

Биоорганическая химия, 2021, T. 47, № 2, стр. 287-296
Проблемы синтеза олигонуклеотидных производных при реализации анхимерного эффекта
Е. С. Дюдеева 1, А. С. Павлова 1, М. С. Купрюшкин 1, Д. В. Пышный 1, 2, И. А. Пышная 1, *
1 ФГБУН “Институт химической биологии и фундаментальной медицины” СО РАН
630090 Новосибирск, просп. акад. Лаврентьева, 8, Россия
2 Новосибирский научно-исследовательский государственный университет
630090 Новосибирск, ул. Пирогова, 1, Россия
* E-mail: pyshnaya@niboch.nsc.ru
Поступила в редакцию 27.08.2020
После доработки 09.09.2020
Принята к публикации 11.09.2020
Аннотация
Установлено, что наличие свободной ОН-группы на 3'-концевом остатке замещенного этиленгликолевого фрагмента в составе производного фосфорилгуанидинового олигодезоксирибонуклеотида является фактором, определяющим нестабильность структуры целевого олигонуклеотидного продукта в условиях стандартного протокола деблокирования. Показано, что основными побочными продуктами реализации такого анхимерного эффекта ОН-группы являются продукты реакции трансэтерификации фосфорилгуанидинового (ФГ) звена, несущего остаток O-замещенного этиленгликоля. Данные масс-спектрометрического анализа показывают, что в щелочных условиях происходит накопление производных, лишенных остатка N,N,N ',N '-замещенного гуанидина (1,3-диметилимидазолидин-2-имин, DMI), либо всего 3'-концевого ФГ-содержащего ненуклеотидного звена.
ВВЕДЕНИЕ
На сегодняшний день широкий спектр научно-исследовательских и прикладных задач тем или иным образом задействует аналоги нуклеиновых кислот (НК) – синтетические олигонуклеотиды (ON), которые при помощи различных химических модификаций наделяют необходимыми дополнительными функциональными свойствами. Введение химических модификаций в ON позволяет “регулировать” их свойства в соответствии с решаемой задачей [1, 2]. Например, стабильность ON по отношению к нуклеазам достигается при введении фосфотиоатных [3], 2'-ОМе- и 2'-F-нуклеотидных звеньев [4]. Для повышения сродства олигонуклеотида к комплементарной НК-мишени используют различные модификации углеводофосфатного остова (например, “замкнутые” нуклеотидные звенья [5]). Наличие пептидилнуклеиновых остатков [6], морфолиновых нуклеотидов [7] или фосфорилгуанидиновых остатков [8, 9] в составе ON приводит к эффективной гибридизации ON с НК-мишенью в условиях растворов с низкой ионной силой. Введение различных ненуклеотидных вставок в состав ON направленно изменяет их гибридизационные свойства [10]. Олигонуклеотиды, несущие остаток биотина [11], широко используют для получения фунционализированных поверхностей биосенсоров, биочипов и т.д.
Следует заметить, что синтез производных ON, содержащих сразу несколько различных модификаций, часто является нетривиальной задачей, тем более в условиях стандартного автоматического амидофосфитного синтеза олигонуклеотидов: использование коммерчески доступных нуклеотидных мономеров и полимерных носителей накладывает свои ограничения. Так, мы обнаружили, что выход целевого продукта синтеза фосфорилгуанидинового олигодезоксирибонуклеотида (ФГО), в составе которого присутствует 3'-концевая ненуклеотидная вставка, содержащая этиленгликолевый линкер (рис. 1), значительно ниже такового для нативного олигонуклеотида. Ранее мы провели детальный анализ превращений, реализующихся при получении ФГО в рамках автоматического ДНК-синтеза [12]. С помощью модельных химических соединений было установлено, что нестабильной формой фосфорилгуанидина является заряженный триэфирный фосфорилгуанидиниевый фрагмент, перевод которого в электронейтральную диэфирную форму обеспечивает стабильность ФГ-звена в условиях деблокирования, повышая выход целевого ON.
Рис. 1.
(а) – Обозначения и нуклеотидная последовательность исследуемых олигодезоксирибонуклеотидов, где “o” – нативная фосфатная группа, “*” – фосфорилгуанидиновая группа; (б) – основные структурные элементы исследуемых олигонуклеотидов. Base – азотистые основания, CPG – полимерный носитель, стекло с контролируемым размером пор.

Данная работа направлена на исследование проблемы существенного уменьшения выхода целевого продукта синтеза ФГО, содержащего 3'-концевой ненуклеотидный фрагмент, и выявление факторов, обеспечивающих результативность синтеза этих производных. С использованием методов масс-спектрометрического анализа, SDS-гель-электрофореза и обращенно-фазовой хроматографии (ОФХ) установлено, что ФГ-звено, находящееся вблизи ненуклеотидной вставки на основе замещенного фрагмента этиленгликоля (рис. 1), нестабильно в условиях стандартного протокола деблокирования ON, т.е. при обработке концентрированным водным раствором аммиака.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Чтобы разобраться в проблеме существенного снижения выхода 3'-функционализированных производных ФГО на автоматическом ДНК/РНК-синтезаторе, мы синтезировали 3'-биотиновые производные 26-звенного олигодезоксирибонуклеотида, остов которого содержал либо только заряженные фосфатные группы (N26o, рис. 1а), либо только ФГ-остатки (N26*, рис. 1а). Синтез проводили по амидофосфитному протоколу, описанному в работах [8, 12, 13].
Все олигонуклеотиды были синтезированы с использованием коммерчески доступного CPG-носителя (см. “Эксперим. часть”). В состав данного носителя входит ненуклеотидное звено, содержащее биотиновый остаток на триэтиленгликолевом линкере (рис. 1б), и в ходе автоматического синтеза ДНК к нему присоединяются нуклеотиды, образуя необходимую последовательность. Разрыв связи полученных олигонуклеотидов с CPG-носителем и дальнейшее деблокирование защитных групп олигонуклеотида осуществляли в условиях стандартного протокола деблокирования ON, т.е. при обработке концентрированным водным раствором аммиака. Целевой продукт синтеза, в состав которого входило ненуклеотидное биотиновое звено в виде, приведенном на рис. 1б (R = ОH), выделяли методом ВЭЖХ на обращенной фазе в линейном градиенте ацетонитрила. Характеризацию выделенных производных олигонуклеотидов проводили методом масс-спектрометрии с электроспрей-ионизацией (ESI MS). Немодифицированный олигомер N26o детектировали в виде полианионов. В случае ФГО N26* в анализируемый образец добавляли муравьиную кислоту и детектировали образуемые поликатионы. Стоит отметить, что ФГ-остатки, в отличие от амидофосфатных аналогов, стабильны в кислых и щелочных условиях [8, 13]. Масс-спектры олигонуклеотидов N26o и N26* приведены на рис. 2.
Рис. 2.
Результаты анализа олигодезоксирибонуклеотидов N26o (а) и N26* (б) методом ESI MS; (в) – значения m/z, соответствующие основным продуктам деструкции N26*– утере ненуклеотидного звена, одного или двух остатков DMI.

В случае нативного N26o полученный профиль масс-спектра содержал группу пиков, соответствующих различным зарядовым формам (z = –15…–9) данного олигонуклеотида (рис. 2а). Сопоставимых по интенсивности пиков, соответствующих побочным продуктам, в спектре не обнаруживали. Масс-спектр N26* имел сложный вид (рис. 2б). Для каждой зарядовой формы мы обнаружили группы пиков, отличающихся по значениям m/z. Рассмотрим одну из таких групп более подробно. Прежде всего, видно, что ФГО детектируется как поликатион, содержащий в т.ч. катионы триэтиламмония (“TEA”, компонент буферной смеси). За счет этого мы наблюдали смещение m/z в сторону бóльших значений. Например, два пика с отношением m/z 1313.8 и 1326.1 являются своего рода “аналогами” пика m/z 1301.2. Таким образом, сигналы m/z 1301.2, 1313.8 и 1326.1 отвечают одной и той же зарядовой форме z = +8 одной молекулы, но с различными “видами” катионов, вошедших в структуру детектированного поликатиона: [M + H]+8, [M + TEA + H]+8, [M + 2TEA + H]+8 соответственно. В спектре N26* присутствовали также и пики с меньшими значениями m/z, что свидетельствовало, вероятно, о деградации целевого продукта синтеза. При этом мы не обнаружили значения массы молекулярных ионов, соответствующих полноразмерному продукту синтеза, в котором содержались бы все 25 остатков N,N,N ',N '-замещенного гуанидина (1,3-диметилимидазолидин-2-имин, DMI): все наблюдаемые пики соответствовали утере либо хотя бы одного остатка DMI, либо всего биотин-содержащего ненуклеотидного звена (рис. 2в).
Дополнительно был проанализирован в режиме детекции поликатионов олигонуклеотид N26o. В полученном спектре отсутствовали пики, свидетельствующие о деградации этого немодифицированного олигонуклеотида (данные не приведены). Поскольку процедуры пробоподготовки и самого масс-спектрометрического анализа не приводили к деградации биотинилированного олигонуклеотида, то, вероятно, в случае N26* именно непосредственная близость ФГ-группы и ненуклеотидного звена на 3'-конце ON приводит к разрушению целевого продукта на каком-то из этапов синтеза и/или выделения олигонуклеотида. Чтобы проверить это предположение, был синтезирован олигодезоксирибонуклеотид N25*1o (рис. 1а), в котором между первым (с 3'-конца последовательности) нуклеотидным и ненуклеотидным звеньями был сохранен немодифицированный фосфодиэфирный остаток. Постсинтетическую обработку и выделение целевого продукта проводили согласно приемам олигонуклеотидного синтеза, описанным выше. Полученный продукт N25*1o был также охарактеризован методом масс-спектрометрии, и в спектре не было обнаружено пиков, отвечающих “утере” ненуклеотидного звена (данные не приведены). С помощью метода SDS-гель-электрофореза, адаптированного нами ранее для анализа частично заряженных и электронейтральных аналогов олигонуклеотидов [14], было проведено сравнение продуктов синтеза N26* и N25*1o (рис. 3).
Рис. 3.
Результат электрофоретического анализа биотиновых ФГО в 15%-ном денатурирующем SDS-ПААГ: 1 – N26*, 2 – N25*1o.

Для полностью ФГ-замещенного N26* (рис. 3, дорожка 1) мы наблюдали три отдельных пятна. Вероятно, они соответствуют обнаруженным с помощью масс-спектров продуктам распада, т.е. либо утере биотин-содержащего звена с сохранением всех остатков DMI, либо, наоборот, утере одного или двух остатков DMI с сохранением биотинового звена (рис. 2б). Действительно, при отсутствии ненуклеотидного звена олигонуклеотид N26* сохраняет свою электронейтральность, и верхнее пятно (a) в дорожке 1 может соответствовать ON с незаряженным остовом. В случае утери остатков DMI остов N26* приобретает отрицательный заряд, и это должно привести к увеличению электрофоретической подвижности. Соответственно, пятна (b) и (c) могут быть продуктами деструкции, в которых сохранено ненуклеотидное звено, но утерян один или два остатка DMI. В дорожке 2, где присутствует аналогичный по нуклеотидной последовательности ФГО N25*1o, есть только одно пятно, и его электрофоретическая подвижность полностью совпадает с таковой для пятна (b) в дорожке 1. Таким образом, видно, что при наличии ФГ-группы в 3'-концевой позиции вблизи ненуклеотидного звена N26* деградирует с образованием трех продуктов в сопоставимых количествах. Принимая во внимание этот факт, а также химическую структуру терминального ненуклеотидного звена (рис. 1б), мы предположили, что, вероятно, реализуется “анхимерное содействие”, согласно которому деструкция ФГ-группы ускоряется соседней ОН-группой линкера на основе замещенного этиленгликолевого остатка, входящего в состав ненуклеотидного звена.
Модель деструкции 3'-функционализированных производных ФГО. ФГ-группа в межнуклеозидном положении в целом является электронейтральной после проведения деблокирования ON, но в то же время вероятно, что в ФГ-группе может реализовываться распределение электронной плотности согласно резонансным структурам, приведенным на рис. 4а.
Рис. 4.
(а) – Резонансные структуры, возможные для ФГ-звена, расположенного в межнуклеозидной позиции; (б) – схема внутримолекулярного образования циклического эфира фосфорной кислоты.

Остаток DMI в данной схеме может выступать в роли донора электронов по мезомерному эффекту, стабилизируя положительный заряд, возникший на атоме фосфора в результате поляризации двойной связи P=O. Таким образом, возможно, что в ФГ-группе двойная связь P=O ионизована в бóльшей степени, чем в составе немодифицированной фосфатной группы. В том случае, когда ФГ-группа находится вблизи ненуклеотидного звена (рис. 4б), становится возможной внутримолекулярная атака свободной гидроксигруппы, входящей в состав линкера, по атому фосфора. Это может привести к образованию циклического эфира фосфорной кислоты, который в дальнейшем может деградировать с расщеплением одной из связей P–O или P–N, что и привело бы к утере либо остатка DMI, либо ненуклеотидного звена. Аналогичный эффект гидролиза связей P–O или P–N при реализации анхимерного эффекта карбоксильной и гидроксигрупп описан в работах [15, 16].
Для проверки этой гипотезы мы синтезировали модельные гомо- и гетеротринуклеотиды, содержащие 3'-концевое ненуклеотидное звено и остатки DMI в различных позициях (рис. 5а). Все указанные тринуклеотиды были получены с использованием стандартного автоматического фосфитамидного метода синтеза олигонуклеотидов, обработаны концентрированным раствором аммиака (2 ч, 56°С), и каждая реакционная смесь (без предварительного разделения компонентов реакционной смеси) была проанализирована методом масс-спектрометрии.
Рис. 5.
(а) – Последовательности и теоретические молекулярные массы модельных биотиновых гомо- и гетеротринуклеотидов с различным расположением ФГ-звена; (б) – результаты анализа биотиновых тритимидилатов T*T*To-[Bio] и ToToT*-[Bio] методом ESI MS.
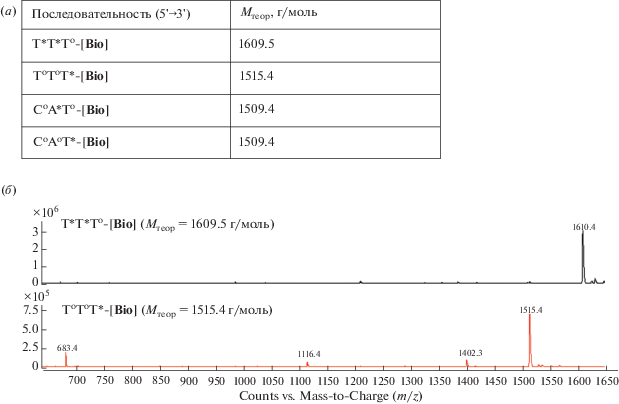
На примере гомотимидилатных олигонуклеотидов T*T*To-[Bio] и ToToT*-[Bio] рассмотрим результаты масс-спектрометрического анализа в режиме детектирования анионов (рис. 5б). Для данных производных тритимидилатов возможны только две зарядовые формы (z = –1 или –2), поскольку структуры содержат лишь одну или две фосфатные группы, способные нести отрицательный заряд. В случае, когда остатки DMI находятся только между нуклеотидными звеньями, в профиле масс-спектра наблюдается пик, соответствующий полноразмерному продукту синтеза T*T*To-[Bio]. Других значений m/z, свидетельствующих о деградации целевого продукта, в спектре не наблюдали.
Во втором случае, когда ФГ-группа находится на 3'-конце рядом с ненуклеотидной вставкой, в полученном масс-спектре присутствует как пик, соответствующий целевому продукту ToToT*-[Bio] (m/z 1515.4, z = –1), так и другие пики с меньшими значениями m/z. Ни один из них не является второй зарядовой формой целевого продукта с z = –2, поскольку в этом случае отношение m/z имело бы значение ~756.7. Следовательно, другие пики также соответствуют зарядовому числу z = –1, но с меньшей молекулярной массой, т.е. снова наблюдается деградация целевого продукта.
Согласно описанной выше гипотезе, тринуклеотид ToToT*-[Bio] мог бы образовать циклический эфир фосфорной кислоты. В процессе обработки концентрированным водным раствором аммиака возможен разрыв одной из связей Р–О или P–N. На рис. 6 приведены возможные продукты деградации в зависимости от того, какая связь была разорвана (пути A, B, C, D). Также на рис. 6 указаны теоретические молекулярные массы биотин-содержащих продуктов распада.
Рис. 6.
Схема деструкции циклического эфира фосфорной кислоты с разрывом одной из связей A, B, C, D. Приведены теоретические молекулярные массы биотин-содержащих продуктов (I–IV).

Весьма вероятно, что образующиеся по пути C или D циклические триэфир (III) и фосфорилгуанидин (IV) могут быть также гидролизованы с образованием ациклических форм, что следует учитывать при анализе масс-спектров.
Так, помимо пика, соответствующего целевому продукту (II) (или его изомерной форме (I), образующейся в соответствии со схемой по пути B), наблюдается пик со значением m/z 1402.3 (рис. 5б). Данное значение может соответствовать циклическому фосфату (III), образовавшемуся по пути C вследствие разрыва связи P–N и утери остатка DMI. Также в полученном спектре было обнаружено значение m/z 683.4, что может соответствовать образованному в случае распада по пути D продукту (IV) в ациклической форме.
Таким образом, основываясь на данных масс-спектрометрического анализа реакционных смесей, полученных в результате синтеза T*T*To-[Bio] и ToToT*-[Bio], можно утверждать, что непосредственная близость ФГ-группы и ненуклеотидного звена, содержащего свободную гидроксигруппу в составе этиленгликолевого линкера, приводит к деструкции целевого продукта синтеза ToToT*-[Bio]. Значения масс, найденные для продуктов распада целевого ON, не противоречат предложенной схеме, основанной на внутримолекулярном образовании циклического эфира фосфорной кислоты при участии прилегающей гидроксигруппы ненуклеотидного звена. Для гетеротринуклеотидов CoA*To-[Bio] и CoAoT*-[Bio] результаты анализа ESI MS ничем принципиально не отличались: деструкция наблюдалась только тогда, когда возможна внутримолекулярная нуклеофильная атака гидроксигруппы по ФГ-группе (с образованием пятичленного цикла).
На примере тринуклеотидов CoA*To-[Bio] и CoAoT*-[Bio], содержащих одну ФГ-группу, дополнительно рассматривали различные условия деблокирования (удаления с полимерного носителя) при 56°С, варьируя время (2 ч или ночь) обработки концентрированным раствором аммиака. Кроме того, опробовали подход, предложенный в работе Bazhenov et al. [12]: последовательно обрабатывали ON безводным 50%-ным раствором триэтиламина в ацетонитриле (1 ч, 56°С), после чего удаляли триэтиламин и обрабатывали ON раствором аммиака (2 ч, 56°С). Такой подход позволяет провести селективное β-элиминирование цианэтильной защитной группы, не удаляя олигонуклеотид с полимерного носителя CPG, что, согласно данным работы Bazhenov et al. [12], “стабилизирует” ФГ-группу при дальнейшей обработке аммиаком. Все реакционные смеси анализировали методом ОФХ (рис. 7).
Рис. 7.
Профили ОФХ, полученные в результате обработки гетеротринуклеотидов CoA*To-[Bio] и CoAoT*-[Bio] водным раствором аммиака в течение 2 ч (а, в) или ночи (б, г) при 56°С.

Видно, что олигонуклеотид CoA*To-[Bio], в котором ФГ-группа расположена между нуклеотидными звеньями, практически не содержит побочных продуктов: на профиле хроматограммы присутствует один выраженный пик, время удерживания ~7.8 мин (рис. 7а). Профили хроматограмм идентичны как в случае двухчасовой обработки олигонуклеотида аммиаком, так и в случае обработки в течение ночи (рис. 7б). Таким образом, деблокирование CoA*To-[Bio] происходит без деструкции целевого продукта, как и ожидалось согласно предложенной схеме (рис. 4).
В случае CoAoT*-[Bio] после 2-часовой обработки аммиаком на хроматограмме наблюдаются три сопоставимых по величине пика (рис. 7в). Один из них также имеет время удерживания ~7.9 мин, что соответствует полноразмерному продукту синтеза, остальные обладают меньшим временем удерживания. В случае последовательной обработки безводным раствором триэтиламина (для удаления β-цианэтильной защитной группы [12]) и затем аммиаком профиль хроматограммы в целом не изменялся (данные не приведены). Длительная обработка CoAoT*-[Bio] аммиаком приводила к почти полному исчезновению пика на 7.9 мин (рис. 7г). Вероятно, это свидетельствует о том, что само по себе наличие остатка DMI возле атома фосфора благоприятствует внутримолекулярной нуклеофильной атаке гидроксигруппы, и дальнейшая деградация циклического эфира происходит необратимо, не приходя к равновесному состоянию. Данный факт накладывает ограничение на химические структуры, в состав которых возможно ввести остаток DMI: непосредственная близость ФГ-группы и заместителей, обладающих нуклеофильными свойствами, может привести к деградации целевого продукта. Например, весьма вероятно, что деградация будет происходить в случае синтеза ФГ-РНК, поскольку 2'-гидроксигруппа рибозы также может атаковать по межнуклеотидному атому фосфора, и введение остатков DMI, по-видимому, будет провоцировать этот процесс.
Таким образом, на примере модельных тринуклеотидов было получено экспериментальное подтверждение того, что непосредственная близость ФГ-группы и нуклеофильных заместителей в структуре олигонуклеотида приводит к разрушению целевого продукта в стандартных условиях деблокирования (обработки водным раствором аммиака). Для точного подтверждения механизма требуются дополнительные эксперименты, поскольку использованные в данной работе методы – масс-спектрометрия и ОФХ – не позволяют судить о конкретных структурных преобразованиях внутри молекулы. Тем не менее, имеющиеся экспериментальные данные согласованно и воспроизводимо указывают на нестабильность ФГ-звена в том случае, если возможна внутримолекулярная нуклеофильная атака по атому фосфора, и подтверждают анхимерное содействие ОН-группы. Эту особенность необходимо учитывать на стадии дизайна функциональных ON, содержащих остатки DMI в своей структуре, поскольку для 2'-OMe ФГ-содержащего 26-звенного биотинилированного олигонуклеотида, содержащего такой же этиленгликолевый линкер в составе ненуклеотидного звена, все описанные выше эффекты сохранялись (данные не приведены).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы. В работе были использованы ацетонитрил, триэтиламин (Fluka, Швейцария); мочевина, акриламид, уксусная кислота, персульфат аммония, ацетон, додецилсульфат натрия (ДиаэМ, Россия); N,N '-метиленбисакриламид, бромфеноловый синий, борная кислота, ЭДТА, N,N-диметилформамид, ксиленцианол, тетраметилэтилендиамин (Sigma, США); реактивы и растворители квалификации х.ч. и ос.ч. отечественного и импортного производства. Абсолютизирование растворителей проводили стандартными методами с последующим выдерживанием их над молекулярными ситами или гидридом кальция.
Синтез олигонуклеотидов. Синтез олигонуклеотидов проводили на автоматическом синтезаторе ASM800 (Биосcет, Россия) согласно стандартному протоколу 2-цианэтильного фосфитамидного метода, используя коммерческие дезоксирибонуклеозидные мономеры и биотин-содержащее пористое стекло (Biotin CPG; Primetech, Республика Беларусь). Олигонуклеотиды, содержащие ФГ-звенья, синтезировали, используя протокол, описанный нами ранее [8, 13].
Деблокирование олигонуклеотидов. 26-Звенные биотиновые олигонуклеотиды удаляли с полимерного носителя, обрабатывая полимер концентрированным водным раствором аммиака в течение ночи при нагревании до 56°С. Для модельных биотиновых тритимидилатов обработку проводили в течение 2 ч при нагревании до 56°С. Гетеротринуклеотиды (CoA*To-[Bio] и CoAoT*-[Bio]) обрабатывали в течение 2 ч при 56°С, затем (если необходимо) выдерживали в концентрированном водном аммиаке в течение ночи. В независимом эксперименте тринуклеотид CoAoT*-[Bio] обрабатывали сначала безводным раствором 50%-ного триэтиламина в ацетонитриле (1 ч, 56°С), затем удаляли раствор триэтиламина и обрабатывали CoAoT*-[Bio] водным раствором аммиака (2 ч, 56°С).
Хроматография. Препаративное выделение (DMTr-on) 26-звенных биотиновых олигонуклеотидов осуществляли методом обращенно-фазовой высокоэффективной жидкостной хроматографии (офВЭЖХ) на хроматографе Agilent 1200 (Agilent, США). Использовали колонку (4.6 × × 150 мм) с сорбентом Eclipse XDB-C18 (5 мкм; Agilent, США), элюировали в линейном градиенте концентрации ацетонитрила (0–90%) в 0.02 М растворе триэтиламмоний ацетата за 40 мин, скорость потока 1.5 мл/мин.
Для анализа реакционных смесей биотиновых гетеротринуклеотидов проводили аналитическую ОФХ на хроматографе Милихром A02 (Эконова, Россия) с использованием колонки (2 × 75 мм) с ProntoSIL 120-5-C18 (Эконова, Россия), элюировали в линейном градиенте концентрации ацетонитрила (0–55%) в 0.02 М водном растворе триэтиламмоний ацетата за 15 мин, скорость потока 150 мкл/мин, температура термостата 35°С. Выход хроматографических пиков детектировали по интенсивности оптического поглощения на длине волны 260, 280 и 300 нм.
Масс-спектрометрия. Молекулярные массы олигонуклеотидов определяли методом ESI MS на приборе Agilent G6410A LCMS/MS (Agilent, США). Для анализа олигонуклеотидов образцы растворяли в 20 мМ триэтиламмоний ацетате в 60%-ном водном ацетонитриле (до концентрации 0.1 мМ и объема 10 мкл). Анализ проводили с использованием 80%-ного водного ацетонитрила в качестве элюента при скорости потока 0.1 мл/мин в режиме регистрации отрицательно заряженных ионов. Для анализа ФГО образец растворяли в 20 мМ муравьиной кислоте в 60%-ном водном ацетонитриле (до концентрации 0.1 мМ и объема 10 мкл), анализ проводили в режиме регистрации положительно заряженных ионов.
Гель-электрофорез. Электрофоретический анализ в денатурирующих условиях проводили в 15%-ном ПААГ, содержавшем SDS (акриламид : N,N '-метиленбисакриламид (29 : 1), 5 М мочевина, 0.05% SDS), в буфере TBE-SDS (89 мМ Трис-борат, pH 8.3, 2 мM Na2EDTA, 0.05% SDS) при напряжении 50 В/см. Для нанесения олигонуклеотидных образцов в гель использовали раствор, содержавший 5 М мочевину, 0.025%-ный ксиленцианол и 0.025%-ный бромфеноловый синий, 0.05% SDS. Результаты электрофоретического разделения визуализировали, помещая гель на флуорофор-содержащую пластинку TLC Silica gel 60 F254 (Merck, США) и облучая УФ-светом (265 нм). Олигонуклеотидный материал проявляется в виде “теней” на фоне флуоресцирующей подложки.
Список литературы
Glazier D.A., Liao J., Roberts B.L., Li X., Yang K., Stevens C.M., Tang W. // Bioconjugate Chem. 2020. V. 31. P. 1213–1233. https://doi.org/10.1021/acs.bioconjchem.0c00060
Bennett C.F. // Annu. Rev. Med. 2019. V. 70. P. 307–321. https://doi.org/10.1146/annurev-med-041217-010829
Eckstein F. // Nucleic Acid Ther. 2014. V. 24. P. 374–387. https://doi.org/10.1089/nat.2014.0506
Rettig G.R., Behlke M.A. // Mol. Ther. 2012. V. 20. P. 483–512. https://doi.org/10.1038/mt.2011.263
Braasch D.A., Corey D.R. // Chem. Biol. 2001. V. 8. P. 1–7. https://doi.org/10.1016/S1074-5521(00)00058-2
Nielsen P.E. // Chem. Biodivers. 2010. V. 7. P. 786–804. https://doi.org/10.1002/cbdv.201000005
Du L., Gatti R.A. // J. Immunol. Methods. 2011. V. 365. P. 1–7. https://doi.org/10.1016/j.jim.2010.12.001
Kupryushkin M.S., Pyshnyi D.V., Stetsenko D.A. // Acta Naturae. 2014. V. 6. P. 116–118.
Dyudeeva E.S., Kupryushkin M.S., Lomzov A.A., Pyshnaya I.A., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 709–718. https://doi.org/10.1134/S1068162019060153
Pyshnaya I.A., Lomzov A.A., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 677–683. https://doi.org/10.1134/S1068162019060335
Kor K., Turner A., Zarei K., Atabati M., Beni V., Mak W.C. // Anal. Bioanal. Chem. 2016. V. 408. P. 1475–1485. https://doi.org/10.1007/s00216-015-9250-9
Bazhenov M.A., Shernyukov A.V., Kupryushkin M.S., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 699–708. https://doi.org/10.1134/S1068162019060074
Stetsenko D.A., Kupryushkin M.S., Pyshnyi D.V. // Patent WO2016028187A1, 2016.
Pavlova A.S., Dyudeeva E.S., Kupryushkin M.S., Amirkhanov N.V., Pyshnyi D.V., Pyshnaya I.A. // Electrophoresis. 2018. V. 39. P. 670–674. https://doi.org/10.1002/elps.201700415
Maiti M., Michielssens S., Dyubankova N., Maiti M., Lescrinier E., Ceulemans A., Herdewijn P. // Chemistry. 2012. V. 18. P. 857–868. https://doi.org/10.1002/chem.201102279
Kupryushkin M.S., Konevetz D.A., Vasilyeva S.V., Kuznetsova A.S., Stetsenko D.A., Pyshnyi D.V. // Nucleosides Nucleotides Nucleic Acids. 2013. V. 32. P. 306–319. https://doi.org/10.1080/15257770.2013.787147
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия