

Биоорганическая химия, 2020, T. 46, № 3, стр. 261-268
Трехдоменные антитела против фактора некроза опухоли: исследование биологической активности in vitro
Е. А. Василенко 1, Е. Н. Горшкова 1, И. В. Астраханцева 1, С. А. Недоспасов 2, 3, В. В. Мохонов 4
1 Национальный исследовательский Нижегородский государственный университет им. Н.И. Лобачевского
603950 Нижний Новгород, пр. Гагарина, 23, Россия
2 ФГБУН Институт молекулярной биологии им. В.А. Энгельгардта РАН
119991 Москва, ул. Вавилова, 32, Россия
3 Московский государственный университет имени М.В. Ломоносова
119991 Москва, ул. Ленинские Горы, 1, Россия
4 ФБУН “Нижегородский НИИ эпидемиологии и микробиологии им. академика И.Н. Блохиной”
603950 Нижний Новгород, ул. Малая Ямская, 71, Россия
Поступила в редакцию 01.11.2019
После доработки 08.11.2019
Принята к публикации 12.12.2019
Аннотация
Целью данной работы было изучение свойств новых трехдоменных антител, состоящих из двух доменов, специфичных к фактору некроза опухоли человека (hTNF), и третьего домена, отвечающего за связывание с клетками миелоидного ряда. Дополнительный hTNF-связывающий домен был введен в конcтрукцию антител для увеличения их биологической активности. Способность этих белков связывать hTNF на поверхности макрофагов и нейтрализовать его биологическую активность была исследована в экспериментах in vitro.
ВВЕДЕНИЕ
Мультиспецифические антитела на основе вариабельных доменов неканонических антител мозоленогих, так называемых наноантител (VHH), имеют большой потенциал в качестве диагностических и терапевтических препаратов для лечения заболеваний различной природы. Специфичность антител по отношению к двум и более типам молекул позволяет точно локализовать терапевтическую мишень, тем самым повысить эффективность борьбы с резистентными или быстро мутирующими штаммами вирусов, например, вирусом гриппа [1]. Кроме того, обеспечивая направленное связывание целевой молекулы (например, опухолевого маркера) и эффекторной клетки, можно повысить эффективность терапии онкологических заболеваний за счет активации механизмов клеточного лизиса [2]. Структура наноантител открывает широкие возможности для их форматирования путем слияния как идентичных, так и различных мономерных блоков для увеличения авидности или же сопутствующей блокировки нескольких неперекрывающихся эпитопов [3]. Это позволяет увеличить эффективность мультивалентных комплексов по сравнению с моновалентными наноантителами [4, 5]. Так, например, перспективными для терапии аутоиммунных заболеваний могут быть биспецифические наноантитела, одновременно связывающие два различных провоспалительных цитокина, например, TNF и IL‑17 [6] или IL-1 и IL-17A [7], что усиливает терапевтический эффект по сравнению с антителами против одного цитокина.
Помимо цитокинов мишенью для второй специфичности в составе мультиспецифичных конструкций могут быть различные рецепторы на клетках, участвующих в развитии воспалительного процесса [8], например, поверхностные молекулы клеток, обеспечивающих максимальный вклад в усиление воспалительного ответа в ходе развития того или иного заболевания. Ранее было показано, что основным источником провоспалительного цитокина фактора некроза опухоли (tumor necrosis factor, TNF) в модели аутоиммунного артрита являются миелоидные клетки, при этом подавление Т-клеточного TNF приводило к усугублению заболевания. Плейотропные свойства данного цитокина, обеспечивающие как защитное, так и “патогенное” воздействие на функции организма, легли в основу концепции о необходимости селективной нейтрализации TNF [9–11]. На ее основе были разработаны биспецифические антитела, позволяющие селективно нейтрализовать TNF из определенных клеточных источников, не препятствуя при этом реализации защитных функций данного цитокина, тем самым, увеличивая эффективность анти-TNF-терапии. Так, созданные на основе наноантител биспецифические агенты, связывающие и блокирующие человеческий TNF на поверхности миелоидных клеток за счет взаимодействия с маркерной молекулой F4/80, показали высокую эффективность на культуре макрофагов in vitro и в модели острого септического шока in vivo на гуманизированных по гену TNF мышах [12]. Однако перенос данной модели на человека осложнен тем фактом, что человеческий ортолог F4/80 (EMR1) экспрессируется не только моноцитами/макрофагами, но в большей степени гранулоцитарными клетками [13]. Поэтому важной задачей для создания новых биспецифических антител является поиск дополнительных молекул-мишеней на миелоидных клетках. В качестве одной из таких молекул-мишеней была рассмотрена молекула CD11b [14], ортолог которой (Mac-1) характерен для популяции моноцитов/макрофагов человека и не продуцируется эозинофилами. Однако ранее полученные анти-TNF антитела, способные связываться с молекулой CD11b, обладали более низкой эффективностью нейтрализации TNF по сравнению с анти-F4/80 VHH-содержащими белками [15], поэтому для увеличения эффективности биспецифических антител в настоящей статье мы рассмотрели возможность введения в их доменный состав дополнительного TNF-узнающего модуля (Vhh41), связывающего TNF с высокой аффинностью [16]. Таким образом, нами были созданы и изучены трехдоменные антитела, обладающие специфичностью в отношении TNF за счет наличия двух различных анти-TNF VHH-доменов: ahTNFVHH [4] и Vhh41 [16], а также в отношении поверхностных маркеров миелоидных клеток F4/80 и CD11b за счет наличия aF4/80 VHH, либо aCD11bVHH доменов соответственно.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В ходе работы были сконструированы генетические конструкции для продукции трехдоменных антител TFV и TCV в бактериальной системе, структура которых схематически представлена на рис. 1. Дизайн генетических конструкций и получение белков подробно описаны в разделе “Экспериментальная часть”. Анализ гомогенности полученных белков проводили с помощью денатурирующего электрофореза в ПААГ (рис. 2а, 2б) и вестерн-блоттинга (рис. 2в).
Рис. 1.
Схема доменного состава биспецифических белков ahTNFVHH–aF4/80VHH–Vhh41 (TFV) и ahTNFVHH–aCD11bVHH–Vhh41 (TCV).
Рис. 2.
Примеры типичной электрофореграммы (SDS-ПААГ) и вестерн-блоттинга трехдоменных антител: М – маркер молекулярных масс (Page Ruler Unstained Protein Ladder, кат. № 26614, Thermo Fisher Scientific). а – образцы с разных стадий очистки белков на примере TFV методом металл-хелатной аффинной хроматографии: 1 – лизат клеток, 2 – фракция не связавшихся в ходе хроматографии белков, 3 – элюируемый белок TFV. б – образцы очищенных белков TCV (дорожка 1) и TFV (дорожка 2). в – вестерн блоттинг образцов белков TFV (дорожка 1) и TCV (дорожка 2).

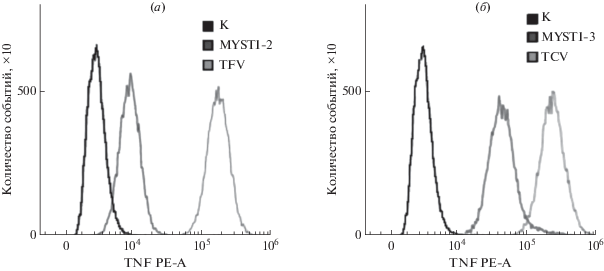
Эффективность связывания полученных трехдоменных антител с молекулами F4/80 и CD11b на поверхности макрофагов оценивали с помощью метода проточной цитофлуориметрии. В эксперименте использовали макрофаги, полученные из костного мозга гуманизированных по гену TNF мышей, у которых человеческий TNF (hTNF) продуцируется вместо мышиного. В ходе эксперимента анализировали уровень флуоресцентного сигнала от макрофагов, проинкубированных с трех- или с аналогичными им двухдоменными белками, затем с рекомбинантным hTNF, и, наконец, с детектирующими флуоресцентно-меченными антителами к hTNF. Интенсивность флуоресцентного сигнала была пропорциональна количеству макрофагов, связанных с белками-блокаторами. Таким образом, уровень сигнала косвенно характеризовал эффективность связывания белков с ФНО и поверхностными белками на макрофагах. Было показано, что введение дополнительного модуля способствует повышению эффективности взаимодействия антител TFV и TCV с макрофагами по сравнению с MYSTI-2 и MYSTI-3, в 10 и 6 раз соответственно (рис. 3).
Рис. 3.
Эффективность связывания биспецифических белков-блокаторов TNF с макрофагами гуманизированных по гену TNF (huTNFKI) мышей. Уровень флуоресцентного сигнала пропорционален количеству связавшихся с клетками анализируемых белков. Cравнительный анализ флуоресценции клеток, инкубированных с: TFV или MYSTI-2 (а), TCV или MYSTI-3 (б). К – уровень автофлуоресцентного сигнала от клеток, обработанных мечеными PE-антителами против человеческого TNF.
Способность трехдоменных антител удерживать на поверхности мышиных макрофагов продуцируемый ими TNF оценивали, анализируя количество свободного TNF в культуральной среде клеток методом иммуноферментного анализа. Эффективность удержания цитокина связанными с клетками биспецифическими антителами в данном эксперименте обратно пропорциональна его уровню в супернатанте (рис. 4). В случае контрольных образцов (культуральной среды макрофагов без добавления биспецифических антител) при инкубации с липополисахаридом происходило резкое увеличение концентрации TNF в среде (рис. 4). Для всех исследуемых белков наблюдался более низкий уровень TNF по сравнению с контролем, причем количество TNF в среде повышалось с понижением концентрации биспецифических белков (рис. 4б). Кроме того, трехдоменные блокаторы TNF оказались способны удерживать этот цитокин на поверхности макрофагов с эффективностью, сравнимой с эффективностью двухдоменного белка MYSTI-2 и превышающей эффективность белка MYSTI-3.
Рис. 4.
Сравнение количества свободного TNF в культуральной среде макрофагов, проинкубированных с 1 мкМ (а) и 0.25 мкМ (б) биспецифических белков и липополисахаридом. На диаграмме представлены средние значения со стандартными отклонениями. * ‒ статистически значимые отличия (p < 0.05), n.s. ‒ статистически не значимые отличия.

Биологическую активность белков in vitro исследовали в ходе цитотоксического (МТТ) теста на клетках линии WEHI-164 (рис. 5). Наиболее показательными характеристиками биологической активности в данном эксперименте являются значения IC50, которые для белков MYSTI-3 и TCV составили: 3.451 и 0.536 нМ, а для MYSTI-2 и TFV – 4.5 и 21.9 нМ соответственно. Таким образом, эффективность трехдоменного белка, содержащего aCD11b VHH-домен, оказалась выше эффективности его двухдоменного аналога.
Рис. 5.
Графики зависимости выживаемости клеток WEHI-164 от концентрации биспецифических антител, нейтрализующих действие экзогенного TNF в ходе цитотоксического (МТТ) теста. Сравнительный анализ TNF-нейтрализующей активности белков TFV и MYSTI-2 (а), а также белков TCV и MYSTI-3 (б).

Таким образом, введение дополнительного TNF-узнающего модуля (Vhh41) в доменный состав биспецифических белков позволило существенно увеличить эффективность удержания фактора некроза опухоли на поверхности макрофагов гуманизированных мышей, и, кроме того, в составе anti-CD11b VHH-содержащего антитела увеличило TNF-нейтрализующий потенциал мультиспецифической конструкции по сравнению с его двухдоменным аналогом MYSTI-3 в тестах in vitro.
В целом, результаты исследования показали, что идея конструирования биспецифических антител с увеличенным количеством TNF-связывающих участков является перспективной: эта особенность трехдоменных антител впоследствии может быть использована для создания прототипов лекарств для терапии воспалительных заболеваний, связанных с гиперэкспрессией данного цитокина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы. Штамм DH5α E. coli с генотипом F– φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(${\text{r}}_{{\text{K}}}^{ - },\,\,{\text{m}}_{{\text{K}}}^{ + }$) phoA supE44 λ– thi-1 gy‑rA96 relA1, Rosetta2(DE3)ΔSlyD/X E. coli с генотипом F– ompT hsdSB(${\text{r}}_{{\text{B}}}^{ - }\,\,{\text{m}}_{{\text{B}}}^{ - }$) gal dcm ΔSlyD/X (DE3) pRARE2 (CamR), плазмидный вектор pET28b+ (Novagen), бактотриптон (Amresco, США); хлорид натрия (Helicon, Россия); хлорид никеля (Helicon, Россия); дрожжевой экстракт (Amresco, США); бакто-агар “Difco” (США); агароза (Helicon, Россия); β-меркаптоэтанол (Helicon, Россия); Triton X-100 (Panreac Applichem, Германия); акриламид и метиленбисакриламид (Helicon, Россия); ИПТГ (Helicon, Россия); ЭДТА (Serva, Германия); SDS (Panreac Applichem, Германия); имидазол (Panreac Applichem, Германия); диметилсульфоксид (Panreac Applichem, Германия); персульфат аммония (Panreac Applichem, Германия); ТЕМЕD (Panreac Applichem, Германия); глицерин – “PanReac” (Германия); канамицин (ПАО “Биохимик”, Россия); хлорамфеникол (Sigma-Aldrich, США); смола для металл-аффинной хроматографии – “Ni-NTA Superflow” фирмы “GE Healthcare” (Великобритания); Tris (Amresco, США); HEPES (Amresco, США); L-аргинин (Диаэм), гуанидина гидрохлорид (Amresco, США); глутатион окисленный (Amresco, США); глутатион восстановленный (Amresco, США); бычий сывороточный альбумин (Panreac Applichem, Германия); сухое обезжиренное молоко (Nonfat dried milk powder, Panreac Applichem); наборы стандартов молекулярного веса ДНК (“Thermo Scientific”, США); наборы стандартов молекулярного веса белков (“Thermo Scientific”, США); набор для проведения клонирования по методу Гибсона Gibson assambly cloning kit (NEB), набор реактивов для секвенирования: BigDye™ Terminator v3.1 Cycle Sequencing Kit (“Thermo Scientific”, США); набор для иммуноферментного анализа Human TNFα ELISA Ready-SET-Go! (eBioscience, США); anti-4His-HRP антитела (MACS, Miltenyi Biotec, Германия); наборы West Dura Extended Duration Substrate (SuperSignal, США) и ECL Western Blotting Substrate (Promega, США); DPBS (1×) (Gibco), RPMI Medium 1640 (1×) (Gibco), DMEM (1×) (Gibco), HS (hourse serum, Gibco), L-глутамин (ПанЭко, Россия), смесь антибиотиков пенициллин/стрeптомицин (ПанЭко, Россия).
Дизайн генетических конструкций TCV и TFV, кодирующих блокаторы TNF с трехдоменной структурой, осуществляли с использованием пакета программ DNA Star Lasergene (DNA Star Madison, WI, USA) (рис. 1). Получение ДНК-конструкций целевых белков проводили с помощью стандартных методов молекулярной биологии. Экспрессионные плазмиды были получены на основе вектора pET28b (Novagene). Последовательности олигонуклеотидных праймеров (табл. 1) были синтезированы компанией ООО “ДНК-синтез” (г. Москва). Анализ последовательностей ДНК отдельных клонов клеток, трансформированных продуктом реакции, проводили с помощью электрофореза в 1.5% агарозном геле с последующим секвенированием плазмидной ДНК (ABI Prism 310 Genetic Analyzer). Полученные генетические конструкции использовали для трансформации клеток экспрессионного штамма Rosetta2(DE3)ΔSlyD/X E. coli [17].
Таблица 1.
Используемые в работе олигонуклеотидные праймеры
| Праймер | Последовательность |
|---|---|
| hTNF GC_F | TTAACTTTAAGAAGGAGATATAATGTCCCAAGTTCAGCTTCAAG |
| hTNF GC_R | CGCCTCCAGAGCCTCCACCGCCAGAGCCACCTCCTCCCGCGCCGGCAGAAGAAACAGTCAC |
| VHH41 GC_F | CCGCAACCACAACCAAAACCACAACCAAAACCTGAACCAGAAAGCGGATCCATGGCTCAGGTGCAGCTGGTG |
| VHH41 GC_R | CGATCCTCGGCCGGAAGCGTAGTCCGGAACGTCGTACGGGTAGCGGCCGCTGGAGAC |
| pET28 GC_F | TCCGGACTACGCTTCCGGCCGAGGATCGCTCGAGCACCACCACCACCACCACTGA |
| pET28 GC_R | CTTGAAGCTGAACTTGGGACATTATATCTCCTTCTTAAAGTTAA |
| F4/80 GC_F | GGCTCTGGCGGTGGAGGCTCTGGAGGCGGTGGCTCCATGGCCCAGGTGCAGCTGCAG |
| F4/80 GC_R | GTTGTGGTTTTGGTTGTGGTTGCGGTTGTGGTTGTGGTTTTGGTTGTGGTTGGGGTATCTTGGGTTCTGAGGAGACGGTGACCTGGGTCC |
| CD11b GC_F | TCTGGCGGTGGAGGCTCTGGAGGCGGTGGCTCCATGAGCCAGGTGCAGCTGCAGGAGTC |
| CD11b GC_R | GTTGTGGTTTTGGTTGTGGTTGCGGTTGTGGTTGTGGTTTTGGTTGTGGTTGGGGTATCTTGGGTTCGCCACCGGTTTCCGGCAGTCCTC |
Получение белков. Колонии Rosetta2(DE3)ΔSlyD/ X E.coli культивировали в жидкой среде LB, содержащей селективные антибиотики, в течение ночи при 37°С и 150–200 об./мин, затем переносили в соотношении 1 : 10 в свежую среду LB и культивировали в течение 1.5 ч при 37°С и 150 об./мин до достижения оптической плотности суспензии 0.6–0.7 ОЕ при 600 нм. Индукцию экспрессии проводили изопропил-β-D-1-тиогалактопиранозидом (0.5 мМ) в течение 19–20 ч при 20°С и 150 об./мин. Затем культуру осаждали центрифугированием при 9000 об./мин в течение 15 мин и проводили выделение белков. Полученный осадок ресуспендировали в буфере, содержащем 25 мМ HEPES (pH 7.0), 500 мМ хлорида натрия, 6 М гидрохлорида гуанидина, 10% глицерина, 1% TritonX-100, 5 мМ имидазола, 1 мМ 2-меркаптоэтанола. Полученную суспензию дезинтегрировали с помощью QSonica Q55 (QSonica sonicators, США), далее центрифугировали лизат в течение 20–30 мин при 9000 об./мин и проводили очистку методом аффинной хроматографии с помощью сорбента Ni-NTA Superflow (GEHealthcare, Великобритания). Для удаления несвязавшихся с хроматографическим матриксом белков использовали раствор, содержащий 15 мМ имидазола. Далее осуществляли рефолдинг белков на колонке путем формирования ступенчатого градиента гидрохлорида гуанидина от 6 М до 0 М с использованием буферных растворов, содержащих 25 мМ HEPES (pH 7.0), 500 мМ хлорида натрия, 10% глицерина, 0.4 М L-аргинина, 1 мМ восстановленного глутатиона и 0.1 мМ окисленного глутатиона. После полного удаления хаотропного агента проводили элюцию белков, используя буферный раствор состава: 25 мМ HEPES (pH 7.0), 500 мМ хлорида натрия, 10% глицерина, 0.4 М L-аргинина, 500 мМ имидазола, 1 мМ 2-меркаптоэтанола. Элюат диализовали против буфера, содержащего 25 мМ HEPES (pH 7.0), 150 мМ хлорида натрия, 5% глицерина, 1 мМ 2-меркаптоэтанола. Препараты белков после диализа концентрировали с помощью концентраторов Amicon Ultra 15 (Merck Millipore Ltd) с MWCO 5000 Da. Белковые препараты анализировали стандартным методом электрофореза в 12% полиакриламидном геле с последующей окраской раствором CoomassieR250 [16], а также методом Вестерн-блоттинга с использованием антител anti-4His-HRP (MACS, Miltenyi Biotec, Германия). Для визуализации результатов использовали West Dura Extended Duration Substrate (SuperSignal, США).
Оценка связывания антител с лигандами методом проточной цитофлуориметрии. Макрофаги, полученные из костного мозга (BMDM) гуманизированных по гену TNF мышей [18, 19], культивировали в чашках Петри в течение 10 дней в среде DMEM, содержащей 20% (об./об.) лошадиной сыворотки, 30% (об./об.) кондиционированной среды от клеток L929, L-глутамин (292 мкг/мл), пенициллин (100 ед./мл) и стрептомицин (100 мкг/мл). Клетки промывали и удаляли с поверхности чашек ледяным фосфатно-солевым буфером (PBS), подсчитывали количество клеток с помощью камеры Горяева (погибшие клетки отличали от живых окрашиванием трипановым синим). Блокировку Fcγ-рецепторов осуществляли специфическими антителами TruStain FcX™ (anti-mouse CD16/32) Antibody (кат. № 101 319, Biolegend, США), затем клетки последовательно инкубировали (30 мин, 4°C) с полученными антителами (4 мкМ), рекомбинантным человеческим TNF (100 мкг/мл) (30 мин, 4°C) и c мечеными фикоэритрином (PE) антителами против человеческого TNF-α (Biolegend, США) (30 мин, 4°C). Образцы анализировали на проточном цитофлуориметре Cytoflex S (Beckman Coulter, США) с использованием программного обеспечения CytExpert 2.0 (Beckman Coulter, США).
Оценка удержания TNF активированными макрофагами. Макрофаги, полученные описанным выше методом, культивировали в 96-луночных планшетах (Corning® 96-WellMicroplates, Corning, США) из расчета 105 клеток на лунку и инкубировали в течение ночи при 37°С и 5% СО2. После промывания полной средой DMEM клетки инкубировали с анализируемыми белками (2 мкМ) или DMEM (30 мин, 37°C, 5% CO2) в качестве контроля. Затем клетки стимулировали (4 ч, 37°C, 5% CO2) липополисахаридом (LPS, Sigma-Aldrich, L2630) в дозе 100 нг/мл для продукции клетками фактора некроза опухоли (TNF). После этого супернатант переносили в лунки 96-луночного планшета (Corning Costar, Corning, США) и измеряли концентрацию TNF с помощью набора HumanTNF-αELISAReady-SET-Go® (eBioscience, США) согласно протоколу производителя.
Цитотоксический МТТ-тест. Исследование TNF-ингибирующей активности полученных рекомбинантных белков проводили с помощью цитотоксического МТТ-теста на клеточной линии WEHI-164, чувствительной к действию человеческого TNF [20]. Клетки переносили в 96-луночные культуральные планшеты (Corning® 3788 96-Well Polystyrene Microplates, Corning, США) из расчета 2 × 103 клеток на лунку. Рекомбинантный hTNF добавляли в концентрации 100 ед./мл, исследуемые антитела – в серийных разведениях 1 мМ–2 пМ. После 24 ч инкубации добавляли 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромид (MTT) (Sigma-Aldrich, США) в концентрации 4 мкг/мл и инкубировали в течение 4 ч, после чего кристаллы формазана растворяли в 10% (мас./об.) SDS в ДМСО с добавлением уксусной кислоты, и измеряли оптическую плотность (OD) при 570 нм с помощью спектрофотометра Multiskan EX (ThermoFisher Scientific, США). Далее рассчитывали процент живых клеток и отображали полученные значения на кривых нелинейной регрессии с использованием программного обеспечения Prism 5 (GraphPad, США).
Статистическая обработка результатов. Статистический анализ полученных результатов проводили с помощью U-критерия Манна–Уитни. Статистически значимыми считали различия между группами при уровне значимости p < 0.05.
Список литературы
Ibanez L.I., de Filette M., Hultberg A., Verrips T., Temperton N., Weiss R.A., Vandevelde W., Schepens B., Vanlandschoot P., Saelens X. // J. Infect. Dis. 2011. V. 203(8). P. 1063–1072.
Wu J., Fu J., Zhang M., Liu D. // J. Hematol. Oncol. 2015. V. 8(1). P. 96.
Василенко Е.А., Мохонов В.В., Горшкова Е.Н., Астраханцева И.В. // Молекулярная биология. 2018. Т. 52(3). С. 380–393. [Vasilenko E.A., Mokhonov V.V., Gorshkova E.N., Астраханцева I.V. // Molecular Biology. 2018. V. 52(3). P. 323–334.]
Coppieters K., Dreier T., Silence K., de Haard H., Lauwereys M., Casteels P., Beirnaert E., Jonckheere H., Van de Wiele C., Staelens L., Hostens J., Revets H., Remaut E., Elewaut D., Rottiers P. // Arthritis & Rheumatism. 2006. V. 54. P. 1856–1866.
Bradley M.E., Dombrecht B., Manini J., Willis J., Vlerick D., De Taeye S., Van den Heede K., Roobrouck A., Grot E., Kent T.C., Laeremans T., Steffensen S., Van Heeke G., Brown Z., Charlton S.J., Cromie K.D. // Molecular Pharmacology. 2015. V. 87. P. 251–262.
Qi J., Kan F., Ye X., Guo M., Zhang Y., Ren G., Li D. // International Immunopharmacology. 2012. V. 14(4). P. 770–778.
Liu M., Xie M., Jiang S., Liu G., Li L., Liu D., Yang X. // J. Biotechnology. 2014. V. 186. P. 1–12.
Kruglov A.A., Lampropoulou V., Fillatreau S., Nedospasov S.A. // J. Immunology. 2011. V. 187(11). P. 5660–5670. https://doi.org/10.4049/jimmunol.1100663
Grivennikov S.I., Tumanov A.V., Liepinsh D.J., Kruglov A.A., Marakusha B.I., Shakhov A.N., Murakami T., Drutskaya L.N., Förster I., Clausen B.E., Tessarollo L., Ryffel B., Kuprash D.V., Nedospasov S.A. // Immunity. 2005. V. 22. P. 93–104.
Winsauer C., Kruglov A.A., Chashchina A.A., Drutskaya M.S., Nedospasov S.A. // Cytokine Growth Factor Rev. 2014. V. 25. P. 115–123.
Ефимов Г.А., Вахрушева О.А., Сазыкин А.Ю., Муфазалов И.А., Круглов А.А., Купраш Д.В., Недоспасов С.А. // Российский иммунологический журнал. 2009. Т. 3(12). С. 23–29. [Efimov G.A., Vakhrusheva O.A., Sazykin A.Y., Mufazalov I.A., Kruglov A.A., Kuprash D.V., Nedospasov S.A. // Russian Journal of Immunology. 2009. V. 3(12). P. 23–29.]
Efimov G.A., Kruglov A.A., Khlopchatnikova Z.V., Rozov F.N., Mokhonov V.V., Rose-John S., Scheller J., Gordon S., Stacey M., Drutskaya M.S., Tillib S.V., Nedospasov S.A. // Proc. Natl Acad. Sci. USA. 2016. V. 113(11). P. 3006–3011.
Hamann J., Koning N., Pouwels W., Ulfman L.H., van Eijk M., Stacey M., Lin H.H., Gordon S., Kwakkenbos M.J. // Eur. J. Immunol. 2007. V. 37(10). P. 2797–2802.
Мохонов В.В., Шилов Е.С., Корнеев К.В., Атретханы К.-С.Н., Горшкова Е.А., Жданова А.А., Василенко Е.А., Горяйнова О.С., Купраш Д.В., Тиллиб С.В., Друцкая М.С., Ефимов Г.А., Недоспасов С.А. // Российский иммунологический журнал. 2016. Т. 10(19). С. 378–385. [Mokhonov V.V., Shilov E.S., Korneev K.V., Atretkhany K.-S.N., Gorshkova E.A., Zhdanova A.A., Vasilenko E.A., Goryainova O.S., Kuprash D.V., Tillib S.V., Drutskaya M.S., Efimov G.A., Nedospasov S.A. // Russian Journal of Immunology. 2016. V. 10(19). P. 378–385.]
Laemmli U. // Nature 1970. V. 227. 680e685.
Ефимов Г.А., Хлопчатникова З.В., Сазыкин А.Ю., Друцкая М.С., Круглов А.А., Шилов Е.С., Кучмий А.А., Недоспасов С.А., Тиллиб С.В. // Российский иммунологический журнал. 2012. Т. 6(15). С. 337–345. [Efimov G.A., Khlopchatnikova Z.V., Sazikin A.Yu., Drutskaya M.S., Kruglov A.A., Shilov E.S., Kuchmiy А.А., Nedospasov S.А., Tillib S.В. // Russian Journal of Immunology. 2012. V. 6(15). P. 337–345.]
Mokhonov V.V., Vasilenko E.A., Gorshkova E.N., Astrakhantseva I.V., Novikov D.V., Novikov V.V. // Biochem. Biophys. Res. Commun. 2018. V. 499(4). P. 967–972. https://doi.org/10.1016/j.bbrc.2018.04.029
Olleros M.L., Chavez-Galan L., Segueni N., Bourigault M.L., Vesin D., Kruglov A.A., Drutskaya M.S., Bisig R., Ehlers S., Aly S., Walter K., Kuprash D.V., Chouchkova M., Kozlov S.V., Erard F., Ryffel B., Quesniaux V.F., Nedospasov S.A., Garcia I. // Infect. Immun. 2015. V. 83(9). P. 3612–3623.
Atretkhany K.N., Mufazalov I.A., Dunst J., Kuchmiy A., Gogoleva V.S., Andruszewski D., Drutskaya M.S., Faustman D.L., Schwabenland M., Prinz M., Kruglov A.A., Waisman A., Nedospasov S.A. //Proc. Natl Acad. Sci. USA. 2018. V. 115(51). P. 13051–13056. https://doi.org/10.1073/pnas.1807499115
Espevik T., Nissen-Meyer J. // J. Immunol. Methods. 1986. V. 95. P. 99–105.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия