характеристикой, определяющей возможность применения аналога хромофора в качестве красителя, является положение максимумов абсорбции и эмиссии. В связи с тем, что область поглощения большинства биологических объектов лежит в коротковолновой части спектра, получение соединений, демонстрирующих существенный батохромный сдвиг, представляет актуальную задачу.
Как известно, подобного смещения часто удается добиться путем увеличения размера сопряженной π-системы молекулы красителя. Так, для различных производных хромофора GFP (5-(4-гидроксибензилиден)-3,5-дигидро-4Н-имидазол-4-она) (схема 1) подобная модификация чаще всего производится по второму положению имидазолонового цикла. В частности, введение в это положение стирольного фрагмента приводит к образованию производных хромофора белка Kaede (схема 1), обладающих заметным батохромным смещением [6]. Эти соединения находят применение в качестве красителей, а также могут использоваться как сенсибилизирующие краски в солнечных батареях [7].
В настоящей работе мы предложили альтернативный вариант увеличения системы сопряженных связей производного хромофора GFP для смещения положений максимумов абсорбции и эмиссии – модификацию по бензилиденовому фрагменту (схема 1).
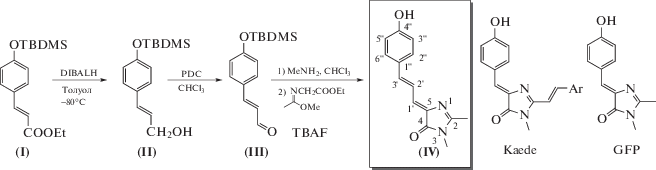
Схема 1. Схема синтеза производного (IV).
Синтез соединения (IV) был проведен с использованием производного коричного альдегида (схема 1), так как одним из наиболее универсальных методов синтеза бензилиденимидазолонов является взаимодействие оснований Шиффа, полученных из альдегидов, с карбоксиимидатом [8]. Необходимый альдегид (III) был получен с достаточно хорошим выходом из коммерчески доступного силилированного производного этилфенилакрилата (I) восстановлением и последующим окислением (схема 1). Удивительным образом образование основания Шиффа не было затруднено побочными реакциями α,β-непредельного альдегида. Из-за наличия сопряженных двойных связей конечное соединение (IV) было получено в виде смеси Е- и Z-изомеров по обеим двойным связям, которую, однако, легко удалось перевести в индивидуальный изомер, а именно E- по положению 3' и Z- по положению 1' аллилиденового остатка (схема 1) в ходе медленного переосаждения из метанола, что хорошо подтверждается данными ЯМР: константа спин-спинового взаимодействия между атомами водорода при связи 2'-3' составляет 11.4 Гц, что соответствует транс-конфигурации, а положение сигнала протона при атоме 1' соответствует типичному положению для Z-изомеров других производных хромофора GFP, у которых эта конфигурация является наиболее стабильной [9].
Рис. 1.
Нормализованные спектры абсорбции (слева) и эмиссии (справа) растворов хромофора GFP (черный) и соединения (IV) (серый) в воде при pH 10.

Изучение оптических свойств полученного соединения (IV) (рис. 1, табл. 1), а также сравнение их со свойствами GFP показало, что введение увеличенной сопряженной π-системы в безилиденовой части молекулы приводит к батохромному смещению максимумов абсорбции и эмиссии.
Таким образом, нами был получен новый аналог хромофора GFP из производного коричного альдегида. Максимумы абсорбции и эмиссии данного соединения лежат в более длинноволновой области, чем у аналога хромофора GFP, за счет увеличения сопряженной системы π-связей в бензилиденовом фрагменте.
Таблица 1.
Положения максимумов поглощения и испускания хромофора GFP и соединения (IV)
| Растворитель | GFP | (IV) | ||
|---|---|---|---|---|
| λабс, нм | λэм, нм | λабс, нм | λэм, нм | |
| Диоксан | 369 | 435 | 388 | 482 |
| Этилацетат | 367 | 420 | 386 | 491 |
| Ацетонитрил | 368 | 438 | 385 | 508 |
| Метанол | 370 | 436 | 397 | 513 |
| Вода (нейтральная форма) | 367 | 453 | 395 | 458 |
| Вода (депротонированная форма) | 425 | 491 | 458 | 600 |
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры ЯМР (δ м.д., J Гц) зарегистрированы на приборе Bruker Avance III (700 МГц, США) в DMSO-d6 и дейтерохлороформе (внутренний стандарт – Me4Si). Спектры поглощения в УФ и видимом диапазонах регистрировали на спектрофотометре Varian Cary 100 Bio (США). Спектры флуоресценции регистрировали на спектрофлуориметре Varian Cary Eclipse (США). Температуры плавления определены на приборе SMP 30 (Великобритания) и не исправлены.
(E)-3-(4-((трет-Бутилдиметилсилил)окси)фенил)проп-2-ен-1-ол (II). Раствор 2.97 г (10 ммоль) этил-(E)-3-(4-((трет-бутилдиметилсилил)окси)-фенил)акрилата) (I) [10] в 60 мл толуола охлаждали до –80°С. Добавляли гидрид диизобутилалюминия (DIBALH) (10.6 мл, 60 ммоль), выдерживали 20 ч при –80°С. Затем в реакционную смесь добавляли 400 мл этилацетата и 150 мл раствора тартрата калия, перемешивали в течение 1 ч. Далее водную фазу экстрагировали этилацетатом (2 × 150 мл), органические фракции объединяли, промывали насыщ. раствором NaCl (2 × × 100 мл). Раствор высушивали над безводным Na2SO4, упаривали, и продукт очищали с помощью колоночной хроматографии (EtOAc–гексан, 1 : 3). Выход продукта (II) 2.14 г (81%). 1H-ЯМР: δ 7.28 (д, J2 8.7, 2Н, H2'', H6''), 6.80 (д, J2 8.7, 2Н, H3'', H5''), 6.56 (д, J2 16.0, 1H, H3'), 6.25 (дт, J2 15.8, 6.0, 1Н, Н2'), 4.30 (т, J2 5.2, 2H, СН2), 1.00 (с, 9Н, t-Bu), 0.21 (с, 6Н, 2 СН3). Соответствует литературе [11].
(E)-3-(4-((трет-Бутилдиметилсилил)окси)фенил)акрилальдегид (III). К раствору спирта (II) в 50 мл хлороформа добавляли 20 экв. дихромата пиридиния. Раствор перемешивали в течение 1.5 ч при комнатной температуре, за протеканием реакции следили ТСХ в системе EtOAc-гексан, 1 : 3. Реакционную смесь отфильтровывали через 1 см силикагеля, упаривали. Выход 823 мг (39%). 1H-ЯМР: δ 9.67 (д, J2 7.7, 1Н, СНО), 7.48 (д, J2 8.5, 2Н, H2'', H6''), 7.42 (д, J2 15.8, 1Н, H3'), 6.89 (д, J2 8.5, 2Н, H3'', H5''), 6.62 (дд, J2 15.8, 7.7, 1Н, Н2'), 1.00 (с, 9Н, t-Bu), 0.25 (с, 6Н, 2 СН3). Соответствует литературе [12].
5-([1Z,3E]-4-(4-Гидроксифенил)аллилиден)-2,3-диметил-3,5-дигидро-4H-имидазол-4-он (IV). 1) К раствору 262 мг (1 ммоль) альдегида (III) в 20 мл хлороформа, добавляли 0.3 мл 40% водного раствора метиламина и безводный сульфат натрия (2 г). Смесь перемешивали в течение 48 ч при комнатной температуре, затем органическую фазу отделяли, высушивали над безводным Na2SO4 и упаривали.
2) Полученный имин (1 ммоль) смешивали с этил-2-((1-метоксиэтилиден)амино)ацетатом (175 мг, 1.1 ммоль) и перемешивали при комнатной температуре в течение 36 ч. Затем в реакционную смесь добавляли 50 мл этилацетата, промывали водой (2 × 5 мл) и насыщ. раствором NaCl (2 × 5 мл). К органической фазе добавляли 10 экв. фторида тетрабутиламмония, перемешивали 10 мин и добавляли 2 мл ледяной уксусной кислоты. Реакционную смесь промывали водой (5 × 5 мл) и насыщ. раствором NaCl (3 × 5 мл). Раствор высушивали над безводным Na2SO4, упаривали и очищали с помощью колоночной хроматографии (градиент EtOAc–EtOAc : i-PrOH, 1 : 4). Индивидуальный изомер (E по положению 3' и Z- по положению 1') был получен путем медленного переосаждения из метанола. Выход 88 мг (36%) желт. крист., т. пл. около 300°C с разложением. 1H-ЯМР: δ 9.88 (с, 1Н, ОН), 7.44 (д, J2 8.2, 2Н, H2'', H6''), 7.29 (дд, J2 15.4, 11.6, 1Н, H2'), 7.11 (д, J2 15.6, 1Н, H1'), 6.84 (д, J2 11.4, 1Н, H3'), 6.79 (д, J2 8.2, 2Н, H3'', H5''), 3.06 (с, 3Н, 3-CH3), 2.29 (с, 3Н, 2-CH3). 13C-ЯМР: δ 168.7 (C4), 161.0 (C2), 158.9 (C4''), 141.6 (C3'), 138.3 (C1'), 129.1 (C2'', C6''), 127.4 (C1''), 127.3 (C5), 120.0 (C2'), 115.9 (C3'', C5''), 26.1 (3-CH3), 15.0 (2-CH3).
ФОНДОВАЯ ПОДДЕРЖКА
Работа поддержана грантом РФФИ 17-00-00401_КОМФИ. Работа выполнена с использованием оборудования ЦКП ИБХ, поддержанного Минобрнауки России, идентификатор соглашения RFMEFI62117X0018.
СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ
Настоящая статья не содержит каких-либо исследований с участием людей в качестве объектов исследований и с использованием животных в качестве объектов.
КОНФЛИКТ ИНТЕРЕСОВ
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Walker C.L., Lukyanov K.A., Yampolsky I.V., Mishin A.S., Duraj-Thatte A.M., Bahareh A., Tolbert L.M., Solntsev K.M. // Curr. Opin. Chem. Biol. 2015. V. 27. P. 64–74.
Yuan L., Lin W., Chen H., Zhu S., He L. // Angew. Chem. 2013. V. 52. P. 10018–10022.
Povarova N.V., Bozhanova N.G., Sarkisyan K.S., Gritcenko R., Baranov M.S., Yampolsky I.V., Lukyanov K.A., Mishin A.S. // J. Mater. Chem. C. 2016. V. 4. P. 3036–3040.
Baldridge A., Solntsev K.M., Song C., Tanioka T., Kowalik J., Tolbert L.M. // Chem. Commun. 2010. V. 46. P. 5686–5688.
Olsen S., Baranov M.S., Baleeva N.S., Antonova M.M., Johnsond K.A., Solntsev K.M. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 26703–26711.
Yampolsky I.V., Kislukhin A.A., Amatov T.T., Shcherbo D., Potapov V.K., Lukyanov S., Lukyanov K.A. // Bioorg. Chem. 2008. V. 36. P. 96–104.
Chuang W.T., Chen B.S., Chen K.Y., Cheng-Chih H. // Chem. Commun. 2009. V. 45. P. 6982–6984.
Baldridge A., Kowalik J., Tolbert L.M. // Synthesis. 2010. № 14. P. 2424–2436.
Bozhanova N.G., Baranov M.S., Sarkisyan K.S., Gritcenko R., Mineev K.S., Golodukhina S.V., Baleeva N.S., Lukyanov K.A., Mishin A.S. // ACS Chem. Biol. 2017. V. 12. P. 1867–1873.
Boschi D., Tron G. C., Lazzarato L., Chegaev K., Cena C., Di Stilo A., Giorgis M., Bertinaria M., Fruttero R., Gasco A. // J. Med. Chem. 2006. V. 49. P. 2886–2897.
Hiroyuki A., Kawahara E., Kishida M., Kato K. // J. Mol. Catal. B: Enzym. 2006. V. 40. P. 8–15.
Satoshi I., Rika O., Chihiro M., Masahiro K., Kazutoshi H., Mitsuhiro N., Yoshiharu A., Satoshi K., Takashi H., Shojiro M., Haruki N. // Tetrahedron. 2013. V. 69. P. 3847–3856.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия