

Биоорганическая химия, 2021, T. 47, № 1, стр. 162-172
Противоопухолевая активность монасникотиновой кислоты, выделенной из гриба Aspergillus cavernicola
Г. К. Рысцов 1, 2, Т. В. Антипова 1, К. В. Зайцев 3, М. Ю. Земскова 1, 2, *
1 Институт биохимии и физиологии микроорганизмов им. Г.К. Скрябина РАН − обособленное подразделение ФГБУН “Федеральный исследовательский центр “Пущинский научный центр биологических исследований” РАН
142290 Московская обл., Пущино, пр. Науки, 5, Россия
2 Московский государственный областной университет
141014 Московская обл., Мытищи, ул. Веры Волошиной, 24, Россия
3 Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: mzemskova@ibpm.pushchino.ru
Поступила в редакцию 30.06.2020
После доработки 11.07.2020
Принята к публикации 13.07.2020
Аннотация
Обнаружено, что монасникотиновая кислота (MНК), выделенная из гриба Aspergillus cavernicola ВKM F-906, снижает пролиферацию и миграцию клеток рака простаты человека LnCaP и ингибирует сигнальные пути AKT–mTORC1 и FAK–Src. Однако MНК также повышает уровень HSP60 и фосфорилирование c-Jun – факторов, стимулирующих жизнедеятельность раковых клеток. МНК-индуцированные клеточные ответы появляются только через 48 ч, что подразумевает участие либо продуктов метаболизма МНК, либо таутомеров, возникающих при протонировании МНК в результате роста клеточных культур и утилизации соединения клетками человека. Эти результаты позволяют предположить, что MНК является перспективным соединением для получения различных производных, которые будут ингибировать онкогенные сигнальные пути без стимуляции факторов, обеспечивающих выживание опухолевых клеток.
Азафилоновые соединения, продуцируемые грибами рода Monascus, традиционно используются для получения пищевых добавок и натуральных красителей, которые оказывают положительное воздействие на здоровье человека [1]. По спектрам поглощения эти соединения традиционно классифицируются как красные, оранжевые и желтые пигменты. В настоящее время у Monascus было идентифицировано около сотни различных азафилоновых пигментов [2]. Несколько подобных пигментов были обнаружены у грибов Penicillium, Talaromyces и др. [3]. Эти соединения обладают антиоксидантными, гиполипидемическими, противовоспалительными, антимикробными и противоопухолевыми свойствами [2, 4]. Исследование монапурпиридина А показало, что это соединение вызывает апоптоз клеток рака молочной железы MCF-7 [5].
Штамм Aspergillus cavernicola VKM F-906 продуцирует большое количество красных пигментов, подобных монаскорубрамину [6]. Недавно из этого штамма нами был выделен новый метаболит [7]. Структура соединения была определена с помощью масс-спектрометрии, 1D и 2D ЯМР-спектроскопии и рентгеноструктурного анализа; соединение было описано как 4-[(1E)-2-ацетил-5-метил-4-оксогепт-1-ен-1-ил]-6-[(1Z)-проп-1-ен-1-ил]пиридин-3-карбоновая кислота (рис. 1). Из-за сходства его молекулярной структуры с монасникотинатами [8] и монапурпиридином А [5], ранее обнаруженными у грибов рода Monascus, соединение было названо монасникотиновой кислотой (МНК). MНК представляет особый интерес для изучения ее биологической активности как нового соединения из этого класса природных продуктов.

В статье мы приводим данные об исследовании противоопухолевой активности и некоторых аспектов молекулярного механизма действия МНК на клетки рака предстательной железы человека. С помощью современной технологии xCELLigence, позволяющей проводить анализ клеток в реальном времени, мы определяли влияние МНК на пролиферацию и миграцию клеток рака простаты LnCaP. Для идентификации клеточных мишеней, на которые действует МНК, был применен анализ фосфорилирования ключевых киназ, контролирующих гомеостаз опухолевых клеток. Используя преимущества этих подходов, нам удалось раскрыть потенциальный механизм ингибирования пролиферации и миграции опухолевых клеток при воздействии МНК вследствие снижения активностей сигнальных путей AKT–mTORC1 и FAK–Src.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
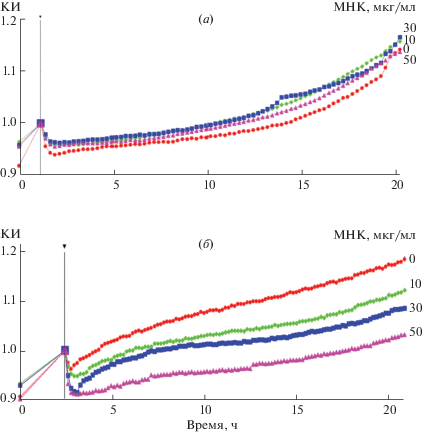
Биосенсорная технология xCELLigence (RTCA) разработана для непрерывного мониторинга адгезионных свойств клеток in vitro в режиме реального времени. В данной технологии используются специально разработанные планшеты (Е- и CIM-планшеты), дно лунок планшетов покрыто золотыми электродами, к которым прикрепляются клетки-мишени. Вследствие размножения клеток происходят изменения электрического сопротивления, которые преобразуются в значения клеточного индекса (КИ). Показатели КИ увеличиваются, когда клетки прикрепляются к электродам и размножаются. Следовательно, любое воздействие, которое вызывает изменения в количестве клеток (их пролиферация или гибель) или в их движении (миграция или инвазия), может быть исследовано посредством измерений КИ с применением Е- и CIM-планшетов соответственно. Используя преимущества технологии xCELLigence, мы определяли способность МНК ингибировать размножение клеток рака предстательной железы человека LnCaP, выявляли, когда это происходит и какова природа клеточного ответа. Клетки высевали в лунки E-планшета и через 24 ч, когда клетки прикреплялись к электродам и начинали размножаться, добавляли либо контрольное соединение (DMSO), либо МНК в различных концентрациях и измеряли значения КИ в течение 86 ч. Как видно на рис. 2а, существенных изменений в адгезии и росте клеток не наблюдалось в течение первых часов обработки (т.е. КИ клеток, обработанных MНК, был идентичен КИ контрольных клеток в течение 24 ч после добавления соединения). Затем, однако, наблюдалось статистически значимое снижение значений КИ, которое проявлялось примерно через 48 ч после добавления MНК к клеткам и зависело от количества вносимого вещества и времени инкубации (рис. 2а и 2б). Полумаксимальная ингибирующая концентрация MНК (IC50) для клеток LnCaP через 48 ч обработки была определена как 34.71 мкг/мл (105 мкМ).
Рис. 2.
Цитостатическое действие МНК на клетки рака простаты человека LnCaP в зависимости от концентрации МНК и времени культивирования. Эксперимент проведен с использованием технологии xCELLigence: (а) – инкубация с МНК, добавленной к клеткам перед началом измерения клеточного индекса (КИ) (опыт 1); (б) – гистограмма значений КИ в опыте 1; (в) – инкубация с МНК, внесенной к клеткам, предварительно обработанным МНК в течение 96 ч (опыт 2); (г) – гистограмма значений КИ в опыте 2. Черными стрелками на графиках КИ отмечено время добавления МНК, белые стрелки указывают на различия в кривых КИ в опытах 1 и 2. Временные точки на гистограммах (б) и (г) показаны в соответствии с временным периодом, указанным для кривых КИ (а) и (в). * Статистически значимые различия значений КИ (p < 0.05) клеток, обработанных МНК, по сравнению с контрольными клетками, обработанными DMSO (контроль – 0). Показаны результаты одного репрезентативного измерения из трех разных экспериментов.

Поскольку MНК-индуцированный ответ не был очевиден сразу, мы изучили пролиферацию клеток в условиях более длительной обработки соединением. Клетки LnCaP предварительно обрабатывали МНК в течение 96 ч при дозе IC50 и затем анализировали с помощью технологии xCELLigence в течение последующих 86 ч. Как показано на рис. 2в, у предварительно обработанных клеток наблюдался замедленный рост по сравнению с клетками без предварительной обработки (рис. 2а), хотя в обоих экспериментах в лунки Е-планшетов добавляли равное количество клеток и инкубировали в течение первых 24 ч в среде, не содержавшей МНК. Эти данные свидетельствуют о том, что индуцированное МНК ингибирование пролиферации клеток сохраняется даже в отсутствие препарата. Добавление МНК к предварительно обработанным клеткам вызвало снижение их КИ по сравнению с КИ как контрольных клеток, так и клеток без предварительной обработки соединением (рис. 2б и 2г). Это статистически значимое уменьшение наблюдалось уже через 24 ч после внесения препарата, с последующим понижением значений КИ с увеличением времени инкубации и концентрации соединения (рис. 2в и 2г). После 48 ч воздействия МНК для предварительно обработанных клеток значение IC50 составило 19 мкг/мл (57 мкМ), что примерно в 2 раза меньше, чем IC50 для клеток LnCaP без предварительной инкубации с соединением.
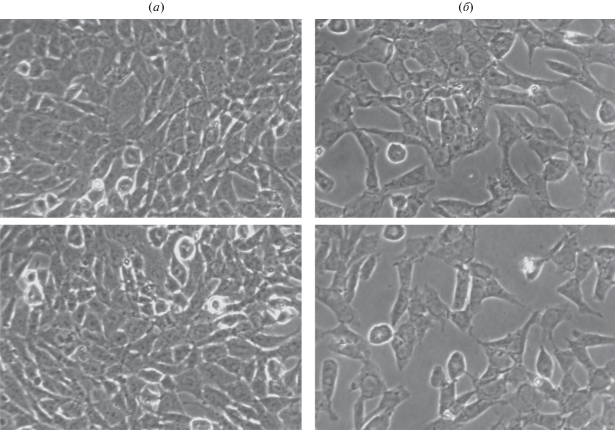
Анализ кривых КИ показывает, что МНК не вызывает гибель клеток, поскольку не наблюдалось значительного уменьшения значений КИ, вызванного падением сопротивления вследствие открепления гибнущих клеток от электродов-датчиков. Этот вывод был также подтвержден микроскопическими наблюдениями (рис. 3). В клетках, обработанных МНК, наблюдалось снижение количества прикрепленных клеток, однако изменений размера, формы, уменьшения клеточной адгезии и плавающих мертвых клеток не обнаружено (рис. 3б). Следовательно, MНК ингибирует пролиферацию клеток рака предстательной железы линии LnCaP и, таким образом, обладает цитостатическими свойствами без ярко выраженного цитотоксического эффекта.
Рис. 3.
Микрофотографии двух случайно выбранных полей клеток LnCaP, инкубированных либо с DMSO, либо с МНК (35 мкг/л) в течение 96 ч: (а) – контроль (DMSO); (б) – МНК. Увеличение 40×.
Одной из характеристик опухолевых клеток является их способность к миграции, приводящая в условиях организма к образованию метастаз в удаленных от опухоли органах. Мы оценили влияние MНК на миграцию клеток LnCaP в направлении градиента ростовых факторов, используя CIM-планшеты и технологию xCELLigence, которая позволяет измерять количество клеток, мигрирующих через мембрану. Как показано на рис. 4а, МНК не подавляла миграцию клеток в направлении градиента сыворотки в течение 20 ч после добавления соединения в различных концентрациях. Такое отсутствие активности MНК согласуется с результатами, представленными на рис. 2а, когда индуцированное MНК ингибирование пролиферации клеток также не наблюдалось в течение первых 24 ч воздействия препарата. Для того чтобы определить, приведет ли увеличение времени обработки к снижению миграции, клетки LnCaP инкубировали в присутствии МНК (35 мкг/мл) в течение 96 ч и затем помещали в CIM-планшеты для дальнейшего анализа миграции в присутствии различных концентраций соединения. Как показано на рис. 4б, в течение 3–20 ч МНК индуцировала значительное ингибирование миграции клеток LnCaP по сравнению с контролем. Было установлено, что через 15 ч обработки IC50 МНК составляет 7.8 мкг/мл (23.8 мкМ).
Рис. 4.
Влияние MНК на миграцию клеток LnCaP при различных концентрациях МНК относительно клеток, обработанных DMSO (контроль – 0). Данные получены с использованием технологии xCELLigence. Представлены профили миграции клеток LnCaP через мембрану по направлению к градиенту сыворотки в условиях, когда DMSO или соединение добавляли непосредственно перед началом измерения КИ (а) и при добавлении либо DMSO (контроль), либо МНК к клеткам, предварительно обработанным в течение 96 ч DMSO или МНК в дозе 35 мкг/мл соответственно (б).
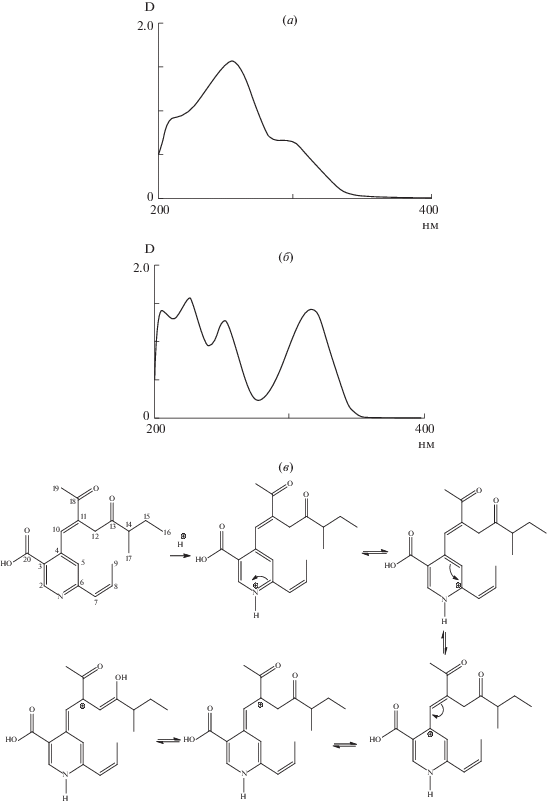
В совокупности наши данные указывают на то, что МНК обладает умеренной ингибирующей активностью в отношении пролиферации и миграции клеток LnCaP, и для проявления этой активности требуется относительно длительное время. Это наблюдение предполагает, что биологическая активность данного соединения проявляется либо в том случае, когда критические концентрации достигаются после накопления МНК опухолевыми клетками, либо соединение сначала должно метаболизироваться клетками, и только продукты метаболизма MНК обладают ингибирующим эффектом. Существует еще одно возможное объяснение замедленного клеточного ответа на введение соединения. МНК содержит карбонильные группы и подвижные протоны у С12, С14 и С19 [7], что приводит к кето-енольной таутомерии (рис. 5). При подкислении атом азота пиридинового кольца протонируется, и в результате таутомерии происходит перераспределение заряда и увеличение сопряжения в молекуле. Именно это изменение и может оказывать влияние на биологическую функцию соединения. Из-за физиологического подкисления культуральной среды вследствие роста и метаболизма клеток нельзя исключать возможность того, что с увеличением времени культивирования енольная форма МНК начинает преобладать, и, по-видимому, этот таутомер обладает более сильными ингибирующими способностями, чем кето-форма МНК. Это допущение поддерживается экспериментальными данными, указывающими на большую биологическую активность енолятов относительно кето-форм в различных физиологических реакциях [9]. Такое наблюдение дает основание для развития нового подхода в рациональной разработке лекарств в медицинской химии.
Рис. 5.
УФ- (MeOH) спектры MНК при нормальном (а) и кислом значениях pH (б) имеют полосы поглощения с λмакс. 254, 298 (пл.) нм, а при 0.05 н. HCl в MeOH наблюдается батохромный сдвиг с λмакс. 212, 250, 306, 433 нм, и раствор метанола становится желтым, что указывает на возможную кето-енольную таутомерию молекулы МНК при кислых значениях рН (в).
Анализ жизнедеятельности клеток в реальном времени показал, что, в отличие от монапурпиридина A [6], MНК не вызывает гибель клеток. Однако обе молекулы имеют близкую структуру и отличаются наличием сложноэфирной этиловой группы в монапурпиридине А, а также длиной и структурой алкильного фрагмента. Существует вероятность того, что введение различных заместителей в карбоксильную группу МНК может усилить противоопухолевые свойства этого соединения. Известно, что использование соединений, в которых варьируют заместители в карбоксильной группе (свободная СООН и ее комплексы, сложные эфиры), позволяет изменять полярность молекулы лекарственного соединения, улучшает проявление фармакологической активности и замедляет процесс биодекарбоксилирования [10]. Для производных пиридина показана разнообразная биологическая активность, в том числе антибактериальная и противогрибковая [11].
Чтобы определить клеточные мишени для МНК в период времени, когда наблюдалось ингибирование пролиферации и миграции клеток, клетки LnCaP обрабатывали МНК в течение 96 ч при IC50, а затем клеточные лизаты анализировали для определения фосфорилирования клеточных киназ и уровней белков, регулирующих гомеостаз опухолевых клеток. Как видно на рис. 6, значительное снижение фосфорилирования Ser473 (S473) и Thr308 (T308) было обнаружено для всех трех изоформ AKT-киназы в клетках, обработанных MНК, по сравнению с контролем. Как известно, каскадное фосфорилирование этих аминокислотных остатков необходимо для активации AKT [12]. Ингибирование активности AKT имело функциональные последствия для фосфорилирования хорошо известных субстратов этой киназы, таких как GSK3α/β (S21/S9) [13], PRAS40 (T246) [14] и WNK1 (T60) [15], и для фосфорилирования непрямых мишеней AKT, таких как киназы mTOR (S2448) и p70S6 (T421/ S424). Следовательно, MНК может нарушать активацию сигнального пути AKT–mTOR, который является одним из основных регуляторов пролиферации и миграции клеток [12]. Данные фосфопротеомного анализа также указывают на МНК-индуцированное ингибирование фосфорилирования тирозинкиназы Src, продукта онкогена c-src, и фокальной адгезионной киназы FAK (рис. 6). Оба белка демонстрировали снижение уровней фосфорилирования тирозина Y397 для FAK и Y419 для Src. Как было показано, фосфорилирование остатков тирозина Y397 для FAK и Y419 для Src необходимо для взаимодействия киназ FAK–Src и активации комплекса для индукции различных клеточных ответов, включая клеточную адгезию и миграцию [16–20]. Следовательно, МНК может ингибировать миграцию клеток, влияя на активность комплекса FAK–Src.
Рис. 6.
МНК модулирует уровни фосфорилирования клеточных киназ и уровень шаперона HSP60 в клетках LnCaP. (а) – Репрезентативное изображение мембран фосфопротеомного анализа, где антитела, иммобилизованные на мембране и специфичные к фосфорилированным аминокислотным остаткам определенных белков, представлены в двух экземплярах. Интенсивность каждого сигнала соответствует уровню фосфорилирования или уровню общего белка (в случае HSP60). 1 – Src Y419, 2 – FAK Y397, 3 – HSP60, 4 – c-Jun S63, 5 – PRAS40 T246, 6 – GSK-3 S21/S9, 7 – AKT1/2/3 S473, 8 – AKT1/2/3 T308, 9 – p70S6 киназа T421/S424, 10 – WNK1 T60, 11 – mTOR S2448, RS – контрольные точки; (б) – денситометрический анализ интенсивности сигналов, представленных на мембранах и полученных из лизатов клеток, обработанных либо DMSO, либо МНК. Гистограммы показывают среднюю плотность пикселей, которые отражают уровень интенсивности сигналов. Планки погрешностей представляют ± s.d.

Однако в клетках, обработанных МНК, наблюдалось увеличение уровня белка теплового шока HSP60 (в 1.75 раза) и активация протоонкогена c-Jun (в 2.5 раза) (рис. 6). Известно, что оба этих фактора увеличивают рост и миграцию клеток и предотвращают их гибель [21, 22]. Фосфорилирование Ser63 c-Jun (S63) необходимо для стабильности белка и его транскрипционной активности [21, 23]. Известно, что c-Jun играет важную роль в стимуляции пролиферации клеток; этот факт позволяет предположить, что относительно слабая активность МНК в ингибировании роста клеток может быть опосредована активацией c-Jun. Шаперон HSP60 организует широкую программу выживания клеток посредством увеличения количества антиапоптотических белков Bcl-2 и Bcl-xl, уменьшения количества проапоптотических Bax-содержащих комплексов [24], стабилизации митохондриальных белков [25] и ограничения функционирования p53 [26]. Таким образом, индуцированное MНК повышение уровня HSP60 может действовать как защитный механизм против цитотоксического действия соединения.
Чтобы подтвердить результаты фосфопротеомного анализа клеточных мишеней, мы оценили уровни экспрессии AKT1 и HSP60 в клеточных лизатах LnCaP, обработанных МНК в течение 96 ч, посредством иммуноблоттинга. В клетках, инкубированных в присутствии МНК, наблюдалось снижение уровней как фосфорилированной формы киназы (фосфо-АКТ1), так и общего белка AKT1 в 3.4 и в 1.5 раза соответственно, тогда как уровень HSP60 повышался примерно в 2 раза в клетках, обработанных MНК, по сравнению с контролем (рис. 7). Эти данные подтверждают результаты фосфопротеомного анализа. Известно, что mTOR активирует трансляцию белка посредством фосфорилирования критических компонентов клеточного трансляционного аппарата [27]. Таким образом, различия в уровнях общего белка AKT1 между обработанными MНК клетками и контролем (рис. 7а–7в) могут быть объяснены MНК-индуцированным ингибированием пути AKT1–mTORC1, приводящим к снижению скорости синтеза киназы. Однако следует отметить, что при снижении общего уровня киназы в клетках не наблюдалось тотального ингибирования трансляции, о чем свидетельствует выявленное двукратное повышение уровня HSP60 (рис. 7г, 7д) в МНК-обработанных клетках по сравнению с контрольными клетками. Следовательно, МНК обладает избирательным действием относительно АКТ1 и не подавляет тотальный синтез белков.
Рис. 7.
Иммуноблот-анализ, документирующий изменения экспрессии белков в клетках LnCaP, обработанных МНК в концентрации IC50 (35 мкг/мл) в течение 96 ч. Методом иммуноблоттинга были определены количества фосфо-АКТ1 (S473), общего АКТ1 (а) и уровня общего белка HSP60 (г). В качестве контроля равного количества белков в каждой пробе использовали идентификацию β-актина. (б), (в), (д) – Денситометрический анализ сигналов иммуноблоттинга фосфо-АКТ1 (S473), общего АКТ1 и HSP60, нормализованных к уровню β-актина. Гистограммы представляют кратные изменения уровней экспрессии фосфо-АКТ1 (S473), общего АКТ1 и HSP60 в клетках, обработанных МНК, относительно контрольных клеток, обработанных DMSO. Планки погрешностей представляют ± s.d.

Таким образом, в настоящем исследовании охарактеризована биологическая активность нового соединения – монасникотиновой кислоты (МНК), выделенной из гриба A. сavernicola. Кроме того, мы выяснили механизм, по которому МНК может контролировать рост и миграцию клеток рака предстательной железы человека посредством регуляции сигнальных путей AKT–mTOR, Src–FAK, c-Jun и HSP60. Эти данные закладывают основу для дальнейшего конструирования и синтеза производных MНК с оптимальными противоопухолевыми свойствами, которые будут сохранять способность ингибировать онкогенный путь AKT–mTOR без активации сигналов выживания опухолевых клеток.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Условия выращивания штамма гриба и выделение МНК. Штамм гриба A. сavernicola ВКМ F-906 был получен из фонда Всероссийской коллекции микроорганизмов (ВКМ) Института биохимии и физиологии микроорганизмов им. Г.К. Скрябина РАН. Штамм выращивали в среде следующего состава (г/л дистиллированной воды): маннит – 50.0; янтарная кислота – 5.4; MgSO4 · 7H2O – 0.3; KH2PO4 – 1.0; ZnSO4 · 7H2O – 0.0044, рН доводили 25%-ным раствором NH4OH до значения 5.4. Засев сред осуществляли водной споровой суспензией (1–2 × 106 конидий/мл) 14-суточных культур, выращенных на поверхности скошенного мальц-агара. Штамм культивировали при (24 ± 1)°С в колбах объемом 750 мл в 150 мл жидкой среды в течение 11 сут. Выделение МНК проводили, как описано ранее [7]. Для изучения биологической активности MНК на клеточных культурах человека кристаллизованный метаболит растворяли в стерильном DMSO (Sigma, Германия) в концентрациях 10–50 мг/мл.
Клетки. Клетки рака предстательной железы человека LnCaP культивировали в среде RPMI 1640 с добавлением 10% фетальной бычьей сыворотки (FBS), 50 мкг/мл стрептомицина и пенициллина и 0.3 г/л глутамина (ПанЭко, Россия). Клетки инкубировали при 37°С во влажной атмосфере в присутствии 5% СО2. Микроскопические наблюдения клеток проводили на микроскопе MICROMED E-LUME (Микромед, Россия).
Антитела. Моноклональные антитела мыши (анти-АКТ1 (Santa Cruz Biotechnology, CША) и анти-β-актин, конъюгированные с пероксидазой (Sigma, Германия)) и антитела кролика (анти-фосфо-АКТ1 (S473) (D9E) (Cell Signaling Technology, CША) и анти-HSP60 (Cloud Clone Corp., США)) использовали в качестве первичных антител для иммуноблоттинга. Конъюгированные с пероксидазой антитела против иммуноглобулинов мыши (H&L, goat) и кролика (H&L, goat) (Rockland, США) использовали в качестве вторичных антител для визуализации белка методом вестерн-блоттинга.
Анализ клеточной пролиферации. Все эксперименты проводили с использованием инструмента xCELLigence DP (ACEA Biosciences, США). Сначала 100 мкл ростовой среды RPMI 1640 добавляли в лунки E-планшетов 16 (ACEA Biosciences, США) и помещали в прибор xCELLigence DP (ACEA Biosciences, США) для измерения фонового сопротивления среды. Затем клетки LnCaP (10 000 клеток/лунку) высевали в Е-планшеты. Планшеты помещали в прибор xCELLigence для непрерывной регистрации сопротивления на электродах. Через 24 ч к клеткам добавляли 100 мкл ростовой среды RPMI, содержавшей либо DMSO (контроль), либо МНК в указанных концентрациях. Электрическое сопротивление регистрировали каждые 15 мин в течение 86 ч. Поскольку известно первоначальное количество клеток, внесенных в лунки Е-планшетов, любое изменение их числа, возникающее во время инкубации, вызывает перемену электрического сопротивления. Относительное число клеток, прикрепленных к золотым электродам на дне лунок, представлено в виде КИ. Данные КИ также использовали для определения значений IC50 MНК.
В экспериментах по воздействию МНК в течение длительного времени клетки LnCaP (2 × × 105 клеток/лунку в 6-луночных планшетах) инкубировали с MНК (35 мкг/мл) или с DMSO (контроль) в течение 48 ч. Затем клетки высевали в свежую питательную среду с добавлением тех же концентрацией МНК или DMSO и инкубировали в течение следующих 48 ч. После общего времени обработки 96 ч клетки анализировали с помощью технологии xCELLigence DP (ACEA Biosciences, США), как описано выше.
Мониторинг миграции клеток. Скорость миграции клеток контролировали в режиме реального времени с использованием технологии xCELLigence (CIM-планшеты, ACEA Biosciences, США). Клетки LnCaP (30 000 клеток) помещали в бессывороточную среду, содержавшую DMSO или МНК в указанных концентрациях, и высевали в каждую лунку верхней части камеры CIM-планшета. Среду RPMI, содержавшую 5% FBS, вносили в каждую лунку нижней камеры. Сыворотку использовали в качестве хемоаттрактанта, который стимулирует миграцию клеток через пористую мембрану. Полное сопротивление каждой лунки автоматически контролировалось каждые 10 мин в течение 24 ч и выражалось в виде значений КИ, отражающих количество клеток, которые мигрировали из верхней части камеры через мембрану в нижнюю часть камеры в среду, содержащую хемоаттрактант.
Определение клеточных мишеней действия МНК. Для идентификации клеточных сигнальных путей, на которые действует МНК, мы использовали набор ARY003 Proteome Profiler™ (R&D Systems, США), позволяющий одновременно определять относительный уровень фосфорилирования 46 киназ и белков, контролирующих жизнедеятельность клеток человека. Клетки LnCaP (2 × 105) обрабатывали MНК (35 мкг/мл) или DMSO (контроль) в течение 48 ч. Затем клетки высевали в свежую питательную среду с такими же концентрациями веществ. После 96 ч обработки клетки трижды промывали охлажденным фосфатным буфером, лизировали с использованием прилагаемого в наборе буфера для лизиса и определяли концентрацию белка с использованием стандартного метода анализа поглощения (BCA, Thermo Fisher Scientific, США). Клеточные лизаты, полученные из обработанных МНК или контрольных клеток и содержавшие 300 мкг белка в 1 мл, наносили на мембраны с иммобилизованными антителами, специфичными к определенным клеточным белкам, и инкубировали в соответствии с инструкцией производителя. Интенсивность сигналов, соответствующих количеству связанных с антителами фосфорилированных белков, определяли после обработки мембран хемилюминесцентным реагентом и авторадиографии при помощи системы Fusion Fx (Vilber Lourmat, Франция). Программное обеспечение Studio Image Lite, версия 5.2 (LI-COR Biosciences, США) использовали для количественной оценки интенсивности сигналов.
Иммуноблот-анализ. Клетки LnCaP инкубировали в течение 96 ч в условиях, описанных выше для анализа клеточных мишеней. После обработки клетки промывали холодным фосфатным буфером и лизировали в присутствии 200 мкл буфера для лизиса (20 мМ Трис-HCl, pH 7.5, 1% NP-40, 100 мМ NaCl, 5 мМ EDTA) с добавлением ингибиторов протеаз (cocktail V, Calbiochem, Германия) и фосфатаз (10 мМ бета-глицерофосфат, 10 мМ ортованадат натрия, 10 мМ пирофосфат натрия и 5 мМ фторид натрия). Концентрацию белка измеряли с использованием набора BCA (Thermo Fisher Scientific, США). Клеточные белки (30 мкг) разделяли посредством 10%-ного SDS-PAGE и переносили на нитроцеллюлозные мембраны (Bio-Rad Laboratories, США). Затем мембраны инкубировали с реагентом, блокирующим неспецифическое связывание (5% обезжиренного молока в буфере TBST), и инкубировали с первичными антителами (разведение 1 : 1000 в растворе 5%-ного BSA в буфере TBST) в течение 16 ч. По окончании обработки мембраны промывали буфером TBST и инкубировали со вторичными антителами, конъюгированными с пероксидазой (разведение 1 : 5000 в растворе 5%-ного обезжиренного молока в буфере TBST). Иммунореактивность определяли с использованием хемилюминесцентного субстрата (Thermo Fisher Scientific, США), сигналы специфических белков визуализировали с помощью станции Fusion Fx (Vilber Lourmat, Франция). Относительные уровни экспрессии белка определяли денситометрией сигналов с использованием программного обеспечения Studio Image Lite (версия 5.2).
Статистическая обработка результатов. Эксперименты по анализу пролиферации и миграции клеток проводили в трех повторностях с использованием двух независимых выделений МНК. Статистическую значимость различий между двумя группами оценивали с использованием двустороннего критерия Стьюдента; все данные представлены как среднее ± s.d. Отличия считали статистически значимыми при р < 0.05.
Список литературы
Hyde K.D., Xu J., Rapior S., Jeewon R., Lumyong S., Niego A.G.T., Abeywickrama P.D., Aluthmuhandiram J.V.S., Brahamanage R.S., Brooks S., Chaiyasen A., Chethana K.W.T., Chomnunti P., Chepkirui C., Chuankid B., de Silva N.I., Doilom M., Faulds C., Gentekaki E., Gopalan V., Kakumyan P., Harishchandra D., Hemachandran H., Hongsanan S., Karunarathna A., Karunarathna S.C., Khan S., Kumla J., Jayawardena R.S., Liu J.-K, Liu N., Luangharn T., Macabeo A.P.G., Marasinghe D.S., Meeks D., Mortimer P.E., Mueller P., Nadir S., Nataraja K.N., Nontachaiyapoom S., O’Brien M., Penkhrue W., Phukhamsakda C., Ramanan U.S., Rathnayaka A.R., Sadaba R.B., Sandargo B., Samarakoon B.C., Tennakoon D.S., Siva R., Sriprom W., Suryanarayanan T.S., Sujarit K., Suwannarach N., Suwunwong T., Thongbai B., Thongklang N., Wei D., Wijesinghe S.N., Winiski J., Yan J., Yasanthika E., Stadler M. // Fungal Diversity. 2019. V. 97. P. 1–136. https://doi.org/10.1007/s13225-019-00430-9
Chen W., Chen R., Liu Q., He Y., He K., Ding X., Kang L., Guo X., Xie N., Zhou Y., Lu Y., Cox R.J., Molnár I., Li M., Shaoa Y., Chen F. // Chem. Sci. 2017. V. 8. P. 4917–4925. https://doi.org/10.1039/c7sc00475c
Lebeau J., Venkatachalam M., Fouillaud M., Petit T., Vinale F., Dufossé L., Caro Y. // J. Fungi. (Basel). 2017. V. 28. P. E34. https://doi.org/10.3390/jof3030034
Patakova P. // J. Ind. Microbiol. Biotechnol. 2013. V. 40. P. 169–181.https://doi.org/10.1007/s10295-012-1216-8
Hsu L.C., Hsu Y.W., Liang Y.H., Liaw C.C., Kuo Y.H., Pan T.M. // Molecules. 2012. V. 17. P. 664–673. https://doi.org/10.3390/molecules17010664
Samson R.A., Varga J., Meijer M., Frisvad J.C. // Studies in Mycology. 2011. V. 69. P. 81–97. https://doi.org/10.3114/sim.2011.69.06
Antipova T.V., Zaitsev K.V., Zherebker A.Ya., Tafeenko V.A., Baskunov B.P., Zhelifonova V.P., Ivanushkina N.E., Kononikhin A.S., Nikolaev E.N., Kozlovsky A.G. // Mendeleev Commun. 2018. V. 28. P. 55–57. https://doi.org/10.1016/j.mencom.2018.01.018
Wu M.D., Cheng M.J., Yech Y.J., Chen Y.L., Chen K.P., Chen I.S., Yang P.H., Yuan G.F. // Molecules. 2011. V. 16. P. 4719–4727. https://doi.org/10.3390/molecules16064719
Negi A., Gill B.S. // PharmaTutor. 2013. V. 1. P. 45–53.
Soldatenkov A., Koladina N.M., Shendrik I.V. // Fundamentals of Organic Chemistry of Medicinal Substances. M: Chemistry, 2001. 192 p.
Zaki R.M., Kamal El-Dean, Radwan S.M., Ammar M.A. // Russ. J. Bioorg. Chem. 2020. V. 46. P. 85–96. https://doi.org/10.1134/S1068162020010148
Manning B.D., Toker A. // Cell. 2017. V. 169. P. 381–405. https://doi.org/10.1016/j.cell.2017.04.001
Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. // Nature. 1995. V. 378. P. 785–789. https://doi.org/10.1038/378785a0
Kankanamalage S.G., Karra A.S., Cobb M.H. // Cell Commun. Signal. 2018. V. 16. P. 72. https://doi.org/10.1186/s12964-018-0287-1
Gibbons J.J., Abraham R.T., Yu.K. // Semin. Oncol. 2009. V. 36. P. 3–17. https://doi.org/10.1053/j.seminoncol.2009.10.011
Calalb M.B., Polte T.R., Hanks S.K. // Mol. Cell Biol. 1995. V. 15. P. 954–963. https://doi.org/10.1128/MCB.15.2.954
Martin G.S. // Nat. Rev. Mol. Cell Biol. 2001. V. 2. P. 467–475. https://doi.org/10.1038/35073094
Cox B.D., Natarajan M., Stettner M.R., Gladson C.L. // J. Cell Biochem. 2006. V. 99. P. 35–52. https://doi.org/10.1002/jcb.20956
Summy J.M., Gallick G.E. // Cancer Metastasis Rev. 2003. V. 22. P. 337–358.
Bolós V., Gasent J.M., López-Tarruella S., Grande E. // Onco Targets Ther. 2010. V. 3. P. 83–97. https://doi.org/10.2147/OTT.S6909
Shaulian E., Karin M. // Oncogene. 2001. V. 20. P. 2390–2400. https://doi.org/10.1038/sj.onc.1204383
Cappello F., Conway de Macario E., Marasà L., Zummo G., Macario A.J.L. // Cancer Biol. Therapy. 2008. V. 7. P. 801–809. https://doi.org/10.4161/cbt.7.6.6281
Dunn C., Wiltshire C., MacLaren A., Gillespie D.A. // Cell Signal. 2002. V. 14. P. 585–593. https://doi.org/10.1016/S0898-6568(01)00275-3
Shan Y.X., Liu T.J., Su H.F., Samsamshariat A., Mestril R., Wang P.H. // J. Mol. Cell. Cardiol. 2003. V. 35. P. 1135–1143.
Kim W., Ryu J., Kim J. // Int. J. Mol. Sci. 2019. V. 20. P. 131. https://doi.org/10.3390/ijms20010131
Ghosh J.C., Dohi T., Kang B.H., Altieri D.C. // J. Biol. Chem. 2008. V. 283. P. 5188–5194. https://doi.org/10.1074/jbc.M705904200
Holz M.K., Ballif B.A., Gygi S.P., Blenis J. // Cell. 2005. V. 123. P. 569–580. https://doi.org/10.1016/j.cell.2005.10.024
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия