

Биоорганическая химия, 2019, T. 45, № 6, стр. 563-575
Применение природных полисахаридов в фармацевтике
М. В. Хвостов 1, 2, *, Т. Г. Толстикова 1, 3, 2, С. А. Борисов 1, А. В. Душкин 3
1 Новосибирский институт органической химии им. Н.Н. Ворожцова СО РАН
630090 Новосибирск, просп. Академика Лаврентьева, 9, Россия
2 Новосибирский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
3 Институт химии твердого тела и механохимии СО РАН
630128 Новосибирск, ул. Кутателадзе, 18, Россия
* E-mail: khvostov@nioch.nsc.ru
Поступила в редакцию 14.05.2019
После доработки 14.05.2019
Принята к публикации 06.06.2019
Аннотация
Приведены современные данные об использовании в фармацевтике природных полисахаридов – пуллулана, альгинатов, гиалуроновой кислоты, декстранов, пектина, хитозана и арабиногалактана, которые уже активно применяются или могут быть использованы в будущем для оптимизации различных лекарственных форм путем повышения их растворимости в воде и улучшения биодоступности. Акцент на природные полисахариды сделан в связи с их хорошей биосовместимостью, биодеградируемостью, гидрофильностью, хорошей стабильностью, безопасностью, отсутствием токсичности и адгезивными свойствами. Указанные параметры являются важными и необходимыми для агентов, используемых для доставки лекарственных средств в организм человека.
ВВЕДЕНИЕ
Перечень лекарств, имеющихся в распоряжении современной медицины, насчитывает тысячи наименований [1]. Разработка новых лекарственных средств (ЛС) становится очень дорогим и длительным процессом, проходящим от синтеза такой молекулы до внедрения ее в медицинскую практику [2]. В сложившейся ситуации на передний план выходят новые подходы, состоящие в модификации физико-химических либо фармакологических свойств уже известных лекарственных продуктов [3]. Основными целями таких модификаций ЛС является повышение их эффективности и безопасности. Как известно, многие ЛС плохо растворимы в воде, и это является ограничением для их более широкого клинического применения. Согласно Биофармацевтической Классификационной Системе, такие ЛС относятся ко 2-му классу (плохая растворимость в воде/хорошее проникновение) или к 4-му классу (плохая растворимость в воде/плохое проникновение) ЛС [4]. Используя различные подходы, можно повысить растворимость в воде таких ЛС без изменения их липофильных свойств, что в итоге приведет к их лучшему проникновению через биологические мембраны. К таким подходам относятся: уменьшение размера частиц, образование солей и твердых дисперсий, экструзия расплавов, сушка распылением, комплексообразование, образование микроэмульсий, липосом и применение не водных растворителей [5]. Разрабатываемые новые формы доставки способны улучшать биодоступность, замедлять или регулировать высвобождение ЛС, доставлять их более целенаправленно к органам-мишеням независимо от способа введения.
Одним из перспективных направлений для улучшения биодоступности ЛС является создание водорастворимых супрамолекулярных комплексов включений по типу “гость-хозяин”, где в качестве “хозяина” используют различные соединения, способные к образованию липофильных полостей, а в качестве “гостя” – уже зарегистрированные лекарственные молекулы. Как правило, между молекулами ЛС и молекулами “хозяина?” не образуется ковалентных связей, а лишь водородные и Ван-дер-вальсовые взаимодействия. Такая слабaя связь не требует больших затрат энергии на отрыв молекулы ЛС от молекулы “хозяина”, тем самым обеспечивается быстрая диссоциация таких систем с последующей абсорбцией ЛС организмом. В качестве “хозяина” могут быть использованы вещества природного и не природного происхождения: полисахариды, полимеры, липосомы, наносферы, микросферы, мицеллы [6]. Вещества, используемые для доставки ЛС в живой организм, должны быть не токсичными, биосовместимыми и биодеградируемыми и не вызывать иммунного ответа [7]. Среди всех природных полимеров полисахариды наиболее интересны качестве носителей ЛС ввиду своей биосовместимости, биодеградируемости, гидрофильности, хорошей стабильности, безопасности, не токсичности и адгезивным свойствам [8].
Среди полисахаридов, в современной фармацевтической индустрии в большей степени нашли свое применение пуллулан, альгинаты, гиалуроновая кислота, декстраны, пектин и хитозан. Перспективным кандидатом является арабиногалактан.
ПУЛЛУЛАН
Пуллулан (PUL) – полисахарид, состоящий из мальтотриозных единиц, который впервые был выделен Бернье (Bernier) в 1958 году. В его структуре три глюкозных единицы в мальтотриозе соединены (1α→4)-гликозидной связью, тогда как последовательные мальтотриозные единицы соединены друг с другом гликозидной связью (1α→6). Молекулярная масса находится в диапазоне от 5 до 900 кДа. Пуллулан получают путем ферментации крахмала грибом Aureobasidium pullulans [9].
На протяжении длительного времени этот полисахарид использовали в Японии в качестве пищевой добавки и вспомогательного вещества при производстве лекарств. Кроме того, пуллулан признан безопасным и разрешен к применению в США. Полностью безопасной, считается доза до 10 г в сутки, при приеме больших количеств могут наблюдаться незначительные симптомы со стороны ЖКТ [9].
В сухом состоянии PUL представляет собой белый порошок без запаха и вкуса, а при растворении в воде в концентрации 5–10% образует не гигроскопичные вязкие растворы. Распад полимера начинается при температуре 250°С, а при температуре 280°С происходит обугливание. PUL очень хорошо растворим в воде, биодеградируем [9]. Возможно получение различных полусинтетических производных PUL, c большей активностью, что и расширяет область его применения [9].
В пищевой промышленности PUL применяется как источник пищевых волокон. Он не подвержен воздействию амилаз млекопитающих и оказывает стимулирующий эффект на рост полезных бифидобактерий [10].
При высыхании раствора PUL (обычно 5–10%) образуются прозрачные и не проницаемые для кислорода воздуха пленки, которые обладают отличными механическими свойствами. При попадании таких пленок в воду или ротовую полость происходит их быстрое растворение. Благодаря таким свойствам они могут быть применены в качестве защитных оболочек для пищевых продуктов для предотвращения преждевременного окисления жиров и витаминов.
В фармацевтической промышленности PUL и его производные могут быть использованы как водорастворимый стоматологический адгезив, а также в качестве стабилизатора окраски при производстве таблеток или гранул, покрытых сахарсодержащей оболочкой. Кроме этого, применение PUL совместно с циклодекстринами позволяет улучшать стабильность, биосовместимость и биодеградируемость образуемых ими микросфер [11]. Производные PUL могут быть использованы в качестве конъюгатов при производстве вакцин. Вирус, ковалентно связанный с PUL, достоверно усиливает выработку иммуноглобулинов G и М и снижает количество иммуноглобулинов Е, при этом сам вирус надежно инактивируется. PUL с молекулярной массой в диапазоне 30–90 кДа может быть использован в качестве кровезаменителя. Также этот полисахарид может применяться в качестве ингредиента в различных косметических средствах [12]. Поскольку PUL, как и другие полисахариды, обладает сродством к лектиноподобным рецепторам клеток печени [13, 14], он может быть использован для адресной доставки лекарственных средств в этот орган. Авторами работы [15] был синтезирован конъюгат PUL с интерфероном и проведено изучение его активности in vitro и in vivo. В ходе экспериментов было установлено, что после внутривенного введения конъюгата интерферон концентрируется в печени и проявляет более длительный противовирусный эффект. Авторами работы [16] был получен конъюгат PUL с доксорубицином и изучен его цитотоксический эффект. На клеточных линиях KB и MCF7 было показано, что значения IC50 конъюгата превышает таковое для свободного доксорубицина. Такое различие объясняется более длительным высвобождением ЛС из супрамолекулярной системы.
В большей степени для адресной доставки различных лекарств изучают различные синтетические производные PUL: PUL с холестерином, пуллулан ацетат, карбоксиметил-PUL, PUL с диэтилентриаминпентауксусной кислотой, сукцинат PUL и др. При этом применяются различные виды частиц PUL: наночастицы, наногели, липосомы, микросферы и конъюгаты. Перечень лекарственных средств также обширен и включает инсулин, доцетаксел, адриамицин, доксорубицин, клоназепам, эпирубицин, индометацин и др. [17]. Большинство опубликованных работ по этой тематике, как правило, описывает эксперименты in vitro.
АЛЬГИНАТЫ
Альгинаты (ALG) это общее названия различных природных не разветвленных полианионных полисахаридов состоящих из повторяющихся единиц β-D-маннуроновой кислоты (М) и α-L-гулуроновой кислоты (G) связанных между собой 1→4-гликозидными связями. Молекулярный вес этих полисахаридов может варьировать в широком диапазоне, от 32 до 400 кДа. Основным источником ALG являются различные виды бурых водорослей Macrocystis pyrifera, Laminaria hyperborea, Ascophyllum nodosum [18]. Структуру альгинатов составляют гомополимерные блоки ММ или GG с вкраплением гетерополимерных блоков MG или GM. Соотношение этих блоков различны в зависимости от источника получения ALG. При добавлении воды альгиновая кислота за счет межмолекулярного взаимодействия образует вязкий “кислотный гель”. Механизм гелеобразования объясняется моделью “яичной коробки”, где один двухвалентный катион взаимодействует с четырьмя COOH-группами.
После гелеобразования молекулы воды физически захватываются внутрь альгиновой матрицы, но при этом сохраняют подвижность и способность покинуть ее. Это свойство на протяжении более трех десятилетий используется для инкапсулирования различных ЛС, белков, генов и клеток. ALG обладают мукоадгезивными свойствами, полностью биосовместимы и не вызывают раздражения при попадании в живой организм. Их используют для создания лекарственных форм в составе ранозаживляющих повязок, а также для остановки кровотечений в качестве гемостатика. Коммерчески доступными являются более 200 видов ALG [19].
ALG является гидрофильным полимером, поэтому высвобождение инкапсулированных в него молекул ЛС может происходить разными путями. Водорастворимые вещества, как правило, покидают матрицу путем диффузии, тогда как плохорастворимые высвобождаются путем разрушения матрицы ALG. Высвобождение маленьких молекул происходит быстро, поскольку диаметр пор матрицы альгинатов составляется примерно 5 нм [20]. Однако, при необходимости, возможно создание физического или химического связывания молекулы ЛС с матрицей, что позволит пролонгировать его высвобождение [18].
Несмотря на широкое использование в биологии и медицине, не утихают споры о таких нежелательных побочных эффектах ALG, как иммуногенность. Она может быть вызвана как качеством и недостаточной чистотой биоматериала, так и благодаря непосредственной иммуногенности ALG, в связи с возможным наличием в составе конечного продукта следов тяжелых металлов, эндотоксинов, белков или полифенольных соединений [21].
Сами по себе ALG не усваиваются млекопитающими, поскольку для этого у них нет необходимых ферментов (например, альгиназы), которые могли бы гидролизовать цепочки полимера, однако ионные поперечно сшитые альгинатные гели могут быть растворены посредством высвобождения двухвалентного иона, обеспечивающего сшивание цепочек. Этот процесс происходит путем реакции его обмена с окружающей средой на одновалентный катион, такой как Na+. Тем не менее, даже при этом средняя молекулярная масса большинства коммерчески доступных ALG остается большой для преодоления почечного барьера и полного выведения с мочой [22]. Для повышения биодеградации ALG используют частичное окисление его цепей. В таком виде ALG может разрушаться в водной среде и использоваться в качестве носителя ЛС и клеток [21].
ГИАЛУРОНОВАЯ КИСЛОТА
Гиалуроновая кислота (GLA) – природный линейный полисахарид, состоящий из повторяющихся единиц N-ацетил-D-глюкозамина и D-глюкуроновой кислоты, связанных посредством чередующихся β-1,3- и β-1,4-гликозидных связей [23]. Поскольку значения рКа карбоксильных групп GLA составляют 3–4, то в условиях физиологического рН 7.4 эти функциональные группы находятся в ионизированном состоянии, а сама GLA является полианионом и обозначается как гиалуронан [24]. Молекулярные массы GLA могут находиться в достаточно широком диапазоне от 20 до 4000 кДа. В растворах цепи GLA образуют произвольные спирали. В таких условиях полисахарид обладает выраженной гидрофильностью и окружен молекулами воды, которые связаны с ним при помощи водородных связей. Благодаря таким физико-химическим свойствам растворы GLA очень вязкие и эластичные [25]. В природе GLA в основном встречается во внеклеточном матриксе соединительной ткани и в большом количестве в стекловидном теле глазного яблока. Кроме этого, она играет важную роль в функционировании клетки, поскольку оказывает влияние на пролиферацию, миграцию и модулирование внутриклеточных сигналов [26].
Для фармацевтики ГК представляет интерес в связи с тем, что является биодеградируемым, биосовместимым, не токсичным и не иммуногенным полимером. Кроме этого GLA может быть химически модифицирована путем реакций перекрестного сшивания или конъюгации по своим функциональным группам [27]. В медицине GLA применяют для лечения остеоартритов (Hyalgan®, Artz®, Orthovisc®, Healon® и др.), в глазной и пластической хирургии (Bionect®, Connettivina®, Jossalind®), а также в тканевой инженерии [28]. В качестве носителя GLA используют для рецепторопосредованной доставки ЛС в противоопухолевой терапии (конъюгаты с доксорубицином, паклитакселом, цисплатином), для доставки белков, пептидов и нуклеотидов, противовоспалительных средств (конъюгаты с метотрексатом, дексаметазоном, метилпреднизалоном), а также для доставки контрастных агентов, поскольку GLA способна специфически опознавать рецепторы, экспрессия которых повышена в различных нездоровых клетках [29].
Большое внимание GLA привлекает в качестве системы доставки лекарств в кожу. Как известно, кожные покровы покрыты роговым слоем, непроницаемым барьером для различного рода веществ, в том числе и для ЛС [28]. На мировом фармацевтическом рынке существует препарат под торговой маркой Solaraze®, который представляет собой гель, содержащий гиалуронат натрия и 3%-й диклофенак натрия. Основным показанием для его применения является актинический кератоз – заболевание кожи, вызванное длительным воздействием солнечного излучения. Как правило, оно появляется на коже головы, лица, спины и рук у людей со светлой кожей и в возрасте более 50 лет. В США это заболевание является третьим по распространенности среди кожных болезней [28]. Точный механизм действия нестероидных противовоспалительных средств (НПВС) на данный вид карциномы не известен, однако предполагают, что он связан со снижением синтеза простагландина E2. Тем не менее, авторами работ [30–32] in vitro было установлено, что GLA достоверно усиливает проникновение диклофенака в кожу человека и способствует его накоплению в эпидермисе. Наблюдаемый эффект был подтвержден другими исследованиями, где также было отмечено депонирование диклофенака в эпидермисе [33, 34]. Аналогичный эффект GLA оказывает и на другие ЛС – ибупрофен, клиндамицин фосфат, циклоспорин [32, 35–37].
Местное применение ЛС на поверхности глаза является еще одной областью успешного использования GLA в качестве мукоадгезивного вязкоэластичного носителя. Во многих исследованиях показано, что GLA способна пролонгировать прекорниальную экспозицию и/или увеличить биодоступность пилокарпина [38–40], тропикамида [40, 41], тимолола [42], гентамицина [43, 44], тобрамицина [45].
Использование мукоадгезивных свойств GLA позитивно влияет на биодоступность ЛС, применяемых интраназально. В качестве примера можно привести ксилометазалин, вазопрессин (увеличение в 2 раза) и 1-дезамино-[8-D-аргинин] вазопрессин (увеличение в 1.6 раз). Наблюдаемый эффект зависит от молекулярной массы GLA; увеличение биодоступности проявляется при массе более 300 кДа, а при более низких значениях (55 кДа) увеличение отсутствует [28]. Кроме этого, добавление GLA в структуру микросфер приводит к усилению их мукоадгезивных свойств. Вследствие этого, происходит увеличение биодоступности молекул ЛС, заключенных в таких микросферах [46]. Авторами работы [46] было показано увеличение на 23.3% биодоступности гентамицина при его интраназальном введении в микросферах на основе GLA. В работах [47, 48] GLA была использована для улучшения абсорбции инсулина при его ингаляционном введении. Было показано, что применение GLA приводит к лучшему всасыванию гормона и более выраженному снижению уровню глюкозы у собак.
Помимо перечисленного, GLA широко применяют для модификации липосом, совместно с другим полисахаридом хитозаном, а также для доставки молекул ДНК [28].
ДЕКСТРАНЫ
Декстраны (DEX) это полимеры глюкозы, которые в медицине применяются уже более 50 лет в качестве заменителя плазмы и антитромболитического средства [49]. DEX могут быть получены микробиологическим синтезом из глюкозы или синтетически. Структурно декстраны состоят преимущественно из единиц D-глюкозы соединенной по α1,6-положениям, а боковые цепи присоединены связями α1,3. Степень разветвленности сильно зависит от источника DEX и может варьировать от 0.5 до 60% [50, 51]. Также источник декстрана определяет и его молекулярную массу, что наряду со степенью разветвленности оказывает значительное влияние на физико-химические свойства полимера [52]. При этом, декстраны с разветвленностью более 43% (связь α1,3) не растворимы в воде, поэтому все коммерчески используемые DEX имеют очень низкую степень разветвленности (0.5%) и хорошо растворяются в воде [51].
Другим важным свойством, от которого зависит поведение DEX in vivo, является степень полидисперсности полимера. Природные декстраны обладают высокой степенью полидисперсности, поэтому для получения полимеров с относительно узким молекулярно-массовым распределением, нативные соединения подвергаются частичной деполимеризации или фракционированию [51].
Благодаря наличию большого количества гидроксильных групп, DEX являются хорошими объектами для конъюгирования. В слабокислых или слабощелочных средах полимеры DEX остаются стабильными.
Биодоступность декстранов после перорального введения ничтожно мала. Авторами работы [53] было установлено, что для DEX с молекулярной массой 4, 20, 40 кДа биодоступность составляет менее 0.004. Однако декстраны могут попасть в системный кровоток путем рецепторопосредованной абсорбции, по аналогии с глюкозой [54].
Метаболизируются декстраны в нижних отделах пищеварительного тракта различными α-гликозидазами (декстраназы), при этом такие ферменты также присутствуют в печени, селезенке и почках [51].
В литературе описано большое количество конъюгатов декстранов с различными ЛС, преимущественно это противоопухолевые препараты (дауномицин, доксорубицин, метотрексат, митомицин, цисплатин). Во всех случаях отмечается увеличение периода полувыведения и биодоступности ЛС, конъюгированных с DEX [51, 55].
Ввиду особенности локализации метаболизма, а именно – в толстой кишке, конъюгаты с декстраном используют для таргетной доставки ЛС в этот отдел ЖКТ. Так, конъюгат с будесонидом проявил выраженное местное действие при язвенном колите и способствовал улучшениям, выявляемым как макроскопически, так и микроскопически. Будесонид, введенный в виде обычной суспензии, не проявил столь выраженного эффекта [55, 56]. Кроме этого, в литературе описаны конъюгаты DEX с 5-аминосалициловой кислотой, кетопрофеном, селекоксибом [57].
При помощи конъюгирования с DEX удалось снизить ульцерогенный побочный эффект некоторых НПВС. В работах [58, 59] были получены конъюгаты с кетаролаком и флюрбипрофеном и установлено, что при сохранении анальгетической и противовоспалительной активностей происходит заметное снижение ульцерогенного действия, присущего свободным ЛС. Другими авторами [60] были получены гидрогели на основе декстрана с ибупрофеном и ацетилсалициловой кислотой, было показано, что основное место высвобождения этих НПВС является толстая кишка, при этом в кислой среде желудка высвобождение ЛС не наблюдалось. Благодаря этому снижается вероятность местного ульцерогенного действия указанных ЛС в желудке и ДПК.
ПЕКТИН
Пектин является линейным полисахаридом, состоящим преимущественно из единиц D-галактуроновой кислоты, соединенных посредством α1→4-гликозидных связей. Кроме галактуронановых сегментов в структуре пектина могут встречаться и нейтральные сахара – рамноза, арабиноза, галактоза или ксилоза [61]. Источником пектина являются высшие растения, в клеточных стенках которых его содержание может доходить более чем до 30% в пересчете на сухое вещество. Наибольшее количество пектина находится в срединной ламелле клеточной стенки [62]. Несмотря на то, что пектин содержится практически во всех тканях растений, число природных источников, пригодных для его промышленного получения, ограничены. Это связано с большой вариабельностью молекулярной массы и степени этерификации, что отражается на способности пектина к гелеобразованию. В настоящее время коммерчески доступный пектин получают исключительно из яблочного жмыха или кожуры цитрусовых фруктов (апельсинов, лимонов, лайма), которые являются отходами производства соков. Содержание пектина в яблочном жмыхе составляет 10–15% от сухой массы, в то время как в кожуре цитрусовых до 20–30%. В качестве альтернативных источников могут быть использованы отходы от сахарной свеклы, манго, подсолнуха, бобовых, бананов, кабачков, моркови и помело [61].
Важной характеристикой пектина является степень этерификации его карбоксильных групп метиловым спиртом. В природе этот показатель варьирует от 60 до 90%. В зависимости от этого изменяются желирующие свойства пектина. Пектин со степенью этерификации более 50% обозначается как HM (high metoxy), а со степенью этерификации менее 50% – LM (low metoxy) [63].
Основным свойством пектина, благодаря которому он широко применяется в пищевой и фармацевтической промышленности, является способность образовывать гели. В основе этого лежит образование водородных связей и гидрофобные взаимодействия между молекулами полимера [64]. НМ-пектин образует гели в кислой среде в присутствии сахарозы, а LM-пектин для гелеобразования требует наличия ионов кальция или других мультивалентных катионов. Однако, не все катионы способствуют гелеобразованию LM-пектина. Показано, что для этого неподходящим является Mg2+ [65]. Гелеобразование пектина с низкой степенью этерификации протекает по принципу “яичной коробки” [61], характерной также для альгинатов.
Благодаря своим мукоадгезивным свойствам, биосовместимости, низкой стоимости и отсутствию токсичности пектин может быть использован для контролируемой доставки ЛС – энтеральной, интраназальной либо вагинальной [66, 67]. При пероральном введении пектин выступает в качестве защиты от преждевременного метаболизма для веществ, находящихся в его матрице. Как правило, в роли таких веществ выступают различные полипептиды, высвобождение которых начинается только в толстой кишке после того, как матрица пектина начинает деградировать под воздействием кишечной микрофлоры. Ферментативное разложение пектина в толстой кишке усиливается при наличии ионов кальция и уменьшается при повышении степени этерификации полимера (НМ-пектин). При пероральном введения у композиций, содержащих пектин, есть один недостаток, они могут набухать в физиологических условиях, что может привести к преждевременному высвобождению ЛС. Для минимизирования этого нежелательного эффекта в такие композиции дополнительно добавляют другие полимеры: гидроксипропилметилцеллюлозу, зеин или акриловые полимеры [66].
Интраназальный и вагинальный пути доставки ЛС с помощью пектина рассматриваются как альтернатива инъекционным способам, поскольку слизистая в обоих случаях имеет высокую степень васкуляризации, отсутствует эффект первого прохождения через печень, что в совокупности способствует быстрому проникновению молекул ЛС в системный кровоток [67, 68]. Мукоадгезивные свойства пектина во многом зависят от его молекулярной массы, вязкости, местного рН и функциональных групп в структуре полимера [67, 69].
Кроме непосредственной доставки молекул ЛС, пектин способен усиливать их абсорбцию путем открытия плотных клеточных контактов в эпителии. Этим путем могут проникать в системный кровоток лекарства, для которых возможен парацеллюлярный транспорт; как правило, это гидрофильные молекулы [70]. Авторами работы [71] было показано, что после добавления к клеточной культуре MDCK (клетки почки собаки майдин-дэрби) 1- и 3%-ных растворов пектина с низкой степенью этерификации (5 и 35% соответственно) происходит достоверное уменьшение трансэпителиального электрического сопротивления (TEER) более чем на 80%. Кроме этого, в базальном отделении (под мембраной с эпителиальными клетками) после добавления указанных растворов пектина увеличивается концентрации. В контрольной группе, маннитол практически не проникает через клеточный монослой. Оба этих эффекта свидетельствуют об открытии плотных клеточных контактов.
Массовому распространению пектина в качестве носителя ЛС препятствует нестабильность его композиций при хранении. Так, вязкость растворов пектина может значительно снижаться через шесть месяцев хранения при температуре 25 и 40°С. В свою очередь, это приводит к снижению прочности геля при добавлении ионов кальция. Такое изменение вязкости является результатом деполимеризации пектина с течением времени [66]. С другой стороны, в работе [72] показано, что такое снижение вязкости не влияет на высвобождение ЛС из пектинового геля in vitro. Чаще всего пектин используется как составная часть композиций, включающих в себя различные полимеры [73–75].
ХИТОЗАН
Хитозан является природным линейным биополиаминосахаридом, получаемым путем щелочного дезацетилирования хитина, который, по распространeнности в природе, является вторым полисахаридом после целлюлозы [76]. Хитин – это основной компонент защитной кутикулы ракообразных, таких как крабы, креветки, лобстеры, и клеточной стенкой некоторых грибов, таких как Aspergillus и Mucor. Хитин представляет собой прямолинейный гомополимер, состоящий из β1→4-соединенных N-ацетилглюкозаминовых единиц, тогда как хитозан это сополимер глюкозамина и N-ацетилглюкозамина [77, 78]. Благодаря хорошей доступности аминогрупп в хитозане, он несет положительный заряд, что позволяет ему вступать в реакции с различными отрицательно заряженными поверхностями или полимерами, а также подвергаться хелатированию с ионами металлов, такими как кобальт [79, 80]. Это свойство хитозана используется для разделения металлов.
Хитозан является слабым основанием и не растворим в воде и органических растворителях. Однако, он растворим в разбавленных кислых водных растворах с pH < 6.5, которые могут перевести его глюкозаминовые единицы в растворимую форму R-NH3+ [81]. Он выпадает в осадок в щелочных растворах или при взаимодействии с полианионами и образует гели при низких значениях pH. Его также используют в качестве флоккулирующего агента для обработки сточных вод [82]. Коммерчески доступный хитозан может находиться в виде сухих хлопьев, раствора и мелкого порошка. Он имеет молекулярную массу в диапазоне от 3800 до 2000 кДа и может быть дезацетилирован от 66 до 95% [83]. На фармацевтические свойства композиций, включающих хитозан, влияют степень его дезацетилирования, молекулярная масса, размер частиц, плотность, вязкость.
Хитозан обладает низкой токсичностью и биосовместимостью, биодеградируем. Кроме этого обладает следующими свойствами: гипобилирубинемическим, гипохолестеринемическим, антацидным, противоязвенным, ранозаживляющим, противоожоговым. Также он способен иммобилизировать ферменты и живые клетки [76]. Хитозан обладает мукоадгезивным свойством, которое опосредовано молекулярными силами притяжения, формируемыми электростатическим взаимодействием между положительно заряженным хитозаном и негативно зараженной слизистой поверхностью. Этому способствуют: наличие групп, образующих прочные водородные связи (–OH, –COOH), заряд, большая молекулярная масса, достаточная гибкость цепей, свойства энергии поверхности, благоприятствующие распределению хитозана внутрь слизистого слоя [76]. Кроме этого, хитозан способен открывать плотные клеточные контакты в эпителиальных тканях. В работе [71] было показано, что добавление 0.5% раствора хитозана к клеткам MDCK происходит значительное уменьшение трансэпителиального электрического сопротивления, практически до 0. Кроме этого, происходит значительное увеличение концентрации маннитола в базальном отделении (под мембраной с эпителиальными клетками) после добавления указанного раствора пектина. В контрольной группе, маннитол практически не проникает через клеточный монослой. Оба этих эффекта свидетельствуют об открытии плотных клеточных контактов, достаточное не только для тока ионов, но я для проникновения молекул ЛС.
В фармацевтической промышленности хитозан нашел применение в качестве носителя при производстве таблеток методом прямого прессования, в качестве дезинтегрирующего агента, в качестве связующего и гранулирующего агента, в качестве носителя для ЛС длительного высвобождения, а также как дополнительный растворитель для увеличения растворимости и биодоступности нерастворимых в воде ЛС [71]. Так, например, инкапсулирование ретинола в наночастицы хитозана приводит к 1600-кратному увеличению его растворимости в воде [84].
Самыми распространeнными системами доставки ЛС на основе хитозана являются микросферы. Они применяются для контролируемого высвобождения антибиотиков, антигипертензивных агентов, противоопухолевых и противовоспалительных ЛС, белков, пептидных ЛС, вакцин и др. [76, 85]. Такие микросферы позволяют высвобождать молекулы ЛС в строго положенном месте и в необходимой концентрации, таким образом осуществляется мишеньориентированная терапия. Для “настройки” микросфер используют различные комбинации ЛС-полимер, также можно менять количество ЛС в микросфере и варьировать скорость их высвобождения. Получают микросферы хитозана путем перекрестного сшивания его молекул при добавлении строго определенных количеств мультивалентного аниона. Этот процесс может происходить в среде с любой кислотностью – от кислой до щелочной [76]. На эффективность захвата лекарственных молекул микросферами хитозана влияют множество факторов, как, например, природа ЛС, скорость перемешивания, соотношение ЛС-полимер, концентрация хитозана. При этом известно, что при малых концентрациях полимера происходит недостаточный захват молекул ЛС, а при высокой концентрации хитозан образует очень вязкие растворы, с которыми сложно работать. Высвобождение молекул ЛС из микросфер зависит также от многих факторов: количество ЛС, физическое состояние (молекулярные дисперсии, твердые растворы, кристаллы и др.), степень перекрестного сшивания (чем она больше, тем медленнее происходит высвобождение ЛС), наличие вспомогательных компонентов, pH среды и др. [76].
АРАБИНОГАЛАКТАН
Арабиногалактаны (AG) – это класс полисахаридов, обнаруживаемый в большом количестве растений. Арабиногалактаны подразделяют на 2 типа: арабино-4-галактаны (тип I) и арабино-3,6-галактаны (тип II). Арабиногалактаны II типа наиболее распространены и имеют существенное практическое значение. Они составляют основу камедей покрытосеменных растений, например акации, а также голосеменных, особенно лиственницы (p. Larix) [86, 87]. Камедь акации и арабиногалактан лиственницы составляют значительную часть их биомассы. Так, ядровая древесина некоторых видов лиственницы содержит до 35% AG, а одно дерево акации может ежегодно продуцировать более 2 кг камеди [86]. Для промышленного получения AG в мире в основном используют древесину западной и даурской лиственницы (Larix occidentalis и L. dahurica). Последняя в России больше известна под названием лиственницы Гмелина (L. gmelinii) и произрастает в основном на территории Восточной Сибири и Забайкалья. Также к промышленным видам лиственницы в Сибири относится сибирская лиственница – L. sibirica [86]. Древесина этих двух видов лиственницы содержит 15% AG [88]. Макромолекула арабиногалактана из древесины лиственницы имеет высоко разветвленное строение. Главная цепь ее состоит из звеньев галактозы, соединенных гликозидными связями β1→3, а боковые цепи со связями β1→6 – из звеньев галактозы и арабинозы, из единичных звеньев арабинозы, а также уроновых кислот, в основном глюкуроновой. Имеются сведения о том, что звенья арабинозы присутствуют также в основной цепи макромолекулы [86, 89]. Соотношение звеньев галактозы и арабинозы примерно 6 : 1, при этом 1/3 звеньев арабинозы находится в пиранозной форме, а 2/3 – в фуранозной [86, 89]. Эти соотношения, а также молекулярная масса AG могут варьировать как в зависимости от вида лиственницы, так в пределах одного вида. Состав макромолекул AG варьирует также в зависимости от условий его выделения из древесины и молекулярной массы [86]. Соотношение галактозных и арабинозных фрагментов во фракциях AG из древесины западной лиственницы увеличивается от 2.33 : 1 до 6.99 : 1 с увеличением молекулярной массы от 3 до 79 кДа [86].
Фармакокинетика AG после перорального приема человеком до конца не изучена. Также неизвестна абсолютная концентрация AG в плазме крови после абсорбции из ЖКТ. Однако, известно, что AG подвергается ферментации кишечной микрофлорой до низкомолекулярных продуктов и стимулирует рост полезных анаэробных бактерий: бифидобактерий и лактобактерий [90, 91].
Разветвленное строение, отличающее его от уже используемых в фармацевтике полисахаридов, позволило применить AG в качестве молекулы “хозяина” для лекарственных средств и создания комплексов-включений на его основе. Эти работы были начаты в Новосибирском институте органической химии им. Н.Н. Ворожцова под руководством академика Г.А. Толстикова в содружестве с другими институтами СО РАН [92]. Помимо нового для фармацевтики комплексообразователя в этих работах был применен механохимический твердофазный способ получения таких систем доставки, основанный на исследованиях Nakai и соавторов и школы академика В. В. Болдырева [93, 94]. Такой способ позволяет отказаться от применения растворителей и получать фармацевтические дисперсии в одну стадию.
При механохимической обработке ЛС с арабиногалактаном происходит их аморфизация и, вероятно, молекулярное диспергирование ЛС в избыток твердой фазы носителя с образованием твердых растворов, либо межмолекулярных комплексов. При последующем растворении в воде происходит образование супрамолекулярных комплексов-включений по типу “гость-хозяин”, что подтверждается увеличением растворимости молекул ЛС в воде и уменьшением времени их протонной релаксации в десятки раз, обнаруживаемым методом ЯМР-релаксации [95, 96].
В экспериментах на животных было показано, что при использовании такого подхода происходит повышение биодоступности ЛС из различных фармакологических групп при пероральном введении: ацетилсалициловой кислоты, ибупрофена, напроксена, аторвастатина, симвастатина, нифедипина, альбендазола, празиквантела [97–108]. В большинстве случаев это способствовало снижению их эффективных доз. Кроме этого аналогичный эффект был получен при введении куркумина – биологически активного метаболита куркумы [109]. Для диазепама, клозапина, медазепама, дигидрокверцетина, кверцетина, бета-каротина, варфарина, кантаксантина, карбендазима была значительно повышена растворимость в воде [100, 110–114] (табл. 1). Среди всех изученных ЛС в комплексе с AG имеется одно исключение – варфарин, обладающий стопроцентной биодоступностью, в отличие от всех других ЛС. Было показано, что его биодоступность снижается при комплексообразовании с полисахаридом [114].
Таблица 1.
Увеличение растворимости в воде лекарственных средств из твердых дисперсий с арабиногалактаном (соотношение 1 : 10 по массе) и повышение их биодоступности при введении per os*
| Лекарственное средство | Увеличение растворимости, % | Увеличение биодоступности per os (по величине AUC), разы | Снижение дозы, разы |
|---|---|---|---|
| Ацетилсалициловая кислота | 8.6 | Н.д. | 2 |
| Ибупрофен | 60 | 1.4 | 2 |
| Напроксен | 28.5 | Н.д. | 2 |
| Аторвастатин | 218 | 1.3 | – |
| Симвастатин | 650 | 2 | 2 |
| Альбендазол | 3400 | Н.д. | 5 |
| Празиквантел | 280 | 1.25 | Н.д. |
| Нифедипин | 510 | 5.2 | 2 |
| Куркумин | 958 | 7.8 | Н.д. |
| Диазепам | 139.5–4712** | Н.д. | Н.д. |
| Клозапин | 1950–10700** | Н.д. | Н.д. |
| Медазепам | 1810–13950* | Н.д. | 5 |
| Дигидрокверцетин | 477 | Н.д. | Н.д. |
| Кверцетин | 1005 | Н.д. | Н.д. |
| Бета-каротин | 264 900 | Н.д. | Н.д. |
| Варфарин | 428.5 | Cнижение в 4.2 | Н.д. |
| Кантаксантин | 263 900 | Н.д. | Н.д. |
| Карбендазим | 1522.2 | Н.д. | Н.д. |
Улучшению биодоступности ЛС из комплексов с арабиногалактаном способствует не только повышение растворимости в воде, но и адгезия молекул AG к энтероцитам. Это было установлено in vitro на клеточной линии Caco-2, которая является стандартной моделью для оценки проницаемости лекарственного вещества через стенку ЖКТ in vivo [115, 116]. В этих экспериментах 2.5% раствор AG увеличивал уровень трансэпителиального электрического сопротивления на 20%, что отражает адгезию молекул AG. Такая концентрация полисахарида аналогична применяемой дозе комплекса при пероральном введении животным. На этой же клеточной линии было обнаружено более чем двукратное повышение абсорбции молекул аторвастатина через монослой, что может быть обусловлено ингибированием P-гликопротеина, т.к. аторвастатин является его субстратом [117].
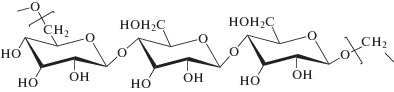


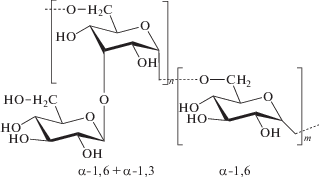
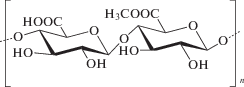
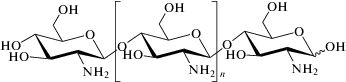

Рис. 8.
Лекарственные средства, использованные для получения твердых дисперсий с арабиногалактаном. 1 – ацетилсалициловая кислота, 2 – ибупрофен, 3 – напроксен, 4 – аторвастатин, 5 – симвастатин, 6 – альбендазол, 7 – празиквантел, 8 – нифедипин, 9 – куркумин, 10 – диазепам, 11 – клозапин, 12 – медазепам, 13 – дигидрокверцетин, 14 – кверцетин, 15 – варфарин, 16 – бета-каротин, 17 – кантаксантин, 18 – карбендазим.

ЗАКЛЮЧЕНИЕ
Рассмотренные в обзоре данные показывают, что полисахариды являются эффективными компонентами для оптимизации состава различных лекарственных форм, что позволяет улучшать профили фармакологических эффектов и фармацевтических параметров, снижать дозировки активных ингредиентов, решать проблему недостаточной растворимости гидрофобных лекарственных соединений. Использование фармакопейных ионных полисахаридов, например, катионного хитозана или анионного альгината, позволяет эффективно комплексовать лекарственные вещества с альтернативными зарядами. Кроме этого, необходимо отметить, что, пока не используемый в производстве лекарств полисахарид из лиственницы, арабиногалактан, представляется весьма перспективным носителем для плохо растворимых в воде ЛС и их пероральной доставки.
Список литературы
URL. http://www.rlsnet.ru.
DiMasi J.A., Hansen R.W., Grabowski H.G. // J. Health Econ. 2003. V. 22. P. 151–185.
Tiwari G., Tiwari R., Sriwastawa B., Bhati L., Pandey S., Pandey P., Bannerjee S.K. // Int. J. Pharm. Investig. 2012. V. 2. P. 2–11.
Amidon G.L., Lennernäs H., Shah V.P., Crison J.R. // Pharm. Res. 1995. V. 12. P. 413–420.
Loftsson T., Jarho P., Másson M., Järvinen T. // Expert Opinion on Drug Deli. 2005. V. 2. P. 335–351.
Arima H., Hayashia Y., Higashi T., Motoyama K. // Expert Opinion Drug Deliv. 2015. V. 12. P. 1425–1441.
Bonnet V., Gervaise C., Djedaïni-Pilard F., Furlan A., Sarazin C. // Drug Discov. Today. 2015. V. 20. P. 1120–1126.
Liu Z., Jiao Y., Wang Y., Zhou C., Zhang Z. // Adv. Drug Deliv. 2008. V. 60. P. 1650–1662.
Prajapati V.D., Jani J.K., Khanda S.M. // Carbohydr. Polymers. 2013. V. 95. P. 540–549.
Yoneyama M., Okada K., Mandai T., Aga H., Sakai S., Ichikawa T. // J. Japan. Soc. Starch Sci. 1990. V. 37. P. 123–127.
Fundueanu G., Constantin M., Mihai D., Bortolotti F., Cortesi R., Ascenzi P. // J. Chromatogr. B. 2003. V. 791. P. 407–419.
Singh R.S., Saini J.K., Kennedy J.F. // Carbohydrate Polymers. 2008. V. 73. P. 515–531.
Seymour L.W., Dunkan R., Chytry V., Strohalm J., Ulbrich K., Kopeček J. // J. Contr. Rel. 1991. V. 16. P. 255–262.
Toth C.A., Thomas P., Broitman S.A., Zamcheck N. // Cancer Res. 1985. V. 45. P. 392–397.
Xi K., Tabata Y., Uno K., Yoshimoto M., Kishida T., Sokawa Y., Ikada Y. // Pharm. Res. 1996. V. 13. P. 1846–1850.
Scomparina A., Salmasoa S., Bersania S., Satchi-Fainarob R., Caliceti P. // Eur. J. Pharm. Sci. 2011. V. 42. P. 547–558.
Singh R.S., Kaur N., Kennedy J.F. // Carbohydrate Polymers. 2015. V. 123. P. 190–207.
Sosnik A. // ISRN Pharmaceutics. 2014. V. 2014. Article ID 926157. 17 p.
Tønnesen H.H., Karlsen J. // Drug Dev. and Industr Pharm. 2002. V. 28. P. 621–630.
Boontheekul T., Kong H.J., Mooney D.J. // Biomat. 2005. V. 26. P. 2455–2465.
Lee K.Y., Mooney D.J. // Progress Poly. Sci. 2012. V. 37. P. 106–126.
Al-Shamkhani A., Duncan R. // J. Bioact. Compat. Polym. 1995. V. 10. P. 4–13.
Meyer K., Palmer J.W. // J. Biol. Chem. 1934. V. 107. P. 629–634.
Laurent T.C., Fraser J.R.E. // FASEB J. 1992. V. 6. P. 2397–2404.
Schante C.E., Zuber G., Herlin C., Vandamme T.F. // Carbohydr. Polym. 2011. V. 85. P. 469–489.
Raemdonck K., Martens T.F., Braeckmans K., Demeester J., De Smedt S.C. // Adv. Drug Deliv. Rev. 2013. V. 65. P. 1123–1147.
Tripodo G., Trapani A., Torre M.L., Giammona G., Trapani G., Mandracchia D. // Eur. J. Pharm. and Biopharm. 2015. V. 97. P. 400–416.
Liao Y.-H., Jones S.A., Forbes B., Martin G.P., Brown M.B. // Drug Deliv. 2005. V. 12. P. 327–342.
Tripodo G., Zhang H., Huang S., Yang X., Zhai G. // Eur. J. Med. Chem. 2014. V. 86. P. 310–317.
Brown M.B., Marriott C., Martin G.P. // Int. J. Tissue Reactions – Exp. Clin. Aspects. 1995. V. 17. P. 133–140.
Brown M.B., Forbes B., Martin G.P. The use of hyaluronan in topical drug delivery // Hyaluronan: Biomedical, Medical and Clinical Aspects / Eds. Kennedy J., Phillips G.O., Williams P.A., Hascall V. Cambridge: Woodhead Publishers, 2002. P. 249–256.
Brown M.B., Martin G.P. // Int. J. Pharm. 2001. V. 225. P. 113–121.
McEwan L.E., Smith J.G. // Aust. J. Derm. 1997. V. 38. P. 187–189.
Wolf J.E., Taylor J.R., Tschen E., Kang S.W. // Int. J. Dermatol. 2001. V. 40. P. 709–713.
Amr S.K., Brown M.B., Martin G.P., Forbes B. // Proc. Millennial World Congress of Pharm. Sci. A60. International Pharmaceutical Federation, San Francisco, USA. 2000.
Brown M.B., Moore A. The effects of a novel formulation of cyclosporine on antibody and cell–mediated immune reactions in the pleural cavity of rats // Hyaluronan in Drug Delivery / Ed. Willoughby D.A. London: Royal Society of Medicine Press, 1996. P. 121–131.
Nazir T., Martin G.P., Brown M.B. // Pharm. Sci. 2001. Suppl. 3. P. 1429.
Gurny R., Ibrahim H., Aebi A., Buri P., Wilson C.G., Washington N., Edman P., Camber O. // J. Control. Rel. 1987. V. 6. P. 367–373.
Bucolo C., Mangiafico P. // J. Ocul. Pharmacol. Ther. 1999. V. 15. P. 567–573.
Saettone M.F., Chetoni P., Torracca M.T., Burgalassi S., Giannaccini B. // Int. J. Pharm. 1989. V. 51. P. 203–212.
Herrero-Vanrell R., Fernandez-Carballido A., Frutos G., Cadorniga R. // J. Ocul. Pharmacol. Ther. 2000. V. 16. P. 419–428.
Bucolo C., Spadaro A., Mangiafico S. // Ophthalmic Res. 1998. V. 30. P. 101–106.
Moreira C.A., Armstrong D.K., Jelliffe R.W., Moreira A.T., Woodford C.C., Liggett P.E., Trousdale M.D. // Acta Ophthalmol. (Copenh). 1991. V. 69. P. 45–49.
Moreira C.A., Armstrong D.K., Jelliffe R.W., Moreira A.T., Woodford C.C., Liggett P.E., Trousdale M.D. // Acta Ophthalmol. (Copenh). 1991. V. 69. P. 50–56.
Gandolfi S.A., Massari A., Orsoni J.G. // Graefes Arch. Clin. Exp. Ophthalmol. 1992. V. 230. P. 20–23.
Lim S.T., Forbes B., Berry D.J., Martin G.P., Brown M.B. // Int. J. Pharm. 2002. V. 231. P. 73–82.
Morimoto K., Metsugi K., Katsumata H., Iwanaga K., Kakemi M. // Drug Dev. Ind. Pharm. 2001. V. 27. P. 365–371.
Surendrakumar K., Martyn G.P., Hodgers E.C.M., Jansen M., Blair J.A. // J. Control. Rel. 2003. V. 91. P. 385–394.
Thoren L. // Devel. Biol. Stand. 1981. V. 48. P. 157–167.
Suflet D.M., Chitanu G.C., Desbrieres J. // Carbohydr. Polym. 2010. V. 82. P. 1271–1277.
Mehvar R. // Journal of Controlled Release. 2000. V. 69. P. 1–25.
Walker G.J. Dextrans // Biochemistry of Carbohydrates II, V. 16 / Ed. Manners D. Baltimore: University Park Press, 1978. P. 75–125.
Mehvar R., Shepard T.L. // J. Pharm. Sci. 1992. V. 81. P. 908–912.
Koyama Y., Miyagawa T., Kawaide A., Kataoka K. // J. Control. Rel. 1996. V. 41. P. 171–176.
Varshosaz J., Emami J., Tavakoli N., Fassihi A., Minaiyan M., Ahmadi F., Dorkoosh F. // Int. J. Pharm. 2009. V. 365. P. 69–76.
Varshosaz J., Emami J., Fassihi A., Tavakoli N., Minaiyan M., Ahmadi F., Mahzouni P., Dorkoosh F. // Int. J. Colorectal Dis. 2010. V. 25. P. 1159–1165.
Varshosaz J. // Expert Opin. Drug Deliv. 2012. V. 9. P. 509–523.
Vyas S., Trivedi P., Chaturvedi S.C. // Acta Pharm. 2007. V. 57. P. 441–450.
Shrivastava S.K., Jain D.K., Trivedi P. // Pharmazie. 2003. V. 58. P. 389–391.
Feeney M., Giannuzzo M., Paolicelli P., Casadei M.A. // Drug Delivery. 2007. V. 14. P. 87–93.
Sriamornsak P. // Expert Opin. Drug Deliv. 2011. V. 8. P. 1009–1023.
Endress H.U., Christensen S.H. Pectins // Phillips GO. Handbook of Hydrocolloids. 2nd ed. / Eds. Williams P.A. Cambridge: Woodhead Publishers Ltd. 2009. P. 274–297.
Sriamornsak P. // Silpakorn. Univ. Int. J. 2003. V. 3. P. 206–228.
Oakenfull D.G. The chemistry of high–methoxyl pectins // The Chemistry and Technology of Pectin / Ed. Walter R.H. New York: Academic Press, 1991. P. 87–108.
Rinaudo M. Polyelectrolytes derived from natural polysaccharides // Monomer, Polymers and Composites from Renewable Resources / Eds. Belgacem M., Gandini A. Amsterdam: Elsevier, 2008. P. 495–516.
Morris G, Kök S, Harding S, Adams G. // Biotechnol. Genet. Eng. Rev. 2010. V. 27. P. 257–284.
Liu L., Fishman, M.L., Hicks, K.B. // Cellulose. 2007. V. 14. P. 15.
Baloglu E., Özyazici M., Hizarcioglu S.Y., Senyigit T. // Pharmaceutical Development and Technology. 2006. V. 11. P. 477–484.
Thirawong N., Kennedy R.A., Sriamornsak P. // Carbohydrate Polymers. 2008. V. 71. P. 170–179.
Gaucher G., Satturwar P., Jones M.-Ch., Furtos A., Leroux J.-Ch. // European Journal of Pharmaceutics and Biopharmaceutics. 2010. V. 76. P. 147–158.
Charlton S.T., Davis S.S., Illum L. // Journal of Controlled Release. 2007. V. 118. P. 225–234.
Chelladurai S., Mishra M., Mishra B. // Chemical and Pharmaceutical Bulletin. 2008. V. 56. P. 1596–1599.
Zhang W., Mahuta K.M., Mikulski B.A., Harvestine J.N., Crouse J.Z., Lee J.C., Kaltchev M.G., Tritt C.S. // Pharm. Dev. Technol. 2016. V. 21. P. 127–130.
Xing P., Shi Y, Dong C., Liu H., Cheng Y., Li J., Sun D., Li M., Sun K., Feng D. // AAPS PharmSciTech. 2017. V. 8. P. 1095–1103.
Saha N.R., Sarkar G., Roya I., Rana D., Bhattacharyya A., Adhikari A., Mukhopadhyay A., Chattopadhyay D. // Carbohydrate Polymers. 2016. V. 136. P. 1218–1227.
Sinha V.R., Singla A.K., Wadhawan S., Kaushik R., Kumria R., Bansal K., Dhawan S. // International Journal of Pharmaceutics. 2004. V. 274. P. 1–33.
Kato Y., Onishi H., Machida Y. // Curr. Pharm. Biotechnol. 2003. V. 4. P. 303–309.
Singla A.K., Chawla M. // J. Pharm. Pharmacol. 2001. V. 53. P. 1047–1067.
Fukuda H. // Bull. Chem. Soc. Jpn. 1980. V. 53. P. 837–840.
Onsoyen E., Skaugrud O. // J. Chem. Technol. Biotechnol. 1990. V. 49. P. 395–404.
Chandy T., Sharma C.P. // Artif. Cells Artif. Organs. 1990. V. 18. P. 1–24.
Demarger-Andre S., Domard A. New properties of chitosan in lipid dispersions // Chitin World / Eds. Karnicki Z.S., Wojtaso Pajak A., Breziski M.M., Bylowski P.J. Germany: Bremerhauser. 1994. P. 153–158.
Kas H.S. // J. Microencapsul. 1997. V. 14. P. 689–711.
Kim D., Jeong Y., Choi C., et al. // Int. J. Pharm. 2006. V. 319. P. 130–138.
Ray S.D. // Acta Pol. Pharm. 2011. V. 68. V. 619–622.
Медведева Е.Н., Бабкин В.А., Остроухова Л.А. // Химия растительного сырья. 2003. Т. 1. С. 27–37.
Odonmazig P., Ebringerova A., Machova E., Alfoldi J. // Carbohydr. Res. 1994. V. 252. P. 317–324.
Антонова Г.Ф., Тюкавкина Н.А. // Химия древесины. 1983. Т. 2. С. 89–96.
Антонова Г.Ф., Усов А.И. // Биоорганическая химия. 1984. Т. 10. С. 1664–1669.
Vince A.J., McNeil N.I., Wager J.D., Wrong O.M. // Br. J. Nutr. 1990. V. 63. P. 17–26.
Kaneo Y., Ueno T., Tanaka T., Iwase H., Yamaguchi Y., Uemura T. // International Journal of Pharmaceutics. 2000. V. 201. P. 59–69.
Душкин А.В., Метелева Е.С., Толстикова Т.Г., Толстиков Г.А., Поляков Н.Э., Неверов Н.А., Медведева Е.Н., Бабкин В.А. // Известия АН Сер. хим. 2008. № 6. С. 1274–1282.
Nakai Y., Fukuoka E., Nakajima S., Yamamoto K. // Chem. Pharm. Bull. 1977. V. 25. P. 3340–3345.
Ягодин А.Ю., Душкин А.В., Болдырев В.В. // Фармация. 1991. № 3. С. 69–71.
Apanasenko I.E., Selyutina O.Yu., Polyakov N.E., Suntsova L.P., Meteleva E.S., Dushkin A.V., Vachali P., Bernstein P.S. // Archives of Biochemistry and Biophysics. 2015. V. 572. P. 58–65.
Polyakov N.E., Leshina T.V., Meteleva E.S., Dushkin A.V. et al. // J. Phys. Chem. B. 2009. V. 113. P. 275–282.
Душкин А.В. Чистяченко Ю.С., Толстикова Т.Г., Хвостов М.В., Поляков Н.Э., Ляхов Н.З., Толстиков Г.А. // Доклады Академии наук. 2013. Т. 451. С. 107–109.
Chistyachenko Yu. S., Dushkin A.V., Polyakov N.E., Khvostov M.V., Tolstikova T.G., Tolstikov G.A., Lyakhov N.Z. // Drug Delivery. 2015. V. 22. P. 400–407.
Khvostov M.V., Tolstikova T.G., Borisov S.A., Zhukova N.A., Dushkin A.V., Chistyachenko Yu.S., Polyakov N.E. // Current Drug Delivery. 2016. V. 13. P. 582–589.
Dushkin A.V., Tolstikova T.G., Khvostov M.V., Tolstikov G.A. Complexes of polysaccharides and glycyrrhizic acid with drug molecules. Mechanochemical synthesis and pharmacological activity // The Complex World of Polysaccharides / Ed. Karunaratne D.N. Croatia: InTech., 2012. P. 573–602.
Khvostov M.V., Borisov S.A., Tolstikova T.G., Dushkin A.V., Tsyrenova B.D., Chistyachenko Yu.S., Polyakov N.E., Dultseva G.G., Onischuk A.A., An’kov S.V. // Eur. J. Drug Metabol. and Pharmacokin. 2017. V. 42. P. 431–440.
Борисов С.А., Хвостов М.В., Толстикова Т.Г., Душкин А.В., Чистяченко Ю.С. // Сибирский научный медицинский журнал. 2017. Т. 37. С. 19–25.
Kong R., Zhu X., Meteleva E.S., Chistyachenko Yu.S., Suntsova L.P., Polyakov N.E., Khvostov M.V., Baev D.S., Tolstikova T.G., Yu J., Dushkin A.V., Su W. // Int. J. Pharmac. 2017. V. 534. P. 108–118.
Kong R., Zhu X., Meteleva E.S., Polyakov N.E., Khvostov M.V., Baev D.S., Tolstikova T.G., Dushkin A.V., Su W. // Drug Delivery and Translational Research. 2018. V. 8. P. 1200–1213.
Толстикова Т.Г., Хвостов М.В., Брызгалов А.О., Толстиков Г.А., Душкин А.В., Метелева Е.C., Нифантьев Н.Э. Патент РФ 2391980, заявл. 22.10.2008, опубл. 20.06.2010. Бюл. № 17.
Чистяченко Ю.С., Хвостов М.В., Белоусов А.И., Жукова Н.А., Пахарукова М.Ю., Катохин А.В., Халиков С.С., Толстикова Т.Г., Душкин А.В., Мордвинов В.А., Ляхов Н.З. // Докл. АН. 2014. Т. 456. P. 741–744.
Душкин А.В, Метелева Е.С., Ляхов Н.З., Чистяченко Ю.С., Халиков С.С., Толстикова Т.Г., Хвостов М.В. и др. Патент РФ 2545797, заявл. 03.10.2013, опубл. 10.04.2015. Бюл. № 10.
Ляхов Н.З., Душкин А.В., Метелева Е.С. Патент РФ 2681649, заявл. 27.12.2017, опубл. 12.03.2019. Бюл. № 8.
Zhang Q., Suntsova L., Chistyachenko Yu.S., Evseenko V., Khvostov M.V., Polyakov N.E., Dushkin A.V., Su W. // International Journal of Biological Macromolecules. 2019. V. 128. P. 158–166.
Ляхов Н.З., Душкин А.В., Толстикова Т.Г. // Известия АН. Серия химическая. 2013. № 1. С. 233–239.
Душкин А.В., Толстиков Г.А., Толстикова Т.Г., Метелёва Е.С. Патент РФ 2337710 заявл. 05.12.2006, опубл. 10.11.2008. Бюл. № 31.
Tolstikova T.G., Khvostov M.V., Lifshits G.I., Dushkin A.V., Meteleva E.S. // Letters in Drug Design and Discovery. 2011. V. 8. P. 201–204.
Khvostov M.V., Tolstikova T.G., Chernonosov A.A., Fedorova O.S., Lifshits G.I. // Asian Journal of Pharmaceutical and Clinical Research. 2012. V. 5. P. 251–252.
Khvostov M.V., Chernonosov A.A., Tolstikova T.G., Kasakin M.F., Fedorova O.S., Dushkin A.V. // BioMed Research International. 2013. V. 2013. Article ID 156381. 4 p.
Artursson P., Palm R., Luthman K. // Advanced Drug Delivery Reviews. 2001. V. 46. P. 27–43.
Pade V., Stavchansky S. // Journal of Pharmaceutical Sciences. 1998. V. 87. P. 1604–1607.
Date A.A., Nagarsenker M.S. // JPP. 2007. V. 59. P. 1583–1584.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия