

Биоорганическая химия, 2020, T. 46, № 1, стр. 3-17
Основы биологии микроРНК: строение, биогенез и регуляторные функции (Обзорная статья)
И. А. Запорожченко *, **, Е. Ю. Рыкова *, **, П. П. Лактионов *, **
* Институт химической биологии и фундаментальной медицины СО РАН, Россия, 630090, Новосибирск, просп. акад. Лаврентьева 8
** ФГБУ Национальный медицинский исследовательский центр имени E.Н. Мешалкина Министерства здравоохранения РФ, Россия, 630055, Новосибирск, Новосибирская обл., Речкуновская ул. 15
Поступила в редакцию 08.07.2019
После доработки 31.07.2019
Принята к публикации 09.09.2019
Аннотация
Эпигенетическая регуляция экспрессии играет ключевую роль в контроле многих клеточных процессов. Важными участниками такой регуляции являются микроРНК, представляющие собой короткие некодирующие РНК. Впервые микроРНК были обнаружены в 1993 г., однако их активное исследование началось лишь с 2000-х годов. Современные данные указывают на то, что микроРНК могут контролировать экспрессию как минимум половины генов человека. Будучи вовлеченными в регуляцию большого количества генов-мишеней, отвечающих за жизнедеятельность клетки, микроРНК необходимы для нормального развития и функционирования организма, а нарушение их функций сопутсвует развитию многих патофизиологических процессов. В настоящем обзоре описаны основные стадии “жизненного цикла” молекул микроРНК в клетках человека. Отдельные разделы посвящены происхождению, созреванию, функциям и регуляции активности микроРНК. Обзор рассчитан в первую очередь на читателей, желающих впервые познакомиться с особенностями биологии этих РНК, однако может быть полезен и специалистам в этой области в качестве справочного материала.
ВВЕДЕНИЕ
В 1993 г. Ли с соавторами обнаружили у Caenorhabditis elegans короткую РНК, продукт гена lin-4, обладавшую антисмысловой комплементарностью к РНК гена lin-14 [1]. Это первое свидетельство существования микроРНК не вызвало большого резонанса – находке не придали особого значения и приписали статус артефакта. Спустя почти десять лет, в 2000 г., Рейнхарт с коллегами обнаружили короткую некодирующую РНК let-7, которая обладала комплементарностью к участку 3' нетранслируемой области (3'-НТО) продукта гена lin-41 и, посредством взаимодействия с ней, регулировала ход поздних стадий развития C. elegans [2]. Эта вторая находка послужила отправной точкой интереса научного сообщества к таким молекулам и вскоре исследователями было обнаружено огромное многообразие молекул со сходными свойствами. Из-за небольшой длины им дали название микроРНК (microRNA, miRNA) и выделили в отдельный класс малых РНК [3–5]. Представители этого класса обнаружены у большинства животных и растений. Последняя редакция базы аннотированных микроРНК (miRBase, http://www.mirbase.org) содержит информацию о более чем 28 000 потенциальных микроРНК у почти 100 биологических видов, в том числе 2400 микроРНК человека, из которых около 1000 определено с высоким уровнем доверия (high confidence) [6–8]. За последние два десятилетия проделана огромная работа по изучению биологии этих РНК – исследованы особенности структуры микроРНК, механизмы их созревания, определены основные функции микроРНК в клетке и спектр ассоциированных с ними белковых факторов.
ПРОИСХОЖДЕНИЕ И СОЗРЕВАНИЕ микроРНК
На данный момент известно несколько разных классов коротких регуляторных РНК, например, малые ядрышковые РНК (snoRNA, small nucleolar RNA) [9], piwi-взаимодествующие РНК (piRNA, piwi-interacting RNA) [10, 11], эндогенные малые интерферирующие РНК (endo-siRNA, endogenous small interfering RNA) [11, 12], короткие шпилечные РНК (shRNA, short hairpin RNA) [13], инициаторные РНК (tiRNA, transcription initiation RNA) [14]. Представителей класса микроРНК от других некодирующих РНК отличает совокупность особых характеристик зрелой молекулы, характер процесса созревания и особенности предшественников, а также ряд их характерных функций в клетке [15]. Зрелая молекула микроРНК представляет собой одноцепочечную молекулу РНК длиной 18–24 нт, выбираемую в процессе созревания из состава асимметричного РНК-дуплекса [16]. Вместе с одним из белков из семейства Аргонавтов (Argonaute, AGO) и вспомогательными белками, эта цепь микроРНК образует RISC – РНК-индуцируемый комплекс выключения гена (RNA-Induced Silencing Complex; в литературе можно также встретить понятия miRISC – miRNA-loaded RISC, и mature RISC, обозначающие функционально активный RISC-комплекс, содержащий в составе зрелую микроРНК). Этот рибонуклеиновый комплекс способен связывать и регулировать экспрессию мРНК мишеней, имеющих соответствующие цис-регуляторные элементы [17].
Номенклатура микроРНК. Для наименования микроРНК используется стандартизованная система имеющая вид hsa-miR-(N)-3/5p (табл. 1) [15]. Первые три буквы указывают видовую принадлежность микроРНК (в данном примере Homo sapiens) и во многих контекстах часто опускаются. Следующий трехбуквенный блок указывает, идет ли речь о зрелой одноцепочечной микроРНК (miR), ее предшественнике (mir или mir) или гене, кодирующем эту микроРНК (MIR или MIR). Далее идет порядковый номер (N), присваиваемый при аннотации, например, miR-17, после которого может следовать латинская буква (a, b, c и т.д.), используемая для указания близкого родства последовательностей микроРНК. Так, последовательности зрелых hsa-miR-19a-3p и hsa-miR-19b-3p отличаются лишь одним основанием. Дополнительные копии генов (или шпилечных предшественников), экспрессирующие идентичные зрелые микроРНК, обозначаются дополнительным порядковым номером – hsa-mir-121-1 и hsa-mir-121-2. Для зрелой микроРНК после номера через дефис следует индикатор цепи шпилечного предшественника, из которой происходит микроРНК (3р для 3', 5р для 5'). В литературе также можно встретить устаревший вариант обозначения цепей микроРНК, основанный на уровне экспрессии каждой цепи. При такой записи для указания направляющей или ведущей (guide) цепи, которая предпочтительно включается в RISC, дополнительных символов не используется, а комплементарная ей пассажирская (passenger) цепь отмечается звездочкой (*), например, miR-17*.
Таблица 1.
Структура наименования микроРНК
| Организм | Тип | Номер | Цепь (копия для гена) |
||||
|---|---|---|---|---|---|---|---|
| Ген | hsa | – | MIR | – | 19a | – | 1 |
| Предшественник | hsa | – | mir | – | 19a | – | 1 |
| Зрелая микроРНК | hsa | – | miR | – | 19a | – | 3p |
Следует отметить, что для обозначения ряда исключительных микроРНК, например, lin-4 и let-7, используются первоначально присвоенные им тривиальные названия, но при этом к ним применяются остальные правила нотации, описанные выше, к примеру, hsa-let-7b-5p.
Кодирование микроРНК в геноме. Последняя редакция базы аннотированных микроРНК miRBase (http://www.mirbase.org) содержит информацию о более чем 28 000 потенциальных микроРНК у почти 100 биологических видов [7]. В том числе в miRBase представлены записи о 2400 отдельных микроРНК человека, из которых около 1000 определено с высоким уровнем доверия (high confidence) [8]. Корреляция между числом микроРНК и разнообразием клеточного состава организма позволяет предположить, что развитие этой системы регуляции может быть одним из ключевых эволюционных факторов усложнения организации. В совокупности с другими некодирующими РНК, существование микроРНК может отчасти объяснять отсутствие корреляции размера генома и числа пристутствующих в нем белок-кодирующих генов с фенотипической сложностью организма (так называемые С- и G-парадоксы) [18, 19]. У сложных многоклеточных организмов количество генов микроРНК может исчисляться сотнями, в то время как у одноклеточных эукариот, например дрожжей Saccharomyces cerevisiae, они отсутствуют полностью [18, 20].
Исторически первые обнаруженные микроРНК являлись продуктами процессинга транскриптов специализированных микроРНК-кодирующих генов [1, 2, 4]. Фромм с соавторами с высокой степенью достоверности выявили 519 канонических генов микроРНК человека, которые расположены на всех аутосомах и X-хромосоме человека и часто сгруппированы в кластеры [21–23]. Гены в пределах одного кластера часто кодируют гомологичные микроРНК со схожим набором регулируемых мишеней, и образуют семейства микроРНК. При этом специализированные гены являются не единственным источником микроРНК – до 50% микроРНК млекопитающих происходит из транскриптов других генов [24, 25].
Основным источником “внутригенных” микроРНК являются интроны (такие микроРНК часто называют миртроны, miRNA + intron = mirtron), но при помощи биоинформатического анализа предсказано существование генов микроРНК в экзонах и регуляторных областях генов, соответствующих 3'- и 5'-нетранслируемым областям (НТО) [18, 26–30]. Часть интронных микроРНК (25–30%) имеет собственные промоторы, находящиeся в предшествующих интронах, транскрипция с которых независима от транскрипции гена-хозяина [29, 31, 32]. Внутригенные микроРНК могут транскрибироваться и процессироваться вместе с геном-хозяином, и в отдельных случаях показана корреляция между их экспрессией [24, 33], но в других исследованиях такая зависимость отсутствовала [30]. Кроме того, ряд авторов предполагают, что источником микроРНК могут служить другие некодирующие РНК, например, малые ядрышковые РНК (мяРНК) и транспортные РНК (тРНК), мобильные элементы генома и продукты процессированных псевдогенов [34–39].
Факт наличия у большинства животных явных гомологов основных белков, участвующих в созревании и функционировании микроРНК позволяет предполагать общее филогенетическое происхождение микроРНК-опосредованной регуляции у животных [20]. Это предположение подтверждается и тем, что многие микроРНК высоко консервативны, и их гомологи встречаются у видов, филогенетически далеко отстоящих друг от друга. Сравнительный анализ малых РНК метазоа показал, что miR-100 встречается у всех билатеральных животных, а также у кишечнополостного Nematostella vectensis [20]. Последовательность функциональных участков микроРНК эволюционно наиболее жестко закреплена, и эта консервативность снижается в ряду: 5'-концевой участок узнавания мишени; 3'-последовательность, содержащая нуклеотиды, участвующие в компенсаторном спаривании; центральная часть основной цепи микроРНК; остальные части микроРНК-дуплекса [20, 40–42]. При этом регуляторные области мРНК, в составе которых есть сайты связывания микроРНК, также находятся под эволюционным давлением [43–45].
Созревание микроРНК. Механизм созревания микроРНК являлся одним из их классообразующих признаков [15]. В основе механизма созревания микроРНК лежит серия последовательных расщеплений незрелых молекул предшественников при помощи специфической или неспецифической РНК-гидролизующей активности белковых комплексов с образованием незрелой двуцепочечной молекулы микроРНК, одна из цепей которой затем включается в состав RISC [46]. Первоначально был предложен один универсальный механизм созревания микроРНК, характерный для специализированных генов микроРНК, однако впоследствии были обнаружены альтернативные источники микроРНК, и в связи с этим круг известных механизмов созревания микроРНК также расширился. В современной научной литературе принято деление путей процессинга микроРНК на канонический и ряд исключений, формирующих гетерогенную группу неканонических путей созревания микроРНК.
Канонический путь созревания микроРНК (рис. 1) начинается в клеточном ядре с транскрипции гена микроРНК РНК-полимеразой II (Pol II) с образованием при-микроРНК (pri-miRNA, primary miRNA), часто достигающей нескольких тысяч нуклеотидов в длину и подвергающейся 5'-кэпированию и, часто, 3'-полиаденилированию [47–49]. Исключение составляют гены, прилегающие к Alu-повторам, которые транскрибирует РНК-полимераза III (Pol III) [50]. Кластеры микроРНК часто транскрибируются с образованием длинных, вплоть до нескольких тысяч нуклеотидов, полицистронных молекул, которые служат источником предшественников нескольких разных микроРНК [33, 51]. При-микроРНК обладают сложной вторичной структурой с множеством шпилек, и могут дополнительно образовывать сложную трехмерную структуру, параметры которой определяют эффективность узнавания и процессинга белковыми факторами [52, 53].
Рис. 1.
Канонический путь созревания микроРНК. На рисунке схематично представлены стадии превращения предшественников микроРНК – от транскрипции до образования активного RISC. Пояснения в тексте.

На следующем шаге при-микроРНК подвергается в ядре гидролизу микропроцессорным комплексом или микропроцессором (microprocessor complex), состоящим из РНКазы III класса Drosha и белка DGCR8 (DiGeorge syndrome critical region gene) [54–56]. За счет сродства к двуцепочечной РНК микропроцессор узнает в при-микроРНК шпильки, обладающие характерной структурой и несущие определенные нуклеотидные мотивы [57–62]. В результате гидролиза высвобождаются шпилечные молекулы РНК длиной ~70 нт, называемые предшественниками микроРНК или пре-микроРНК (precursor miRNA, pre-miRNA) и состоящие из петлевого одноцепочечного участка и двуцепочечного стебля (не всегда обладающего идеальной комплементарностью оснований), заканчивающегося выступающим 3'-концом длиной в 2 нт [55, 63]. Эти структурные элементы необходимы для узнавания экспортином-5 (exportin-5, karyopherin-5), который при участии GTP-азы RanGTP переносит пре-микроРНК в цитоплазму клетки [64–66].
В цитоплазме пре-микроРНК передается комплексу белков, состоящему из РНКазы III класса Dicer, TRBP (transactivation response RNA-binding protein), PACT (protein kinase R activating protein), и одного из белков семейства Argonaute (AGO1–AGO4) [67]. Dicer узнает характерную структуру пре-микроРНК (неспаренные 5'- и 3'-концы, стебель и одноцепоченый петлевой участок [68–71]) и вырезает из нее петлевой участок. Это приводит к образованию асимметричных двуцепочечных молекул РНК длиной 19–24 п.о. с выступающими 3'-концами размером 2 нт – незрелых микроРНК (immature miRNA) [72, 73]. Гидролиз происходит на фиксированном расстоянии в ~22 п.о. от конца дуплекса, которое определяется взаимным пространственным расположением доменов белка, вследствие чего опознавание пре-микроРНК белком Dicer часто сравнивают с молекулярной “линейкой” [70, 74].
Завершающим этапом канонического пути созревания микроРНК является “загрузка” одной из цепей дуплекса на белок AGO с образованием RISC [75, 76]. Структура AGO высоко консервативна и состоит из четырех доменов: N-концевого, PAZ (PIWI-Argonaute-Zwille), MID (middle) и PIWI, попарно объединенных в субъединицы N‑PAZ и MID-PIWI, которые соединены подвижным шарнирным участком [77, 78]. MID и PAZ домены содержат сайты связывания 5'- и 3'-концов микроРНК, соответственно [79, 80]. У человека существует четыре паралога AGO (AGO1–AGO4), не имеющих заметных функциональных и структурных различий [81]. Единственным исключением является эндонуклеазная активность AGO2. Наборы микроРНК, связанные с разными паралогами AGO различаются мало, но относительная избирательность загрузки микроРНК все же присутствует [82, 83].
В процессе загрузки микроРНК в RISC происходит перенос и связывание микроРНК белком AGO, выбор ведущей цепи, расплетание дуплекса и удаление второй цепи. Весь процесс загрузки обеспечивается работой шаперонов, в том числе комплекса Hsc70/Hsp90 [84–86]. Незрелый микроРНК-дуплекс передается на MID домен белка AGO сразу после гидролиза Dicer. Связывание микроРНК происходит асимметрично, и предпочтение обычно отдается концу дуплекса (нуклеотиды 1–4 с 5'-конца цепи) с наименьшей термодинамической стабильностью спаривания [87–89]. Ведущей становится цепь, 5'-конец которой участвует в таком спаривании. После успешного связывания конформационные изменения в AGO “утапливают” микроРНК во внутреннее пространство белка, где происходит связывание 3'-конца ведущей цепи, после чего молекула микроРНК изгибается и укладывается в специальный канал, разделяющий две субъединицы белка [77, 78, 82, 90]. Изгибание дуплекса приводит к его расплетению, в то время как N-концевой домен AGO выполняет роль клина, который разделяет цепи микроРНК, в результате чего пассажирская цепь, не имеющая связей с белком, вытесняется из комплекса [77, 78, 82, 91]. При загрузке на AGO2 в пассажирскую цепь также вносится одноцепочечный разрыв, что дополнительно облегчает ее высвобождение из RISC. За счет связывания белком 3'-конца и сахарно-фосфатного остова ведущей цепи микроРНК остается прочно закрепленной внутри AGO и выполняет роль “стержня”, стабилизируя активную конформацию всего рибонуклеинового комплекса.
Неканонические пути созревания микроРНК отличаются тем, что в процессинге принимают участие не все белки канонического пути. Такие альтернативные пути принято делить на Drosha- и Dicer-независимые. Известно несколько Drosha-независимых путей созревания микроРНК, в большинстве из которых предшественники микроРНК являются побочными продуктами процессинга других РНК (например, мяРНК и тРНК) и не требуют гидролиза при помощи микропроцессорного комплекса [92]. Так процессируются и предшественники ряда интронных микроРНК. Линеаризованные интроны могут формировать шпилечные структуры, которые затем процессируются при помощи Dicer [93–95]. Ярким примером Dicer-независимого процессинга является процесс созревания miR-451 – консервативной микроРНК, принимающей активное участие в дифференцировке клеток эритропоэтического ряда у позвоночных [96]. При-микроРНК mir-451 узнается и гидролизуется микропроцессорным комплексом, но получающаяся в итоге шпилечная структура имеет слишком короткий (17 п.о.) стебель для узнавания Dicer. Вместо этого предшественник напрямую загружается на AGO2, который надрезает одну из цепей, а затем полученный промежуточный продукт укорачивается при помощи poly(А)-специфичной рибонуклеазы (PARN) до получения зрелой молекулы длиной 23 нт [97–99].
Необходимо отметить, что различные варианты созревания микроРНК наблюдаются не только на этапах гидролиза предшественников. Например, известно, что 5'-кэпированые предшественники miR-320a и miR-484 переносит в цитоплазму экспортин-1 (exportin-1), а не экспортин-5 [100]. Недавнее исследование показало, что экспортин-1-зависимый транспорт характерен для многих пре-микроРНК в покоящихся клетках человека, и, таким образом, представляет собой важный альтернативный путь процессинга микроРНК [101].
Разнообразие механизмов созревания микроРНК имеет важный биологический смысл. С эволюционной точки зрения одним его преимуществ является частичная заменимость участников созревания микроРНК. Так, потеря или снижение экспрессии основных белков Drosha и Dicer обычно приводит к нарушению развития, и во многих случаях может быть летальной или условно летальной [102, 103] При этом наличие альтернативных путей процессинга позволяет частично избежать полной потери регуляторных функций микроРНК [104, 105]. Схожей цели может служить и видимая избыточность спектра белков AGO.
Несмотря на то что созревание большинства изученных микроРНК проходит по каноническому пути, наличие альтернативных путей биогенеза придает системе микроРНК дополнительную гибкость, а многие микроРНК, процессирующиеся неканонически, играют важную биологическую роль. В любом случае, вне зависимости от пути созревания, в результате образуется активный комплекс RISC, содержащий ведущую цепь микроРНК и готовый к выполнению регуляторных функций.
МЕХАНИЗМЫ ДЕЙСТВИЯ И ФУНКЦИИ микроРНК
Определяющей чертой микроРНК, наряду с созреванием, являются их функции и особенности механизмов, обеспечивающих их реализацию. Известно, что микроРНК участвуют в регуляции множества клеточных процессов и выступают в качестве ключевых звеньев в составе сложных регуляторных путей. Действие микроРНК играет важную роль в регуляции важнейших биологических процессов, в том числе клеточного цикла, роста и дифференцировки клеток, миграции, апоптоза и реакции на стресс [106, 107].
Основной функцией микроРНК считается подавление экспрессии генов на пост-транскрипционном уровне посредством связывания с участками в нетранслируемых областях мРНК, приводящего к деградации или обратимой инактивации последних. Предполагают, что микроРНК могут контролировать экспрессию от 30 до 50% генов человека и выступают микро-регуляторами экспрессии или “скульпторами транскриптома”, образуя своеобразную “страховочную сеть”, препятствующую бесконтрольному изменению экспрессии клеточных белков [108–110]. Такая аналогия связана с тем, что в большинстве случаев результатом влияния микроРНК является только частичное изменение экспрессии гена-мишени – не более чем в два раза на уровне белкового продукта [67, 111–114]. В некоторых случаях микроРНК могут полностью ингибировать экспрессию гена-мишени (например, lin-4 и let-7 C. elegans [1, 2, 115]), однако это скорее исключение из правила. При этом, несмотря на столь “скромный” вклад большинства индивидуальных микроРНК, их суммарное влияние на регуляцию экспрессии нельзя недооценивать. Активность микроРНК из большинства консервативных семейств абсолютно необходима для нормального развития и функционирования организма (подробно описано в работе [108]).
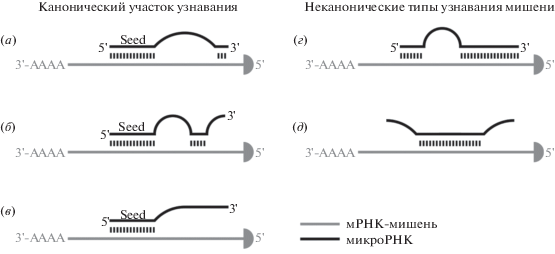
Действие RISC можно разделить на два этапа – распознавание мишени и осуществление регуляторного эффекта. За первое отвечает рибонуклеиновый компонент RISC (за исключением очень редких случаев [116]). Узнавание и связывание обеспечивается комплементарным взаимодействием участка узнавания (seed) микроРНК и узнаваемого участка (seed-match или miRNA response element, MRE), находящегося в мРНК-мишени (рис. 2) [117]. Канонические участки узнавания включают нуклеотиды 2–7 (или 2–8) в составле последовательности микроРНК, иногда подкрепленные неканоническим спариванием 5'-концевого нуклеотида микроРНК с аденозином в мРНК мишени [67, 118–122]. Взаимодействие с мишенью может дополнительно стабилизироваться за счет компенсаторного спаривания одного или нескольких оснований на 3'-конце микроРНК [67]. Описан также ряд неканонических сайтов узнавания, в том числе центральные (centered) участки узнавания, образуемые 4–15 нуклеотидами в составе последовательности микроРНК [42, 123], а также форматы взаимодействия с мишенью, в которых участвуют нуклеотиды, разнесенные по последовательности микроРНК [124, 125]. В комплексах, образованных участком узнавания и мишенью, часто присутствуют ошибочно спаренные и “вывернутые” (bulge) основания, причем предполагается, что это может играть важную функциональную роль в механизме узнавания мишени [123, 126–128]. При этом, взаимодействие микроРНК с мишенью может, в определенной степени, влиять на тип и силу вызываемого регуляторного эффекта [129]. Низкая энергия связывания может негативно влиять на эффективность подавления трансляции [121], а при высокой степени комплементарности мишени микроРНК в комплексе с AGO2 может катализировать гидролиз мишени [130].
Рис. 2.
Примеры взаимодействия микроРНК с мРНК-мишенью. Варианты канонического участка узнавания (seed) с компенсаторным спариванием нуклеотидов на 3'-конце микроРНК (а), (б) и без компенсаторного спаривания (5'-доминантный участок узнaвания мишени) (в). Неканонические типы узнавания мишени: 3'-доминантное связывание с редуцированным участком узнавания (г) и центральный участок узнавания мишени (д).
В силу описанных особенностей узнавания мишеней большинство микроРНК не обладают строгой специфичностью, и способны регулировать до нескольких сотен мишеней [21, 67, 119], причем связывание с множественными мишенями подтверждается экспериментальными данными [131]. При этом в одной мишени также могут присутствовать множественные сайты узнавания (одной или нескольких) микроРНК, которые являются синергистами или антагонистами [114, 132]. Антагонистами по отношению друг к другу могут выступать и сайты связывания, находящиеся на разных мРНК [133]. Гибкость и конкурентность узнавания и связывания мишени приводят к формированию сложных регуляторные сетей, вовлекающих большее число экпрессируемых мРНК [133–135].
Регуляторные эффекты микроРНК обеспечивает белковая составляющая RISC. Помимо AGO важным участником этих событий является белок GW182 (TNRC6A,B,C у человека), который обеспечивает взаимодействие RISC с белками трансляционного аппарата и привлекает дополнительные комплексы, например CCR4-NOT, PAN2-PAN3, DCP1-DCP2 и DDX6 [136–140]. После узнавания и связывания мишени с AGO связывается GW182, что запускает развитие эффекта на мишень. Для подавления трансляции под действием RISC было предложено несколько разных механизмов [17, 141–143]. Известный набор регуляторных механизмов позволяет микроРНК как препятствовать началу трансляции мишени, так и остановить процесс синтеза пептида в процессе элонгации и предотвратить реинициацию за счет деградации мРНК или ограничения ее доступности для рибосом.
Согласно современным взглядам, многие из этих механизмов являются звеньями одного и того же процесса, связанными между собой конкурентными взаимодействиями. Так, если ранее полагалось, что дестабилизация и деградация мРНК является основным механизмом регуляции, то более свежие данные показывают, что подавление трансляции может играть гораздо более важную роль [144]. На основании этих данных рядом авторов была предложена последовательная модель регуляции, обобщающая основные механизмы действия микроРНК (рис. 3) [142, 145, 146]. Согласно этой модели, предотвращение и подавление трансляции является первым, наиболее быстрым и часто основным механизмом действия микроРНК, однако на уровне белка эффект этого механизма относительно невелик [142, 146–148]. Дезаденилирование под действием CCR4-NOT и PAN2-PAN3, декепирование при помощи DDX6 и последующий гидролиз нуклеазами не являются необходимыми для репрессии мРНК, но выступают в качестве вспомогательных механизмов, способных вызывать значительные изменения экспрессии белка [17, 115, 130, 146, 147, 149, 150]. Особняком в этой схеме стоит способность AGO2-содержащих RISC катализировать гидролиз мишени [130].
Рис. 3.
Механизм подавления трансляции под действием RISC. Представлены возможности блока инициации трансляции мРНК и подавления трансляции на стадии элонгации путем обратимой репрессии и последующей деградации мишени. Пояснения в тексте. PABP – poly(A)-связывающий белок. eIF4A, eIF4A2, eIF4E, eIF4G, eIF4E – факторы инициации трансляции.

Интересно, что помимо подавления транскрипции обнаружены и другие механизмы действия микроРНК. Так, микроРНК могут стабилизировать мРНК и усиливать их трансляцию, выполнять функцию “приманки” (decoy), препятствуя взаимодействию белковых факторов с их РНК-мишенями, а также участвовать в процессе созревания других микроРНК [151–153]. В последнее время было показано, что зрелые микроРНК и компоненты RISC обнаружены в клеточном ядре, где они могут принимать участие в регуляции транскрипции и поддержании структуры хроматина [154].
Спектр функций микроРНК постоянно растет, а механизмы, ответственные за их исполнение, постоянно пополняются новыми деталями. Ситуацию дополнительно усложняет наличие сторонних функций у большинства белковых партнеров микроРНК [155]. Например, Dicer и AGO2 участвуют в РНК-интерференции [156]. Drosha и Dicer задействованы в репарации двуцепочечных разрывов, регуляции транскрипции Pol II, сплайсинга и метаболизма рибосомных РНК, а также подавлении транскрипции мобильных элементов [157, 158]. Причем часть этих функций также осуществляется этими белками в тандеме с короткими РНК во многом схожими с микроРНК, что часто мешает достоверной идентификации и аннотации таких молекул.
РЕГУЛЯЦИЯ СОЗРЕВАНИЯ И ФУНКЦИЙ микроРНК
Система регуляции экспрессии под действием микроРНК не существует в изоляции от других клеточных систем. Все стадии “жизненного цикла” микроРНК в клетке регулируются посредством разнообразных механизмов, многие из которых до сих пор не изучены детально или просто неизвестны. В данном разделе будут кратко рассмотрены основные типы факторов, оказывающих влияниe на созревание микроРНК, а также функции и стабильность комплекса RISC.
Регуляция экспрессии и созревания микроРНК. Биогенез микроРНК это сложный многоступенчатый процесс, каждый этап которого контролируется множеством самых разнообразных факторов.
Гены микроРНК могут входить в состав различных геномных локусов, в том числе внутри других генов. Картирование промоторов генов микроРНК человека показало, что они обладают схожими характеристиками с промоторами белок-кодирующих генов, и регулируются классическими транскрипционными факторами, например c-myc, p53, ZEB1/2, MYOD1 и др. [159]. Регуляторами транскрипции микроРНК являются и сигналы, опосредованные стимуляцией ростовых факторов, таких как PDGF, TGF-β и BDNF [159]. Эпигенетическая регуляция также играет важную роль в контроле экспрессии микроРНК, причем многие основные компоненты систем, отвечающих за метилирование и ацетилирование гистонов в свою очередь являются мишенями микроРНК [159, 160].
микроРНК также подвержены активной пост-транскрипционной регуляции. Модификация белков Dicer и Drosha, участвующих в биогенезе микроРНК, и их партнеров, а также взаимодействие с ними регуляторных белковых факторов может модулировать скорость и специфичность созревания предшественников и влиять на уровни зрелых микроРНК в клетке [160]. Мишенями регуляторных белков могут служить и сами предшественники микроРНК. Примером такой регуляции может служить участие HNRNPA1, KSRP и LIN28A/B в созревании let-7, а также способность SRp20/SRSF3 и геликазы DDX17 активировать созревание ряда микроРНК [160]. Активность терминальных уридилтрансфераз TUT7, TUT4 и TUT2 по отношению к предшественникам играет важную роль в регуляции процессинга ряда микроРНК, например let-7 и miR-122 [161, 162]. Показана также роль третичной и четвертичной структуры, образуемой предшественниками микроРНК, на эффективность их процессинга [53, 163].
Клеточная популяция микроРНК микрогетерогенна – в один и тот же момент в клетке могут присутствовать разные изоформы микроРНК, или изомиры (isomiRs). Такие молекулы происходят из одного источника, но отличаются несколькими основаниями (обычно 5'- и 3'-концовыми нуклеотидами) [164–166]. Частично такие различия являются следствием модуляции процессинга [167, 168], но их причиной также может являться укорачивание концов зрелых микроРНК под действием AGO2 [169], а также редактирование последовательности микроРНК (уридинилирование, метилирование, превращение аденозина в инозин под действием РНК-зависимой аденозиндезаминазы ADAR) [160, 165, 170–173]. Современные данные показывают, что редактирование может играть важную роль в регуляции созревания микроРНК [174].
Отдельной проблемой является вопрос выбора цепи микроРНК. Во многих случаях роль каждой цепи микроРНК (ведущая или пассажирская) жестко не закреплена, а иногда в клетке наблюдается паритет включения обеих цепей микроРНК в RISC [175–177]. Выбор цепи является динамическим параметром и зависит от ткани, фазы клеточного цикла, возраста и ряда факторов внешней среды [178–180]. Смена активной цепи микроРНК может быть ответом на активацию других процессов в клетке. Например, появление избытка мишени также способно увеличивать экспрессию пассажирской цепи [181]. Был определен ряд закономерностей последовательности ведущих и пассажирских цепей микроРНК. Так, в 5'-концевом положении ведущей цепи часто стоит урацил, а в пассажирской цепи – цитозин [182]. Кроме того, в последовательности ведущей цепи преобладают пурины, в то время как пассажирские цепи обогащены пиримидиновыми основаниями. Такая статистика коррелирует с данными о роли термодинамической стабильности в асимметричности связывания микроРНК-дуплекса с AGO [87–89]. Мутации и редактирование последовательности микроРНК могут приводить к изменению ряда ключевых нуклеотидов в последовательности микроРНК, в том числе концевых нуклеотидов, и влиять на выбор цепи для включения в RISC [174, 180].
Регуляция действия и стабильности RISC. Доступность белков AGO является одним из основных лимитирующих факторов действия микроРНК [183]. Стабильность AGO регулируется стандартными клеточными механизмами через фосфорилирование, гидроксилирование, poly(ADP-рибозил)-ирование и убиквитинирование, и стабилизируются загрузкой микроРНК [160]. Недавно было обнаружено, что с AGO также могут быть связаны интронные РНК длиной около 100 нт, названные аготронами (agotrons), основной функцией которых может быть регуляция доступности или стабильности AGO в отсутствие микроРНК [184–187]. Недавно были получены данные, что количество зрелых микроРНК в клетках человека может существенно (~ в 14 раз) превышать число молекул AGO [188–190]. Это может означать, что большая часть зрелых микроРНК не находится в RISC, что дополнительно повышает значимость доступности AGO в определении функций микроРНК. Роль такого пула “избыточных” молекул микроРНК в клетке остается неясной, однако показано, что в поддержании концентрации и стабилизации микроРНК участвует ряд РНК-связывающих белков, например, HuR, AUF1 и др. [191]. Хотя эти данные и требуют дополнительного подтверждения, они как минимум подтверждают, насколько мало доподлинно известно о биологии микроРНК.
Активность RISC также зависит от наличия в клетке мишеней микроРНК, входящей в состав комплекса. Избыток сайтов связывания микроРНК может снижать эффективность подавления трансляции [121]. При этом доступность мишеней и сайтов связывания может изменяться под действием мутагенеза, регуляции процессинга мРНК (альтернативный сплайсинг, использование альтернативных сайтов полиаденилирования), в процессе развития организма, в ответ на стресс, в зависимости от факторов среды и внеклеточных сигналов [142].
Известно, что ряд белковых факторов могут влиять на эффективность действия микроРНК кооперативно (PUM1 и PUM2) или конкурентно (DND1, CRD-BP, HNRNP E2 и HuR) (подробно освещено в работе [192]). Недавно также был обнаружен новый вариант регулирования активности микроРНК – феномен конкурентных эндогенных РНК (competing endogenous RNA, ceRNA). Конкурентные РНК содержат множественные сайты связывания микроРНК, благодаря чему они могут эффективно связывать микроРНК, снижая их доступность и способность действовать на целевые мишени [193]. Из-за такой способности “впитывать” микроРНК такие конкурентные РНК часто называют микроРНК-губками (miRNA sponges). Среди известных конкурентных РНК присутствуют длинные некодирующие РНК (днРНК, lncRNA/lincRNA), кольцевые РНК (circular RNA, circRNA), являющиеся продуктами бэксплайсинга кодирующих генов, и даже некоторые белок-кодирующие транскрипты [135, 193–196].
Стабильность микроРНК также может регулироваться за счет механизмов их направленной деградации. Гантье с соавторами показали, что среднее время полужизни микроРНК в клетке в отсутствие Dicer составляет 119 часов, однако этот параметр варьирует более чем в два раза для разных микроРНК [197]. Примерами быстрой направленной деградации микроРНК являются нейроны, в которых блокирование транскрипции при-микроРНК быстро приводит к снижению уровня зрелых микроРНК, а также резкие перестройки спектра микроРНК в ряде тканей, наблюдаемые в процессе развития организма [198–201]. Кроме того, регуляция микроРНК может быть напрямую связана с выполнением ими своей основной функции. Так, у C. elegans существует механизм мишень-опосредованной защиты микроРНК (target-mediated miRNA protection) – в отсутствие мишени микроРНК освобождается из AGO и деградирует [202]. У человека и Drosophila melanogaster обнаружен механизм, работающий по иному принципу – высокая комплементарность микроРНК и мишени приводит к дестабилизации и деградации микроРНК [203, 204]. Недавно также было показано, что стабильность некоторых микроРНК в клетке регулируется за счет полиаденилирования зрелых молекул и их предшественников, которое, в свою очередь, определяется балансом активности poly(А)-полимеразы PAPD5 и РНКазы PARN [205].
ЗАКЛЮЧЕНИЕ
В последние годы микроРНК попали в объектив исследователей в контексте многих важнейших биологических процессов. Были изучены многие особенности структуры микроРНК, их кодирования в геноме ряда организмов, механизмы их экспрессии и созревания; определены основные функции микроРНК в клетке и спектр их основных белковых партнеров. Однако, несмотря на большой прогресс, достигнутый в области изучения микроРНК и некодирующих РНК в целом, многие детали их биогенеза и механизмов действия микроРНК до сих пор остается загадкой. Очевидно, что микроРНК и связанные с ними белковые факторы участвуют в гораздо более широком спектре событий, чем предполагалось изначально. К сожалению, современные данные в области исследования механизмов действия микроРНК неполны и зачастую противоречивы, во многом из-за разнообразия используемых экспериментальных подходов и отсутствия четкого понимания многих основных механизмов действия микроРНК. Поэтому для достижения глубокого понимания всех их функций необходима в первую очередь исчепывающая систематизация современных знаний и методик изучения микроРНК.
Список литературы
Lee R.C., Feinbaum R.L., Ambros V. // Cell. 1993. V. 75. № 5. P. 843–854.
Reinhart B.J., Slack F.J., Basson M., Pasquinelli A.E., Bettinger J.C., Rougvie A.E., Horvitz H.R., Ruvkun G. // Nature. 2000. V. 403. № 6772. P. 901–906.
Lee R.C., Ambros V., Erdmann V.A., Lee R.C., Feinbaum R.L., Ambros V., Reinhart B., Wightman B., Ha I., Ruvkun G., Pasquinelli A.E., Grishok A., Hutvagner G., Sharp P.A., Kent W.J. et al. // Science. 2001. V. 294. № 5543. P. 862–864.
Lagos-Quintana M., Rauhut R., Lendeckel W., Tuschl T., Elbashir S.M., Lendeckel W., Tuschl T., Hamilton A.J., Baulcombe D.C., Hammond S.M., Bernstein E., Beach D., Hannon G.J., Zamore P.D., Tuschl T. et al // Science. 2001. V. 294. № 5543. P. 853–858.
Lau N.C., Lim L.P., Weinstein E.G., Bartel D.P., Lee R.C., Feinbaum R.L., Ambros V., Wightman B., Ha I., Ruvkun G., Reinhart B.J., Moss E.G., Lee R.C., Ambros V., Slack F.J. et al. // Science. 2001. V. 294. № 5543. P. 858–862.
Axtell M.J., Westholm J.O., Lai E.C. // Genome Biol. 2011. V. 12. № 4. P. 221.
Griffiths-Jones S., Saini H.K., Dongen S. van, Enright A.J. // Nucl. Acids Res. 2008. V. 36. Database issue. P. D154–D158.
Kozomara A., Griffiths-Jones S. // Nucl. Acids Res. 2014. V. 42. Database issue. P. D68–D73.
Ying H., Kimmelman A.C., Lyssiotis C.A., Hua S., Chu G.C., Fletcher-Sananikone E., Locasale J.W., Son J., Zhang H., Coloff J.L., Yan H., Wang W., Chen S., Viale A., Zheng H. et al. // Cell. 2012. V. 149. № 3. P. 656–670.
Han B.W., Wang W., Li C., Weng Z., Zamore P.D. // Science. 2015. V. 348. № 6236. P. 817–821.
Hyun S. // Int. J. Mol. Sci. 2017. V. 18. № 5.
Herrmann A., Priceman S.J., Swiderski P., Kujawski M., Xin H., Cherryholmes G.A., Zhang W., Zhang C., Lahtz C., Kowolik C., Forman S.J., Kortylewski M., Yu H. // J. Clin. Invest. 2014. V. 124. № 7. P. 2977–2987.
Kang S., Kang D., Bakhtiar Ul Islam S.M., Je S., Kim J.-H., Song J.J. // Cell. Signal. 2015. V. 27. № 6. P. 1214–1224.
Taft R.J., Hawkins P.G., Mattick J.S., Morris K.V. // Epigenetics Chromatin. 2011. V. 4. P. 13.
Ambros V., Bartel B., Bartel D.P., Burge C.B., Carrington J.C., Chen X., Dreyfuss G., Eddy S.R., Griffiths-Jones S., Marshall M., Matzke M., Ruvkun G., Tuschl T. // RNA. 2003. V. 9. № 3. P. 277–279.
Bartel D.P. // Cell. 2004. V. 116. № 2. P. 281–297.
Fabian M.R., Sonenberg N., Filipowicz W. // Annu. Rev. Biochem. 2010. V. 79. № 1. P. 351–379.
Berezikov E. // Nat. Rev. Genet. 2011. V. 12. № 12. P. 846–860.
Taft R.J., Pheasant M., Mattick J.S. // BioEssays. 2007. V. 29. № 3. P. 288–299.
Grimson A., Srivastava M., Fahey B., Woodcroft B.J., Chiang H.R., King N., Degnan B.M., Rokhsar D.S., Bartel D.P. // Nature. 2008. V. 455. № 7217. P. 1193–1197.
Griffiths-Jones S., Grocock R.J., Dongen S. van, Bateman A., Enright A.J. // Nucleic Acids Res. 2006. V. 34. Database issue. P. D140–D144.
Umu S.U., Langseth H., Bucher-Johannessen C., Fromm B., Keller A., Meese E., Lauritzen M., Leithaug M., Lyle R., Rounge T.B. // RNA Biol. 2017. P. 1–9.
Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E., Peterson K.J. // Annu. Rev. Genet. 2015. V. 23. № 49. P. 213–242.
Rodriguez A., Griffiths-Jones S., Ashurst J.L., Bradley A. // Genome Res. 2004. V. 14. № 10A. P. 1902–1910.
Saini H.K., Enright A.J., Griffiths-Jones S. // BMC Genomics. 2008. V. 9. P. 564.
Campo-Paysaa F., Semon M., Cameron R.A., Peterson K.J., Schubert M. // Evol. Dev. 2011. V. 13. № 1. P. 15–27.
Colaiacovo M., Lamontanara A., Bernardo L., Alberici R., Crosatti C., Giusti L., Cattivelli L., Faccioli P. // Biol. Direct. 2012. V. 7. P. 15.
Saini H.K., Griffiths-Jones S., Enright A.J. // Proc. Natl. Acad. Sci. U. S. A. 2007. V. 104. № 45. P. 17719–17724.
Corcoran D.L., Pandit K. V., Gordon B., Bhattacharjee A., Kaminski N., Benos P. V. // PLoS One. 2009. V. 4. № 4. P. e5279.
Ramalingam P., Palanichamy J.K., Singh A., Das P., Bhagat M., Kassab M.A., Sinha S., Chattopadhyay P. // RNA. 2014. V. 20. № 1. P. 76–87.
Ozsolak F., Poling L.L., Wang Z., Liu H., Liu X.S., Roeder R.G., Zhang X., Song J.S., Fisher D.E. // Genes Dev. 2008. V. 22. № 22. P. 3172–3183.
Monteys A.M., Spengler R.M., Wan J., Tecedor L., Lennox K.A., Xing Y., Davidson B.L. // RNA. 2010. V. 16. № 3. P. 495–505.
Baskerville S., Bartel D.P. // RNA. 2005. V. 11. № 3. P. 241–247.
Yuan Z., Sun X., Liu H., Xie J. // PLoS One. 2011. V. 6. № 3. P. e17666.
Friedländer M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N. // Nucleic Acids Res. 2012. V. 40. № 1. P. 37–52.
Ender C., Krek A., Friedländer M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G. // Mol. Cell. 2008. V. 32. № 4. P. 519–528.
Pederson T. // RNA. 2010. V. 16. № 10. P. 1865–1869.
Ono M., Scott M.S., Yamada K., Avolio F., Barton G.J., Lamond A.I. // Nucleic Acids Res. 2011. V. 39. № 9. P. 3879–3891.
Devor E.J. // J. Hered. 2006. V. 97. № 2. P. 186–190.
Lewis B.P., Shih I.-H., Jones-Rhoades M.W., Bartel D.P. // Cell. 2003. V. 115. P. 787–798.
Lim L.P., Glasner M.E., Yekta S., Burge C.B., Bartel D.P. // Science (80-.). 2003. V. 299. № 5612. P. 1540.
Shin C., Nam J.-W., Farh K.K.-H., Chiang H.R., Shkumatava A., Bartel D.P. // Mol. Cell. 2010. V. 38. № 6. P. 789–802.
Farh K.K.-H. // Science. 2005. V. 310. № 5755. P. 1817–1821.
Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M. // Cell. 2005. V. 123. № 6. P. 1133–1146.
Sood P., Krek A., Zavolan M., Macino G., Rajewsky N. // Proc. Natl. Acad. Sci. U. S. A. 2006. V. 103. № 8. P. 2746–2751.
Daugaard I., Hansen T.B. // Trends Genet. 2017. V. 33. № 3. P. 208–219.
Lee Y., Kim M., Han J., Yeom K.-H., Lee S., Baek S.H., Kim V.N. // EMBO J. 2004. V. 23. № 20. P. 4051–4060.
Cai X., Hagedorn C.H., Cullen B.R. // RNA. 2004. V. 10. № 12. P. 1957–1966.
Ballarino M., Pagano F., Girardi E., Morlando M., Cacchiarelli D., Marchioni M., Proudfoot N.J., Bozzoni I. // Mol. Cell. Biol. 2009. V. 29. № 20. P. 5632–5638.
Borchert G.M., Lanier W., Davidson B.L. // Nat. Struct. Mol. Biol. 2006. V. 13. № 12. P. 1097–1101.
Liang Y., Ridzon D., Wong L., Chen C. // BMC Genomics. 2007. V. 8. P. 166.
Chaulk S.G., Thede G.L., Kent O.A., Xu Z., Gesner E., Veldhoen R.A., Khanna S.K., Goping I.S., MacMillan A.M., Mendell J.T., Young H., Fahlman R.P., Glover M.J.N. // RNA Biol. 2011. V. 8. № 6. P. 1105–1114.
Chakraborty S., Mehtab S., Patwardhan A., Krishnan Y. // RNA. 2012. V. 18. № 5. P. 1014–1028.
Denli A.M., Tops B.B.J., Plasterk R.H.A., Ketting R.F., Hannon G.J. // Nature. 2004. V. 432. № 7014. P. 231–235.
Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Rå Dmark O., Kim S., Kim V.N. // Nature. 2003. V. 425. № 6956. P. 415–419.
Roth B.M., Ishimaru D., Hennig M. // J. Biol. Chem. 2013. V. 288. № 37. P. 26785–26799.
Han J., Lee Y., Yeom K.-H., Nam J.-W., Heo I., Rhee J.-K., Sohn S.Y., Cho Y., Zhang B.-T., Kim V.N. // Cell. 2006. V. 125. № 5. P. 887–901.
Maa H., Wub Y., Choia J.-G., Wua H. // Proc. Natl. Acad. Sci. U. S. A. 2013. V. 110. № 51. P. 20687–20692.
Castilla-Llorente V., Nicastro G., Ramos A. // Biochem. Soc. Trans. 2013. V. 41. № 4. P. 861–865.
Chen C.-Z., Li L., Lodish H.F., Bartel D.P. // Science (80-.). 2004. V. 303. № 5654. P. 83–86.
Fang W., Bartel D.P. // Mol. Cell. 2015. V. 60. P. 131–145.
Auyeung V.C., Ulitsky I., McGeary S.E., Bartel D.P. // Cell. 2013. V. 152. № 4. P. 844–858.
Nicholson A.W. // Wiley Interdiscip. Rev. RNA. 2014. V. 5. № 1. P. 31–48.
Zeng Y., Cullen B.R. // Nucleic Acids Res. 2004. V. 32. № 16. P. 4776–4785.
Lund E., Güttinger S., Calado A., Dahlberg J.E., Kutay U. // Science (80-.). 2004. V. 303. № 5654. P. 95–98.
Yi R., Qin Y., Macara I.G., Cullen B.R. // Genes Dev. 2003. V. 17. № 24. P. 3011–3016.
Bartel D.P. // Cell. 2009. V. 136. № 2. P. 215–233.
Park J.-E., Heo I., Tian Y., Simanshu D.K., Chang H., Jee D., Patel D.J., Kim V.N. // Nature. 2011. V. 475. № 7355. P. 201–205.
Gu S., Jin L., Zhang Y., Huang Y., Zhang F., Valdmanis P.N., Kay M.A. // Cell. 2012. V. 151. № 4. P. 900–911.
MacRae I., Zhou K., Li F., Repic A., Brooks A., Cande Z., Adams P., Doudna J. // Science (80-.). 2006. V. 311. № 5758. P. 195–198.
MacRae I.J., Zhou K., Doudna J.A. // Nat. Struct. Mol. Biol. 2007. V. 14. № 10. P. 934–940.
Provost P., Dishart D., Doucet J., Frendewey D., Samuelsson B., Rådmark O. // EMBO J. 2002. V. 21. № 21. P. 5864–5874.
Hammond S.M. // FEBS Lett. 2005. V. 579. № 26. P. 5822–5829.
Zhang H., Kolb F.A., Brondani V., Billy E., Filipowicz W. // EMBO J. 2002. V. 21. № 21. P. 5875–5885.
Kawamata T., Tomari Y. // Trands Biochem. Sci. 2010. V. 35. № 7.
Nakanishi K. // Wiley Interdiscip. Rev. RNA. 2016. V. 7. № 5. P. 637–660.
Schirle N.T., MacRae I.J. // Science. 2012. V. 336. № 6084. P. 1037–1040.
Elkayam E., Kuhn C.-D., Tocilj A., Haase A.D., Greene E.M., Hannon G.J., Joshua-Tor L. // Cell. 2012. V. 150. № 1. P. 100–110.
Jiang H., Sheong F.K., Zhu L., Gao X., Bernauer J., Huang X. // PLOS Comput. Biol. V. 11. № 7. P. e1004404.
Jinek M., Doudna J.A. // Nature. 2009. V. 457.
Swarts D.C., Makarova K., Wang Y., Nakanishi K., Ketting R.F., Koonin E. V, Patel D.J., van der Oost J. // Nat. Struct. Mol. Biol. 2014. V. 21. № 9. P. 743–753.
Faehnle C.R., Elkayam E., Haase A.D., Hannon G.J., Joshua-Tor L., Keck W.M. // Cell Rep. 2013. V. 3. № 6. P. 1901–1909.
Dueck A., Ziegler C., Eichner A., Berezikov E., Meister G. // Nucleic Acids Res. 2012. V. 40. № 19. P. 9850–9862.
Iwasaki S., Kobayashi M., Yoda M., Sakaguchi Y., Katsuma S., Suzuki T., Tomari Y. // Mol. Cell. 2010. V. 39. № 2. P. 292–299.
Iwasaki S., Sasaki H.M., Sakaguchi Y., Suzuki T., Tadakuma H., Tomari Y. // Nature. 2015. V. 521. № 7553. P. 533–536.
Miyoshi T., Takeuchi A., Siomi H., Siomi M.C. // Nat. Struct. Mol. Biol. 2010. V. 17. № 8. P. 1024–1026.
Khvorova A., Reynolds A., Jayasena S.D., Bartel D.P., Aronin N., Zamore P.D., Rappsilber J., Mann M., Dreyfuss G., Mello C.C. et al. // Cell. 2003. V. 115. № 2. P. 209–216.
Schwarz D.S., Hutvágner G., Du T., Xu Z., Aronin N., Zamore P.D. // Cell. 2003. V. 115. № 2. P. 199–208.
Suzuki H.I., Katsura A., Yasuda T., Ueno T., Mano H., Sugimoto K., Miyazono K. // Nat. Struct. Mol. Biol. 2015. V. 22. № 7. P. 512–521.
Nakanishi K., Weinberg D.E., Bartel D.P., Patel D.J. // Nature. 2012. V. 486. № 7403. P. 368–374.
Kwak P.B., Tomari Y. // Nat. Struct. Mol. Biol. 2012. V. 19. № 2.
Yang J.-S., Lai E.C. // Mol. Cell. 2011. V. 43. № 6. P. 892–903.
Berezikov E., Chung W.-J., Willis J., Cuppen E., Lai E.C. // Mol. Cell. 2007. V. 28. № 2. P. 328–336.
Ruby J.G., Jan C.H., Bartel D.P. // Nature. 2007. V. 448. № 7149. P. 83–86.
Okamura K., Hagen J.W., Duan H., Tyler D.M., Lai E.C. // Cell. 2007. V. 130. № 1. P. 89–100.
Rasmussen K.D., Simmini S., Abreu-Goodger C., Bartonicek N., Giacomo M. Di, Bilbao-Cortes D., Horos R., Lindern M. Von, Enright A.J., O’Carroll D. // J. Exp. Med. 2010. V. 207. № 7. P. 1351–1358.
Cheloufi S., Santos C.O. Dos, Chong M.M.W., Hannon G.J. // Nature. 2010. V. 465. № 7298. P. 584–589.
Cifuentes D., Xue H., Taylor D.W., Patnode H., Mishima Y., Cheloufi S., Ma E., Mane S., Hannon G.J., Lawson N.D., Wolfe S.A., Giraldez A.J. // Science. 2010. V. 328. № 5986. P. 1694–1698.
Yoda M., Cifuentes D., Izumi N., Sakaguchi Y., Suzuki T., Giraldez A.J., Tomari Y. // Cell Rep. 2013. V. 5. № 3. P. 715–726.
Kim Y.-K., Kim B., Kim V.N. // Proc. Natl. Acad. Sci. U. S. A. 2016. V. 113. № 13. P. E1881–E1889.
Martinez I., Hayes K.E., Barr J.A., Harold A.D., Xie M., Bukhari S.I.A., Vasudevan S., Steitz J.A., DiMaio D. // Proc. Natl. Acad. Sci. 2017. V. 114. № 25. P. E4961–E4970.
Bernstein E., Kim S.Y., Carmell M.A., Murchison E.P., Alcorn H., Li M.Z., Mills A.A., Elledge S.J., Anderson K.V., Hannon G.J. // Nat. Genet. 2003. V. 35. № 3. P. 215–217.
Davis T.H., Cuellar T.L., Koch S.M., Barker A.J., Harfe B.D., McManus M.T., Ullian E.M. // J. Neurosci. 2008. V. 28. № 17. P. 4322–4330.
Babiarz J.E., Hsu R., Melton C., Thomas M., Ullian E.M., Blelloch R. // RNA. 2011. V. 17. № 8. P. 1489–1501.
Chong M.M.W., Zhang G., Cheloufi S., Neubert T.A., Hannon G.J., Littman D.R. // Genes Dev. 2010. V. 24. № 17. P. 1951–1960.
Ambros V. // Nature. 2004. V. 431. № 7006. P. 350–355.
Kloosterman W.P., Plasterk R.H.A. // Dev. Cell. 2006. V. 11. № 4. P. 441–450.
Bartel D.P. // Cell. 2018. V. 173.
Almeida M.I., Reis R.M., Calin G.A. // Mutat. Res. Mol. Mech. Mutagen. 2011. V. 717. № 1–2. P. 1–8.
Bartel D.P., Chen C.-Z. // Nat. Rev. Genet. 2004. V. 5. № 5. P. 396–400.
Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. // Nature. 2008. V. 455. № 7209. P. 58–63.
Baek D., Villén J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. // Nature. 2008. V. 455. № 7209. P. 64–71.
Mukherji S., Ebert M.S., Zheng G.X., Tsang J.S., Sharp P.A., Oudenaarden A. Van // Nat. Genet. V. 43. № 9. P. 854–859.
Fang Z., Rajewsky N. // PLoS One. 2011. V. 6. № 3. P. e18067.
Bagga S., Bracht J., Hunter S., Massirer K., Holtz J., Eachus R., Pasquinelli A.E. // Cell. 2005. V. 122. P. 553–563.
Leoni G., Tramontano A. // Nucleic Acids Res. 2016. V. 44. № 9. P. e82.
Afonso-Grunz F., Müller S. // Cell. Mol. Life Sci. 2015. V. 72. № 16. P. 3127–3141.
Lewis B.P., Burge C.B., Bartel D.P. // Cell. 2005. V. 120. № 1. P. 15–20.
Friedman R.C., Farh K.K.-H., Burge C.B., Bartel D.P. // Genome Res. 2009. V. 19. № 1. P. 92–105.
Royo F., Zuñiga-Garcia P., Sanchez-Mosquera P., Egia A., Perez A., Loizaga A., Arceo R., Lacasa I., Rabade A., Arrieta E., Bilbao R., Unda M., Carracedo A., Falcon-Perez J.M. // J. Extracell. Vesicles. 2016. V. 5. P. 29497.
Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P. // Nat. Struct. Mol. Biol. 2011. V. 18. № 10. P. 1139–1146.
Grimson A. // Nat. Methods. 2010. V. 7. № 10. P. 795–797.
Chi S.W., Hannon G.J., Darnell R.B. // Nat. Struct. Mol. Biol. 2012. V. 19. № 3. P. 321–327.
Helwak A., Kudla G., Dudnakova T., Tollervey D. // Cell. 2013. V. 153. № 3. P. 654–665.
Moore M.J., Scheel T.K.H., Luna J.M., Park C.Y., Fak J.J., Nishiuchi E., Rice C.M., Darnell R.B. // Nat. Commun. 2015. V. 6. P. 8864.
Loeb G.B., Khan A.A., Canner D., Hiatt J.B., Shendure J., Darnell R.B., Leslie C.S., Rudensky A.Y. // Mol. Cell. 2012. V. 48. № 5. P. 760–770.
Hafner M., Lianoglou S., Tuschl T., Betel D. // Methods. 2012. V. 58. № 2. P. 94–105.
Martin H.C., Wani S., Steptoe A.L., Krishnan K., Nones K., Nourbakhsh E., Vlassov A., Grimmond S.M., Cloonan N. // Genome Biol. 2014. V. 15. № 3. P. R51.
Grimson A., Farh K.K.-H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. // Mol. Cell. 2007. V. 27. № 1. P. 91–105.
Valencia-Sanchez M.A. // Genes Dev. 2006. V. 20. № 5. P. 515–524.
Chou C.H., Chang N.W., Shrestha S., Hsu S. Da, Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J., Tsai T.R., Ho S.Y., Jian T.Y., Wu H.Y., Chen P.R. et al. // Nucleic Acids Res. 2016. V. 44. № D1.
Gam J.J., Babb J., Weiss R. // Nat. Commun. 2018. V. 9. № 1. P. 2430.
Jens M., Rajewsky N. // Nat. Rev. Genet. 2015. V. 16. № 2. P. 113–126.
Guo L., Zhao Y., Yang S., Zhang H., Chen F. // Biomed Res. Int. 2014. V. 2014. P. 1–8.
Rong D., Sun H., Li Z., Liu S., Dong C., Fu K., Tang W., Cao H. // Oncotarget. 2017.
Yao B., Li S., Chan E.K.L. // Ten Years Prog. GW/P Body Res. Adv. Exp. Med. Biol. 2013. V. 768. P. 71–96.
Huntzinger E., Kuzuoǧlu-Öztürk D., Braun J.E., Eulalio A., Wohlbold L., Izaurralde E. // Nucleic Acids Res. 2013. V. 41. № 2. P. 978–994.
Collart M.A., Panasenko O.O. // Gene. 2012. V. 492. № 1. P. 42–53.
Wolf J., Passmore L.A. // Biochem. Soc. Trans. 2014. V. 42. № 1. P. 184–187.
Presnyak V., Coller J. // Biochim. Biophys. Acta – Gene Regul. Mech. 2013. V. 1829. № 8. P. 817–823.
Morozova N., Zinovyev A., Nonne N., Pritchard L.-L., Gorban A.N., Harel-Bellan A. // RNA. 2012. V. 18. № 9. P. 1635–1655.
Wilczynska A., Bushell M. // Cell Death Differ. 2015. V. 22. № 1. P. 22–33.
Fukaya T., Tomari Y. // Mol. Cell. 2012. V. 48. P. 825–836.
Larsson O., Nadon R. // Translation. 2013. V. 1. № 1. P. e24557.
Fabian M.R., Mathonnet G., Sundermeier T., Mathys H., Zipprich J.T., Svitkin Y. V, Rivas F., Jinek M., Wohlschlegel J., Doudna J.A., Chen C.Y.A., Shyu A. Bin, Yates J.R., Hannon G.J., Filipowicz W. et al. // Mol. Cell. 2009. V. 35. № 6. P. 868–880.
Djuranovic S., Nahvi A., Green R. // Science (80-.). 2012. V. 336. № 6078. P. 237–240.
Eichhorn S.W., Guo H., Mcgeary S.E., Rodriguez-Mias R.A., Shin C., Baek D., Hsu S.-H., Ghoshal K., Villén J., Bartel D.P. // Mol Cell. 2014. V. 56. № 1. P. 104–115.
Meijer H.A., Kong Y.W., Lu W.T., Wilczynska A., Spriggs R. V, Robinson S.W., Godfrey J.D., Willis A.E., Bushell M. // Science. 2013. V. 340. № 6128. P. 82–85.
Rehwinkel J. // RNA. 2005. V. 11. № 11. P. 1640–1647.
Fukaya T., Tomari Y. // EMBO J. 2011. V. 30426. P. 4998–5009.
Yang Z., Wang L. // Cell Biosci. 2011. V. 1. № 1. P. 31.
Finnegan E.F., Pasquinelli A.E. // Crit. Rev. Biochem. Mol. Biol. 2013. V. 48. № 1. P. 51–68.
Eiring A.M., Harb J.G., Neviani P., Garton C., Oaks J.J., Spizzo R., Liu S., Schwind S., Santhanam R., Hickey C.J., Becker H., Chandler J.C., Andino R., Cortes J., Hokland P. et al. // Cell. 2010. V. 140. № 5. P. 652–665.
Pitchiaya S., Heinicke L.A., Park J.I., Cameron E.L., Walter N.G. // Cell Rep. 2017. V. 19. № 3. P. 630–642.
Pong S.K., Gullerova M. // FEBS Lett. 2018. V. 592. № 17. P. 2973–2986.
Czech B., Hannon G.J. // Nat. Rev. Genet. 2011. V. 12. № 1. P. 19–31.
Rybak-Wolf A., Jens M., Murakawa Y., Herzog M., Landthaler M., Rajewsky N. // Cell. 2014. P. 1153–1167.
Burger K., Gullerova M. // Nat. Rev. Mol. Cell Biol. 2015. V. 16. № 7. P. 417–430.
Davis-Dusenbery B.N., Hata A. // J. Biochem. 2010. V. 148. № 4. P. 381–392.
Ha M., Kim V.N. // Nat. Rev. Mol. Cell Biol. 2014. V. 15. № 8. P. 509–524.
Lee H., Han S., Kwon C.S., Lee D. // Protein Cell. 2016. V. 7. № 2. P. 100–113.
Kim B., Ha M., Loeff L., Chang H., Simanshu D.K., Li S., Fareh M., Patel D.J., Joo C., Kim N. // EMBO J. 2015. V. 34. P. 1801–1815.
Fernandez N., Cordiner R.A., Young R.S., Hug N., MacIas S., Cáceres J.F. // Nat. Commun. 2017. V. 8.
Guo L., Chen F. // Gene. 2014. V. 544. № 1. P. 1–7.
Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., Blelloch R., Schroth G.P., Nusbaum C., Bartel D.P. // Genes Dev. 2010. V. 24. № 10. P. 992–1009.
Kim B., Jeong K., Kim V.N. // Mol. Cell. 2017. V. 66. № 2. P. 258–269.
Humphreys D.T., Hynes C.J., Patel H.R., Wei G.H., Cannon L., Fatkin D., Suter C.M., Clancy J.L., Preiss T. // PLoS One. 2012. V. 7. № 2. P. e30933.
Starega-Roslan J., Krol J., Koscianska E., Kozlowski P., Szlachcic W.J., Sobczak K., Krzyzosiak W.J. // Nucl. Acids Res. 2011. V. 39. № 1. P. 257–268.
Juvvuna P.K., Khandelia P., Lee L.M., Makeyev E. V. // Nucl. Acids Res. 2012. V. 40. № 14. P. 6808–6820.
Liu X., Zheng Q., Vrettos N., Maragkakis M., Alexiou P., Gregory B.D., Mourelatos Z. // Mol. Cell. 2014. V. 55. P. 868–879.
Heale B.S.E., Keegan L.P., O’Connell M.A. // Adv. Exp. Med. Biol. 2010. V. 700. P. 76–84.
Morin R.D., O’Connor M.D., Griffith M., Kuchenbauer F., Delaney A., Prabhu A.-L., Zhao Y., McDonald H., Zeng T., Hirst M., Eaves C.J., Marra M.A. // Genome Res. 2008. V. 18. № 4. P. 610–621.
Lee L.W., Zhang S., Etheridge A., Ma L., Martin D., Galas D., Wang K. // RNA. 2010. V. 16. № 11. P. 2170–2180.
Li L., Song Y., Shi X., Liu J., Xiong S., Chen W., Fu Q., Huang Z., Gu N., Zhang R. // Genome Res. 2018. V. 28. № 1. P. 132–143.
Yang J.-S., Phillips M.D., Betel D., Mu P., Ventura A., Siepel A.C., Chen K.C., Lai E.C. // RNA. 2011. V. 17. № 2. P. 312–326.
Almeida M.I., Nicoloso M.S., Zeng L., Ivan C., Spizzo R., Gafà R., Xiao L., Zhang X., Vannini I., Fanini F., Fabbri M., Lanza G., Reis R.M., Zweidler-McKay P.A., Calin G.A. // Gastroenterology. 2012. V. 142. № 4. P. 886–896.
Yang X., Du W.W., Li H., Liu F., Khorshidi A., Rutnam Z.J., Yang B.B. // Nucleic Acids Res. 2013. V. 41. № 21. P. 9688–9704.
Kato M., Chen X., Inukai S., Zhao H., Slack F.J. // RNA. 2011. V. 17. № 10. P. 1804–1820.
Potla R., Singh I.S., Atamas S.P., Hasday J.D. // RNA. 2015. V. 21. № 7. P. 1261–1273.
Guo L., Lu Z. // PLoS One. 2010. V. 5. № 6.
Kang S.-M., Choi J.-W., Hong S.-H., Lee H.-J. // Int. J. Mol. Sci. 2013. V. 14. № 7. P. 13231–13240.
Meijer H.A., Smith E.M., Bushell M. // Biochem. Soc. Trans. 2014. V. 42. № 4. P. 1135–1140.
Grimm D., Wang L., Lee J.S., Schürmann N., Gu S., Börner K., Storm T.A., Kay M.A. // J. Clin. Invest. 2010. V. 120. № 9. P. 3106–3119.
Hansen T.B., Venø M.T., Jensen T.I., Schaefer A., Damgaard C.K., Kjems J. // Nat. Commun. 2016. V. 7. P. 11538.
Stagsted L.V.W., Daugaard I., Hansen T.B. // BioEssays. 2017. V. 39. № 4. P. 1600239.
Smibert P., Yang J.-S., Azzam G., Liu J.-L., Lai E.C. // Nat. Struct. Mol. Biol. 2013. V. 20. № 7. P. 789–795.
Martinez N.J., Gregory R.I. // RNA. 2013. V. 19. № 5. P. 605–612.
Janas M.M., Wang B., Harris A.S., Aguiar M., Shaffer J.M., Subrahmanyam Y.V.B.K., Behlke M.A., Wucherpfennig K.W., Gygi S.P., Gagnon E., Novina C.D. // RNA. 2012. V. 18. № 11. P. 2041–2055.
Stalder L., Heusermann W., Sokol L., Trojer D., Wirz J., Hean J., Fritzsche A., Aeschimann F., Pfanzagl V., Basselet P., Weiler J., Hintersteiner M., Morrissey D.V., Meisner-Kober N.C. // EMBO J. 2013. V. 32. № 8. P. 1115–1127.
Flores O., Kennedy E.M., Skalsky R.L., Cullen B.R. // Nucleic Acids Res. 2014. V. 42. № 7.
Zealy R.W., Wrenn S.P., Davila S., Min K.W., Yoon J.H. // Wiley Interdiscip. Rev. RNA. 2017. V. 8. № 5. P. 1–8.
Ciafrè S.A., Galardi S. // RNA Biol. 2013. V. 10. № 6. P. 935–942.
Kartha R. V., Subramanian S. // Front. Genet. 2014. V. 5. № JAN. P. 1–9.
Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M., Loewer A., Ziebold U., Landthaler M., Kocks C., Noble F. le et al. // Nature. 2013. V. 495. № 7441. P. 333–338.
Denzler R., Agarwal V., Stefano J., Bartel D.P., Stoffel M. // Mol. Cell. 2014. V. 54. No 5. P. 766–776.
Thomson D.W., Dinger M.E. // Nat. Rev. Genet. 2016. V. 17. № 5. P. 272–283.
Gantier M.P., McCoy C.E., Rusinova I., Saulep D., Wang D., Xu D., Irving A.T., Behlke M.A., Hertzog P.J., MacKay F., Williams B.R.G. // Nucleic Acids Res. 2011. V. 39. № 13. P. 5692–5703.
Krol J., Busskamp V., Markiewicz I., Stadler M.B., Ribi S., Richter J., Duebel J., Bicker S., Fehling H.J., Schübeler D., Oertner T.G., Schratt G., Bibel M., Roska B., Filipowicz W. // Cell. 2010. V. 141. № 4. P. 618–631.
Okamura K., Phillips M.D., Tyler D.M., Duan H., Chou Y., Lai E.C. // Nat. Struct. Mol. Biol. 2008. V. 15. № 4. P. 354–363.
Avril-Sassen S., Goldstein L.D., Stingl J., Blenkiron C., Quesne J. Le, Spiteri I., Karagavriilidou K., Watson C.J., Tavaré S., Miska E.A., Caldas C. // BMC Genomics. 2009. V. 10. № 1. P. 548.
Kato M., Lencastre A. de, Pincus Z., Slack F.J. // Genome Biol. 2009. V. 10. № 5. P. R54.
Chatterjee S., Fasler M., Büssing I., Großhans H. // Dev. Cell. 2011. V. 20. № 3. P. 388–396.
Bitetti A., Mallory A.C., Golini E., Carrieri C., Carreño Gutiérrez H., Perlas E., Pérez-Rico Y.A., Tocchini-Valentini G.P., Enright A.J., Norton W.H.J., Mandillo S., O’Carroll D., Shkumatava A. // Nat. Struct. Mol. Biol. 2018.
Ameres S.L., Horwich M.D., Hung J.-H., Xu J., Ghildiyal M., Weng Z., Zamore P.D. // Science (80-.). 2010. V. 328. № 5985. P. 1534–1539.
Shukla S., Bjerke G.A., Muhlrad D., Yi R., Parker R. // Mol. Cell. 2019. V. 73. № 6. P. 1204–1216.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия