

Биоорганическая химия, 2020, T. 46, № 6, стр. 563-579
Особенности функционирования белка MutL из системы репарации “мисматчей” различных организмов
М. В. Монахова 1, М. А. Милакина 1, Р. М. Трикин 2, Т. С. Орецкая 1, *, Е. А. Кубарева 1
1 ФГБОУ ВО МГУ имени М.В. Ломоносова, химический факультет и НИИ физико-химической биологии имени А.Н. Белозерского
119991 Москва, Ленинские горы, 1, Россия
2 МИРЭА – Российский технологический университет, Институт тонких химических технологий
имени М.В. Ломоносова
119571 Москва, просп. Вернадского, 86, Россия
* E-mail: oretskaya@belozersky.msu.ru
Поступила в редакцию 24.04.2020
После доработки 28.04.2020
Принята к публикации 30.04.2020
Аннотация
Во всех живых организмах существует система репарации неканонических пар нуклеотидов (“мисматчей”), получившая название MMR (mismatch repair system). Нарушение функционирования MMR на одном из этапов исправления “ошибок” в ДНК приводит к накоплению мутаций в клетках и снижает их жизнеспособность. В большинстве организмов ключевую роль в удалении “мисматчей” выполняет белок MutL, обладающий способностью вносить одноцепочечный разрыв в “дочернюю” цепь ДНК. В обзоре рассматриваются особенности репарации неканонических пар нуклеотидов в эукариотах и прокариотах, структура белка MutL, обладающего эндонуклеазной функцией, его взаимодействие с ДНК и другими белками MMR.
ВВЕДЕНИЕ
В живых клетках некомплементарные нуклеотиды или “мисматчи” появляются в результате различных биологических процессов, например, “ошибок” при репликации ДНК, окислительных повреждений и рекомбинации неидентичных цепей [1, 2]. Система репарации “мисматчей” ДНК (MMR) важна для поддержания жизнедеятельности клетки и обнаружена в большинстве живых организмов. MMR исправляет “ошибки”, допущенные ДНК-полимеразой в процессе репликации, тем самым повышая точность копирования ДНК на два–три порядка [3, 4]. Дефекты в генах белков системы репарации приводят к повышенному числу мутаций и генетической нестабильности. В клетках человека такие дефекты могут являться причиной возникновения синдрома Линча, также известного как неполипозный рак прямой кишки. Люди, страдающие синдромом Линча, имеют высокий риск развития других онкологических заболеваний [5].
Исследования на модельных организмах и клетках человека показали, что MMR выполняет множество функций в метаболизме ДНК, помимо исправления “ошибок” репликации. Известно, что белки системы MMR участвуют в связанной с транскрипцией эксцизионной репарации нуклеотидов, клеточном цикле, мейотической рекомбинации и апоптозе [6]. Несмотря на важность системы репарации “мисматчей” для организмов, а также наличие большого числа исследований, посвященных этому вопросу [7–9], до сих пор остается множество “темных пятен”, касающихся механизма действия MMR. Так, на сегодняшний день не выяснено, как происходит процесс дискриминации “дочерней” цепи в ДНК. Эта проблема решена только в одном частном случае для клеток E. coli [10]. Таким образом, анализ публикаций, освещающих различные аспекты функционирования системы репарации “мисматчей”, является актуальной задачей. Данный обзор обобщает имеющуюся информацию о предполагаемом механизме действия всех участников системы MMR, а также более детально описывает строение и функции одного из ее ключевых белков – MutL.
СИСТЕМА РЕПАРАЦИИ “МИСМАТЧЕЙ” В E. coli
Система репарации “мисматчей” весьма консервативна и включает в себя четыре основные стадии: узнавание повреждения; распознавание “дочерней” цепи с ошибочно встроенным нуклеотидом; деградация фрагмента ДНК, содержащего “ошибку”; повторный синтез цепи ДНК и лигирование. Исторически лучше всего изучена система MMR в E. coli [10]. Она полностью реконструирована in vitro из выделенных компонентов [11]. В этой системе для узнавания “мисматчей” и инициации процесса репарации требуются три белка – MutS, MutL и MutH.
MMR “запускается” в момент узнавания неканонической пары нуклеотидов или небольших инсерционных/делеционных петель “сенсорным” белком MutS. MutS узнает почти все некомплементарные пары, которые затем исправляются системой MMR, однако сродство белка к ДНК и эффективность репарации могут зависеть от природы “мисматча” и нуклеотидной последовательности вокруг него [12].
Белки MutS входят в состав ABC-суперсемейства АТФаз. Связывание и гидролиз молекулы ATP необходимы для активации дальнейшего процесса – привлечения белка MutL [13, 14]. Несмотря на то, что бактериальные белки MutS представляют собой гомодимеры, каждая из субъединиц функционирует по-разному по аналогии с гетеродимерными белками. Методом рентгеноструктурного анализа (РСА) было показано, что только одна из субъединиц белка участвует во взаимодействии с “мисматчем” [12, 15]. Способность связывать и гидролизовать молекулу ATP также отличается для двух субъединиц [16, 17]. Благодаря такой асимметричности белок может приобретать множество конформаций, что необходимо для правильного формирования комплексов белка на ДНК и передачи сигнала для дальнейшего протекания репарации. Процессы связывания и гидролиза ATP белком MutS до сих пор уточняются, и современные представления об этих процессах описаны в работе [18]. Согласно полученным на сегодняшний день данным, одна из субъединиц MutS, находясь в поиске “мисматча”, постоянно связывает и гидролизует ATP. После нахождения “мисматча” белком происходят конформационные изменения как в белке, так и в ДНК, что вызывает уменьшение сродства MutS к ADP и быстрое связывание двух молекул ATP. Затем белок претерпевает значительные конформационные перестройки и переход в так называемый “скользящий зажим”. Согласно данным РСА, изменение конформации MutS приводит к формированию поверхности, необходимой для правильного взаимодействия с белком MutL. Если MutS не переходит в “скользящий зажим”, комплекс MutS–MutL не образуется, и процесс репарации не реализуется [19].
Следующий ключевой участник системы репарации “мисматчей” – MutL. Бактериальные белки MutL представляют собой гомодимерные АТФазы, принадлежащие к семейству GHKL. В состав семейства входят ДНК-гиразы, белки теплового шока Hsp90, гистидинкиназы и белки MutL (gyrase, Hsp90, histidine kinase и MutL) [20]. Основная роль MutL – координация белок–белковых взаимодействий в процессах узнавания повреждения и удаления фрагмента одной из цепей ДНК, поэтому этот белок был исторически классифицирован как “молекулярный посредник” [21]. Формирование тройного комплекса между MutS, MutL и ДНК, содержащей “мисматч”, достаточно для того, чтобы активировать эндонуклеазу MutН E. coli.
MutН – сайт-специфическая никующая эндонуклеаза, которая вносит одноцепочечный разрыв (“ник”) в неметилированную цепь монометилированного участка 5'-Gm6ATC-3'/3'-CTAG-5' [22]. Неметилированная (вновь синтезированная) и метилированная (на основе которой осуществлялась репликация) цепи далее будут названы “дочерней” и “материнской” соответственно. Низкая индивидуальная эндонуклеазная активность белка MutH увеличивается в 20–70 раз при взаимодействии с комплексом MutS–MutL. Внесение одноцепочечного разрыва в “дочернюю” цепь монометилированного участка 5'-Gm6ATC-3'/3'-CTAG-5' обеспечивает взаимодействие с ДНК хеликазы II (UvrD) и белка, связывающего одноцепочечную ДНК (SSB – single-strand binding protein). Таким образом, главная задача MutH заключается в том, чтобы гарантировать, что репарация нацелена исключительно на вновь синтезированную цепь [5]. Такая репарация называется метилзависимой.
Репарация “мисматчей” ДНК бинаправлена, так как монометилированные участки 5'-Gm6ATC-3'/ 3'-CTAG-5' могут находиться по обе стороны от “мисматча”. Повреждения корректируются независимо от того, где во вновь синтезированную цепь белок MutH внес разрыв: в 5'- или в 3'-направлении по отношению к повреждению [23]. Если одноцепочечный разрыв локализован в 5'-направлении от “мисматча”, MutH “привлекает” экзонуклеазы RecJ или ExoVII для удаления вновь синтезированной цепи. Экзонуклеазы ExoVII или ExoX участвуют в процессе репарации, если белок MutH гидролизовал ДНК в 3'-направлении от “мисматча”. Затем в обоих случаях хеликаза UvrD расплетает двойную спираль ДНК по направлению к повреждению [24]. Как только цепь, содержащая ошибку, удалена, белок SSB стабилизирует одноцепочечный фрагмент так, чтобы ДНК-полимераза III могла синтезировать правильную последовательность и ДНК-лигаза могла ликвидировать разрыв в олигонуклеотидной цепи [25].
РЕПАРАЦИЯ МИСМАТЧЕЙ В ОТСУТСТВИЕ БЕЛКА MutH
Гомологи белка MutH обнаружены только в некоторых γ-протеобактериях; в большинстве организмов эндонуклеазную функцию выполняет белок MutL. Гомологи MutL в таких организмах имеют консервативный эндонуклеазный мотив, который обеспечивает внесение одноцепочечного разрыва в “дочернюю” цепь ДНК [5] (см. далее). Бактериальные системы MMR, не содержащие MutH и, следовательно, метилнезависимые, мало изучены. Бактериальные MutL с эндонуклеазной функцией были выделены и охарактеризованы только для нескольких микроорганизмов: Aquifex aeolicus, Bacillus subtilis, Neisseria gonorrhoeae, Thermus thermophiles, Pseudomonas aeruginosa [26–30]. Нами был клонирован и выделен белок MutL из Rhodobacter sphaeroides и получены первые сведения о его свойствах [31]. У белков MutL с эндонуклеазной функцией отсутствует сайт-специфичность, и они способны гидролизовать ДНК независимо от нуклеотидной последовательности [32, 33]. Именно то обстоятельство, что бактериальные MutL являются гомодимерами, определяет необходимость четкой регуляции их действия во избежание внесения ими двуцепочечных разрывов в ДНК. Однако вопрос о том, как происходит узнавание “дочерней” цепи в большинстве бактерий, до сих пор остается нерешенным.
Следует отметить, что значительный прогресс в понимании механизма метилнезависимой репарации “мисматчей” был достигнут в случае эукариот. В эукариотах, в отличие от прокариот, гомологи MutS и MutL – гетеродимеры. Важнейшими компонентами системы MMR эукариот являются белки MutSα (гетеродимер MSH2–MSH6) и MutLα (в клетках человека: гетеродимер MLH1–PMS2; в клетках дрожжей S. cerevisiae: гетеродимер MLH1–PMS1) [34–36]. MutSα взаимодействует с “мисматчем” и короткими инсерционными/делеционными петлями. При возникновении в ДНК более длинных петель (2–12 пар нуклеотидов, п.н.) вместо MutSα в узнавании дефекта ДНК участвует MutSβ (гетеродимер MSH2–MSH3) [37, 38]. Белок MutLα является эндонуклеазой; после формирования комплекса MutSα–MutLα или MutSβ–MutLα он вносит одноцепочечный разрыв в “дочернюю” цепь ДНК.
В дополнение к основным белкам, в эукариотической системе MMR участвуют и многие другие белки: экзонуклеаза EXO1 [39–42]; фактор процессивности ДНК-полимеразы δ (PCNA) и репликативный фактор С (RFC), необходимый для посадки PCNA на ДНК [43–47]; репликативный белок А (RPA), связывающий одноцепочечные участки ДНК [46, 48, 49]; ДНК-полимераза δ (Pol δ) [45, 50–53]; эндонуклеаза MutLγ (гетеродимер MLH1–MLH3), функции которой до конца не ясны [54–56]; ДНК-лигаза I [50] и РНКаза H2 [57, 58]. Также было показано взаимодействие белков системы репарации “мисматчей” с поли(ADP-рибоза)полимеразой 1 (PARP1) [59] и продемонстрировано влияние белкового комплекса CAF-1 [60] и метилирования гистона H3K36 [61] на функционирование MMR.
Ключевой особенностью MMR является ее специфичность, которая гарантирует исправление “мисматча” в “дочерней” цепи, а не в “материнской”. Удаление нуклеотида, находящегося в “материнской” цепи, из некомплементарной пары превращает “ошибку” репликации в мутацию. Одним из сигналов дискриминации “дочерней” цепи ДНК в эукариотах являются одноцепочечные разрывы, возникающие при репликации ДНК. Процесс репликации ДНК по своей природе является ассиметричным и протекает по разным механизмам в случае “лидирующей” и “отстающей” цепей. Синтез “лидирующей” цепи ведется непрерывно, в то время как “отстающая” цепь синтезируется короткими фрагментами (фрагменты Оказаки). Наличие разрывов между фрагментами Оказаки в “дочерней” цепи считается основным фактором дискриминации цепей при репарации “мисматчей” в “отстающей” цепи ДНК [62].
Полагают, что одноцепочечные разрывы, направляющие действие MMR, возникают в “лидирующей” цепи в ходе эксцизионной репарации рибонуклеотидов (RER – ribonucleotide excision repair). Показано, что ДНК-полимеразы ε, δ и α дрожжей встраивают один рибонуклеотид на 1250, 500 и 625 нуклеотидных остатков (н.о.) соответственно. Эти рибонуклеотиды удаляются РНКазой H2 на первой стадии процесса RER. Одноцепочечные разрывы, образующиеся при этом, находятся только в “дочерней” цепи, что может быть сигналом для направленного действия MMR [62].
PCNA и β-“зажим” как факторы активации эндонуклеазной функции белков MutL
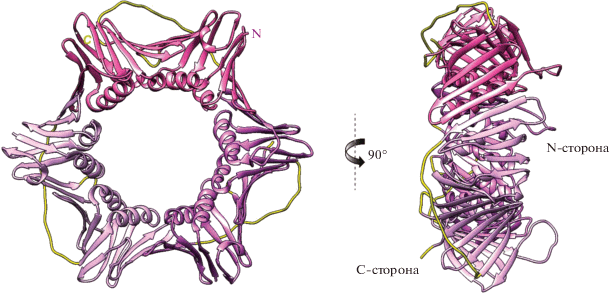
Согласно современным воззрениям, в дискриминации “дочерней” цепи в эукариотических клетках принимает участие белок PCNA [63]. PCNA является гомотримером и по форме напоминает кольцо, которое имеет две неэквивалентные стороны: C и N (рис. 1). “Посадка” PCNA на двуцепочечную ДНК в месте образования “ника” осуществляется белком RFC таким образом, что C-сторона PCNA обращена к свободному 3'-концу ДНК, при этом PCNA приобретает форму кольца, которое скользит по ДНК [64]. Продемонстрировано, что C-сторона PCNA имеет гидрофобную область, с которой контактирует большинство белков-партнеров [65]. На сегодняшний день показано взаимодействие с PCNA белков MSH(2/3/6) и обеих субъединиц MutLα человека и дрожжей [43, 66, 67]. Субъединицы MutLα содержат консервативный мотив, отвечающий за связывание с С-стороной PCNA (QXXIXP). Считается, что взаимодействие MutLα с PCNA обеспечивает ориентацию MutLα, необходимую для внесения одноцепочечного разрыва селективно в “дочернюю” цепь ДНК. Так, в недавней работе [67] продемонстрировано, что аминокислотные замены в мотиве 721-QRLIAP белка PMS2 (723-QKLIIP для дрожжевого PMS1) приводят к снижению эффективности взаимодействия PCNA c MutLα, и, как следствие, к потере способности MMR исправлять “мисматчи”. Замены в PCNA-связывающем мотиве субъединицы MLH1 (562-QILIYDF) мало влияют на образование комплекса PCNA–MutLα и не “выключают” MMR, хотя и снижают эффективность процесса.
Рис. 1.
Кристаллическая структура PCNA человека (PDB: 1VYM) в двух проекциях. Желтым обозначены гидрофобные участки С-стороны белка, участвующие во взаимодействии с белковыми партнерами.
PCNA также вовлечен в активацию эндонуклеазной функции MutLα. На сегодняшний день существуют две альтернативные модели этого процесса [8]. Первая модель предполагает первоначальное образование комплекса между MutLα и находящимся около разрыва PCNA (рис. 2, слева). После формирования комплекса MutLα–PCNA MutLα распознает “дочернюю” цепь по наличию в ней одноцепочечных разрывов. Затем комплекс MutLα–PCNA скользит по ДНК и контактирует с MutSα, который активирует MutLα [68].
Рис. 2.
Схема возможных механизмов активации MutL, внесения белком одноцепочечного разрыва в “дочернюю” цепь ДНК с 5'-стороны от некомплементарной пары и последующей EXO1-зависимой MMR в клетках человека. Предложено согласно [8].
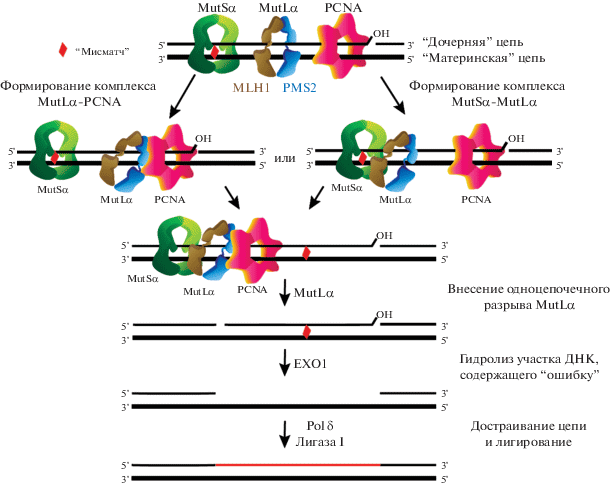
Другая модель предполагает, что первоначально формируется комплекс MutSα и MutLα на ДНК, содержащей “мисматч” (рис. 2, справа). Этот комплекс перемещается по ДНК. MutL в его составе взаимодействует с PCNA, что приводит к образованию тройного комплекса MutSα–MutLα–PCNA. Когда этот комплекс достигает места одноцепочечного разрыва, MutLα распознает “никованную” цепь и принимает активную конформацию. Затем комплекс MutSα–MutLα–PCNA возобновляет движение по ДНК, и MutLα гидролизует “дочернюю” цепь. Несмотря на то, что обе модели согласуются с существующими экспериментальными данными, фактические механизмы активации MutLα до сих пор не ясны.
Бактериальным аналогом PCNA является β-“зажим” (β-субъединица ДНК-полимеразы III) [69]. Структурные мотивы, обеспечивающие контакт с β-“зажимом”, были найдены во многих бактериальных белках, вовлеченных в репликацию и репарацию ДНК [70]. Так, β-“зажим” стимулирует эндонуклеазную активность MutL бактерии Bacillus subtilis (bsMutL) [69]. В работе [27] показано, что регуляторный субдомен С-концевого домена bsMutL содержит мотив QEMIVP, взаимодействующий с β-“зажимом” (рис. 3). Аминокислотные замены в этом мотиве MutL инактивируют функционирование системы MMR in vivo [69, 71]. Для объяснения стимулирующего эффекта эндонуклеазной активности белков MutL при взаимодействии с β-“зажимом” была предложена модель, согласно которой скопление отрицательно заряженных аминокислотных остатков (а.о.) на поверхности С-концевого домена bsMutL препятствует связыванию фермента с ДНК. Взаимодействие bsMutL с β-“зажимом” нейтрализует эти отрицательные заряды, облегчая формирование белково–нуклеинового комплекса [27]. MutL из B. subtilis относят к β-“зажим”-зависимым MutL, образующим подсемейство I этих белков. Несмотря на то, что бактериальные эндонуклеазы MutL являются гомодимерами, их взаимодействие с β-“зажимом” происходит аналогично взаимодействию MutLα с PCNA. Только один из двух мотивов С-концевого домена bsMutL контактирует с β-“зажимом” [72].
Рис. 3.
Сравнительный анализ аминокислотных последовательностей эукариотических и бактериальных белков MutL. Розовой рамкой показан консервативный мотив MutL, участвующий во взаимодействии с PCNA или β-“зажимом”. Три подсемейства бактериальных белков MutL указаны справа.

Кроме подсемейства I, среди бактериальных MutL выделены подсемейства II и III, которые не имеют характерного β-“зажим”-связывающего мотива и отрицательно заряженного кластера, а их эндонуклеазная активность не зависит от присутствия β-“зажима”. Примерами таких белков являются MutL из T. thermophilus (ttMutL) – представитель подсемейства II, у которого β-“зажим”-связывающий мотив присутствует, но не отличается консервативностью; и MutL из A. aeolicus (aaMutL) – представитель подсемейства III, у которого такой мотив отсутствует (рис. 3) [73].
Авторами [73] показано, что β-“зажим” не влияет на эффективность внесения одноцепочечного разрыва в ДНК белками aaMutL и ttMutL; гидролиз ДНК происходит даже в отсутствие β-“зажима”. В работе [74] была реконструирована in vitro система с использованием белков MutL и MutS из T. thermophilus. Однако ttMutL гидролизовал обе цепи ДНК-дуплекса, несмотря на наличие “ников” в одной из цепей, β-“зажима” и белка, осуществляющего “посадку” β-“зажима” на ДНК (ДНК-полимераза III, субъединицы γ, τ, δ и δ'), то есть дискриминация цепей в этом случае отсутствовала.
Таким образом, β-“зажим” по-разному влияет на эндонуклеазную активность бактериальных белков MutL. Это свидетельствует не только о различиях в механизме дискриминации цепей системами MMR бактерий и эукариот, но и о различиях среди самих бактериальных систем MMR, содержащих MutL с эндонуклеазной функцией.
EXO1-зависимая и EXO1-независимая MMR в клетках человека
Основным механизмом ММR у эукариот считается EXO1-зависимая репарация. Белок EXO1 является 5'-3'-экзонуклеазой, поэтому наличие “ника” с 5'-стороны от “мисматча” используется EXO1 в качестве отправной точки для 5'-3'-направленного удаления фрагмента “дочерней” цепи, содержащей “ошибку” (рис. 2). Однако, как и в клетках E. coli, в эукариотах MMR бинаправлена, но в данном случае ход процесса определяется тем, с какой стороны от “мисматча” (5' или 3') локализованы одноцепочечные разрывы. Если “ник” находится с 3'-конца от “мисматча”, то активированная эндонуклеаза MutLα гидролизует такую ДНК, внося разрывы с 5'-стороны от “мисматча”, таким образом обеспечивая EXO1-зависимую репарацию [75]. В этом случае белок RFC, помимо посадки PCNA на ДНК, регулирует активность EXO1, тем самым защищая ДНК от нежелательной деградации [46]. Наличие “ника” с 5'-конца от “мисматча” достаточно для инициации MMR, и внесение дополнительного разрыва в ДНК белком MutLα не требуется. В этом случае EXO1 может непосредственно контактировать с MutSα, что стимулирует экзонуклеазную функцию фермента [8].
В отсутствие EXO1 система MMR может обойтись без стадии удаления фрагмента ДНК (EXO1-независимый механизм) [53]. Для реализации этого механизма необходимы белки MutSα, MutLα, PCNA, RFC и Pol δ, а также наличие “ника” в “дочерней” цепи. В этом случае MutLα гидролизует “дочернюю” цепь ДНК, внося одноцепочечный разрыв с 5'-стороны от “мисматча”. Pol δ синтезирует новый фрагмент ДНК, используя в качестве стартовой позиции образующийся в месте разрыва 3'-конец. За счет синтеза новой цепи холоферментом Pol δ происходит вытеснение цепи, содержащей “ошибку”. В ходе процесса происходит активация белка RPA, который связывает одноцепочечную ДНК, препятствуя образованию ДНК-дуплекса. Показано, что скорость EXO1-независимой MMR значительно ниже, чем скорость репарации с участием EXO1, поэтому в клетках, вероятнее всего, процесс репарации “мисматчей” проходит по EXO1-зависимому механизму [53].
СТРУКТУРА БЕЛКА MutL И ЕГО ФУНКЦИИ
Кроме MMR, белки MutL участвуют в других клеточных процессах [8, 76, 77]. Наряду с MutLα, в эукариотах присутствуют другие гетеродимеры – MutLβ (MLH1–PMS1) и MutLγ (MLH1–MLH3) – роль которых в клетке до конца не выяснена. Субъединица MLH3 в MutLγ обладает эндонуклеазной активностью. Несмотря на то, что MutLγ может частично заменять MutLα в процессе репарации “мисматчей”, показано, что основной его функцией является участие в поздней стадии мейотической рекомбинации, которая заключается в “процессировании” соединенных между собой молекул (таких как структуры Холлидея) в кроссоверные ДНК [78, 79]. Третий гетеродимер, MutLβ, не обладает эндонуклеазной активностью, однако он также играет роль в мейотической рекомбинации. MutLβ взаимодействует с мейотической хеликазой Mer3, привлекая ее к местам рекомбинации независимо от распознавания “мисматчей” [80]. Недавние исследования показали участие MutLβ и MutLγ в экспансии тринуклеотидных повторов [81–87]. Также было продемонстрировано, что эндонуклеазная функция MutLγ активируется только в присутствии MutSβ и инициирует собственный механизм удаления повреждения [88].
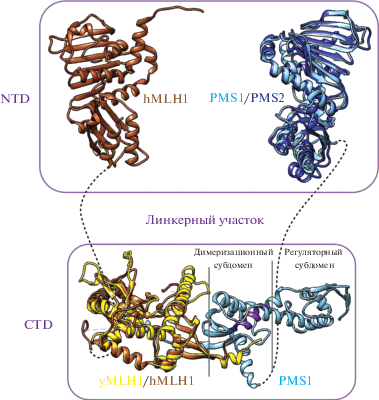
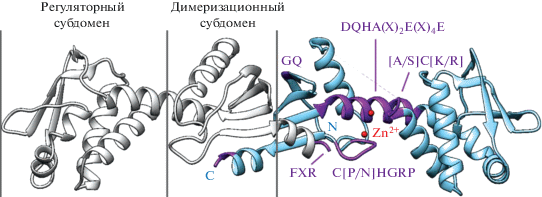
Несмотря на различные роли, которые выполняют белки MutL из разных организмов, все гомологи этих белков имеют два домена – N-концевой и C-концевой, соединенные вариабельным линкером (рис. 4). Первичная структура N-концевого домена MutL является высоко консервативной, в то время как первичная структура С-концевого домена MutL может быть вариабельной. Однако локализация и укладка в пространстве основных элементов вторичной структуры являются строго определенными [89]. N-Концевая область белка содержит АТФазный домен. Также в N-концевой области находится ДНК-связывающий центр MutL. С-Концевая область MutL содержит субдомен, участвующий в димеризации, а также внешний субдомен, обращенный к растворителю, который также называют регуляторным. Между димеризационным и внешним субдоменами расположен металлсвязывающий мотив, играющий важную роль в гидролизе ДНК (эндонуклеазный субдомен).
Рис. 4.
Совмещение кристаллических структур белков MutLα дрожжей (PDB: 4E4W и 3H41) и человека (PDB: 3RBN, 3NA3 и 1H7S). Субъединицы MLH1 дрожжей (yMLH1) и человека (hMLH1) обозначены желтым и коричневым цветами соответственно; субъединицы PMS1 и PMS2 – голубым и синим соответственно. Рамками выделены N- и C-концевые домены белков (NTD и CTD). Фиолетовым цветом обозначен эндонуклеазный мотив DQHA(X)2E(X)4E. Пунктиром обозначены линкерные участки. Предложено согласно [119].
Описанная выше архитектура белков MutL необходима и достаточна для их функционирования. Она предполагает существенные конформационные перестройки белковой молекулы, которые происходят под действием различных факторов и требуются для протекания процесса MMR.
АТФазная функция MutL
Получены кристаллические структуры N-концевых доменов MutL из E. coli и A. aeolicus, а также белков MLH1, PMS2 и PMS1 человека и дрожжей [90–94]. Эти домены состоят из двух субдоменов. Первый субдомен имеет в своем составе 5 консервативных мотивов (VVKELVENALDAGA, DNGXGM, TLGFRGEALXS, GT и NGR), отвечающих за связывание и гидролиз молекулы ATP [31, 95]. Нарушение АТФазной функции белка может приводить к нарушениям в работе системы MMR. Так, а.о. N38 и D63 (MLH1 человека) и N45 и D70 (PMS2) из мотивов VVKELVENALDAGA, DNGXGM необходимы для связывания ATP. Аминокислотами, играющими ключевую роль в гидролизе ATP, являются E34 (MLH1 человека) и E31 (PMS2) из мотива VVKELVENALDAGA [91]. Введение замен N38A, D63N в MLH1, а также N45A, D70N в PMS2 приводит к инактивации системы MMR [96]. При этом мутантные формы с единичной заменой MLH1(E34A) и PMS2(E41A) не препятствуют протеканию MMR. Двойная замена MLH1(E34A) и PMS2(E41A) в гетеродимере MLH1–PMS2 приводит к полному “выключению” репарации “мисматчей”, блокируя способность MutLα вносить разрыв в ДНК в присутствии MutSα, PCNA, RFC, ATP, Mg2+ [96].
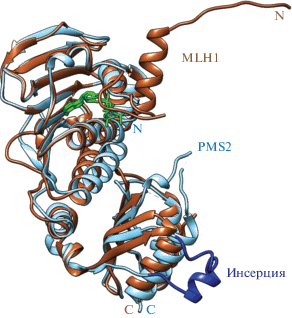
Второй субдомен N-концевого домена MutL, предположительно, отвечает за ДНК-связывающую активность белка [93]. При сравнении полученных кристаллических структур для белков MLH1, PMS2 и PMS1 обнаружено схожее взаимное расположение двух субдоменов [5]. Стоит отметить, что паралоги PMS обладают заметно меньшим сродством к ATP по сравнению с белками MLH [5]. Такое различие, возможно, объясняется особенностями структуры АТФазного домена: белок PMS2 человека (и его гомолог PMS1 в дрожжах) имеет уникальные инсерции, которые находятся во втором субдомене (рис. 5, обозначены синим цветом). Инсерции могут влиять на взаимное расположение субдоменов и, как результат, оказывать воздействие на способность белка связывать ATP.
Рис. 5.
Совмещение кристаллических структур А-ТФазных доменов белков MLH1 и PMS2 в комплексе с ATP и АTPγS соответственно (PDB: 3NA3, 1H7U). Синим цветом обозначена инсерция пептида в белке PMS2, зеленым – нуклеотидные кофакторы ATP и АTPγS.
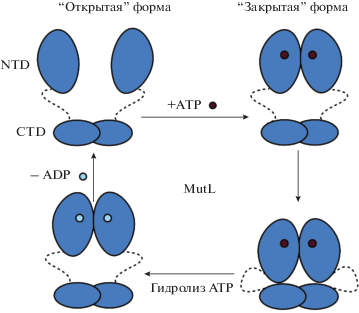
Большинство членов семейства GHKL формируют димеры, которые претерпевают значительные конформационные изменения при связывании или гидролизе ATP [97] (рис. 6). В апо-состоянии C-концевые домены находятся в димерной форме, в то время как N-концевые домены остаются в виде мономеров (“открытая” форма). Связывание ATP приводит к димеризации двух АТФазных доменов и образованию более компактной “закрытой” формы. Последующий гидролиз ATP и диссоциация ADP из комплекса с белком возвращают NTD в мономерное состояние. Интересно, что для эукариотических клеток методом атомно-силовой микроскопии зафиксированы ассиметричные конформационные изменения, связанные с различным сродством двух субъединиц MutLα к ATP [98]. Цикл конформационных перестроек MutL сравнивают с аналогичным циклом для другого белка семейства GHKL, Hsp90, N-концевые домены которого тоже димеризуются при связывании ATP [99]. Конформационные изменения и стабилизация N-концевого домена Hsp90 в результате связывания нуклеотидов позволяют ему взаимодействовать с другими белками. Похожая модель функционирования предложена и для белка MutL, формирующего комплексы с MutS и другими белками MMR [100]. Конформационные изменения при связывании и гидролизе молекулы ATP белками MutL обусловливают регуляцию эндонуклеазной функции их C-концевого домена [28]. Эндонуклеазная активность MutLα человека и дрожжей стимулируется присутствием ATP [75]. С другой стороны, по данным литературы нельзя сделать однозначный вывод о влиянии ATP на эффективность гидролиза ДНК белками MutL из различных бактерий. Способность белков MutL из T. thermophilus и N. gonorrhoeae (ttMutL и ngMutL) вносить одноцепочечный разрыв в ДНК ингибируется добавлением ATP в реакционную смесь [28, 30]. Показано, что эндонуклеазная функция aaMutL может и активироваться [101], и ингибироваться [30] в зависимости от концентрации ATP и ионов Mn2+. Низкие концентрации ATP стимулируют гидролиз ДНК белком bsMutL, а высокие – ингибируют [27]. В работе [31] показано, что присутствие ATP в реакционной смеси снижает степень расщепления суперскрученной ДНК белком MutL из R. sphaeroides (rsMutL). Таким образом, различия во влиянии ATP на эндонуклеазную активность белков MutL из различных организмов могут говорить об отличающихся механизмах активации белка, которые еще предстоит установить.
СВЯЗЫВАНИЕ MutL C ДНК
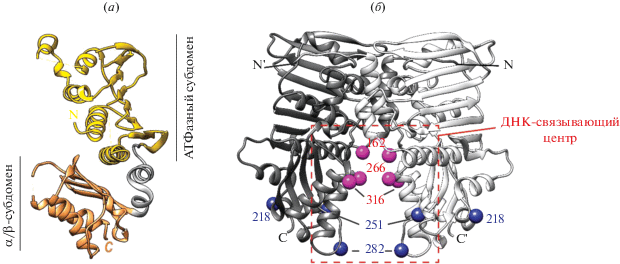
Кристаллическую структуру комплекса полноразмерного MutL с ДНК получить не удается из-за высокой конформационной подвижности белка. Известно, что белки MutL взаимодействуют с ДНК с низкой эффективностью. ДНК-связывающий центр MutL локализован в N-концевом домене белка [102]. Полагают, что ДНК связывается в участке MutL, который богат положительно заряженными аминокислотными остатками и сформирован АТФазным субдоменом и α/β-субдоменом N-концевого домена (рис. 7). Так, в дрожжевых гомологах MutL замены аминокислот Arg273 и Arg274 (в MLH1) в α/β-субдомене или Lys197 и Arg198 (в PMS1) в АТФазном субдомене снижают эффективность комплексообразования MutL с ДНК и приводят к понижению способности системы MMR исправлять “мисматчи” [94, 103, 104]. Двойная замена R273E/R274E в дрожжевом белке MLH1 блокирует MMR [1]. В работе [90] был обнаружен положительно заряженный участок в N-концевом домене MutL из Aquifex aeolicus, и было продемонстрировано, что а.о. Arg156, Lys158, Arg259 и Lys265 являются необходимыми для связывания белком ДНК. В случае белка MutL из E. coli также показано, что замена Arg162, Arg266 и Arg316, находящихся в А-ТФазном и α/β-субдоменах, на Glu приводит к полной потере способности белка образовывать комплексы с ДНК [19, 93, 105, 106]. Недавно нами с помощью комбинации методов “кросслинкинга” и FRET было продемонстрировано, что положения 218, 251 и 282 в MutL из E. coli являются сближенными с ДНК в процессе функционирования белка [107].
Рис. 7.
Строение N-концевого домена MutL из E. coli. (а) Кристаллическая структура мономера MutL-NTD (PDB: 1BKN). Желтым цветом отмечен АТФазный субдомен, коричневым – α/β-субдомен. (б) Кристаллическая структура димера MutL-NTD в комплексе с AMP-PNP (PDB: 1B63). Розовыми сферами обозначены остатки Arg, необходимые для связывания ДНК согласно [19], синими – остатки, сближенные с ДНК в процессе функционирования белка согласно [107]. Красной пунктирной рамкой указана предполагаемая ДНК-связывающая область MutL.
Показано, что N-концевой домен гомологов белка MutL из E. coli, человека и дрожжей способен связывать ДНК самостоятельно. Однако наличие С-концевого домена увеличивает сродство N-концевого домена к ДНК [93, 94].
Белок MutL и его гомологи связывают как одноцепочечную, так и двуцепочечную ДНК независимо от ее нуклеотидной последовательности [38] и наличия в ней “мисматча” [32, 33]. Эффективность комплексообразования MutL с ДНК зависит от многих факторов: ионной силы раствора, длины ДНК и наличия кофакторов (ADP, ATP, негидролизуемых аналогов ATP). Показано, что наиболее стабильные комплексы c ДНК белки MutL формируют при низкой ионной силе раствора [108]. Эффективность образования комплексов с ДНК растет по мере увеличения длины последней. Так, при первом исследовании ДНК-связывающей активности белка MutL из E. coli было обнаружено, что эффективнее всего белок образует комплексы с ДНК длиной более 100 п.н. [32]. С развитием методов детекции было зафиксировано связывание MutL из E. coli с 41-звенной ДНК [19]. Кроме того, недавно были впервые получены ковалентно связанные комплексы белка с 17-звенной ДНК [109]. В случае белка MutL из A. aeolicus, который содержит удивительно короткий линкерный участок (5 а.о.), авторами работы [71] была предложена модель взаимодействия MutL с ДНК, а также было показано, что “место посадки” такого белка на ДНК составляет 30 п.н.
ДНК-связывающая функция гомологов MutL влияет на их АТФазную активность, как и в случае белков MutS [14, 110]. Например, наличие одноцепочечной и двуцепочечной ДНК в реакционной смеси может значительно повышать АТФазную активность MutL [93]. В то же время эффективность комплексообразования MutL с ДНК практически не изменяется при связывании белком ATP. Показано, что ATP не требуется для образования комплекса MutL с ДНК, но необходим для формирования комплекса MutS–MutL [19]. В недавней работе убедительно продемонстрировано на системе из E. coli, что основная функция связывания ATP белком MutL – это образование “скользящего зажима” на ДНК [7]. Образующийся стабильный долгоживущий комплекс MutS–MutL “скользит” по ДНК в поисках места гидролиза белком MutH. Кроме того, авторы предполагают, что “скользящий” по ДНК комплекс включает в себя не только белки MutS и MutL, но и MutH. Таким образом, связывание и гидролиз ATP необходимы белку MutL для активации дальнейших стадий репарации.
Эндонуклеазная функция белка MutL
Кристаллографический анализ C-концевых доменов (CTD) бактериальных и эукариотических гомологов MutL показал, что каждый домен состоит из регуляторного и димеризационного субдоменов [8, 27, 111]. Сравнение аминокислотных последовательностей белков MutL выявило наличие 5 консервативных мотивов (DQHA(X)2E(X)4E, [A/S]C[K/R], CPHGRP, FXR и GQ), образующих каталитический центр (рис. 8) [95]. Примечательно, что в работе [31] с помощью биоинформатического анализа была выявлена группа бактериальных белков MutL, у которых присутствует только мотив DQHA(X)2E(X)4E и отсутствуют все остальные эндонуклеазные мотивы. Например, к этой группе относятся белки MutL из организмов, принадлежащих классам Chlamydiales и Bacteroidia. Авторы предполагают, что белки MutL с одним консервативным эндонуклеазным мотивом способны гидролизовать ДНК, однако эта гипотеза требует экспериментальной проверки.
Рис. 8.
Кристаллическая структура C-концевого домена bsMutL (PDB: 3KDK). Консервативные мотивы эндонуклеазного центра белка указаны фиолетовым цветом. Ионы цинка обозначены красными сферами. Предложено согласно [27].
Моделирование структуры C-концевого домена MutL из B. subtilis (bsMutL-CTD) с ДНК показало, что мотив DQHA(X)2E(X)4E и фосфодиэфирная связь в ДНК находятся на расстоянии, которое позволяет карбоксильной группе остатка Asp в первой позиции этого мотива катализировать гидролиз ДНК [27]. Замена Asp в этом мотиве на Asn в MutL из B. subtilis и T. thermophilus инактивирует их эндонуклеазную функцию и, соответственно, препятствует протеканию MMR [27, 30]. В работе [28] получено несколько мутантных форм, содержащих замены аминокислотных остатков в DQHA(X)2E(X)4E-мотиве MutL из N. gonorrhoeae (ngMutL). Мутантные формы c одной заменой D491N и E497Q оказались активными на 70% по сравнению с “диким типом”; белок ngMutL с двойной заменой D491N/E497Q в C-концевом домене сохранял эндонуклеазную активность лишь на 15%, что может дополнительно свидетельствовать о важности остатков Asp и Glu для осуществления белком гидролитической функции.
В случае эукариотических гомологов MutL замены в консервативных мотивах, формирующих эндонуклеазный центр белка, также приводят к нарушению функции катализа гидролиза ДНК. Так, замены D699N и E705K в мотиве 699‑DQHA(X)2E(X)4E-710 в субъединице PMS2 белка MutLα человека приводят к полной потере эндонуклеазной активности [53, 75]. Подобная замена в дрожжевом белке PMS1(E707K) не только нарушает способность MutLα гидролизовать ДНК, но и подавляет связанную с MMR гомологическую рекомбинацию [112]. Также при исследовании мотива DQHA(X)2E(X)4E субъединицы PMS2 MutLα человека было показано, что аминокислотный остаток H701 является необходимым для функционирования MutLα, а консервативный остаток E710 – нет [113]. Это может свидетельствовать об участии этой аминокислоты в другой, еще невыясненной, функции MutLα. В целом, исследования гидролитической активности эукариотических белков MutLα показывают, что эндонуклеазная функция необходима не только для реализации процесса MMR, но и для протекания гомологической рекомбинации и апоптоза [114].
Влияние ионов двухвалентных металлов на гидролиз ДНК белком MutL
Несмотря на то, что и бактериальные, и эукариотические белки MutL содержат консервативные мотивы в C-концевом домене, катализ гидролиза ДНК этими белками имеет ряд индивидуальных особенностей, связанных с влиянием ионов двухвалентных металлов на этот процесс. Показано, что MutLα человека и дрожжей проявляют эндонуклеазную функцию только в присутствии ионов Mn2+ [75, 112], в то время как их бактериальные аналоги активны в присутствии ионов разных двухвалентных металлов. Например, MutL из T. thermophilus (ttMutL) и из A. aeolicus (aaMutL) гидролизуют суперскрученную плазмиду в присутствии ионов Mn2+, Ni2+ или Co2+, но не в присутствии ионов Mg2+, Zn2+ или Ca2+ [29, 30]. В работе [28] показано, что ngMutL и его C-концевой домен способны вносить разрыв в плазмидную ДНК в присутствии ионов Mn2+ или Mg2+, но не в присутствии ионов Ca2+. Белок bsMutL проявляет гидролитическую активность только в присутствии ионов Mn2+. В нашей работе [31] установлено, что ионы Mn2+, Mg2+, Co2+ и Cd2+ стимулируют гидролиз плазмидной ДНК белком rsMutL, а присутствие ионов Ni2+, Ca2+ и Zn2+ не влияет на эндонуклеазную функцию белка. Интересно, что добавление в реакционную смесь, содержащую bsMutL и и-оны Mn2+, второго иона металла (Ca2+, Zn2+, Co2+) увеличивает количество “никованной”, а также линейной формы ДНК по сравнению с действием только Mn2+ в тех же условиях [27]. Влияние ионов двухвалентных металлов на эндонуклеазную функцию бактериальных MutL суммировано нами ранее в работе [31].
Важное место среди ионов двухвалентных металлов занимают ионы Zn2+. При анализе кристаллической структуры C-концевого домена bsMutL, как и в случае MutLα дрожжей, показано, что мотивы DQHA(X)2E(X)4E и [A/S]C[K/R] координируют два иона Zn2+ (рис. 9). Основная разница в структурах bsMutL и MutLα дрожжей заключается в характере связывания ионов Zn2+. В случае bsMutL-CTD только один ион Zn2+ (номер 1) полностью координирован а.о. Glu468, Cys604, His606 и молекулой воды (рис. 9а), в то время как в координационную сферу второго иона Zn2+ (номер 2) только частично включены функциональные группы а.о. His464, Glu468, Cys573 [27]. Замены консервативных аминокислотных остатков, входящих в каталитический и предполагаемый Zn2+-связывающий центры, нарушают действие MutL in vitro [112].
Рис. 9.
Структура эндонуклеазных доменов (а) bsMutL (PDB: 3KDK) и (б) MutLα дрожжей (PDB: 4E4W). Красными сферами обозначены ионы цинка; розовым цветом показан мотив DQHA(X)2E(X)4E. Предложено согласно [89].
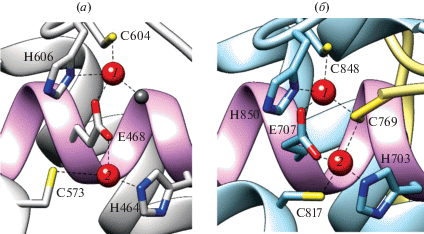
В отличие от bsMutL-CTD, в MutLα дрожжей задействован четвертый консервативный мотив – FXR, который имеет строгую консенсусную последовательность (FERC). В структуре дрожжевого белка MutLα мотив FERC находится в непосредственно близости от каталитического центра PMS1 и координирует два иона Zn2+ (рис. 9б) [111]. В бактериальном MutL этот мотив не имеет консервативного остатка Cys, контактирующего с Zn2+ [113]. Таким образом, в структуре MutLα-CTD оба иона Zn2+ полностью координированы а.о. из четырех консервативных эндонуклеазных мотивов.
Подобная разница в сродстве MutL из разных организмов к ионам Zn2+ может свидетельствовать о том, что ионы металла выполняют в этих белках разную роль. Так, схожий с MutLα механизм хелатирования наблюдается у нескольких нуклеаз, где а.о. из четырех мотивов также координируют два иона металла [115]. Предполагают, что ионы Me2+ входят в каталитический центр белка [111]. В случае bsMutL из-за меньшего сродства белка к ионам Zn2+ считается, что Zn2+-связывающий центр имеет скорее структурную, а не каталитическую роль [27].
В работе [27] bsMutL-CTD был закристаллизован в трех различных формах – в условиях, соответствующих увеличению концентрации Zn2+ в среде. Структурный анализ этих форм показал существенное сближение друг с другом а.о., ответственных за гидролиз ДНК, благодаря комплексообразованию с Zn2+. Связывание MutL с ионами двухвалентных металлов также обеспечивает устойчивую взаимную ориентацию димеризационных и регуляторных субдоменов гомодимера белка, которые в отсутствие ионов металлов более подвижны.
В пользу предположения о структурной, а не каталитической роли ионов Zn2+ свидетельствует и отмеченное в работе [27] сходство первичной и вторичной структур MutL-CTD и белков семейства IdeR (iron-dependent repressors), а также конформационные изменения, вызванные связыванием ионов металлов. Так, IdeR из Mycobacterium tuberculosis имеет два металлсвязывающих центра, один из которых является функциональным и имеет высокое сродство к Fe2+, а второй – структурный – обладает сродством к Zn2+. В работе [116] показано, что при одновременном присутствии ионов Fe2+ и Zn2+ степень связывания ДНК белком IdeR значительно выше, чем в присутствии только ионов Fe2+ в той же концентрации. Таким образом, предполагается, что связывание ионов Zn2+ способствует предварительной организации IdeR, приводящей к конформационному переходу в структуру, обеспечивающую более эффективное связывание Fe2+.
В противоположность данным, полученным для bsMutL, в кристаллической структуре ngMutL-CTD не наблюдались значительные конформационные перестройки при анализе его в присутствии ионов Zn2+, что может свидетельствовать, формировании структуры эндонуклеазного центра без участия этого иона [117].
В работе [118] была решена кристаллическая структура aaMutL-CTD (PDB: 5Z42). В ней визуализировались три иона цинка, расположенные около спиралей α1 и α2 и петли L2, которые включают мотивы DQHAX2EX4E, ACR и CPHGRP (рис. 10а). Ион Zn2+ в позиции 1 координируется а.о. Glu357, Cys402 и His404; в позиции 2 – His353, Glu357 и Cys371 (рис. 10б). Ион Zn2+ в позиции 3 координируется Cys371 спирали α2. Ионы Zn2+ в позициях 1 и 2 в структуре aaMutL-CTD занимают схожую позицию с их локализацией в структуре bsMutL-CTD.
Рис. 10.
Кристаллические структуры C-концевого домена MutL из A. aeolicus (aaMutL-CTD). (а) Кристаллическая структура мономера aaMutL-CTD с тремя ионами цинка (PDB: 5Z42); (б) цинк-связывающий центр aaMutL-CTD; (в) совмещение двух структур aaMutL-CTD (PDB: 5Z42 и 5Z41) – с тремя ионами цинка (зеленый цвет) и с одним ионом цинка (серый цвет). Петля в структуре 5Z41 с единственным ионом цинка и α-спираль, образующаяся при связывании трех ионов цинка в структуре 5Z42, обозначены темно-серым и темно-зеленым цветами соответственно. Предложено согласно [118].
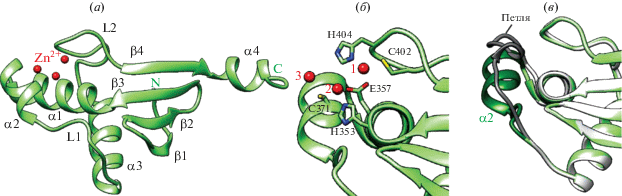
В работе [118] также была предпринята попытка получить кристаллическую структуру aaMutL-CTD в отсутствие ионов металла. Однако этого сделать не удалось; в результате структурного анализа в aaMutL-CTD было обнаружено присутствие одного иона Zn2+ в позиции 1 (PDB: 5Z41). Этот единственный ион Zn2+ координируется а.о. Glu357, Cys371, Cys402 и His404. Отметим, что взаимодействие иона Zn2+ с Cys371 отсутствовало в структуре с тремя ионами металла. При совмещении структур с одним и тремя ионами цинка (PDB: 5Z41 и 5Z42) была замечена значительная разница в организации некоторых участков полипептидной цепи (рис. 10в). Так, а.о. 367–374 в присутствии трех ионов Zn2+ формируют спираль α2, в то время как в присутствии одного иона Zn2+ в этой области образуется петля [118]. Следует отметить, что способность белка aaMutL связывать большее число ионов цинка (по сравнению с другими полученными бактериальными MutL) может являться следствием особенностей строения данного белка (отсутствия регуляторного домена [73]). Предполагают, что связывание нескольких ионов цинка необходимо для формирования правильной структуры каталитического центра aaMutL. В доказательство этого предположения свидетельствует, что способность гидролизовать ДНК у aaMutL, связанного с тремя ионами Zn2+, в три раза превышает активность aaMutL с одним ионом Zn2+.
Наличие в реакционной смеси только ионов Zn2+ не стимулирует гидролиз суперскрученной плазмиды для всех выделенных бактериальных MutL [31]. Очевидно, что различное влияние ионов Zn2+ на структуру белков MutL обусловливает и различное влияние этих ионов на эндонуклеазную функцию белков.
ЗАКЛЮЧЕНИЕ
Несмотря на значительное количество информации о функционировании MMR, которая имеется на сегодняшний день, многие детали механизма этого процесса только предстоит узнать. На основе проведенного нами анализа данных литературы представляется возможным наметить направления дальнейших исследований этой системы репарации.
Так, долгое время полагали, что MutL, главным образом, координирует белок–белковые взаимодействия во время узнавания повреждения и удаления фрагмента одной из цепей ДНК. Открытие эндонуклеазной функции MutLα, а также MutL из бактерий, не содержащих MutH, привело к пониманию ключевой роли MutL в инициации репарации. Механизм этого процесса, как и влияние MutS и факторов процессивности на функционирование MutL, нуждается в дальнейшем исследовании.
В аспектах, касающихся взаимодействия MutL с ДНК, также существует множество невыясненных моментов. Например, не предложен единый механизм, который мог бы объяснить способ распознавания “дочерней” цепи ДНК в тех организмах, где отсутствует эндонуклеаза MutH. Также следует детально изучить процесс внесения одноцепочечного разрыва белком MutL в ходе MMR в бактериальных системах.
Связывание и гидролиз ДНК белком MutL зависят от многих факторов, которые нуждаются в систематизации и уточнении. Так, эндонуклеазная активность MutL модулируется ионами двухвалентных металлов. Тем не менее определенной корреляции между природой металла и гидролитической способностью разных белков MutL не наблюдается. Кроме того, считается, что β-“зажим” способен повышать эндонуклеазную активность MutL, обеспечивая правильную локализацию ДНК в каталитическом центре белка или связывание эндонуклеазного домена MutL с ДНК. Однако есть группа организмов, для которых не выявлено влияние β-“зажима” на эндонуклеазную функцию. Остается неясным, как в таких организмах происходит процесс дискриминации “дочерней” цепи. В дальнейших исследованиях должна быть уточнена роль ATP в функционировании MutL, так как присутствие ATP в некоторых организмах активирует эндонуклеазную функцию MutL, а в других – ингибирует.
В заключение нужно отметить, что система репарации “мисматчей” не только устраняет повреждения в ДНК, но и взаимодействует с другими белковыми комплексами в клетке. Исследование функционирования MutL в разных организмах является ключом к детальному пониманию того, как работает MMR и какие клеточные процессы связаны с этой системой. Эти данные позволят лучше понять природу заболеваний, связанных с накоплением мутаций в ДНК, что может быть использовано в биомедицинских целях.
Список литературы
Kunkel T.A., Erie D.A. // Annu. Rev. Biochem. 2005. V. 74. P. 681–710. https://doi.org/10.1146/annurev.biochem.74.082803. 133243
Modrich P., Lahue R. // Annu. Rev. Biochem. 1996. V. 65. P. 101–133. https://doi.org/10.1146/annurev.bi.65.070196.000533
Jiricny J. // Cold Spring Harb. Perspect. Biol. 2013. V. 5. P. a012 633. https://doi.org/10.1101/cshperspect.a012633
Arana M.E., Kunkel T.A. // Semin. Cancer Biol. 2010. V. 20. P. 304–311. https://doi.org/10.1016/j.semcancer.2010.10.003
Guarné A. // Progress in Molecular Biology and Translational Science. 2012. P. 41–70. https://doi.org/10.1016/B978-0-12-387665-2.00003-1
Peltomäki P. // J. Clin. Oncol. 2003. V. 21. P. 1174–1179. https://doi.org/10.1200/JCO.2003.04.060
Liu J., Hanne J., Britton B.M., Bennett J., Kim D., Lee J.-B., Fishel R. // Nature. 2016. V. 539. P. 583–587. https://doi.org/10.1038/nature20562
Kadyrova L.Y., Kadyrov F.A. // DNA Repair (Amst). 2016. V. 38. P. 42–49. https://doi.org/10.1016/j.dnarep.2015.11.023
Liu J., Lee J.-B., Fishel R. // J. Mol. Biol. 2018. V. 430. P. 4456–4468. https://doi.org/10.1016/j.jmb.2018.05.039
Modrich P. // J. Biol. Chem. 1989. V. 264. P. 6597–6600.
Grilley M., Griffith J., Modrich P. // J. Biol. Chem. 1993. V. 268. P. 11830–11837.
Lamers M.H., Perrakis A., Enzlin J.H., Winterwerp H.H.K., Wind N. de, Sixma T.K. // Nature. 2000. V. 407. P. 711–717. https://doi.org/10.1038/35037523
Lahue R., Au K., Modrich P. // Science. 1989. V. 245. P. 160–164. https://doi.org/10.1126/science.2665076
Acharya S., Foster P.L., Brooks P., Fishel R. // Mol. Cell. 2003. V. 12. P. 233–246. https://doi.org/10.1016/S1097-2765(03)00219-3
Obmolova G., Ban C., Hsieh P., Yang W. // Nature. 2000. V. 407. P. 703–710. https://doi.org/10.1038/35037509
Bjornson K.P., Modrich P. // J. Biol. Chem. 2003. V. 278. P. 18557–18562. https://doi.org/10.1074/jbc.M301101200
Antony E., Hingorani M.M. // Biochemistry. 2004. V. 43. P. 13115–13128. https://doi.org/10.1021/bi049010t
Hingorani M.M. // DNA Repair (Amst). 2016. V. 38. P. 24–31. https://doi.org/10.1016/j.dnarep.2015.11.017
Groothuizen F.S., Winkler I., Cristóvão M., Fish A., Winterwerp H.H., Reumer A., Marx A.D., Hermans N., Nicholls R.A., Murshudov G.N., Lebbink J.H., Friedhoff P., Sixma T.K. // Elife. 2015. V. 4. e06 744. https://doi.org/10.7554/eLife.06744
Dutta R., Inouye M. // Trends Biochem. Sci. 2000. V. 25. P. 24–28. https://doi.org/10.1016/S0968-0004(99)01503-0
Sancar A., Hearst J. // Science. 1993. V. 259. P. 1415–1420. https://doi.org/10.1126/science.8451638
Au K.G., Welsh K., Modrich P. // J. Biol. Chem. 1992. V. 267. P. 12 142–12 148.
Welsh K.M., Lu A.L., Clark S., Modrich P. // J. Biol. Chem. 1987. V. 262. P. 15 624–15 629.
Hall M.C. // EMBO J. 1998. V. 17. P. 1535–1541. https://doi.org/10.1093/emboj/17.5.1535
Lopez de Saro F.J., O’Donnell M. // Proc. Natl. Acad. Sci. 2001. V. 98. P. 8376–8380. https://doi.org/10.1073/pnas.121009498
Correa E.M.E., Martina M.A., Tullio L. De, Argaraña C.E., Barra J.L. // DNA Repair (Amst). 2011. V. 10. P. 1106–1113. https://doi.org/10.1016/j.dnarep.2011.08.007
Pillon M.C., Lorenowicz J.J., Uckelmann M., Klocko A.D., Mitchell R.R., Chung Y.S., Modrich P., Walker G.C., Simmons L.A., Friedhoff P., Guarné A. // Mol. Cell. 2010. V. 39. P. 145–151. https://doi.org/10.1016/j.molcel.2010.06.027
Duppatla V., Bodda C., Urbanke C., Friedhoff P., Rao D.N. // Biochem. J. 2009. V. 423. P. 265–277. https://doi.org/10.1042/BJ20090626
Iino H., Kim K., Shimada A., Masui R., Kuramitsu S., Fukui K. // Biosci. Rep. 2011. V. 31. P. 309–322. https://doi.org/10.1007/s00792-015-0745-2
Fukui K., Nishida M., Nakagawa N., Masui R., Kuramitsu S. // J. Biol. Chem. 2008. V. 283. P. 12 136–12 145. https://doi.org/10.1074/jbc.M800110200
Монахова М.В., Пенкина А.И., Павлова А.В., Лящук А.М., Кучеренко В.В., Алексеевский А.В., Лунин В.Г., Фридхофф П., Клуг Г., Орецкая Т.С., Кубарева Е.А. // Биохимия. 2018. V. 83. P. 281–293. https://doi.org/10.1134/S0006297918030082
Bende S.M., Grafström R.H. // Nucleic Acids Res. 1991. V. 19. P. 1549–1555. https://doi.org/10.1093/nar/19.7.1549
Drotschmann K., Hall M.C., Shcherbakova P.V., Wang H., Erie D.A., Brownewell F.R., Kool E.T., Kunkel T.A. // Biol. Chem. 2002. V. 383. P. 969–975. https://doi.org/10.1515/BC.2002.103
Drummond J., Li G., Longley M., Modrich P. // Science. 1995. V. 268. P. 1909–1912. https://doi.org/10.1126/science.7604264
Li G.M., Modrich P. // Proc. Natl. Acad. Sci. 1995. V. 92. P. 1950–1954. https://doi.org/10.1073/pnas.92.6.1950
Strand M., Prolla T.A., Liskay R.M., Petes T.D. // Nature. 1993. V. 365. P. 274–276. https://doi.org/10.1038/365274a0
Harfe B.D., Jinks-Robertson S. // Annu. Rev. Genet. 2000. V. 34. P. 359–399. https://doi.org/10.1146/annurev.genet.34.1.359
McCulloch S.D., Gu L., Li G.-M. // J. Biol. Chem. 2003. V. 278. P. 3891–3896. https://doi.org/10.1074/jbc.M210687200
Bowen N., Smith C.E., Srivatsan A., Willcox S., Griffith J.D., Kolodner R.D. // Proc. Natl. Acad. Sci. 2013. V. 110. P. 18472–18477. https://doi.org/10.1073/pnas.1318971110
Szankasi P., Smith G. // Science. 1995. V. 267. P. 1166–1169. https://doi.org/10.1126/science.7855597
Tishkoff D.X., Boerger A.L., Bertrand P., Filosi N., Gaida G.M., Kane M.F., Kolodner R.D. // Proc. Natl. Acad. Sci. 1997. V. 94. P. 7487–7492. https://doi.org/10.1073/pnas.94.14.7487
Genschel J., Bazemore L.R., Modrich P. // J. Biol. Chem. 2002. V. 277. P. 13 302–13 311. https://doi.org/10.1074/jbc.M111854200
Umar A., Buermeyer A.B., Simon J.A., Thomas D.C., Clark A.B., Liskay R.M., Kunkel T.A. // Cell. 1996. V. 87. P. 65–73. https://doi.org/10.1016/S0092-8674(00)81323-9
Johnson R.E., Kovvali G.K., Guzder S.N., Amin N.S., Holm C., Habraken Y., Sung P., Prakash L., Prakash S. // J. Biol. Chem. 1996. V. 271. P. 27 987–27 990. https://doi.org/10.1074/jbc.271.45.27987
Almawi A.W., Scotland M.K., Randall J.R., Liu L., Martin H.K., Sacre L., Shen Y., Pillon M.C., Simmons L.A., Sutton M.D., Guarné A. // Nucleic Acids Res. 2019. V. 47. P. 4831–4842. https://doi.org/10.1093/nar/gkz115
Genschel J., Modrich P. // Mol. Cell. 2003. V. 12. P. 1077–1086. https://doi.org/10.1016/S1097-2765(03)00428-3
Dzantiev L., Constantin N., Genschel J., Iyer R.R., Burgers P.M., Modrich P. // Mol. Cell. 2004. V. 15. P. 31–41. https://doi.org/10.1016/j.molcel.2004.06.016
Lin Y.-L., Shivji M.K.K., Chen C., Kolodner R., Wood R.D., Dutta A. // J. Biol. Chem. 1998. V. 273. P. 1453–1461. https://doi.org/10.1074/jbc.273.3.1453
Qiu H., Wang Y. // J. Proteome Res. 2009. V. 8. P. 1983–1991. https://doi.org/10.1021/pr8009319
Zhang Y., Yuan F., Presnell S.R., Tian K., Gao Y., Tomkinson A.E., Gu L., Li G.-M. // Cell. 2005. V. 122. P. 693–705. https://doi.org/10.1016/j.cell.2005.06.027
Longley M.J., Pierce A.J., Modrich P. // J. Biol. Chem. 1997. V. 272. P. 10 917–10 921. https://doi.org/10.1074/jbc.272.16.10917
Constantin N., Dzantiev L., Kadyrov F.A., Modrich P. // J. Biol. Chem. 2005. V. 280. P. 39 752–39 761. https://doi.org/10.1074/jbc.M509701200
Kadyrov F.A., Genschel J., Fang Y., Penland E., Edelmann W., Modrich P. // Proc. Natl. Acad. Sci. 2009. V. 106. P. 8495–8500. https://doi.org/10.1073/pnas.0903654106
Flores-Rozas H., Kolodner R.D. // Proc. Natl. Acad. Sci. 1998. V. 95. P. 12 404–12 409. https://doi.org/10.1073/pnas.95.21.12404
Nishant K.T., Plys A.J., Alani E. // Genetics. 2008. V. 179. P. 747–755. https://doi.org/10.1534/genetics.108.086645
Cannavo E., Marra G., Sabates-Bellver J., Menigatti M., Lipkin S.M., Fischer F., Cejka P., Jiricny J. // Cancer Res. 2005. V. 65. P. 10 759–10 766. https://doi.org/10.1158/0008-5472.CAN-05-2528
Ghodgaonkar M.M., Lazzaro F., Olivera-Pimentel M., Artola-Borán M., Cejka P., Reijns M.A., Jackson A.P., Plevani P., Muzi-Falconi M., Jiricny J. // Mol. Cell. 2013. V. 50. P. 323–332. https://doi.org/10.1016/j.molcel.2013.03.019
Lujan S.A., Williams J.S., Clausen A.R., Clark A.B., Kunkel T.A. // Mol. Cell. 2013. V. 50. P. 437–443. https://doi.org/10.1016/j.molcel.2013.03.017
Liu Y., Kadyrov F.A., Modrich P. // DNA Repair (Amst). 2011. V. 10. P. 1145–1153. https://doi.org/10.1016/j.dnarep.2011.08.012
Kadyrova L.Y., Blanko E.R., Kadyrov F.A. // Proc. Natl. Acad. Sci. 2011. V. 108. P. 2753–2758. https://doi.org/10.1073/pnas.1015914108
Li F., Mao G., Tong D., Huang J., Gu L., Yang W., Li G.-M. // Cell. 2013. V. 153. P. 590–600. https://doi.org/10.1016/j.cell.2013.03.025
Liu D., Keijzers G., Rasmussen L.J. // Mutat. Res. 2017. V. 773. P. 174–187. https://doi.org/10.1016/j.mrrev.2017.07.001
Pluciennik A., Dzantiev L., Iyer R.R., Constantin N., Kadyrov F.A., Modrich P. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. P. 16 066–16 071. https://doi.org/10.1073/pnas.1010662107
Moldovan G.-L., Pfander B., Jentsch S. // Cell. 2007. V. 129. P. 665–679. https://doi.org/10.1016/j.cell.2007.05.003
Mossi R., Jónsson Z.O., Allen B.L., Hardin S.H., Hübscher U. // J. Biol. Chem. 1997. https://doi.org/10.1074/jbc.272.3.1769
Clark A.B., Valle F., Drotschmann K., Gary R.K., Kunkel T.A. // J. Biol. Chem. 2000. V. 275. P. 36 498–36 501. https://doi.org/10.1074/jbc.C000513200
Genschel J., Kadyrova L.Y., Iyer R.R., Dahal B.K., Kadyrov F.A., Modrich P. // Proc. Natl. Acad. Sci. 2017. V. 114. P. 4930–4935. https://doi.org/10.1073/pnas.1702561114
Gradia S., Acharya S., Fishel R. // Cell. 1997. V. 91. P. 995–1005. https://doi.org/10.1016/S0092-8674(00)80490-0
Pillon M.C., Babu V.M.P., Randall J.R., Cai J., Simmons L.A., Sutton M.D., Guarné A. // Nucleic Acids Res. 2015. V. 43. P. 10 746–10 759. https://doi.org/10.1093/nar/gkv918
Friedhoff P., Li P., Gotthardt J. // DNA Repair (Amst). 2016. V. 38. P. 50–57. https://doi.org/10.1016/j.dnarep.2015.11.013
Pillon M.C., Miller J.H., Guarné A. // DNA Repair (Amst). 2011. V. 10. P. 87–93. https://doi.org/10.1016/j.dnarep.2010.10.003
Liu L., Ortiz Castro M.C., Rodríguez González J., Pillon M.C., Guarné A. // DNA Repair (Amst). 2019. V. 73. P. 1–6. https://doi.org/10.1016/j.dnarep.2018.10.003
Fukui K., Baba S., Kumasaka T., Yano T. // J. Biol. Chem. 2016. V. 291. P. 16 990–17 000. https://doi.org/10.1074/jbc.M116.739664
Shimada A., Kawasoe Y., Hata Y., Takahashi T.S., Masui R., Kuramitsu S., Fukui K. // FEBS J. 2013. V. 280. P. 3467–3479. https://doi.org/10.1111/febs.12344
Kadyrov F.A., Dzantiev L., Constantin N., Modrich P. // Cell. 2006. V. 126. P. 297–308. https://doi.org/10.1016/j.cell.2006.05.039
Lyer R.R., Pluciennik A., Burdett V., Modrich P.L. // Chem. Rev. 2006. V. 106. P. 302–323. https://doi.org/10.1021/cr0404794
Manhart C.M., Alani E. // DNA Repair (Amst.). 2016. V. 38. P. 84–93. https://doi.org/10.1016/j.dnarep.2015.11.024
Zakharyevich K., Tang S., Ma Y., Hunter N. // Cell. 2012. V. 149. P. 334–347. https://doi.org/10.1016/j.cell.2012.03.023
Ranjha L., Anand R., Cejka P. // J. Biol. Chem. 2014. V. 289. P. 5674–5686. https://doi.org/10.1074/jbc.M113.533810
Duroc Y., Kumar R., Ranjha L., Adam C., Guérois R., Md Muntaz K., Marsolier-Kergoat M.-C., Dingli F., Laureau R., Loew D., Llorente B., Charbonnier J.-B., Cejka P., Borde V. // Elife. 2017. V. 6. e21 900. https://doi.org/10.7554/eLife.21900
Gomes-Pereira M. // Hum. Mol. Genet. 2004. V. 13. P. 1815–1825. https://doi.org/10.1093/hmg/ddh186
Pluciennik A., Burdett V., Baitinger C., Iyer R.R., Shi K., Modrich P. // Proc. Natl. Acad. Sci. 2013. V. 110. P. 12 277–12 282. https://doi.org/10.1073/pnas.1311325110
Pinto R.M., Dragileva E., Kirby A., Lloret A., Lopez E., Claire J. St., Panigrahi G.B., Hou C., Holloway K., Gillis T., Guide J.R., Cohen P.E., Li G.-M., Pearson C.E., Daly M.J., Wheeler V.C. // PLoS Genet. / ed. Zeitlin S.O. 2013. V. 9. P. e1 003 930. https://doi.org/10.1371/journal.pgen.1003930
Halabi A., Fuselier K.T.B., Grabczyk E. // Nucleic Acids Res. 2018. V. 46. P. 4022–4032. https://doi.org/10.1093/nar/gky143
Zhao X., Zhang Y., Wilkins K., Edelmann W., Usdin K. // PLos Genet. 2018. V. 14. P. e1 007 719. https://doi.org/10.1371/journal.pgen.1007719
Lee J.-M., Correia K., Loupe J., Kim K.-H., Barker D., Hong E.P., Chao M.J., Long J.D., Lucente D., Vonsattel J.P.G., Pinto R.M., Abu Elneel K., Ramos E.M., Mysore J.S., Gillis T., Wheeler V.C., MacDonald M.E., Gusella J.F., McAllister B., Massey T., Medway C., Stone T.C., Hall L., Jones L., Holmans P., Kwak S., Ehrhardt A.G., Sampaio C., Ciosi M., Maxwell A., Chatzi A., Monckton D.G., Orth M., Landwehrmeyer B.G., Paulsen J.S., Dorsey E.R., Shoulson I., Myers R.H. // Cell. 2019. V. 178. P. 887–900. e14. https://doi.org/10.1016/j.cell.2019.06.036
Ciosi M., Maxwell A., Cumming S.A., Hensman Moss D.J., Alshammari A.M., Flower M.D., Durr A., Leavitt B.R., Roos R.A.C., Holmans P., Jones L., Langbehn D.R., Kwak S., Tabrizi S.J., Monckton D.G. // EBioMedicine. 2019. V. 48. P. 568–580. https://doi.org/10.1016/j.ebiom.2019.09.020
Kadyrova L.Y., Gujar V., Burdett V., Modrich P.L., Kadyrov F.A. // Proc. Natl. Acad. Sci. 2020. V. 117. P. 3535–3542. https://doi.org/10.1073/pnas.1914718117
Guarné A., Charbonnier J.-B. // Prog. Biophys. Mol. Biol. 2015. V. 117. P. 149–156. https://doi.org/10.1016/j.pbiomolbio.2015.02.002
Fukui K., Iino H., Baba S., Kumasaka T., Kuramitsu S., Yano T. // Biochim. Biophys. Acta – Proteins Proteomics. 2017. V. 1865. P. 1178–1187. https://doi.org/10.1016/j.bbapap.2017.06.024
Ban C., Yang W. // Cell. 1998. V. 95. P. 541–552. https://doi.org/10.1016/S0092-8674(00)81621-9
Guarné A. // EMBO J. 2001. V. 20. P. 5521–5531. https://doi.org/10.1093/emboj/20.19.5521
Ban C., Junop M., Yang W. // Cell. 1999. V. 97. P. 85–97. https://doi.org/10.1016/S0092-8674(00)80717-5
Arana M.E., Holmes S.F., Fortune J.M., Moon A.F., Pedersen L.C., Kunkel T.A. // DNA Repair (Amst.). 2010. V. 9. P. 448–457. https://doi.org/10.1016/j.dnarep.2010.01.010
Banasik M., Sachadyn P. // Mutat. Res. Mol. Mech. Mutagen. 2014. V. 769. P. 69–79. https://doi.org/10.1016/j.mrfmmm.2014.07.006
Räschle M., Dufner P., Marra G., Jiricny J. // J. Biol. Chem. 2002. V. 277. P. 21 810–21 820. https://doi.org/10.1074/jbc.M108787200
Yang W. // Mutat. Res. Repair. 2000. V. 460. P. 245–256. https://doi.org/10.1016/S0921-8777(00)00030-6
Sacho E.J., Kadyrov F.A., Modrich P., Kunkel T.A., Erie D.A. // Mol. Cell. 2008. V. 29. P. 112–121. https://doi.org/10.1016/j.molcel.2007.10.030
Schopf F.H., Biebl M.M., Buchner J. // Nat. Rev. Mol. Cell Biol. 2017. V. 18. P. 345–360. https://doi.org/10.1038/nrm.2017.20
Polosina Y.Y., Cupples C.G. // BioEssays. 2010. V. 32. P. 51–59. https://doi.org/10.1002/bies.200900089
Mauris J., Evans T.C. // PLoS One / ed. Marinus M.G. 2009. V. 4. P. e7175. https://doi.org/10.1371/journal.pone.0007175
Guarné A., Ramon-Maiques S., Wolff E.M., Ghirlando R., Hu X., Miller J.H., Yang W. // EMBO J. 2004. V. 23. P. 4134–4145. https://doi.org/10.1038/sj.emboj.7600412
Schorzman A.N., Perera L., Cutalo-Patterson J.M., Pedersen L.C., Pedersen L.G., Kunkel T.A., Tomer K.B. // DNA Repair (Amst). 2011. V. 10. P. 454–465. https://doi.org/10.1016/j.dnarep.2011.01.010
Hall M.C., Wang H., Erie D.A., Kunkel T.A. // J. Mol. Biol. 2001. V. 312. P. 637–647. https://doi.org/10.1006/jmbi.2001.4958
Robertson A., Pattishall S.R., Matson S.W. // J. Biol. Chem. 2006. V. 281. P. 8399–8408. https://doi.org/10.1074/jbc.M509184200
Junop M. // DNA Repair (Amst.). 2003. V. 2. P. 387–405. https://doi.org/10.1016/S1568-7864(02)00245-8
Monakhova M., Ryazanova A., Kunetsky V., Li P., Shilkin E., Kisil O., Rao D.N., Oretskaya T., Friedhoff P., Kubareva E. // Biochimie. 2020. V. 171–172. P. 43–54. https://doi.org/10.1016/j.biochi.2020.02.004
Niedziela-Majka A., Maluf N.K., Antony E., Lohman T.M. // Biochemistry. 2011. V. 50. P. 7868–7880. https://doi.org/10.1021/bi200753b
Монахова М.В., Кубарева Е.А., Романова Е.А., Семкина А.С., Набережнов Д.С., Рао Д.Н., Зацепин Т.С., Орецкая Т.С. // Биоорган. химия. 2019. Т. 45. С. 303–314. [Monakhova M.V., Kubareva E.A., Romanova E.A., Semkina A.S., Naberezhnov D.S., Rao D.N., Zatsepin T.S., Oretskaya T.S. // Russ. J. Bioorg. Chem. V. 45. P. 144–154.] https://doi.org/10.1134/S1068162019020079
Bjornson K.P., Allen D.J., Modrich P. // Biochemistry. 2000. V. 39. P. 3176–3183. https://doi.org/10.1021/bi992286u
Gueneau E., Dherin C., Legrand P., Tellier-Lebegue C., Gilquin B., Bonnesoeur P., Londino F., Quemener C., Du M.-H. Le, Márquez J.A., Moutiez M., Gondry M., Boiteux S., Charbonnier J.-B. // Nat. Struct. Mol. Biol. 2013. V. 20. P. 461–468. https://doi.org/10.1038/nsmb.2511
Kadyrov F.A., Holmes S.F., Arana M.E., Lukianova O.A., O’Donnell M., Kunkel T.A., Modrich P. // J. Biol. Chem. 2007. V. 282. P. 37 181–37 190. https://doi.org/10.1074/jbc.M707617200
Kosinski J., Plotz G., Guarné A., Bujnicki J.M., Friedhoff P. // J. Mol. Biol. 2008. V. 382. P. 610–627. https://doi.org/10.1016/j.jmb.2008.06.056
Erdeniz N., Nguyen M., Deschênes S.M., Liskay R.M. // DNA Repair (Amst.). 2007. V. 6. P. 1463–1470. https://doi.org/10.1016/j.dnarep.2007.04.013
Yang W. // Nat. Struct. Mol. Biol. 2008. V. 15. P. 1228–1231. https://doi.org/10.1038/nsmb.1502
Stapleton B., Walker L.R., Logan T.M. // Biochemistry. 2013. V. 52. P. 1927–1938. https://doi.org/10.1021/bi301608p
Namadurai S., Jain D., Kulkarni D.S., Tabib C.R., Friedhoff P., Rao D.N., Nair D.T. // PLoS One. 2010. V. 5. e13 726. https://doi.org/10.1371/journal.pone.0013726
Fukui K., Baba S., Kumasaka T., Yano T. // FEBS Lett. 2018. V. 592. P. 1611–1619. https://doi.org/10.1002/1873-3468.13050
Groothuizen F.S., Sixma T.K. // DNA Repair (Amst.). 2016. V. 38. P. 14–23. https://doi.org/10.1016/j.dnarep.2015.11.012
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия