

Биоорганическая химия, 2021, T. 47, № 1, стр. 153-161
Галлоцианин как флуороген для выявления NADPH-зависимой продукции супероксидного анион-радикала клетками крови
О. М. Панасенко 1, *, В. Е. Реут 2, И. В. Бородина 1, Д. С. Матюшкина 1, В. А. Иванов 1, Д. В. Григорьева 2, И. В. Горудко 2, А. В. Соколов 1, 3, С. Н. Черенкевич 2
1 ФГБУ “Федеральный научно-клинический центр физико-химической медицины” ФМБА России
119435 Москва, ул. Малая Пироговская, 1а, Россия
2 Белорусский государственный университет
220030 Минск, просп. Независимости, 4, Беларусь
3 Институт экспериментальной медицины
197376 Санкт-Петербург, ул. Академика Павлова, 12, Россия
* E-mail: o-panas@mail.ru
Поступила в редакцию 09.04.2020
После доработки 18.04.2020
Принята к публикации 21.04.2020
Аннотация
Методами спектрофотометрии, масс-спектрометрии и спектрофлуориметрии исследовано взаимодействие красителя оксазинового ряда галлоцианина с активными формами кислорода $\left( {^{\centerdot }{\text{О}}_{2}^{ - },\,\,{{{\text{H}}}_{2}}{{{\text{O}}}_{{\text{2}}}}} \right)$ и галогенов (HOCl). Показано, что галлоцианин реагирует с HOCl и $^{\centerdot }{\text{О}}_{2}^{ - }$ (но не с Н2О2) с образованием флуоресцирующих продуктов. С использованием ингибиторного анализа установлено, что как в системе ксантин/ксантиноксидаза, так и в присутствии активированных нейтрофилов крови человека основной вклад в превращение галлоцианина во флуорофор вносит его реакция с $^{\centerdot }{\text{О}}_{2}^{ - }.$ Полученные результаты позволяют заключить, что галлоцианин может выполнять роль флуорогенного хемосенсора и использоваться для оценки активации нейтрофилов, NADPH-зависимой продукции супероксидного анион-радикала нейтрофилами и другими клетками крови, а также для тестирования антиоксидантных препаратов, разрабатываемых с целью коррекции заболеваний, ассоциированных с окислительным стрессом.
ВВЕДЕНИЕ
Нейтрофилы представляют собой самый многочисленный подвид гранулоцитарных лейкоцитов (до ~70%) крови человека, играющий ключевую роль в реализации клеточного звена врожденного иммунитета во многом благодаря продукции активных форм кислорода (АФК) и галогенов (АФГ). Эти соединения не только играют роль медиаторов важных внутриклеточных и межклеточных редокс-чувствительных сигнальных путей, таких как ионная проницаемость, апоптоз, нетоз и рецепторные взаимодействия, но и, будучи высокореакционными, уничтожают патогены (бактерии, вирусы, грибы и др.), выполняя антимикробную функцию [1]. За продукцию АФК и АФГ в нейтрофилах отвечают главным образом ферменты: NADPH-оксидаза (КФ 1.6.3.1), супероксиддисмутаза (SOD, КФ 1.15.1.1) и миелопероксидаза (MPO, КФ 1.11.2.2). Активация нейтрофилов в ответ на провоспалительный стимул сопровождается сборкой NADPH-оксидазного комплекса, который катализирует реакцию восстановления молекулярного кислорода (О2) до супероксидного анион-радикала $\left( {^{\centerdot }{\text{О}}_{2}^{ - }} \right)$ [2]:
(1)
${\text{NADPH}} + 2{{{\text{О}}}_{2}} \to {\text{NAD}}{{{\text{P}}}^{ + }} + {{2}^{\centerdot }}{\text{О}}_{2}^{ - } + {{{\text{Н}}}^{ + }}.$(2)
${{2}^{\centerdot }}{\text{О}}_{2}^{ - } + 2{{{\text{Н}}}^{ + }} \to {{{\text{О}}}_{2}} + {{{\text{Н}}}_{2}}{{{\text{О}}}_{2}},$АФК и особенно АФГ (включая HOCl), образующиеся при активации нейтрофилов и обладающие повышенной реакционной способностью, реагируют со многими биологически важными молекулами (нуклеиновыми кислотами, белками, липидами, антиоксидантами и др.), повреждают клетки и ткани организма, вызывая окислительный/галогенирующий стресс, провоцирующий развитие воспалительных заболеваний: сердечно-сосудистых, нейродегенеративных, онкологических и др. [3–5]. Становится понятно, насколько важно контролировать и оценивать активность ферментов, ответственных за образование АФК и АФГ. Очевидно, что регистрация продукции клетками АФК/АФГ дает возможность разработки новых методов и подходов, направленных на профилактику, диагностику и мониторинг эффективности терапии заболеваний, ассоциированных с окислительным/галогенирующим стрессом. Среди методов, используемых для обнаружения АФК и АФГ (масс-спектрометрия в сочетании с различными вариантами хроматографии, иммуноферментный анализ, хемилюминесценция, спиновые ловушки и др.), флуоресцентный анализ отличается высокой чувствительностью и доступностью [6–8].
В последнее время предложено большое количество низкомолекулярных флуорогенных зондов, позволяющих в том числе в режиме реального времени регистрировать продукцию различных АФК и АФГ как внутри, так и вне клетки: 2',7'-дихлордигидрофлуоресцеиндиацетат, дигидрородамин 123, 10-ацетил-3,7-дигидроксифеноксазин (Amplex Red), аминофенилфлуоресцеин (2-[6-(4'-амино)фенокси-3H-ксантен-3-он-9-ил]бензойная кислота) и др. [6–11]. Однако указанные зонды не всегда являются достаточно чувствительными и специфичными при анализе тех или иных АФК/АФГ, зачастую их синтез весьма трудоемок, что значительно увеличивает стоимость, либо они попросту неудобны в анализе (плохая растворимость в воде, недостаточная стабильность и т.п.). Например, упомянутые 2',7'-дихлордигидрофлуоресцеиндиацетат, дигидрородамин 123 и аминофенилфлуоресцеин одинаково хорошо окисляются свободными радикалами, пероксинитритом и HOCl [6, 9]. Amplex Red, предназначенный для определения Н2О2 в присутствии пероксидаз, легко окисляется под действием АФГ. Более того, такие восстановители, как NADH и глутатион, могут заметно искажать результат анализа [10, 11].
Ранее нами был предложен кинетический спектрофотометрический метод для измерения галогенирующей активности MPO, основанный на обесцвечивании под действием АФГ красителя оксазинового ряда – целестинового синего В [12]. Позднее мы показали, что этот краситель с успехом может быть использован для выявления HOCl, продуцируемой активированными нейтрофилами, методами проточной цитометрии и конфокальной микроскопии [13].
В настоящей работе мы сосредоточили свое внимание на другом красителе оксазинового ряда – галлоцианине (4-гидрокси-7-диметиламино-1-карбоксифеноксазон-3 хлорид). Он – структурный аналог целестинового синего В, но отличается большей фотостабильностью, относительно хорошей растворимостью в фосфатно-солевом буфере и легко синтезируется с выходом, близким к количественному. Благодаря в том числе этим свойствам, галлоцианин уже более полувека используется при гистологическом анализе для окраски ядер [14] и количественного определения нуклеиновых кислот [15]. Нами было исследовано взаимодействие галлоцианина с основными АФК $\left( {^{\centerdot }{\text{О}}_{2}^{ - },\,\,{{{\text{H}}}_{2}}{{{\text{O}}}_{{\text{2}}}}} \right)$ и АФГ (HOCl) с целью использования красителя для выявления продукции этих соединений активированными нейтрофилами.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Взаимодействие галлоцианина с $^{\centerdot }{\mathbf{О}}_{{\mathbf{2}}}^{ - }{\mathbf{.}}$ Известно, что диоксид калия (КО2) в водной среде гидролизуется с образованием короткоживущего супероксидного анион-радикала:
(3)
${\text{K}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {{{\text{H}}}^{ + }} + {\text{KOH}}\,\, + \,{{\,}^{\centerdot }}{\text{О}}_{2}^{ - },$(4)
$2{\text{K}}{{{\text{O}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to 2{\text{KOH}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} + {{{\text{O}}}_{2}}.$Для исследования взаимодействия галлоцианина с $^{\centerdot }{\text{О}}_{2}^{ - }$ к раствору галлоцианина добавляли навеску сухого КО2, перемешивали и регистрировали спектры поглощения в видимой области (400–750 нм). Результаты измерения приведены на рис. 1. Видно, что раствор галлоцианина имеет максимум поглощения при 623 нм. По мере увеличения концентрации добавленного КО2 наблюдается снижение высоты этого пика, одновременно появляется пик поглощения с максимумом при 510 нм, увеличивается его интенсивность, при этом окраска раствора меняется с синей на бледно-фиолетовую. В совокупности это свидетельствует о расходовании галлоцианина и образовании как минимум одного продукта. В контрольных экспериментах мы показали, что добавление к галлоцианину (20 мкМ) раствора Н2О2 (10–1000 мкМ), образующегося по реакции (4), не приводит к изменению спектра поглощения галлоцианина (данные не приведены). Это значит, что галлоцианин не вступает в реакцию с Н2О2. В таком случае можно заключить, что изменение спектра поглощения галлоцианина при добавлении к нему КО2 обусловлено его реакцией с $^{\centerdot }{\text{О}}_{2}^{ - },$ образующимся в качестве интермедиата при гидролизе КО2 по реакции (3).
Рис. 1.
Спектры поглощения галлоцианина до и после добавления к нему KO2. Среда измерения – ФСБ-2, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 20 мкМ. Концентрация КО2 (мМ): 1 – 0; 2 – 0.22; 3 – 0.76; 4 – 0.98; 5 – 1.31; 6 – 1.64.

Этот факт подтверждают изображенные на рис. 2 масс-спектры галлоцианина, зарегистрированные до и после добавления к нему КО2 в различных концентрациях. Масс-спектр галлоцианина (спектр 1 на рис. 2) представляет собой моноизотопное распределение, максимально интенсивный пик (m/z 301.1) соответствует массе катиона [C15H13N2O5]+, состоящего из основных изотопов (12С, 1Н, 14N, 16О). По мере роста концентрации КО2 сначала появляется (спектр 2 на рис. 2), увеличивается (спектры 3 и 4), а затем исчезает (спектр 5) сигнал с m/z 339.1. Параллельно, начиная с концентрации КО2 2.84 мМ, появляется и увеличивается сигнал с m/z 230.0. При концентрации КО2 11.38 мМ (спектр 5 на рис. 2) пик от исходного галлоцианина (m/z 301.1) исчезает полностью и остается только сигнал с m/z 230.0. Такой результат можно объяснить, если предположить, что в реакции галлоцианина с КО2 сначала образуется продукт с m/z 339.1, который в конце концов превращается в соединение с m/z 230.0. Как минимум один из продуктов является флуорофором, что подтверждают результаты, приведенные на рис. 3, из которого видно, что добавление к раствору галлоцианина КО2 приводит к возникновению флуоресценции, которая усиливается по мере увеличения концентрации КО2 (максимумы в спектре возбуждения в области 260–370 нм, испускания – 480–500 нм, рис. 3).
Рис. 2.
Масс-спектры галлоцианина до и после добавления к нему KO2. Среда измерения – ФСБ-2, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 80 мкМ. Концентрация КО2 (мМ): 1 – 0; 2 – 1.14; 3 – 2.84; 4 – 5.69; 5 – 11.38; 6 – масс-спектр матрицы (2,5-дигидроксибензойной кислоты) в отсутствие галлоцианина.

Рис. 3.
Спектры возбуждения и испускания флуоресценции галлоцианина до и после добавления к нему КО2. Среда измерения – ФСБ-1, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 5 мкМ. Концентрация КО2 (мМ): 1 – 0; 2 – 0.7; 3 – 1.6; 4 – 2.7; 5 – 4.1. Возбуждение – при 360 нм, испускание – при 490 нм.
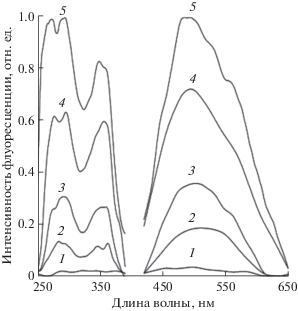
Помимо КО2 для продукции $^{\centerdot }{\text{О}}_{2}^{ - }$ нами был использован фермент ксантиноксидаза, который в присутствии ксантина катализирует восстановление О2 до $^{\centerdot }{\text{О}}_{2}^{ - }$ [16]. Результаты эксперимента приведены на рис. 4. Видно, что в присутствии ксантиноксидазы и ее субстрата ксантина наблюдается увеличение интенсивности флуоресценции при 490 нм (возбуждение при 360 нм) (кривая 1 на рис. 4), тогда как в отсутствие ксантина ферментативная реакция не идет, и прирост интенсивности флуоресценции отсутствует (кривая 2 на рис. 4). Этот результат свидетельствует о взаимодействии синтезирующегося в ферментативной реакции $^{\centerdot }{\text{О}}_{2}^{ - }$ с галлоцианином с образованием флуорофора. Из данных, представленных на рис. 5, видно, что интенсивность флуоресценции (h20) и химическая реакция галлоцианина (v) в системе ксантин/ксантиноксидаза практически полностью подавляются в присутствии SOD. Этот факт свидетельствует о решающем вкладе $^{\centerdot }{\text{О}}_{2}^{ - }$ в химические превращения красителя.
Рис. 4.
Кинетика изменения интенсивности флуоресценции галлоцианина в системе ксантин/ксантиноксидаза (кривая 1). Концентрация галлоцианина – 5 мкМ, ксантина – 1 мМ, ксантиноксидазы – 0.52 мкМ. Среда измерения – ФСБ-1, рН 7.4 (см. “Эксперим. часть”). Кривая 2 – контрольная кривая, полученная в отсутствие ксантина. Возбуждение – при 360 нм, испускание – при 490 нм. На рисунке обозначены определяемые параметры: интенсивность флуоресценции по сравнению с фоновым уровнем через 7 (h7) или 20 (h20) мин после запуска реакции и скорость химического превращения галлоцианина (v).

Рис. 5.
Влияние SOD (1.6 мкМ) на интенсивность (h20) и скорость нарастания флуоресценции (v) в системе галлоцианин + ксантин/ксантиноксидаза. Концентрация галлоцианина – 5 мкМ, ксантина – 1 мМ, ксантиноксидазы – 0.52 мкМ. Среда измерения – ФСБ-1, рН 7.4 (см. “Эксперим. часть”). Возбуждение – при 360 нм, испускание – при 490 нм.

Взаимодействие галлоцианина с HOCl. На рис. 6 приведены спектры поглощения галлоцианина до и после добавления к нему HOCl в различных концентрациях. Видно, что раствор галлоцианина имеет максимум поглощения при 623 нм (спектр 1 на рис. 6а). Рассчитанный из этого спектра молярный коэффициент экстинкции составил ε623 = 28 000 М–1 см–1, что хорошо согласуется с данными литературы [17, 18]. Добавление HOCl к красителю приводило к уменьшению оптической плотности раствора при длине волны 623 нм и появлению менее интенсивной полосы поглощения с максимумом при 475 нм (рис. 6а), что свидетельствует об образовании продукта реакции. Из вставки на рис. 6а видно, что падение концентрации галлоцианина происходит пропорционально концентрации добавленной HOCl так, что коэффициент стехиометрии HOCl : галлоцианин равен ~1.5. Однако, как видно из рис. 6б, дальнейшее увеличение концентрации HOCl приводит к снижению оптической плотности раствора при 475 нм и появлению полосы поглощения при 416 нм. В ходе реакции сине-фиолетовая окраска исходного раствора галлоцианина сначала меняется на оранжевую, а при дальнейшем увеличении концентрации HOCl раствор обесцвечивается. Такой результат говорит о том, что HOCl реагирует с галлоцианином с образованием как минимум двух продуктов, первый из которых, имеющий полосу поглощения при 475 нм, является промежуточным и при более высоких концентрациях HOCl претерпевает превращение в продукт, поглощающий при 416 нм.
Рис. 6.
Спектры поглощения галлоцианина до и после добавления к нему HOCl. Среда измерения – ФСБ-2, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 20 мкМ. Концентрация HOCl (мкМ) (а): 1 – 0; 2 – 7.5; 3 – 15.0; 4 – 22.5; 5 – 40.0; (б): 6 – 45.0; 7 – 52.5; 8 – 60.0; 9 – 67.5; 10 – 75.0. На вставке приведена концентрационная зависимость убыли галлоцианина в растворе при добавлении к нему HOCl, рассчитанная по убыли оптической плотности при 623 нм.

Такой вывод подтверждают и приведенные на рис. 7 масс-спектры галлоцианина, зарегистрированные до и после добавления к нему HOCl в различных концентрациях. Видно, что по мере увеличения концентрации HOCl интенсивность сигнала, соответствующего массе катиона исходного галлоцианина [C15H13N2O5]+ (m/z 301.1), постепенно снижается. Параллельно появляются сигналы с m/z 328.3 и 452.0, по всей вероятности, соответствующие двум продуктам реакции HOCl с галлоцианином. Как минимум один из этих продуктов является флуорофором. Об этом свидетельствуют представленные на рис. 8 спектры возбуждения и испускания флуоресценции раствора галлоцианина после добавления к нему HOCl в различных концентрациях. Видно, что по мере возрастания концентрации добавленной к галлоцианину HOCl наблюдается увеличение интенсивности флуоресценции раствора красителя (максимумы в спектре возбуждения – 269, 292 и 360 нм, испускания – 490 нм, рис. 8), что обусловлено образованием продукта реакции.
Рис. 7.
Масс-спектры галлоцианина до и после добавления к нему HOCl. Среда измерения – ФСБ-2, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 80 мкМ. Концентрация HOCl (мМ): 1 – 0; 2 – 0.4; 3 – 4.0; 4 – 8.0; 5 – 16.0; 6 – масс-спектр матрицы (2,5-дигидроксибензойной кислоты) в отсутствие галлоцианина.

Рис. 8.
Спектры возбуждения и испускания флуоресценции галлоцианина до и после добавления к нему HOCl. Среда измерения – ФСБ-1, рН 7.4 (см. “Эксперим. часть”). Концентрация галлоцианина – 5 мкМ. Концентрация HOCl (мкМ): 1 – 0; 2 – 2.5; 3 – 5.0; 4 – 7.5; 5 – 10.0. Возбуждение – при 360 нм, испускание – при 490 нм.

Таким образом, оба исследованных нами реагента ($^{\centerdot }{\text{О}}_{2}^{ - }$ и HOCl) в бесклеточной среде реагируют с галлоцианином с образованием флуоресцирующих продуктов. Далее мы попытались выяснить, можно ли использовать галлоцианин в качестве флуорогенного хемосенсора для регистрации продукции АФК и/или АФГ нейтрофилами крови человека.
Продукция $^{\centerdot }{\mathbf{О}}_{{\mathbf{2}}}^{ - }$ нейтрофилами. Известно, что среди всех клеток крови нейтрофилы являются основными носителями МРО и NADPH-оксидазы. Стимуляция нейтрофилов приводит, с одной стороны, к сборке и активации NADPH-оксидазного ферментного комплекса, что сопровождается усиленной продукцией $^{\centerdot }{\text{О}}_{2}^{ - }$ [2], а с другой – к дегрануляции клеток, секреции MPO из азурофильных гранул во внеклеточное пространство и, как следствие – к синтезу HOCl [1, 4]. Оба события, как демонстрируют представленные выше результаты, должны способствовать трансформации галлоцианина в его флуорогенную форму. Для того чтобы выяснить, какая из ферментных систем нейтрофилов в большей степени ответственна за превращение галлоцианина во флуорофор, мы активировали нейтрофилы добавлением N-формил-метил-лейцил-фенилаланина (fMLP) или форбол-12-миристат-13-ацетата (PMA) в присутствии галлоцианина и различных веществ, способных как перехватывать и/или элиминировать АФК/АФГ, так и ингибировать их продукцию, регистрируя при этом изменение интенсивности флуоресценции. Результаты экспериментов приведены в табл. 1.
Таблица 1.
Влияние ингибиторов продукции АФК/АФГ и их перехватчиков на параметры, характеризующие химическое превращение галлоцианина в суспензии нейтрофилов, активированных fMLP или РМА
| Агонист | Параметр | SOD, 1.6 мкМ | CP, 1.1 мкМ | 4-АВАН, 100 мкМ | Таурин, 3.2 мМ | Маннитол, 10 мМ |
|---|---|---|---|---|---|---|
| fMLP, 0.5 мкМ | v, % от контроля | 7 ± 5* | 5 ± 4* | 102 ± 29 | 98 ± 27 | 113 ± 20 |
| h7, % от контроля | 14 ± 4* | 19 ± 18* | 129 ± 30 | 92 ± 15 | 107 ± 35 | |
| PMA, 50 нМ | v, % от контроля | 0** | 2 ± 3* | 93 ± 19 | 105 ± 20 | 125 ± 20 |
| h7, % от контроля | 0** | 6 ± 9* | 127 ± 23 | 104 ± 19 | 110 ± 15 |
Из данных табл. 1 видно, что гидразид 4-аминобензойной кислоты (4-АВАН; специфический ингибитор каталитической активности МРО, а значит, и продукции HOCl [19]), таурин (перехватчик HOCl) и маннитол (перехватчик гидроксильного радикала) не оказывали достоверного влияния на параметры окисления галлоцианина в присутствии активированных нейтрофилов. В то же время при добавлении к нейтрофилам SOD или церулоплазмина (СР) значения амплитуды интенсивности флуоресценции (h7) и скорости реакции (v) были значительно ниже контроля (активация нейтрофилов fMLP или PMA без ингибиторов или перехватчиков АФК/АФГ) или не определялись вовсе в результате отсутствия прироста флуоресценции спустя 20 мин после активации нейтрофилов. Как известно, SOD элиминирует $^{\centerdot }{\text{О}}_{2}^{ - },$ катализируя его дисмутацию по реакции (2) до Н2О2, который, как нам удалось установить, не реагирует с галлоцианином (см. выше). Обнаруженный эффект CP, по всей видимости, обусловлен SOD-активностью этого белка [20]. Полученные результаты позволяют предположить, что основной вклад в окисление галлоцианина в суспензии нейтрофилов происходит за счет его реакции с $^{\centerdot }{\text{О}}_{2}^{ - }.$
Следует отметить, что в крови человека подавляющее количество MPO сосредоточено в нейтрофилах и лишь незначительная ее часть – в моноцитах [1, 4]. Что касается NADPH-оксидазы, то различные ее изоформы присутствуют не только в нейтрофилах, но и практически во всех остальных типах клеток крови [2]: моноцитах [21], лимфоцитах [22], тромбоцитах [23], эритроцитах [24].
Таким образом, полученные результаты позволяют заключить, что галлоцианин может выполнять роль флуорогенного хемосенсора и использоваться для оценки NADPH-зависимой продукции супероксидного анион-радикала, по всей вероятности, не только нейтрофилами, но и другими клетками крови, а также для тестирования антиокислительных свойств препаратов, разрабатываемых с целью коррекции заболеваний, ассоциированных с окислительным стрессом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы. В работе использовали следующие реактивы: цитрат натрия, N-формил-метил-лейцил-фенилаланин (fMLP), форбол-12-миристат-13-ацетат (PMA), гипохлорит натрия (NaOCl), галлоцианин, диоксид калия (KО2), ксантиноксидазу, ксантин, гидразид 4-аминобензойной кислоты (4-АВАН), супероксиддисмутазу (SOD), таурин, маннитол, 2,5-дигидроксибензойную кислоту фирмы Sigma (США); декстран Т70 фирмы Roth (Германия); гистопак фирмы Nycomed (Норвегия). Остальные реактивы были производства “Реахим” (Россия) и “Белмедпрепараты” (Беларусь). Церулоплазмин (СР) выделяли из крови здоровых доноров с помощью ионообменной и аффинной хроматографии [20].
Концентрацию гипохлорит-аниона (OClˉ) определяли по его поглощению при 290 нм и рН 12, используя коэффициент молярной экстинкции ε290 = 350 М–1 см–1 [25]. Принимая во внимание тот факт, что рКа HOCl составляет ~7.5 [25] и при физиологических значениях рН примерно половина кислоты находится в молекулярной форме, а остальная часть – в виде аниона, под HOCl мы подразумевали смесь HOCl/OClˉ, присутствующую в исследуемой среде.
В качестве буферного раствора использовали фосфатно-солевой буфер, содержавший 10 мМ Na2HPO4/KН2РО4, 137 мМ NaCl, 2.7 мМ KCl, рН 7.4 (ФСБ-1) либо 10 мМ Na2HPO4/NaН2РО4, рН 7.4 (ФСБ-2).
Спектры поглощения регистрировали на двухлучевых спектрофотометрах РВ 2201 (Solar, Беларусь) и Cary-50 (Varian, США).
Флуоресцентные измерения проводили на спектрофлуориметре CM 2203 (Solar, Беларусь).
Выделение нейтрофилов осуществляли путем центрифугирования в градиенте плотности гистопака из крови здоровых доноров, как описано ранее [26]. Полученный осадок нейтрофилов отмывали в ФСБ-1, содержавшем 2 мг/мл D-глюкозы, при 4°С и сразу использовали в эксперименте.
Продукцию АФК и АФГ нейтрофилами оценивали флуоресцентным методом с использованием галлоцианина. К нейтрофилам (106 кл./мл) в ФСБ-1, содержавшем 1 мМ CaCl2 и 0.5 мМ MgCl2, добавляли галлоцианин (5 мкМ), fMLP (0.5 мкМ) или PMA (50 нМ), ингибиторы продукции АФК/ АФГ или их перехватчики (SOD, 4-АВАН, CP, таурин, маннитол). Регистрацию осуществляли при 37°С и постоянном перемешивании образца. Продукцию $^{\centerdot }{\text{О}}_{2}^{ - }$ в системе ксантин/ксантиноксидаза регистрировали в ФСБ-1 при 25°С, концентрация ксантина – 1 мМ, ксантиноксидазы – 0.52 мкМ, галлоцианина – 5 мкМ. Для регистрации флуоресценции эмпирически были подобраны значения длин волн возбуждения и испускания 360 и 490 нм соответственно. Для характеристики химического превращения красителя были выбраны следующие параметры (рис. 4): тангенс угла наклона начального линейного участка кинетической кривой изменения интенсивности флуоресценции (v), характеризующий скорость реакции, и интенсивность флуоресценции образца по сравнению с фоновым уровнем через 7 (h7) или 20 (h20) мин после запуска активации нейтрофилов, отражающую количество накопившегося флуоресцирующего продукта реакции.
Масс-спектры регистрировали на масс-спектрометре Ultraflex II TOF/TOF (Bruker Daltonics, Германия), оснащенном УФ-лазером (Nd), в режиме положительных ионов с использованием рефлектрона. Время задержки анализатора – 200 нс. Напряжение на электроде ускорителя составляло 25.01 кВ, накапливающем электроде – 21.71 кВ, фокусирующей линзе – 10.01 кВ. Параметры масс-спектрометра были оптимизированы для диапазона m/z 50–1000. Сигнал накапливался как усредненный по 200 одиночным лазерным импульсам с постоянной мощностью лазерного излучения на уровне порогового значения для увеличения разрешения. В качестве матрицы использовали 2,5-дигидроксибензойную кислоту.
Статистическую и графическую обработку данных выполняли с использованием пакета программ OriginPro 2016. Данные представлены как среднее ± стандартная ошибка среднего. Различия считали статистически достоверными при значении p < 0.05, рассчитанном по критерию Стьюдента. Типичные кинетические кривые представляют собой инвариант 3–5 независимых экспериментов.
Список литературы
Klebanoff S.J. // J. Leukoc. Biol. 2005. V. 77. P. 598–625. https://doi.org/10.1189/jlb.1204697
Меньщикова Е.Б., Зенков Н.К. // Усп. совр. биол. 2006. Т. 126. С. 97–112.
Sies H. // Klin. Wochenschr. 1991. V. 69. P. 965–968. https://doi.org/10.1007/BF01645140
Панасенко О.М., Горудко И.В., Соколов А.В. // Усп. биол. химии. 2013. Т. 53. С. 195–244. [Panasenko O.M., Gorudko I.V., Sokolov A.V. // Biochemistry (Moscow). 2013. V. 78. P. 1466–1489.] https://doi.org/10.1134/S0006297913130075
Панасенко О.М., Сергиенко В.И. // Вестник Российской АМН. 2010. № 1. С. 27–39.
Huang J., Milton A., Arnold R.D., Huang H., Smith F., Panizzi J.R., Panizzi P. // J. Leukoc. Biol. 2016. V. 99. P. 541–548. https://doi.org/10.1189/jlb.3RU0615-256R
Tarpey M.M., Fridovich I. // Circ. Res. 2001. V. 89. P. 224–236. https://doi.org/10.1161/hh1501.094365
Kettle A.J., Albrett A.M., Chapman A.L., Dickerhof N., Forbes L.V., Khalilova I., Turner R. // Biochim. Biophys. Acta. 2014. V. 1840. P. 781–793. https://doi.org/10.1016/j.bbagen.2013.07.004
Setsukinai K., Urano Y., Kakinuma K., Majima H.J., Nagano T. // J. Biol. Chem. 2013. V. 278. P. 3170–3175. https://doi.org/10.1074/jbc.M209264200
Votyakova T.V., Reynolds I.J. // Arch. Biochem. Biophys. 2004. V. 431. P. 138–144. https://doi.org/10.1016/j.abb.2004.07.025
Grivennikova V.G., Kareyeva A.V., Vinogradov A.D. // Redox Biol. 2018. V. 17. P. 192–199. https://doi.org/10.1016/j.redox.2018.04.014
Sokolov A.V., Kostevich V.A., Kozlov S.O., Donskyi I.S., Vlasova I.I., Rudenko A.O., Zakharova E.T., Vasilyev V.B., Panasenko O.M. // Free Radic. Res. 2015. V. 49. P. 777–789. https://doi.org/10.3109/10715762.2015.1017478
Козлов С.О., Кудрявцев И.В., Грудинина Н.А., Костевич В.А., Панасенко О.М., Соколов А.В., Васильев В.Б. // Бюллетень ВСНЦ СО РАМН. 2016. Т. 1. № 3 (109). Часть 2. С. 86–91.
Загородний В.В. // Архив патологии. 1982. Т. 44. С. 67–68.
Brown A., Scholtz C.L. // Stain Technol. 1979. V. 54. P. 89–92. https://doi.org/10.3109/10520297909112640
Borges F., Fernandes E., Roleira F. // Curr. Med. Chem. 2002. V. 9. P. 195–217. https://doi.org/10.2174/0929867023371229
Balavoine G.G., Geletii Y.V. // Nitric Oxide. 1999. V. 3. P. 40–54.https://doi.org/10.1006/niox.1999.0206
Kettisen K., Bulow L., Sakai H. // Bioconjug. Chem. 2015. V. 26. P. 746–754. https://doi.org/10.1021/acs.bioconjchem.5b00076
Malle E., Furtmüller P.G., Sattler W., Obinger C. // Br. J. Pharmacol. 2007. V. 152. P. 838–854. https://doi.org/10.1038/sj.bjp.0707358
Sokolov A.V., Kostevich V.A., Varfolomeeva E.Y., Grigorieva D.V., Gorudko I.V., Kozlov S.O., Kudryavtsev I.V., Mikhalchik E.V., Filatov M.V., Cherenkevich S.N., Panasenko O.M., Arnhold J., Vasilyev V.B. // Biochem. Cell Biol. 2018. V. 96. P. 457–467. https://doi.org/10.1139/bcb-2017-0277
Okura Y., Yamada M., Kuribayashi F., Kobayashi I., Ariga T. // J. Clin. Immunol. 2015. V. 35. P. 158–167. https://doi.org/10.1007/s10875-015-0138-4
Bánfi B., Molnár G., Maturana A., Steger K., Hegedûs B., Demaurex N., Krause K.H. // J. Biol. Chem. 2001. V. 276. P. 37 594–37 601. https://doi.org/10.1074/jbc.M103034200
Fuentes E., Gibbins J.M., Holbrook L.M., Palomo I. // Trends Cardiovasc. Med. 2018. V. 28. P. 429–434. https://doi.org/10.1016/j.tcm.2018.03.001
George A., Pushkaran S., Konstantinidis D.G., Koochaki S., Malik P., Mohandas N., Zheng Yi., Joiner C.H., Kalfa T.A. // Blood. 2013. V. 121. P. 2099–2107. https://doi.org/10.1182/blood-2012-07-441188
Morris J.C. // J. Phys. Chem. 1966. V. 70. P. 3798–3805.
Timoshenko A.V., Kayser K., Gabius H.J. // Lectin Methods and Protocols / Eds. Rhodes J.M., Milton J.D. Humana Press, 1998. P. 441–451. https://doi.org/10.1385/0-89603-396-1:441
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия