

Биоорганическая химия, 2020, T. 46, № 1, стр. 18-35
Димерные (олигомерные) производные циклодекстринов как новый класс супрамолекулярных систем. Синтез и соединения включений на их основе (Мини-обзорная статья)
М. К. Грачев *, И. В. Терехова **, Д. А. Шипилов *, Н. В. Кутяшева *, Е. Ю. Емельянова *
* Московский педагогический государственный университет, Институт биологии и химии, Россия, 129164, Москва, ул. Кибальчича 6/2
** Институт химии растворов им. Г.А. Крестова РАН, Россия, 153045, Иваново, ул. Академическая 1
Поступила в редакцию 29.05.2019
После доработки 20.06.2019
Принята к публикации 22.06.2019
Аннотация
Приведен обзор работ по получению и применению димерных (олигомерных) производных циклодекстринов. Благодаря наличию двух и более внутренних циклодекстриновых полостей, их пространственной сближенности и ряду других особенностей, эти производные обладают повышенным, так называемым “кооперативным” эффектом по отношению к включению многочисленных “гостей”, что позволяет определять их как новый класс супрамолекулярных структур. Кратко проанализированы возможности их использования в различных областях органической, аналитической и медицинской химии.
СОДЕРЖАНИЕ
1. Введение. Особые свойства циклодекстриновых димеров (олигомеров)
2. Циклодекстриновые димеры (олигомеры), связанные линкером по первичным гидроксильным группам
3. Циклодекстриновые димеры, связанные линкером по вторичным гидроксильным группам
4. Заключение
5. Список литературы
ВВЕДЕНИЕ. ОСОБЫЕ СВОЙСТВА ЦИКЛОДЕКСТРИНОВЫХ ДИМЕРОВ (ОЛИГОМЕРОВ)
Циклодекстрины (1)–(3) (α-, β- и γ-) (рис. 1) представляют собой регулярно построенные циклические олигосахариды, в которых соответственно 6 (m = 1, n = 6), 7 (m = 2, n = 7) или 8 (m = 3, n = 8) остатков D-глюкопиранозы соединены α-1–4-гликозидными связями. Благодаря своей относительной дешевизне, биоразлагаемости и нетоксичности они нашли широкое применение в различных областях химии, в первую очередь супрамолекулярной химии, тонком органическом синтезе, аналитической, фармацевтической химии, а также в косметической и пищевой промышленности [1–6]. Основной интерес к циклодекстринам обусловлен их циклическим строением и наличием внутренней гидрофобной полости, способной к образованию соединений включения типа “гость–хозяин” с различными органическими субстратами. При этом такие важные свойства циклодекстринов, как растворимость в воде и органических растворителях, способность к образованию соединений включения, могут быть направленно изменены путем селективной модификации их структуры [5–7]. Однако направленная функционализация циклодекстринов является сложной в экспериментальном отношении задачей из-за присутствия в их молекулах трех типов различных по природе гидроксильных групп – один набор первичных (при С6 на узкой стороне) и два набора вторичных гидроксильных групп (при С2 и С3 на широкой стороне циклодекстринового каркаса), склонных к образованию сильных внутри- и межмолекулярных водородных связей [8, 9].

Первоначально, при химической модификации циклодекстринов, речь обычно шла о полном или хаотичном замещении их гидроксильных групп [10], но в дальнейшем, в связи с тем, что в практическом отношении бóльший интерес представляют региоселективно замещенные молекулы, внимание стали привлекать именно такие производные [5–7]. Между тем оказалось, что, несмотря на хорошую разработанность многих методик синтеза применительно к моносахаридам и линейным олигосахаридам, простой их перенос на циклодекстрины оказался невозможен из-за наличия большого количества близких по реакционной способности пространственно сближенных гидроксильных групп, а главное, из-за наличия внутренней полости, обладающей склонностью к образованию соединений включения с реагентами, что, как следствие, приводит к изменению “обычного” порядка протекания реакций [11–14] и ограничивает практическое использование циклодекстринов. Поэтому, несмотря на обилие работ по синтезу производных циклодекстринов [4–6], общие возможности и закономерности их функционализации исследованы ограниченно.
В еще меньшей степени изучено влияние на модификацию циклодекстринов других важных факторов, таких как природа растворителя, мольное отношение реагентов, температура. Кроме этого, следует учитывать и еще одно важное обстоятельство, которое во многом определяет стратегию синтеза и применения циклодекстринов: β-циклодекстрин намного хуже растворим в воде (18.4 г л–1), чем α- и γ-циклодекстрины (145 и 232 г л–1, соответственно). И наоборот, β-циклодекстрин хорошо растворим в таких полярных органических растворителях, как пиридин, DMSO и DMF, тогда как α- и γ-циклодекстрины в них малорастворимы [15].
В ряду циклодекстринов их димерные (олигомерные) производные, т.е. содержащие два или более фрагментов циклодекстрина, занимают особое место. Так, начиная с первых сообщений [16–20], был отмечен так называемый “кооперативный” (не “аддитивный”) эффект у данных соединений, то есть более высокую связывающую способность и молекулярную селективность в случае достаточно близкого расположения двух циклодекстриновых полостей [21]. Это свойство димерных (олигомерных) циклодекстринов определило их использование в качестве “гибких” рецепторов для молекулярного узнавания [22], в гомогенном катализе [23, 24], химиотерапии [25], для изучения мультивалентных взаимодействий [26] с ди- и олигодентатными лигандами.
Интерес к такого рода структурам определяется в первую очередь тем обстоятельством, что в одних случаях, в циклодекстриновых димерах, энергия связывания бидентатных лигандов может быть просто аддитивной [20], а в других – когда фрагменты циклодекстрина относительно жестко фиксированы, связывание с дитопным субстратом становится более сильным (“кооперативным”), чем отвечающее простой аддитивности [27]. Это является результатом выигрыша в энтальпии в сравнении со связыванием двумя монодентатными лигандами [28], что и обеспечивает более прочное хелатирование и компенсирует неблагоприятное действие энтропийной составляющей [25]. Хороший пример – структура (4), у которой линкер несет каталитическую группу и может действовать как дважды связанный лиганд. Например, был изучен гидролиз сложных диэфиров, имеющих на концах гидрофобные группы, способные каждая включаться в полость одного циклодекстринового фрагмента димера (4).

В случае иона металла Cu(II) или Zn(II), привязанного к бипиридину, катализ проходит в 1.4 × 106 раз быстрее, чем без циклодекстринов [29, 30]. Такой эффект достигается, во-первых, за счет того, что два гидролизованных фрагмента становятся более не связаны хелатированием и, поэтому, быстро покидают катализатор и освобождают место для дальнейшего гидролиза следующего субстрата, и во-вторых, за счет благоприятной геометрии связывания дитопного субстрата с циклодекстриновым димером.
Некоторые циклодекстриновые димеры способны эффективно связываться с гидрофобными участками пептидных звеньев. Так, показана специфичность связывания участков пептидов с L-Phe-D-Pro-последовательностью циклодекстриновым димером (5).

Для сравнения эффективности связывания были выбраны циклический пептид (6) и его линейный аналог (7). Найдено, что связывание димера (5) с циклическим пептидом (6) предпочтительнее, чем с его линейным аналогом (7), что, возможно, обусловлено самоагрегацией гидрофобных групп линейного пептида (7), и это было подтверждено лучшим связыванием пептида (7) с мономерным циклодекстрином. Очевидно, что агрегаты (7) разрушаются при проникновении в полость мономерного циклодекстрина, и тогда другой остаток Phe-Pro лучше включается в полость второго мономерного циклодекстрина. Циклический же пептид настолько “жесткий”, что у него нет внутренней агрегации, которую необходимо разрушить включением в циклодекстрин, хотя, с другой стороны, такая жесткость и создает трудности для идеального хелатирования путем “тонкого” подстраивания [31].
Следует отметить, что подобной агрегации протеинов уделяется повышенное внимание, особенно в присутствии агентов, которые могут ее нарушить [32–38]. Многие ферменты действуют только в виде олигомеров, и их агрегация обычно осуществляется за счет гидрофобного связывания неполярных боковых цепей. Ожидается, что связывание циклодекстриновыми димерами этих боковых цепей может нарушить агрегацию и ингибировать ферменты. Например, ВИЧ-протеаза относится именно к ферментам такого типа [35], и этот факт может найти важное медицинское применение.
Агрегация конкретного белка путем ассоциации его гидрофобных участков – важное и широко распространенное явление, которое может происходить также путем связывания двух разных белков, как, например, связывание гормона роста человека со своим рецептором [39]. В развитии такой возможности для медико-биологических исследований в разных направлениях окажется полезным использование не только димерных, но и олигомерных циклодекстринов.
Например, известна так называемая противоопухолевая фотодинамическая терапия, которая заключается в том, что фоточувствительный препарат (фотосенсибилизатор) после фотооблучения опухолевого участка онкобольного генерирует синглетный кислород, который убивает соседние клетки. Проблема, однако, заключается в обеспечении точечной доставки фотосенсибилизатора к опухолевому участку, чтобы максимально повысить эффективность и, главное, минимизировать побочные токсичные эффекты при фотоактивации других частей организма. С этой целью был синтезирован циклодекстриновый димер (8), чей линкер содержит олефиновый фрагмент с двумя атомами серы по краям [40].
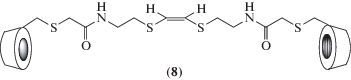


Такой электрононасыщенный олефин легко реагирует с синглетным кислородом, образуя сначала диоксетан, который затем расщепляется, образуя карбонильные группы [34]. Найдено, что димер (8) связывает фталоцианиновый фотосенсибилизатор (9), несущий гидрофобные трет-бутилфенильные группы, и обеспечивает его водорастворимость [40]. Фотооблучение этого комплекса вызывает разрыв линкера синглетным кислородом и сенсибилизатор удаляется. Важно, что высвобождение сенсибилизатора облегчается потерей хелатного связывания у циклодекстриновых фрагментов (10), при этом расщепленные фрагменты линкера сами включаются в циклодекстриновые полости, конкурентно вытесняя сенсибилизатор. Другая перспектива применения циклодекстриновых димеров заключается в том, что их комплексы включения с такими важными противоопухолевыми препаратами как Паклитаксел (Paclitaxel) [41] и Метотрексат (Methotrexate) [42, 43], а также с рядом других лекарственных соединений [44, 45] становятся достаточно водорастворимыми, а их биодоступность возрастает по сравнению с исходным лекарственным средством, в том числе за счет более прочного связывания с димерным, чем с мономерным циклодекстрином.
Еще одна перспектива использования олигомерных циклодекстринов заключается в создании искусственных ферментов, которые будут связывать субстраты и работать с ними с селективностью, определяемой только геометрией комплексов. Эта возможность была реализована при использовании хелатирующего связывания с помощью циклодекстриновых тетрамеров. Так, получены тетрафенилпорфирины, несущие циклодекстриновые фрагменты, и исследованы их Mn(II) комплексы (11) как катализаторы для гидроксилирования связанных стероидов [46, 47]. Стероид (12), который дал наиболее интересные результаты, был получен из андростандиола путем присоединения к каждому гидроксилу сложноэфирной группы, которая несла гидрофобные трет-бутилфенильные группы (для связывания с циклодекстринами) и водосолюбилизирующие сульфонатные группы.



Обнаружено, что субстрат (12) связывается с циклодекстринами на противоположных концах порфиринового фрагмента в комплексе (11) таким образом, что стероидный углерод (С*) находится прямо над Mn(III) в порфирине. Последующая обработка иодозобензолом, в качестве окислителя, вызывает перенос атома кислорода на Mn(III), приводя к гидроксилированию насыщенного атома углерода стероида (С*). Образуется только единственный продукт (13) с количественным выходом, высокой стереоселективностью и примерно со 180 каталитическими циклами.
Малоизученный, но важный аспект применения димерных циклодекстринов заключается в том, что они могут быть представлены как так называемые Болаамфифилы (Bolaamphiphiles) – то есть амфифильные молекулы, которые имеют две гидрофильные группы на концах относительно длинной гидрофобной углеводородной цепи. Присутствие второй гидрофильной “головки” резко повышает растворимость в воде, увеличивает критическую концентрацию мицеллообразования, что позволяет таким болаамфифилам образовывать в воде разнообразные ансамбли: сферы, цилиндры, диски, везикулы и т.п.
ЦИКЛОДЕКСТРИНОВЫЕ ДИМЕРЫ (ОЛИГОМЕРЫ), СВЯЗАННЫЕ ЛИНКЕРОМ ПО ПЕРВИЧНЫМ ГИДРОКСИЛЬНЫМ ГРУППАМ
Выше уже упоминались трудности при регионаправленной функционализации циклодекстринов, главным образом, из-за наличия внутренней гидрофобной полости и ряда других структурных особенностей. Понятно, что в случае димерных (олигомерных) циклодекстринов это обстоятельство должно еще более затруднять их получение и направленную функционализацию.
(R. Breslow) Р. Бреслоу для молекулярного узнавания холестерина синтезировал димер (14) (схема 1 ) и определил
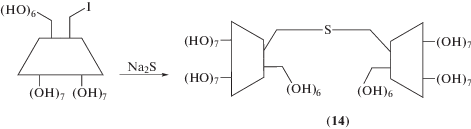
Схема 1 .
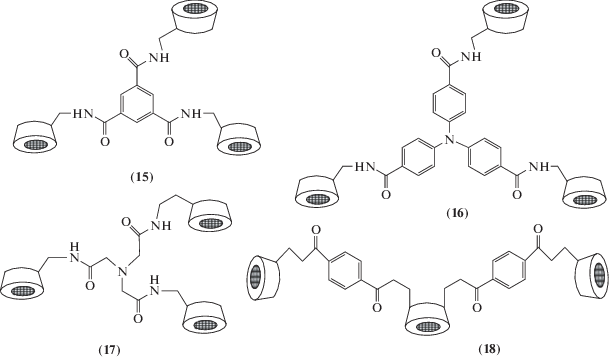
константу связывания, которая оказалась в 200‒300 раз выше, чем константа связывания холестерина с мономерным β-циклодекстрином (2) [48]. Ацилированием 6-амино-6-дезокси-β-циклодекстрина триспентафторфениловыми эфирами соответствующих трикарбоновых кислот получен набор тримеров (15)–(18), тритопное связывание которыми ведет к сильному и избирательному комплексообразованию [49].
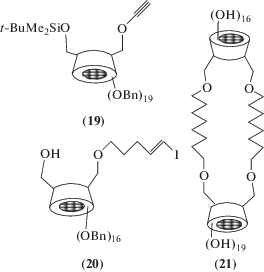
Из пропаргилированного бензилзащищенного β-циклодекстрина (19) и иодоалкенильного α-циклодекстрина (20) впервые получен так называемый циклодекстриновый гетеродуплекс (21) [50], содержащий в своем составе два разных (α- и β-) циклодекстриновых фрагмента. Ранее аналогично получен и гомодуплекс, содержащий два одинаковых остатка α-циклодекстрина [51].
Димерные β-циклодекстрины (22) и (23) с диселеноорганическим мостиком были исследованы для узнавания размера и формы молекул ряда красителей [52] (акридиновый красный, нейтральный красный, метиленовый голубой, метиловый оранжевый и метиловый красный) и найдено, что они обладают повышенной комплексообразующей способностью и возможностью распознавать структурные различия молекул красителей именно благодаря кооперативному связыванию двумя близко расположенными циклодекстриновыми полостями.

Такие же результаты получены и при связывании β-циклодекстриновым димером (24) в водном растворе метил- (R = Me) и этил- (R = Et) оранжа (25) [53], а также при связывании разными димерами (26) этилоранжа [54].

С аналогичной целью синтезированы четыре димера (27)–(30), различающихся длиной связующего мостика, и изучена их комплексообразующуя способность по отношению к шести разным красителям [55].
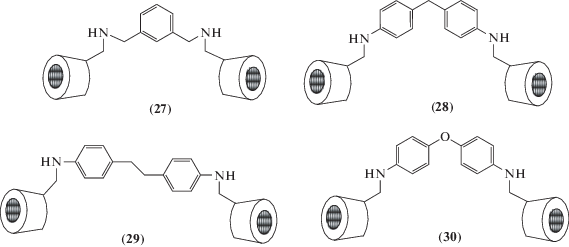
Найдено, что длина линкера между двумя циклодекстринами играет определяющую роль в молекулярном узнавании молекул красителей так, что константы связывания с молекулами “хозяевами” снижаются при увеличении длины линкера.
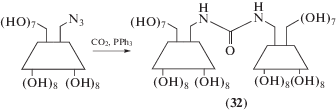
В работе [56] предложен способ получения димерного производного γ-циклодекстрина (31) (схема 2 ) исходя из монотозильного производного и бис(4-нитрофенил)сукцината (4-NPS) или димерного производного (32) (схема 3 ), через азидопроизводное γ-циклодекстрина:

Путем конденсации 6-амино-6-дезокси-α-, β‑циклодекстринов с терефталевой кислотой получены три α,α-, α,β- и β,β-димеры (33) (схема 4 ) [57], которые изучены на предмет образования супрамолекулярных гидрогелей с так называемым виологеновым (viologen) полимером (34).
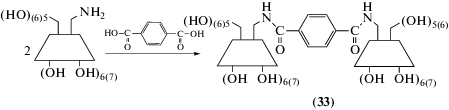
Схема 4 .

Найдено, что только с α,α-димером образуется высокоэластичный супрамолекулярный гидрогель за счет “нанизывания” на метиленовые гидрофобные участки виологенового полимера, а терминальные катионные гидрофильные участки полимера выполняют роль “стопперов”.
Монотозильное производное β-циклодекстрина широко используется для получения димерных диамин-производных (35) (схема 5 ) с различной длиной диаминного мостика [58–60], которые нашли применениe как молекулы “хозяева” для различных лекарственных соединений (схема 6 ) [58, 59]:

Схема 5.
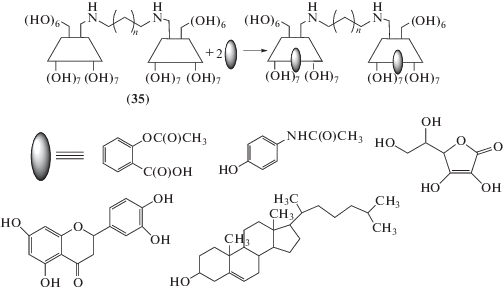
Схема 6 .
Моноиодпроизводное β-циклодекстрина послужило исходным соединением для получения дикатионного (“заряженного”) димера (36) (схем-а 7 ):

Схема 7 .
Методами спектроскопии ЯМР 1Н, капиллярного электрофореза, спектрофотометрии и фазовой растворимости обнаружено, что димер (36) действует как эффективная дитопная молекула “хозяина” по отношению к противоопухолевому соединению Метотрексат, образуя в воде стабильные комплексы включения состава 1 : 2. Изучена термодинамика и геометрия этого комплекса в сравнении с комплексами мономерного и полимерных циклодекстринов и найдено, что благодаря “кооперативному” эффекту двух циклодекстриновых полостей димерный циклодекстрин (36) проявляет существенно более эффективное комплексообразование, чем мономерный и полимерные циклодекстриновые аналоги [43].
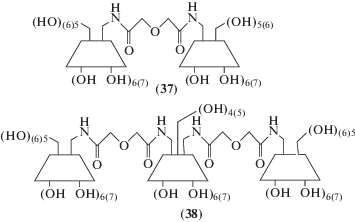
Исходя из моноаминопроизводных α- и β-циклодекстринов получены ди- (37) и тримерные (38) производные, содержащие гибкий диамидный эфирный мостик для лучшего подстраивания при имитации взаимодействия рецептора и лиганда в биологических процессах [61].
Аналогично, из моноаминопроизводного β-циклодекстрина синтезирован новый фотопереключаемый димер (39), несущий диазабензольную группировку в качестве линкера, обладающий высокой водорастворимостью и способностью к цис-, транс-фотоизомеризации при УФ-облучении (схема 8 ) [62].

Схема 8 .
Аналогично диазасоединениям транс-стильбеновые димерные β-циклодекстрины (40) (схема 9 ) открывают путь для получения молекулярных машин, переключаемых светом из транс- в цис-положение.

Схема 9 .
Изучено включение дитопного диадамантанового “гостя” (41) при фотооблучении.

Оказалось, что в стабильной транс-конфигурации димер (40) с “гостем” (41) образует стабильный комплекс 1 : 1 с небольшим количеством супрамолекулярного линейного полимера, тогда как в условиях УФ-облучения изомеризованный цис-изомер образует супрамолекулярные полимеры с высокой молекулярной массой [63].
Для получения супрамолекулярных полимеров также изучены димер (42) (схема 10 ) на основе терефталевой кислоты и дитопный диадамантановый “гость” (43).


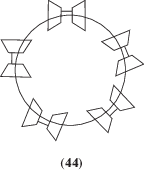
Оказалось, что дитопный “гость” (43) с жестким мостиком (n = 0) между двумя терминальными адамантанами образует с димером (42) высокомолекулярный супрамолекулярный линейный полимер, тогда как с гибким мостиком (n = 2, 3) образуются циклические супрамолекулярные олигомеры типа (44) [64].
Возможна и “прививка” (grafting) циклодекстринового димера (45) (схема 11 ) на азидфункционализированную кварцевую поверхность [65].

Схема 11 .

Схема 12 .
Этот “привитый” димер (46) (схема 12 ) образует комплекс 1 : 1 с флуоресцентным “гостем” (2-анилинонафталин-6-сульфоновая кислота: 2,6-ANS), причем флуоресциирующий гость включается в обе циклодекстриновые полости.
Легко получаемые олигомерные циклодекстриновые наногубки (карбонатные (47) (схема 13 ) и карбоксилатные (48) (схема 14 ) [66]) могут быть использованы как инновационные эксипиенты для наномедицины, для улучшения водорастворимости плохо растворимых в воде лекарственных соединений и защиты их от биоразложения.


Циклодекстриновые димеры (49), (50), образованные длинным мостиком по первичным гидроксильным группам “голова к голове”, обладают неожиданным свойством: в зависимости от жесткости (или гибкости) и от длины соединяющего мостика они могут образовывать псевдоротаксаны путем самовключения одного (49↔51) или двух (50↔52) концов мостика в полость циклодекстрина за счет обратимого поворота вокруг 1,4-межгликозидной связи одного (или двух) глюкозидных фрагментов, несущих мостиковый заместитель. Такой переход зависит от природы растворителя и приводит к образованию димеров с конфигурацией “голова к хвосту” 51 (схема 15 ) или “хвост к хвосту” 52 (схема 16 ) [67–70]. Важно, что такое самовключение ограничивает возможности димера к включению других “гостей”.
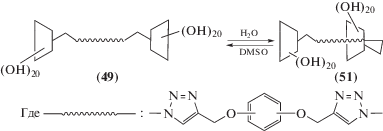
Схема 15.

Схема 16.
Дисульфидный димер (53) (схема 17 ) показал высокие комплексующие свойства по отношению к потенциальному противоопухолевому препарату Cu-BBC адресной доставки (ди-трет-бутил-Cu-циклен) за счет включения гидрофобных трет-бутилфенильных остатков в циклодекстриновые полости димера [71].

Схема 17.
ЦИКЛОДЕКСТРИНОВЫЕ ДИМЕРЫ, СВЯЗАННЫЕ ЛИНКЕРОМ ПО ВТОРИЧНЫМ ГИДРОКСИЛЬНЫМ ГРУППАМ
В литературе такие производные представлены заметно скромнее, что связано с более сложным путем их синтеза из-за необходимости постановки защит на первичных гидроксилах, хотя именно они часто проявляют более широкие способности для хелатирования дитопных лигандов за счет более широкого “входа” со стороны вторичных гидроксильных групп [72]. Обычно их получают либо путем образования 2,3-эпоксида из вторичных гидроксильных групп с последующим алкилированием, либо путем депротонирования наиболее кислотной гидроксильной группы положения 2 глюкозидного остатка с последующим алкилированием образующего карбоксилат аниона. Причем из-за существенной разности в кислотности гидроксильных групп (3-ОН-группы заметно менее кислотные, чем 2-ОН) для получения 2,2'- и 3,3'-димеров применяют разные экспериментальные подходы [25].
Так, например, изучены хелатирующие свойства двух димерных производных β-циклодекстрина (54) по отношению к 6-(п-толуидино)-2-нафталинсульфоновой кислоте (TNS), а димерное производное с бипиридильным мостиком использовали для получения металлокомплексов с Rh(II) [73].
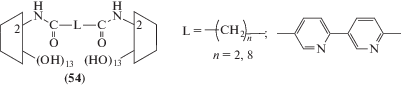
Оказалось, что димер с коротким линкером (n = 2) связывает крепче молекулу TNS, чем димер с более длинным (n = 8) и примерно так же, как димер с сульфидным мостиком по первичным гидроксилам [74].
Интересно, что попытки алкилирования незащищенного β-циклодекстрина, предварительно обработанного гидридом натрия с целью получения бипиридинового мостика для образования металлокомплексов аналогичных димеру (54), привела к сложной смеси трех возможных изомеров (55) (схема 18 ) [74]:

Схема 18.
Аналогично получен димер (56) (схема 19 ), содержащий гибкий пара-диметиленфениленовый мостик [75],

Схема 19.
а с помощью 2-тозилпроизводного β-циклодекстрина получен димер (57), содержащий гибкие сульфидные мостики [76].
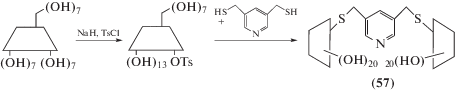
Схема 20.
Обнаружено, что димер (57) (схема 20 ) проявляет исключительно высокие константы связывания по отношению к некоторым азакрасителям, значительно превышающие таковые для мономерного β-циклодекстрина.
Обработкой 2,3-эпоксида γ-циклодекстрина гидроокись аммония, а затем бис(4-нитроенил)сукцинатом (4-NPS) получен 3,3'-диамидосвязанный димер (58) (схема 21 ), представляющий особый интерес для связывания объемных “гостей” (фуллерены, порфирины) [56].

Схема 21.
Более сложная схема получения индивидуальных 2,2'- и 3,3'-циклодекстринов (59) (схема 22 ) включает сначала получение защищенных циклодекстринов с одним свободным гидроксилом в положении 2 или 3, последующую молекулярную сборку путем димеризации двух одинаковых терминальных алкенсодержащих фрагментов и снятие силильных защит [77].

Схема 22.
Недавно были синтезированы два димерных 2,2'-связанных циклодекстрина (60), с жестким линейным спейсером разной длины [42].

Оба димера способны включать каждый по две молекулы противоопухолевого хемотерапевтического соединения метотрексата, причем с константой связывания в 2.4–3.5 большей, чем для мономерного β-циклодекстрина [79]. Аналогичные данные получены и при комплексообразовании метотрексата с димерным циклодекстрином, связанным по первичным гидроксилам [43].
Благодаря кооперативному содействию двух циклодекстриновых полостей, дансил-модифицированный димер (61) (схема 23 ) образует значительно более прочные комплексы со стероидными солями желчи, чем нативный β-циклодекстрин, а флуоресцентная метка служит сенсором для стероидов [78].
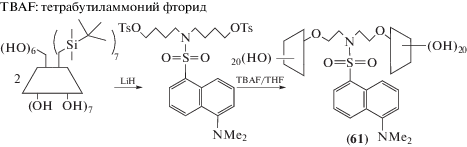
Схема 23 .
Димерный 2,2'-дителлуробис-β-циклодекстрин (62) (схема 24 ) хорошо работает как селективный флуоресцентный сенсор по хиральному разделению дансил-L-фенилаланина (DLP) и дансил-D-фенилаланина (DDP) [79].

Схема 24 .
ЗАКЛЮЧЕНИЕ
В настоящий момент димерные (олигомерные) производные циклодекстринов представляют собой перспективный класс соединений с большими возможностями практического применения в различных областях супрамолекулярной химии, металлокомплексном катализе, биомиметической и фармацевтической химии, биохимических исследованиях и в ряде других междисциплинарных направлениях. Прогресс в развитии этих направлений во многом определяется нашими знаниями о возможностях синтеза таких структур и их способностей к образованию соединений включения. Можно надеяться, что данный обзор будет способствовать пониманию важной роли димерных (олигомерных) производных циклодекстринов и стимулирует новые исследования в этой области.
Список литературы
Лен Ж.-М. Супрамолекулярная химия: концепции и перспективы. Пер. с англ. Новосибирск: Наука. Сиб. Предприятие РАН, 1998. 334 с.
Steed J.W., Atwood J.L. // Supramolecular Chemistry, J. Wiley& Sons Ltd. Chichester. 2000. 745 p.
Dodziuk H. Cyclodextrins and Their Complexes. Chemistry, Analytical Methods, Application. Wiley-VCH, Weinheim, 2006. 489 p.
Chem. Rev. (special issue). 1998. V. 98. № 5.
Beilstein J. Org. Chem. (special issue), 2012. V. 8.
Beilstein J. Org. Chem. (special issue), 2014. V. 10.
Khan A.R., Forgo P., Stine K.J., D’Souza V.T. // Chem. Rev. 1998. V. 98. № 5. P. 1977–1996.
Szejtli J. // Chem. Rev. 1998. V. 98. № 5. P. 1743–1753.
Saenger W., Jacob J., Gessler K., Steiner T., Hoffman D., Sanbe H., Kozumi K., Smith S.M., Tanaka T. // Chem. Rev. 1998. V. 98. № 5. P. 1787–1802.
Croft A.P., Bartsch R.A. // Tetrahedron. 1983. V. 39. № 9. P. 1417–1474.
Clazyrin A.E., Kurochkina G.I., Crachev M.K., Nifant’ev E.E. // Mendeleev Commun. 2001. № 6. P. 218–219.
Грачев М.К., Глазырин А.Е., Курочкина Г.И., Нифантьев Э.Е. // Ж. общ. хим. 2004. Т. 74. Вып. 5. С. 877–878 [Grachev M.K., Glazyrin A.E., Kurochkina G.I., Nifant’ev E.E. // Russ. J. Gen. Chem. 2004. V. 74. P. 808–809].
Глазырин А.Е., Сырцев А.Н., Курочкина Г.И., Кононов Л.О., Грачев М.К., Нифантьев Э.Е. // Изв. АН. Сер. хим. 2003. Вып. 1. С. 225–234 [Glazyrin A.E., Syrtsev A.N., Kurochkina G.I., Kononov L.O., Crachev M.K., Nifant’ev E.E. // Russ. Chem. Bull. 2003. V. 52. P. 237–243].
Грачев M.К. // Усп. хим. 2013. Т. 82. Вып. 11. С. 1034–1046 [Crachev M.K. // Russ. Chem. Rev. 2013. V. 82. P. 1034–1046].
de Miranda J.C., Martins T.E.A., Veiga F., Ferraz H.G. // Braz. J. Pharm. Sci. 2011. V. 47. № 4. P. 665–681.
Harada A., Furue M., Nozakura S.I. // Macromolecules. 1977. V. 10. P. 676–679.
Tabushi I., Kuroda Y., Shimokawa K. // J. Am. Chem. Soc. 1979. V. 101. P. 1614–1615.
Harada A., Furue M., Nozakura S.I. // Polym. 1981. V. 13. P. 777–781.
Fujita K., Ejima S., Imoto T. // Chem. Soc., Chem. Commun. 1984. P. 1277–1278.
Breslow R., Greenspoon N., Guo T., Zarzycki R. // J. Am. Chem. Soc. 1989. V. 111. P. 8296–8297.
Cho E., Jung S. // Molecules. 2015. V. 20. P. 19620–19646.
Liu Y., Chen Y. // Acc. Chem. Res. 2006. V. 39. P. 681–691.
Tilloy S., Binkowski-Machut C., Menuel S., Bricout H., Monflier E. // Molecules. 2012. V. 17. P. 13062–13072.
Blaszkiewicz C., Bricout H., Leonard E., Len C., Landy D., Cezard C., Djedaϊni-Pilard F., Monflier E., Tilloy S. // Chem. Commun. 2013. V. 49. P. 6989–6991.
Breslow R., Belvedere S., Gershell L., Leung D. // Pure Appl. Chem. 2000. V. 72. № 3. P. 333–342.
Beilstein J. Org. Chem. (special issue), 2015. V. 11.
Breslow R., Chung S. // J. Am. Chem. Soc. 1990. V. 112. P. 9659–9660.
Zhang B., Breslow R. // J. Am. Chem. Soc. 1993. V. 115. P. 9353–9354.
Zhang B., Breslow R. // J. Am. Chem. Soc. 1997. V. 119. P. 1676–1681.
Yan J., Breslow R. // Tetrahedron Lett. 2000. V. 41. P. 2059–2062.
Breslow R., Yang Z., Ching R., Trojandt G., Odobel F. // J. Am. Chem. Soc. 1998. V. 120. P. 3536–3537.
Zutschi R., Brickner M., Chmielewski J. // Current Opinion in Chemical Biology. 1998. V. 2. P. 62–66.
Chmielewski J. // Biopolymers. 1998. V. 47. P. 277–283.
Zutshi R., Shultz M.D., Ulysse L., Lutgring R., Bishop P., Schweitzer B., Vogel K., Franciskovich J., Wilson M., Chmielewski J. // Synlett. 1998. V. 10. P. 1040–1044.
Zutshi R., Franciskovich M., Shultz M., Schweitzer B., Bishop M., Wilson M., Chmielewski J. // J. Am. Chem. Soc. 1997. V. 119. P. 4841–4845.
Daugherty D.L., Gellman S.H. // J. Am. Chem. Soc. 1999. V. 121. P. 4325–4333.
Park H.S., Lin Q., Hamilton A.D. // J. Am. Chem. Soc. 1999. V. 121. P. 8–13.
Lin Q., Park H.S., Humaro Y., Lee C.S., Hamilton A.D. // Biopolymers. 1998. V. 47. P. 285–297.
Clackson T., Ultsch M.H., Wells J.A., de Vos A.M. // J. Mol. Biol. 1998. V. 277. P. 1111–1128.
Ruebner A., Yang Z., Leung D., Breslow R. // Proc. U.S. Natl. Acad. Sci. USA. 1999. V. 96. P. 14692–14693.
Liy Y., Chen G.-S., Li L., Zhang H.-Y., Cao D.-X., Yuan Y.-J. // J. Med. Chem. 2003. V. 46. P. 4634–4637.
Aykaҫ A., Martos-Maldonado M.C., Casas-Solvas J.M., Garsiá-Fuentes L., Vargas-Berenguel A. // J. Drug Del. Sci. Tech. 2012. V. 22. P. 270–272.
Kritskiy I., Kumeev R., Volkova T., Shipilov D., Kutysheva N., Grachev M., Terekhova I. // New J. Chem. 2018. V. 42. P. 14559–14567.
Grachev M.K., Malenkovskaya M.A., Vasyanina L.K. // J. Incl. Phenom. and Macrocyclic Chem. 2015. V. 83. № 3–4. P. 209–214.
Маленковская М.А., Васянина Л.К., Грачев М.К. // Ж. общ. хим. 2015. Т. 85. Вып. 7. С. 1161–1165. [Malenkovskaya M.A., Vasyanina L.K., Grachev M.K. // Russ. J. Gen. Chem. 2015. V. 85. P. 1161–1165].
Breslow R., Zhang X., Huang Y. // J. Am. Chem. Soc. 1997. V. 119. P. 4535–4536.
Breslow R., Gabriele B., Yang J. // Tetrahedron Lett. 1998. V. 39. P. 2887–2890.
Breslow R., Zhang B. // J. Am. Chem. Soc. 1996. V. 118. № 35. P. 8495–8496.
Leung D.K., Atkins J.H., Breslow R. // Tetrahedron Lett. 2001. V. 42. № 36. P. 6255–6258.
Bistri O., Lecourt T., Mallet J.-M., Sollogoub M., Sinaӱ P. // Chem. and Biodivers. 2004. V. 1. № 1. P. 129–137.
Lecourt T., Mallet J.-M., Sinaӱ P. // Tetrahedron Lett. 2002. V. 43. P. 5533–5536.
Chen Y., Li L., Liu Y. // Chem. J. Chin. Univ. 2002. V. 23. № 6. P. 1091–1093.
Zheng X.Q., Wang Y.H., Guo Q.X., Yang L., Liu Y.C. // Chin. Chem. Lett. 2003. V. 14. № 8. P. 848–851.
Song L.X. // Chin. Chem. Lett. 2002. V. 13. № 10. P. 1005–1006.
Zhao Y., Liu X.Q., gu Juan., Wang L.-Q., Zhu H.-Y., Huang R., Wang Y.-F., Yang Z.-M. // J. Phys. Org. Chem. 2008. V. 21. № 6. P. 440–448.
Pham D.-T., Ngo H.T., Lincoln S.F., May B.L., Easton C.J. // Tetrahedron. 2010. V. 66. P. 2895–2898.
Takashima Y., Yuting Y., Otsubo M., Yamaguchi H., Harada A. // Beilstein J. Org. Chem. 2012. V. 8. P. 1594–1600.
Маленковская М.А., Левина И.И., Грачев М.К. // Ж. орг. хим. 2014. Т. 50. Вып. 8. С. 1211–1215 [Malenkovskya M.A., Levina I.I., Grachev M.K. // J. Org. Chem. 2014. V. 50. № 8. P. 1211–1215].
Shipilov D.A., Kurochkina G.I., Akhlebinin A.K., Kutyasheva N.V., Grachev M.K. // Macroheterocycles. 2019. V. 12. № 1. P. 94–97.
Tripodo G., Wischke C., Neffe A.T., Lendlein A. // Carbohydr. Res. 2013. V. 381. P. 59–63.
Kurlemann M., Kavoo B.J. // Beilstein J. Org. Chem. 2014. V. 10. P. 2428–2440.
Hamon F., Blaszkiewicz C., Monflier E., Djedaim-Pilard F. // Beilstein J. Org. Chem. 2014. V. 10. P. 2874–2885.
Kuad P., Miyawaki A., Takashima Y., Yamaguchi H., Harada A. // J. Am. Chem. Soc. 2007. V. 129. № 42. P. 12630–12631.
Ohga K., Takashima Y., Takashima Y., Kawaguchi Y., Yamaguchi H., Harada A. // Macromolecules. 2005. V. 38. № 14. P. 5897–5904.
Städe L.W., Nielsen T.T., Duroux L., Wimmer R., Shimizu K., Larsen K.L. // Beilstein J. Org. Chem. 2015. V. 11. P. 514–523.
Trotta F., Zanetti M., Cavelli R. // Beilstein J. Org. Chem. 2012. V. 8. P. 2091–2099.
Maurer M., Hapiot F., Monflier E., Menuel S. // Tetrahedron. 2008. V. 64. P. 7159–7163.
Manuel S., Azaroul N., Landy D., Six N., Hapiot F., Monflier E. // Chem. Eur. J. 2011. V. 17. P. 3949–3955.
Voskuhl J., Schaepc K., Ravoo B.J. // Int. J. Mol. Sci. 2011. V. 12. P. 4637–4646.
Terao J., Konoshima Y., Matono A., Masai H., Fujihara T., Tsuji Y. // Beilstein J. Org. Chem. 2014. V. 10. P. 2800–2808.
Edwards W.B., Reichert D.E., d’Avignon D.A., Welch M.J. // Chem. Commun. 2001. P. 1312–1313.
Casas-Solvas J.M., Quesad-Soriano I., Carreno-Gazquez D., Gimenez-Martinez J.J., Garcia-Fuentes L., Vargas-Berenguel // Langmuir. 2011. V. 27. P. 9729–9737.
Venema F., Baselier C.M., van Dienst E., Ruel B.H.M., Feiters M.C., Endersen J.F.J., Reinhoudt D.N., Nolte R.J.M. // Tetrahedron Lett. 1994. V. 35. № 11. P. 1773–1776.
Yan J., Breslow R. // Tetrahedron Lett. 2000. V. 41. № 13. P. 2059–2062.
vanDienst E., Shellink B.H.M., von Piekartz I., Grote Gansey M.H.B., Venema F., Feiters M.C., Nolte R.J.M., Engbersen J.F.J., Reinhoudt D.N. // J. Org. Chem. 1995. V. 60. P. 6537–6545.
Jiang T., Sukumaran D.K., Soni S.-D., Lawrence D.S. // J. Org. Chem. 1994. V. 59. № 18. P. 5149–5155.
Chiu S.-H., Myles D.C., Garrell R.L., Stoddart J.F. // J. Org. Chem. 2000. V. 65. № 9. P. 2792–2796.
De Jong M.R., Engbersen J.F.J., Huskens J., Reinhoudt D.N. // Chem. Eur. J. 2000. V. 6. P. 4034–4040.
Jia S.-Y., Hao Y.-Q., Li L.-N., Chen K., Wu Y., Liu J., Wu L., Ding Y.-H. // Chem. Lett. 2005. V. 34. № 9. P. 1248–1249.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия