

Биоорганическая химия, 2021, T. 47, № 3, стр. 348-364
Триазиниламидофосфатные олигонуклеотиды: получение и исследование их взаимодействия с клетками и днк-связывающими белками
М. С. Купрюшкин 1, Т. Д. Жарков 1, 2, Е. С. Ильина 1, 2, О. В. Марков 1, А. С. Кочеткова 1, 2, М. М. Ахметова 2, А. А. Ломзов 1, Д. В. Пышный 1, О. И. Лаврик 1, 2, С. Н. Ходырева 1, *
1 ФГБУН “Институт химической биологии и фундаментальной медицины” СО РАН
630090 Новосибирск, просп. акад. Лаврентьева, 8, Россия
2 ФГБОУ ВПО “Новосибирский национальный исследовательский государственный университет”
630090 Новосибирск, ул. Пирогова, 2, Россия
* E-mail: svetakh@niboch.nsc.ru
Поступила в редакцию 19.10.2020
После доработки 26.10.2020
Принята к публикации 28.10.2020
Аннотация
Впервые получены представители класса триазиниламидофосфатных олигонуклеотидных производных, предложена и осуществлена схема введения модифицированного звена на твердофазном носителе при проведении стадии окисления высокореакционным 2-азидо-4,6-дихлор[1,3,5]триазином по реакции Штаудингера с последующим введением различных алифатических остатков при обработке соответствующими аминами. Был синтезирован ряд модельных олигонуклеотидов, содержащих триазиниламидофосфатную модификацию. Исследованы химическая устойчивость, проникновение в клетки и цитотоксичность полученных производных. Изучено влияние типа алифатического заместителя в составе полученных олигонуклеотидов на их устойчивость к ферментативному гидролизу в экстрактах культивируемых клеток человека с различным содержанием ключевых регуляторных белков репарации ДНК, Ku-антигена и поли(АДФ-рибоза)полимеразы 1 (PARP1) – потенциально наиболее эффективных акцепторов модифицированных олигонуклеотидов.
ВВЕДЕНИЕ
Синтетические олигонуклеотиды широко применяются в различных областях молекулярной биологии, биотехнологии и медицины [1, 2]. Одним из наиболее перспективных направлений можно считать использование олигонуклеотидов в качестве терапевтических агентов – не менее 11 препаратов уже одобрены организацией FDA (Food and Drug Administration) и множество препаратов находятся на различных стадиях клинических испытаний [3–5]. В области онкотерапии существует подход, основанный на применении ДНК-повреждающих агентов совместно с синтетическими олигонуклеотидами в качестве ингибиторов систем репарации [6]. Такие олигонуклеотиды имитируют реальные повреждения клеточной ДНК, что позволяет их использовать в качестве эффективных “ловушек”. Ключевые белки репарации ДНК связываются с такими олигонуклеотидами, что вызывает ингибирование репарации. В результате возникшие при химио- или лучевой терапии повреждения клеточной ДНК не репарируются, что приводит к гибели раковой клетки.
В составе каждого олигонуклеотидного препарата присутствуют химические модификации, что позволяет значительно улучшить важные для его биологического применения свойства, такие как эффективность проникновения через клеточную мембрану, химическая и ферментативная устойчивость, стабильность образуемого дуплекса с комплементарным участком, улучшенное биораспределение и т.п. [7]. Не исключена возможность с помощью таких модификаций улучшить эффективность связывания с олигонуклеотидами белков систем репарации.
В структуре олигонуклеотида присутствует ряд положений для модификации – азотистые основания, углеводный компонент сахарофосфатного остова, межнуклеотидная фосфатная группа. Для введения неприродных химических групп межнуклеотидная фосфатная группа выгодно отличается от остальных позиций, поскольку введение модификаций через фосфатный фрагмент остова незначительно влияет на фундаментальное свойство олигонуклеотидов – способность образовывать комплементарные комплексы с клеточными мишенями – нуклеиновыми кислотами. Поскольку в качестве белков-мишеней систем репарации нами были выбраны Ku-антиген и PARP1 (поли(АДФ-рибоза)полимераза 1), взаимодействующие преимущественно с ДНК-дуплексами и имеющие высокое сродство к концам ДНК-дуплексов, имитирующим двойные разрывы, то сохранение стабильности дуплексов имеет большое значение. При этом введение различных групп способно наделить создаваемое соединение широким диапазоном новых свойств.
На данный момент известно множество типов модификаций межнуклеотидной фосфатной группы. К примеру, ранее в Институте химической биологии и фундаментальной медицины СО РАН был предложен новый класс аналогов нуклеиновых кислот – фосфорилгуанидиновые олигонуклеотиды (ФГО), полученные при взаимодействии фосфит-триэфирного производного олигонуклеотида с различными диаминокарбений азидами [8]. Простота и эффективность введения модификации с использованием пока единственного коммерчески доступного азида-модификатора позволяют получать ФГО, обладающие различными свойствами, обусловленными варьированием количества и места введения фосфорилгуанидиновых звеньев, но не их типом. Получение же диаминокарбений азидов, несущих различные функциональные остатки, – отдельная задача синтетической химии для каждого типа вводимых заместителей. В связи с этим актуальна разработка нового типа модификации по фосфатной группе, введение которой позволит легко варьировать различные функциональные группы в составе создаваемого олигонуклеотидного производного, а также при конструировании ДНК-дуплексов, проникающих в клетки и эффективно взаимодействующих с белками систем репарации.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Выбор структуры реагента-модификатора. Учитывая успешный опыт применения реакции Штаудингера при создании олигонуклеотидов, модифицированных по межнуклеотидной фосфатной группе, в качестве реагента-модификатора было принято решение использовать органический азид. При этом для эффективного протекания реакции необходимо, чтобы органический заместитель при азидогруппе обладал выраженными электронно-акцепторными свойствами. С другой стороны, для возможности варьирования вводимых заместителей необходимо наличие в структуре модификатора дополнительных реакционноспособных центров.
После рассмотрения различных вариантов остова в качестве наиболее подходящего был выбран замещенный 2,4,6-замещенный-1,3,5-триазин и, соответственно, цианурхлорид как его коммерчески доступный предшественник.
Триазиновый остов модификации был выбран по нескольким причинам. Во-первых, наличие трех реакционных центров в структуре цианурхлорида обеспечивает, помимо введения необходимой азидогруппы в состав целевого модификатора, возможность введения еще двух функциональных групп. Во-вторых, триазиновый остаток, особенно с электронно-акцепторными заместителями, обеспечит высокую реакционноспособность создаваемому азиду в реакции Штаудингера. В-третьих, получаемая после этапа финального деблокирования триазиниламидофосфатная группа, в отличие от алкиламидофосфатов, не должна быть склонна к кислотному гидролизу.
Для получения 2-азидо-4,6-дихлор-1,3,5-триазина необходимо заместить один из трех эквивалентных атомов хлора в исходном цианурхлориде на азидогруппу. Известно, что последовательные реакции замещения атомов хлора на донорные группы в цианурхлориде протекают при разных температурах – если первое замещение проходит при 0°С, то второе – уже при 25°С, а последнее замещение протекает только при нагревании до 60–70°С [9]. Однако азидогруппа по своим акцепторным свойствам близка к атому хлора, поэтому даже при охлаждении продуктами реакции цианурхлорида с азидом натрия (рис. 1, реакция 1) выступают моно-, ди-, и тризамещенные азидотриазины. В связи с этим требуется тщательно подбирать условия реакции [10], а также дополнительно очищать синтезированный азид от примесей продуктов реакций избыточного замещения.
Рис. 1.
Принципиальная схема синтеза триазиниламидофосфатных производных: 1 – получение 2-азидо-4,6-дихлор-1,3,5-триазина; 2 – введение триазинового остова синтезируемой олигонуклеотидной цепи через реакцию Штаудингера; 3 – введение различных заместителей в состав создаваемой структуры, продолжение олигонуклеотидного синтеза и финальное деблокирование с получением целевого соединения.
Учитывая электронно-акцепторную природу атомов хлора в составе полученного азида, было принято решение использовать именно это соединение в качестве азида-модификатора на этапе окисления образующегося фосфит-триэфира при конденсации очередного звена при синтезе олигонуклеотидов амидофосфитным методом синтеза (рис. 1, реакция 2). При этом с использованием соответствующих аминов атомы хлора триазинового производного будут замещены на функциональные остатки уже в составе получаемого фосфазенового олигонуклеотидного производного (рис. 1, реакция 3). Большое количество разнообразных коммерчески доступных аминов позволит широко варьировать вводимые группировки, что обеспечит необходимую универсальность выбранного подхода.
Таким образом, в рамках выбранной стратегии получения триазиниламидофосфатных производных олигонуклеотидов предполагается использование наиболее реакционноспособного азидотриазина – 2-азидо-4,6-дихлор-1,3,5-триазина, получение общего промежуточного фосфазенового производного и дальнейшая обработка разнообразными аминами для введения в состав получаемого олигонуклеотидного производного интересующих функциональных групп.
Исследование эффективности протекания реакции Штаудингера, получение модельных триазиниламидофосфатных декатимидилатов. В качестве модельных систем для проверки эффективности протекания реакции модификации были выбраны декатимидилаты, в которых модификация вводилась в ближайший к 5'-концу межнуклеотидный фосфат.
Получение модельных олигонуклеотидов осуществляли следующим образом. Синтез олигонуклеотидной цепи проводили на ДНК-синтезаторе вплоть до стадии конденсации звена, чья фосфатная часть будет модифицирована триазиновым остатком. Далее полученный фосфит-триэфир, иммобилизованный на твердофазном носителе, переносили в пробирку и проводили этап окисления добавлением раствора 2-азидо-4,6-дихлор-1,3,5-триазина в ацетонитриле (0.1 М, 15 мин, 25°C). После отмывки полимера от азида-модификатора добавляли раствор бутиламина (1 М в ацетонитриле, 60 мин, 25°C, О2) или додециламина (3 М в пиридине, 30 мин, 55°C, О3) для введения в структуру синтезируемого олигонуклеотида соответствующих остатков. Далее полимерный носитель переносили в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по амидофосфитному протоколу. После завершения олигонуклеотидного синтеза проводили финальное деблокирование концентрированным водным раствором метиламина. В случае остатков метиламина введение соответствующих остатков выполняли непосредственно на этапе финального деблокирования (О1).
Общая схема получения модельных декатимидилатов представлена на рис. 2.
Рис. 2.
Общая схема синтеза модифицированных триазиниламидофосфатных олигонуклеотидов. CPG – полимерный носитель, Base – азотистое основание, DMTr – диметокситритильная защитная группа, R1 и R2 – вводимые в состав олигонуклеотида заместители.

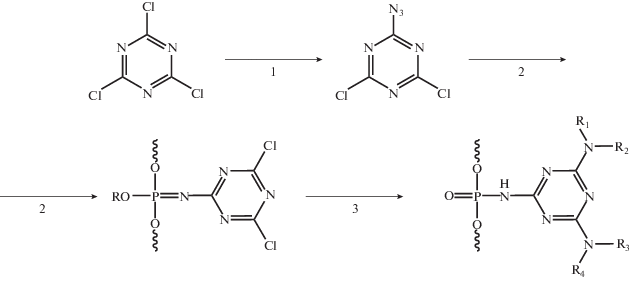
Эффективность модификации для всех заместителей была оценена методом офВЭЖХ реакционных смесей олигонуклеотидов (рис. 3). Из анализа профилей хроматографического разделения реакционных смесей различных представителей триазиниламидофосфатов можно сделать вывод, что выходы целевых продуктов сильно зависят от условий протекания реакций с участием аминов, несущих функциональные остатки. При этом, как и ожидалось, стадия окисления азидотриазином проходит более чем на 90% даже при комнатной температуре.
Рис. 3.
Профиль офВЭЖХ реакционной смеси модельных триазиниламидофосфатных олигонуклеотидов с остатками метиламина (О1, желтый), бутиламина (O2, зеленый) и додециламина (O3, красный) и контрольного олигонуклеотида без модификации (Т10, синий). Градиент: 0–90% CH3CN за 15 мин, λ = 260 нм.
Таким образом, на модельных олигонуклеотидах была исследована эффективность получения триазиниламидофосфатных производных и их устойчивость в рамках протоколов твердофазного автоматического амидофосфитного синтеза ДНК, и, в частности, устойчивость вводимых модификаций к жестким щелочным условиям – обработке концентрированным водным раствором метиламина.
Получение модифицированных олигонуклеотидов, обладающих гетеронуклеотидной последовательностью. При исследовании возможности введения триазиниламидофосфатной модификации в состав синтезируемой олигонуклеотидной последовательности, содержащей звенья, отличные от тимидилатов, после завершения синтеза полноразмерной последовательности в реакционной смеси были обнаружены значительные количества побочных продуктов (данные не приведены). Вероятно, в результате наличия нескольких реакционных центров в 2-азидо-4,6-дихлор-1,3,5-три-азине при проведении этапа окисления происходит и побочная реакция взаимодействия используемого модификатора даже с защищенными экзоциклическими аминогруппами цитозина, гуанина и аденина. Данный факт – ограничение используемого подхода для введения триазин-амидофосфатных модификаций в произвольное положение гетеронуклеотидной последовательности. Однако это не препятствует получению олигонуклеотидов, содержащих модификацию в 3'-концевой области последовательности, где модифицируемый фосфат будет располагаться между звеньями, не содержащими цитозина, гуанина или аденина, например, между двумя тимидилатными звеньями.
В качестве последовательности для дальнейшего изучения свойств создаваемых модифицированных олигонуклеотидов был выбран 16-звенный олигонуклеотид 5'-[FAM]-CTGACTATGAAGTAT*T-3', содержащий на 5'-конце остаток флуоресцеина и триазиниламидофосфатную модификацию в 3'-концевой области. Были синтезированы модифицированные олигонуклеотиды, содержащие бутильные остатки (OSN1F), додецильные (OSN4F), контрольный олигонуклеотид, не содержащий триазиновой модификации (OSNF) и комплементарный ему олигонуклеотид 5'-AATACTTCATAGTCAG-3'.
Стоит отметить, что выбор вводимых заместителей не случаен. Ранее мы показали [11], что олигонуклеотиды, содержащие два и более остатка додецила, способны с определенной эффективностью проникать внутрь клетки. Сравнение эффективности проникновения различных три-азиниламидофосфатных олигонуклеотидов позволит оценить вклад как триазинового остова (OSNF против OSN1F), так и гидрофобности заместителей в остове (OSN1F против OSN4F).
Полученные олигонуклеотиды были очищены и охарактеризованы методами офВЭЖХ и масс-спектрометрии. На рис. 4 представлены профили аналитической офВЭЖХ выделенных олигонуклеотидов. Последовательности олигонуклеотидов и результаты ESI масс-спектрометрии приведены в табл. 1.
Рис. 4.
Профиль офВЭЖХ реакционной смеси триазиниламидофосфатных олигонуклеотидов с остатками бутиламина (OSN1F) додециламина (OSN4F) и контрольного олигонуклеотида без модификации (OSNF). Градиент: 0–90% CH3CN за 15 мин, λ = 260 нм.

Таблица 1.
Последовательности полученных олигонуклеотидов и результаты масс-спектрометрического анализа
| Шифр | Последовательность (5'–3') | Вводимые остатки | Мтеор, г/моль | Мэксп, г/моль |
|---|---|---|---|---|
| OSNF | [FAM]CTGACTATGAAGTATT | – | 5432.3 | 5432.0 |
| OSN1F | [FAM]CTGACTATGAAGTAT*T | Бутил | 5652.5 | 5652.1 |
| OSN4F | [FAM]CTGACTATGAAGTAT*T | Додецил | 5876.8 | 5876.4 |
| ISN | AATACTTCATAGTCAG | – | 4864.3 | 4863.8 |
Представленные профили аналитической офВЭЖХ наглядно демонстрируют зависимость гидрофобности получаемых соединений от типа вводимых заместителей.
Исследование устойчивости триазиниламидофосфатного звена в кислой среде. Исследуемые производные можно отнести к классу амидофосфатных производных. Известно, что амидофосфаты могут подвергаться кислотному гидролизу. Степень гидролиза выше для алкиламидофосфатов и снижается при переходе к ароматическим заместителям [12].
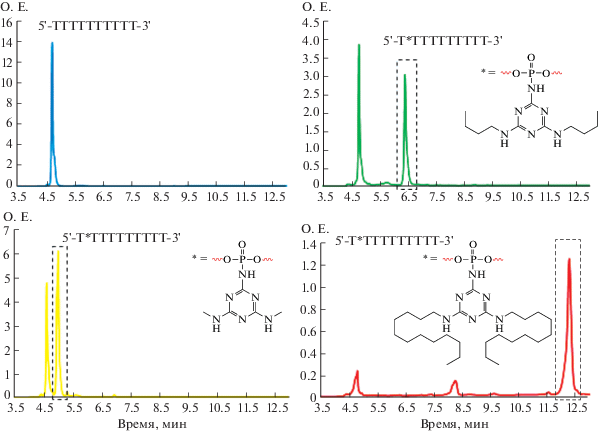
Для сравнения гидролитической активности разных типов модификаций был проведен эксперимент по выдерживанию в кислой среде (0.1 М HCl) олиготимидилатов с триазиниламидофосфатной (O2) и бензиламидофосфатной (Bz) модификациями. Результаты анализа гидролиза с течением времени представлены на рис. 5.
Рис. 5.
Химическая устойчивость триазиниламидофосфатов и бензиламидофосфатов в кислой среде. Содержание негидролизованного соединения оценивали методом офВЭЖХ в пробах через 30, 90, 180 и 780 мин после начала реакции.
Количество олигонуклеотида, содержащего бензиламидофосфатную модификацию (Bz), уже через 3 ч снижалось на 60%, а после 13 ч – более чем на 85%. В то же время количество олигонуклеотида, содержащего триазиниламидофосфатную модификацию (O2), не менялось на протяжении всего времени эксперимента.
Таким образом, можно заключить, что триазиниламидофосфатная модификация, в отличие от алкиламидофосфатных аналогов, обладает высокой степенью устойчивости при низких значениях pH.
Выбор модельных клеточных линий и оценка в них содержания белков репарации. Для изучения возможности применения модифицированных олигонуклеотидов в качестве агентов, сенсибилизирующих клетки к действию химио- или лучевой терапии, необходима информация о взаимодействии олигонуклеотидов с ключевыми белками-инициаторами репарации ДНК, в частности с Ku-антигеном и PARP1. Ku-антиген –ключевой фактор процесса негомологичной рекомбинации, ответственной за исправление двойных разрывов в ДНК [13]. PARP1 регулирует различные механизмы репарации, в том числе репарацию двойных разрывов в ДНК [14]. Для этих целей рационально проводить исследования на модельных клеточных системах с известным содержанием этих белков.
Для оценки в клетках количества ключевых белков репарации был использован метод аффинной модификации фотоактивируемым ДНК-интермедиатом эксцизионной репарации оснований (рис. 6). Анализ содержания целевых белков в экстрактах основан на оценке количества ковалентных комплексов, образованных ими с мечеными ДНК-интермедиатами репарации. Этот метод продемонстрировал свою эффективность при исследовании репарации ДНК в экстрактах клеток млекопитающих, различающихся по содержанию Ku-антигена и PARP1 [15].
Рис. 6.
(а) – Фотоаффинная модификация белков цельноклеточных экстрактов. Наименование клеточных линий приведено под электрофореграммой. Реакционные смеси содержали 1 мг/мл белков экстракта, 100 нМ [32P]-содержащей фотоактивируемой ДНК и другие стандартные компоненты. Схематическое изображение использованной ДНК приведено над рисунком. Красной звездочкой указано положение радиоактивного фосфата, желтой звездочкой – положение фотоактивируемого остатка FAP-dCMP; (б) – структурная формула FAP-dCTP.
При выборе модельных клеточных линий в панель были включены онкотрансформированные клетки разного происхождения: шейка матки (HeLa), молочная железа (MCF7), периферическая кровь (В-лимфобласты (Im9), лимфома Беркита (BJAB)), мозг человека (глиобластома T98G), а также иммортализованные неонкотрансформированные клетки эмбриональной почки человека (HEK293T) и эмбриональные легочные фибробласты человека (MRC5).
Анализ данных показывает, что основными белками-мишенями фотоактивируемых ДНК выступают PARP1, Ku80- и Ku70-субъединицы Ku-антигена (рис. 6). Интересно отметить, что соотношение интенсивностей продуктов мечения, относящихся к PARP1 и Ku-антигену, варьирует в зависимости от экстракта. В экстракте HEK293Т (дорожка 4) содержание Ku-антигена и PARP1 близко к максимальному, а в экстракте T98G (дорожка 3) эти белки практически не детектируются.
Содержание Ku-антигена и PARP1 в клеточных экстрактах было дополнительно оценено методом вестерн-блоттинга (рис. 7). Оценка количеств белков PARP1 и Ku-антигена в экстрактах клеток на основе данных фотоаффинной модификации (рис. 6) хорошо согласуется с результатами вестерн-блот-анализа этих белков в экстрактах (рис. 7). Экстракт T98G существенно обеднен по целевым белкам, а экстракт HEK293T содержит количества этих белков, близкие к максимальным. Интересно отметить, что использованные подходы взаимно дополняют друг друга. Вестерн-блоттинг позволяет оценить содержание целевых полипептидов, но не их активность в связывании ДНК. Аффинная модификация детектирует молекулы белка, связанные с ДНК, и учитывает конкуренцию других ДНК-связывающих белков экстракта. Следует отметить, что HEK293T – часто используемая в работах контрольная (неонкотрансформированная) клеточная линия.
Рис. 7.
Содержание PARP1 и Ku-антигена в экстрактах различных клеток. На дорожках 1–7 нанесены белки клеточных экстрактов (указаны сверху) с суммарным содержанием белка 10 мкг на пробу. На дорожке 8 нанесено 28 нг индивидуального PARP1 или 38 нг индивидуального Ku-антигена.

Таким образом, для дальнейших экспериментов были выбраны клеточные линии HEK293T и T98G со значительным различием по содержанию целевых белков. Кроме того, эти клеточные линии неприхотливы в культивировании.
Проверка устойчивости модифицированных олигонуклеотидов к действию нуклеаз цельноклеточных экстрактов. В эукариотической клетке присутствует более десятка белков для деградации собственной и чужеродной ДНК [16, 17]. В связи с этим для оценки влияния триазиниламидофосфатной группы на устойчивость одно- и двуцепочечных олигонуклеотидов к действию нуклеаз были проведены эксперименты с использованием цельноклеточных экстрактов линий HEK293T и T98G, выбранных нами для дальнейших исследований (рис. 8).
Рис. 8.
Устойчивость одноцепочечных олигонуклеотидов и ДНК-дуплексов к действию нуклеаз клеточных экстрактов HEK293T и T98G. Реакционные смеси, содержащие 1 мг/мл белков экстракта HEK293T или T98G, 100 нМ FAM-содержащих олигонуклеотидов (OSNF, OSNF/ISN, OSN1F, OSN1F/ISN, OSN4F, OSN4F/ISN, схематическое изображение приведено сверху) и другие стандартные компоненты, инкубировали 7.5 или 15 мин при 37°С, затем аликвоты анализировали в ПААГ в присутствии 7 М мочевины. Желтой звездочкой отмечено положение флуоресцентной метки, голубым кружком показано положение триазинового заместителя.

Для модифицированных ДНК-дуплексов (OSN1F/ISN и OSN4F/ISN) и контрольного немодифицированного дуплекса (OSNF/ISN) было установлено, что температуры их плавления (Тпл) в условиях, близких к физиологическим (см. “Эксперим. часть”), лишь незначительно различаются и составляют, соответственно, 49.6, 49.2 и 50°С. Следовательно, введение предложенных модификаций межнуклеотидного фосфата вблизи конца дуплексной структуры слабо влияет на его термическую стабильность. При этом увеличение объема и гидрофобности остатка (OSN4F/ISN, Тпл = 49.2°С) в большей степени дестабилизирует двойную спираль.
Из данных, представленных на рис. 8, можно выявить ряд закономерностей. Во-первых, для обоих экстрактов характерна меньшая устойчивость всех одноцепочечных олигонуклеотидов к действию нуклеаз по сравнению с соответствующими ДНК-дуплексами; во-вторых, эффективность расщепления одноцепочечных олигонуклеотидов несколько выше в экстракте HEK293T, чем в экстракте T98G, причем эта закономерность наблюдается для нативного и модифицированных олигонуклеотидов; в-третьих, наличие и тип модифицирующей группы вблизи 3'-конца вносят значимый вклад в устойчивость олигонуклеотидов. Для немодифицированного олигонуклеотида в одноцепочечном состоянии или в составе дуплекса в экстракте T98G удалось зарегистрировать продукты частичного экзонуклеазного гидролиза в направлении 3'→5', в отличие от экстракта HEK293T, где детектировались только продукты полного гидролиза, обладающие максимальной электрофоретической подвижностью. Следует отметить, что формирование низкомолекулярного продукта может быть дополнительно обусловлено непосредственным расщеплением остова вблизи FAM-группы. Отсутствие продуктов “пошагового” расщепления в экстракте HEK293T, скорее всего, обусловлено защитным эффектом белков экстракта, в частности Ku-антигена. Ku-антиген связывается с ДНК, “надеваясь как бусина на нитку”, локализуется преимущественно вблизи концов и накрывает ~12–14 нт [18]. Ku-антиген способен связываться с одноцепочечной ДНК, хотя и менее эффективно [19]. Косвенно на защитное действие Ku-антигена указывает значительно более выраженный эффект на ДНК-дуплексах, проявляющийся в экстракте HEK293T, который обогащен по этому белку. Ku-антиген способен “нагружаться” на ДНК со стороны тупого конца, не содержащего модификации, в то время как введение объемной флуоресцентной группы в 5'-конец, вероятно, препятствует связыванию Ku-антигена с ДНК с соответствующего тупого конца или затрудняет этот этап. Учитывая размер отрезка ДНК, находящегося в непосредственном контакте с Ku-антигеном, и длину олигонуклеотида (16 звеньев), можно предположить, что возле FAM-модифицированного конца остается достаточное пространство для связывания нуклеаз, которые расщепляют остов ДНК. На возможность такого хода событий указывает отсутствие продуктов “пошагового” 3'→5' экзонуклеазного расщепления в экстракте HEK293T, характерного для экстракта T98G. Хотя Ku-антиген преимущественно локализуется возле концов, он может перемещаться вдоль ДНК-дуплекса без затрат энергии [20], благодаря чему возможно проявление его защитного эффекта в участках ДНК, удаленных от концов.
В том случае, если связывание Ku-антигена затруднено, следует ожидать отсутствия его защитного влияния. Действительно, для дуплексов OSN1F/ISN и OSN4F/ISN с модифицированными олигонуклеотидами характер расщепления в обоих экстрактах схож и резко отличается от картины, характерной для немодифицированного дуплекса OSNF/ISN.
В целом устойчивость олигонуклеотидов к действию клеточных нуклеаз в направлении 3'→5' как для одноцепочечных олигонуклеотидов, так и в составе ДНК-дуплексов, уменьшается в ряду OSN4F > OSN1F > OSNF. Таким образом, введение триазинового производного в область 3'-конца олигонуклеотида увеличивает его устойчивость к действию клеточных нуклеаз, причем увеличение размера заместителей в составе вводимого производного приводит к увеличению этой устойчивости.
Таким образом, было показано, что по сравнению с немодифицированными олигонуклеотидами их триазиниламидофосфатные производные обладают повышенной устойчивостью к ферментативному гидролизу, катализируемому клеточными нуклеазами, что значительно расширяет возможности использования таких олигонуклеотидов в экспериментах с клетками.
Проникновение триазиниламидофосфатных олигонуклеотидов через клеточную мембрану. Для оценки способности олигонуклеотида OSN4F, содержащего два остатка додецила, проникать в клетки в отсутствие трансфектантов с помощью проточной цитофлуорометрии было исследовано внутриклеточное накопление OSN4F в клетках HEK293T и T98G. В качестве отрицательного контроля использовали доставку контрольного олигонуклеотида OSNF в отсутствие трансфектантов, а в качестве положительного контроля – доставку OSNF с помощью Липофектамина 2000. Для определения влияния замены нативной межнуклеотидной фосфатной группы на триазиниламидофосфатное звено с нейтрализацией заряда без увеличения общей гидрофобности были также проведены эксперименты по оценке проникновения олигонуклеотида, содержащего два бутильных остатка (OSN1F). Трансфекцию клеток проводили в течение 4 ч в среде IMDM без сыворотки и антибиотиков, после чего клетки анализировали с помощью проточной цитофлуорометрии (рис. 9). Из представленных данных видно, что олигонуклеотид OSN4F эффективно проникает в клетки HEK293T как с точки зрения количества трансфицированных клеток, так и интенсивности флуоресценции (рис. 9а, 9б), при этом эффективность трансфекции напрямую зависела от концентрации олигонуклеотида в инкубационной среде – до 100% клеток были трансфицированы с интенсивностью флуоресценции от 1.3 × 105 до 4.2 × 105 RFU для 1 и 5 мкМ OSN4F соответственно. При этом уровень трансфекции OSN4F был сопоставим с эффективностью доставки контрольного олигонуклеотида OSNF с помощью коммерческого трансфектанта Липофектамина 2000 (100% трансфицированных клеток, 2.9 × 105–5.8 × 105 RFU).
Рис. 9.
Внутриклеточное накопление олигонуклеотидов в клетках HEK293T (а, б) и T98G (в, г). Процент флуоресцентных клеток (а, в) и среднюю интенсивность флуоресценции (б, г) измеряли с помощью проточной цитофлуорометрии через 4 ч после трансфекции. RFU – относительные флуоресцентные единицы, LF – Липофектамин 2000. Данные представлены как средние значения ± SD.
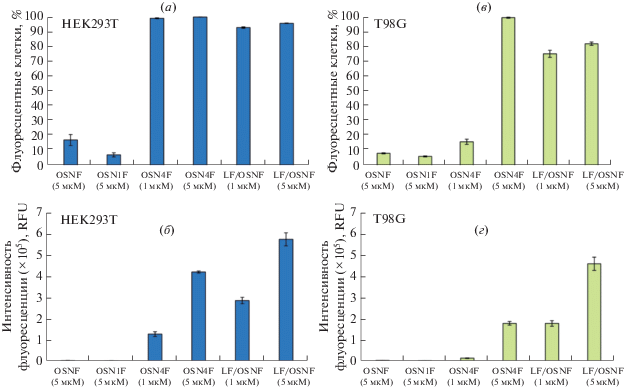
Для клеток глиобластомы T98G было показано, что эффективность накопления как для олигонуклеотида OSN4F, так и комплексов OSNF с Липофектамином 2000 была 2–5 раз ниже по сравнению с клетками HEK293T (рис. 9в, 9г), при этом сохранялась дозозависимая закономерность увеличения эффективности трансфекции. Снижение эффективности трансфекции для данного типа клеток, вероятно, связано с их морфологическими и метаболическими особенностями. Для клеток T98G также следует отметить, что в отличие от HEK293T, разница между эффективностью доставки олигонуклеотида OSN4F и липофекцией OSNF с помощью Липофектамина 2000 была значительнее – интенсивность флуоресценции составила 1.5 × 104–1.8 × 105 для 1 и 5 мкМ OSN4F, 1.8 × 105–4.6 × 105 для 1 и 5 мкМ Липофектамина 2000/OSNF соответственно.
Таким образом, было показано, что олигонуклеотид OSN4F способен эффективно накапливаться в клетках HEK293T и T98G, при этом эффективность его доставки сопоставима (для HEK293T) или в 2 раза ниже (для T98G) по сравнению с эффективностью трансфекции контрольного олигонуклеотида с помощью Липофектамина 2000.
Внутриклеточное накопление дуплексов в клетках HEK293T и T98G. С помощью проточной цитофлуорометрии была также исследована эффективность накопления дуплекса OSN4F/ISN в клетках HEK293T и T98G через 4 ч после начала трансфекции. В качестве отрицательного контроля использовали дуплекс, сформированный из контрольного немодифицированного олигонуклеотида OSNF, не несущего липофильные модификации, и комплементарного олигонуклеотида ISN. Основываясь на результатах эксперимента по доставке одноцепочечного олигонуклеотида, описанного выше, в качестве рабочей была выбрана концентрация 5 мкМ.
Показано, что дуплекс OSN4F/ISN эффективно накапливался в клетках HEK293T (100% трансфицированных клеток, 4.1 × 105 RFU) и в 2 раза менее эффективно – в клетках T98G (100% трансфицированных клеток, 1.9 × 105 RFU) (рис. 10). Следует отметить, что эффективность доставки дуплекса OSN4F/ISN была практически равнозначна эффективности накопления одноцепочечного олигонуклеотида OSN4F (рис. 9 и 10).
Рис. 10.
Внутриклеточное накопление дуплексов OSNF/ISN и OSN4F/ISN олигонуклеотидов в клетках HEK293T (а, б) и T98G (в, г). Процент флуоресцентных клеток (а, в) и среднюю интенсивность флуоресценции (б, г) измеряли с помощью проточной цитофлуорометрии через 4 ч после трансфекции. RFU – относительные флуоресцентные единицы. Данные представлены как средние значения ± SD.

Таким образом, с использованием проточной цитофлуорометрии была продемонстрирована эффективность проникновения через клеточную мембрану как одноцепочечного олигонуклеотида, несущего в своем составе два остатка додецила (OSN4F), так и соответствующего комплементарного комплекса (OSN4F/ISN).
Проверка цитотоксичности триазиниламидофосфатных олигонуклеотидов. Помимо стабильности в биологических средах и эффективности проникновения в клетки важное свойство, определяющее перспективность терапевтического применения исследуемых олигонуклеотидов, – общая цитотоксичность. Исследование цитотоксичности модифицированного олигонуклеотида OSN4F и дуплекса OSN4F/ISN в отношении клеток HEK293T и T98G выполняли in vitro в режиме реального времени с помощью прибора xCELLigence (ACEA Biosciences, США) в течение 24–36 ч. Данные по выживаемости клеток в зависимости от времени инкубации представлены на рис. 11 (левая панель). На основе полученных данных были построены дозозависимые кривые выживаемости клеток и рассчитаны значения IC50 (рис. 11, правая панель). Из представленных данных видно, что олигонуклеотид OSN4F – нетоксичен в отношении клеток HEK293T и T98G (IC50 = 201.8 и 385.8 мкМ соответственно) (рис. 11а, правая панель). Дуплекс OSN4F/ISN оказался на порядок более токсичным в отношении исследованных клеток по сравнению с одноцепочечным олигонуклеотидом OSN4F – IC50 составила 33.1 мкМ для HEK293T и 28.1 мкМ для T98G (рис. 11б, правая панель). Тем не менее, рассчитанные значения IC50 для дуплекса были значительно выше рабочих концентраций олигонуклеотидов, обычно используемых в клеточных экспериментах, т.е. 1–5 мкМ. Было показано, что и олигонуклеотид OSN4F, и дуплекс OSN4F/ISN – нетоксичны для клеток HEK293T и T98G в рабочих концентрациях до 5 мкМ.
Рис. 11.
Цитотоксичность олигонуклеотида OSN4F (а) и дуплекса OSN4F/ISN (б) в отношении клеток HEK293T и T98G. Клетки инкубировали с увеличивающейся концентрацией олигонуклеотида/дуплекса в течение 24–36 ч. Левая панель – данные по выживаемости клеток в режиме реального времени, полученные с помощью прибора xCELLigence (ACEA Biosciences, США). Контроль – клетки HEK293T или T98G, инкубированные в отсутствие олигонуклеотида/дуплекса. Правая панель – дозозависимые кривые выживаемости клеток в конечной точке инкубации. Все экспериментальные точки выполнены в трех повторах для статистического анализа. Данные представлены как средние значения ± SD.

ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы. В работе были использованы нуклеотидные амидофосфиты (Glen Research, США); цианурхлорид, азид натрия, Tween 20, Nonidet P-40, формамид (PanReac AppliChem, Испания); бутиламин (Merck, Германия); додециламин (Acros Organics, США); ТЕМЕД, бис-акриламид (Amresco, США); Трис, бромфеноловый синий (Sigma, США); акриламид, ЭДТА, ксиленцианол, персульфат аммония (Helicon, Россия); реактивы и растворители квалификации х.ч. и о.с.ч.
FAP-dCTP (экзо-N-{2-[N-(4-азидо-2,5-дифтор-3-хлорпиридин-6-ил)-3-аминопропионил]-аминоэтил}-2'-дезоксицитидин-5'-трифосфат) любезно предоставлен В.Н. Сильниковым (Институт химической биологии и фундаментальной медицины СО РАН). [γ-32P]ATP (>3000 Ки/ммоль) произведен в лаборатории биотехнологии Института химической биологии и фундаментальной медицины СО РАН.
Поликлональные антитела кролика к PARP1 любезно предоставлены д.б.н. Г.Л. Диановым (Институт цитологии и генетики СО РАН, Россия); моноклональные антитела кролика к Ku80-субъединице Ku-антигена (Oncogene Research Products, США), Т4 полинуклеотидкиназа (Биосан, Россия) и конъюгат пероксидазы хрена со вторичными антителами к антителам кролика (Биосан, Россия); таблетированный коктейль ингибиторов протеаз Mini EDTA Free (Roche, Германия); маркеры молекулярной массы белков PageRuler (Thermo Scientific, США). Краситель Coomassie Brilliаnt Blue R-250, фильтровальная бумага и нитроцеллюлозная мембрана для вестерн-блот-анализа, буфер Trans-blot Buffer (Bio-Rad, США); субстрат для проявки вестерн-блота Pierce ECL Western Blotting Substrate, Coomassie Protein Assay Reagent (Thermo Scientific, США), сухое обезжиренное молоко (Regilait, Франция). ТЕМЕD, бис-акриламид (Amresco, США); MgCl2, Трис, SDS, DTT, бромфеноловый синий, NaBH4, бычий сывороточный альбумин, дитиотреитол, акриламид, персульфат аммония, ЭДТА, глицин (Sigma, США); глицерин (ICN, США).
Для ТСХ применяли пластинки DC-Alufolien Kieselgel 60 F254 (Merck, Германия). Для колоночной адсорбционной хроматографии использовали колонки объемом 100 и 200 мл с рассчитанным количеством сорбента Kieselgel 60 (размер частиц 0.06–0.20 мм, размер пор 60 Å; Merck, Германия).
Для концентрирования водных и органических растворов использовали ротационные испарители Rotavapor R200, Rotavapor RЕ120 (Buchi, Швейцария) и CentriVap Vacuum Concentrator Systems (Labconco, США) при давлении 10–15 мм рт. ст. и температуре до 40°С. Получаемые соединения высушивали до постоянной массы, выдерживая их в эксикаторе при пониженном давлении (4 мм рт. ст.) над P2O5.
офВЭЖХ реакционных смесей модифицированных олигонуклеотидов проводили на хроматографе Милихром A02 с использованием колонки ProntoSIL-120-5-C18 (2 × 75 мм; Эконова, Россия). Разделение продуктов реакции осуществляли в градиенте ацетонитрила (0–90% за 15 мин) в 0.02 М водном растворе триэтиламмоний ацетата, pH 7 (скорость потока 200 мкл/мин, температура термостата 35°С). Детекцию осуществляли на четырех длинах волн: 260, 280, 300 и 360 нм.
Хроматографическое выделение олигонуклеотидов проводили на хроматографе Agilent 1200 (США) с колонкой Zorbax SB-C18 (5 мкм) 4.6 × × 150 мм в градиенте ацетонитрила 0–90% в 20 мМ ацетате триэтиламмония, рН 7, в течение 30 мин при скорости потока 1.5 мл/мин. Фракции, содержащие целевой продукт, упаривали со смесью H2O/EtOH 1 : 1 в вакууме на установке SpeedVac) (Thermo Fisher Scientific, США). Затем олигонуклеотиды осаждали добавлением 1 мл раствора 1 M LiClO4 в ацетоне, осадок промывали ацетоном и сушили на воздухе в течение 20 мин.
Спектры ESI-MS модифицированных олигонуклеотидов регистрировали на масс-спектрометре Reflex III Autoflex Speed (Bruker, Германия) в варианте отрицательных ионов.
13С-ЯМР-спектры регистрировали на спектрометре Bruker AC 200 (300, 500 МГц; Bruker, Германия) в дейтерированном хлороформе с концентрацией 100 мг/мл. Спектры анализировали с помощью программы SPINWORKS. Спектры регистрировали в Химическом исследовательском центре коллективного пользования СО РАН при Новосибирском институте органической химии им. Н.Н. Ворожцова СО РАН.
Электрофорез в денатурирующих условиях проводили в 20%-ном ПААГ с соотношением акриламида к бис-акриламиду 19 : 1 и 7 М мочевиной в 1× ТВЕ-буфере (20× TBE : 1 М Трис, рН 8.3, 1 М H3BO3, 0.0265 М ЭДТА). Для полимеризации добавляли 10 мкл персульфата аммония (10%) и 1 мкл ТЕМЕD на 1 мл объема. В качестве электродного буфера использовали 1× ТВЕ-буфер. Денатурирующий буфер (5×) содержал формамид, бромфеноловый синий и ксиленцианол.
В работе использовали спектрофотометр U‑0080D (Hitachi, Япония); вакуумную сушку для акриламидных гелей GD 2000 (Hoefer, США); рН-метр OР 211/1 (Radelkis, Венгрия). Распределение флуоресценции в гелях определяли с использованием системы Typhoon (GE Healthcare, США) и программного обеспечения Quantity One (Bio-Rad, США).
Синтез 2-азидо-4,6-дихлор-1,3,5-триазина. К цианурхлориду (3 г, 16.3 ммоль) в 40 мл ацетона добавляли азид натрия (1.06 г, 16 ммоль). Реакцию проводили при интенсивном перемешивании в течение 2 ч при комнатной температуре. Реакционную смесь упаривали, растворяли в 20 мл CH2Cl2, промывали концентрированным раствором NaCl (4 × 20 мл). Органическую фазу сушили над безводным Na2SO4, упаривали до получения белого осадка продукта. Очистку проводили методом колоночной хроматографии в системе CH2Cl2/гексан, 1 : 1. Объединенные фракции упаривали, вещество сушили до постоянной массы. Масса полученного вещества составила 1.016 г (выход 33%). Rf 0.33; система CH2Cl2/гексан, 1 : 1. 13С-ЯМР (500 МГц, CDCl3, δ, м.д): 171.41, 172.61.
Синтез олигонуклеотидов. Синтез олигонуклеотидов проводили на автоматическом синтезаторе ASM800 (Биосcет, Россия) согласно стандартному протоколу 2-цианэтильного амидофосфитного метода, используя стандартные коммерческие дезоксирибонуклеозидные мономеры и соответствующие пористые стекла (Glen Research, США). Для введения триазиниламидофосфатной модификации после конденсации звена, чья фосфатная часть будет модифицирована, синтез останавливали, полимерный носитель переносили в пробирку, образовавшийся фосфит-триэфир окисляли в реакции с 200 мкл 0.1 М раствора 2-азидо-4,6-дихлор-1,3,5-триазина в ацетонитриле в течение 15 мин при 25°C. После промывки полимера от азида-модификатора добавляли раствор амина: 1 М бутиламин в ацетонитриле (60 мин, 25°C); 3 М додециламин в пиридине (30 мин, 55°C). Далее загружали полимерный носитель в реактор и продолжали автоматический синтез по амидофосфитному протоколу на ДНК-синтезаторе.
Присоединение к олигонуклеотидам остатка флуоресцеина [FAM] проводили по стандартному амидофосфитному протоколу синтеза с увеличенным временем (30 мин), объемом (70 мкл) и концентраций подаваемого коммерчески доступного мономера (0.1 М) (6-FAM amidite, LumiProbe, Россия).
Клеточные линии. Клеточные линии, любезно предоставленные сотрудниками Института химической биологии и фундаментальной медицины СО РАН, культивировали, как описано в работе Ilina et al. [21]. Клетки HeLa, MCF7, T98G, HEK293T, MRC5 культивировали в среде IMDM (Gibco, США), клетки Im9 и BJAB культивировали в среде RPMI 1640 (Gibco, США) в присутствии 10% FBS (HyClone, США), 100 ед./мл пенициллина (Invitrogen, США) и 100 г/мл стрептомицина (Invitrogen, США) в атмосфере 5% CO2. Цельноклеточные экстракты получали по методике Biade et al. [22]. Концентрацию белка в клеточных экстрактах определяли методом Бредфорд [23] с бычьим сывороточным альбумином в качестве стандарта. Концентрации суммарного белка в экстрактах выравнивали, замораживали в аликвотах и хранили при −70°C.
Введение [32P]-метки в 5'-конец олигонуклеотида и получение ДНК-дуплексов. Введение радиоактивной метки и получение ДНК-дуплексов осуществляли согласно методике Ilina et al. [21].
Фотоаффинная модификация. Фотоактивируемый аналог dCTP встраивали в ДНК с помощью ДНК-полимеразы β с использованием FAP-dCTP (экзо-N-{2-[N-(4-азидо-2,5-дифтор-3-хлорпиридин-6-ил)-3-аминопропионил]аминоэтил}-2'-дезоксицитидин-5'-трифосфата) в качестве субстрата, как описано в работе Ilina et al. [21]. Фотоаффинную модификацию проводили согласно методике Ilina et al. [21]. Реакционные смеси для облучения содержали 0.1 мкМ фотоактивируемую ДНК и 1 мг/мл белков клеточного экстракта (HeLa, MCF7, T98G, HEK293T, BJAB, Im9 или MRC5). Контрольные пробы вместо белков экстракта содержали 34 или 20 нМ индивидуальные белки Ku и PARP1 соответственно. Гели анализировали с использованием сканера Typhoon (GE Healthcare, США) и программы Quantity One (Bio-Rad, США).
Сравнение количества PARP1 и Ku в клеточных экстрактах с помощью вестерн-блот-анализа. Вестерн-блот-анализ проводили согласно методике Ilina et al. [24]. Кратко, 10 мкг суммарного белка клеточного экстракта (HeLa, MCF7, T98G, HEK293T, BJAB, Im9 или MRC5) или чистые белки PARP1 или Ku разделяли с помощью электрофореза по методу Лэммли [25]. Процедуру переноса проводили с использованием набора Trans-Blot Turbo в условиях, указанных производителем. Нитроцеллюлозную мембрану инкубировали в растворе первичных антител (антитела кролика к PARP1 или Ku80 в разведении 1 : 1000), в растворе конъюгата пероксидазы хрена с вторичными антителами к антителам кролика, затем с коммерческим субстратом для проявки вестерн-блота Super Signal West Pico PLUS (Thermo Scientific, США). Хемилюминесценцию регистрировали на приборе Amersham Imager 600 (GE Healthcare, США).
Проверка устойчивости модифицированных олигодезоксирибонуклеотидов к действию нуклеаз цельноклеточных экстрактов. Реакционные смеси содержали 1 мг/мл белков клеточного экстракта (T98G или HEK293T), 0.1 мкМ ДНК и буферные компоненты: 10 мМ MgCl2, 50 мМ Трис-HCl (pH 8.0), 50 мМ NaCl. Реакцию проводили в течение 7.5 и 15 мин для каждого экстракта при 37°С. Реакцию останавливали введением ЭДТА до конечной концентрации 20 мМ. После этого аликвоты анализировали с помощью электрофореза в ПААГ в денатурирующих условиях по методике Мазина с соавт. [26].
Формирование ДНК-дуплексов и определение температур их плавления. Для получения ДНК-дуплексов OSNF/ISN и OSN4F/ISN соответствующие олигодезоксирибонуклеотиды в концентрации 2.5 мкМ каждого смешивали в эквимолярном соотношении в буфере PBS, смеси нагревали в течение 5 мин при 95°C, а затем медленно охлаждали. Полученные дуплексы использовали для экспериментов сразу после отжига.
Термическую денатурацию осуществляли на установке Cary 300 BioMelt (Varian, Австралия). Температуры плавления (Тпл) комплексов определяли, анализируя профили денатурации и ренатурации дуплексов, зарегистрированные на длинах волн 260 и 270 нм. Скорость нагрева/охлаждения образцов не превышала 0.5°C/мин. Точность определения величин Тпл составила 0.1°C. Термодинамический анализ проводили, как описано в работе Shevelev et al. [27].
Исследование токсичности олигонуклеотидов и их дуплексов. Цитотоксичность олигонуклеотида OSN4F и дуплекса OSN4F/ISN изучали на клеточных культурах HEK293T и T98G в режиме реального времени с помощью прибора xCELLigence (ACEA Biosciences, США) в течение 24–36 ч. Клетки высеивали на 16-луночные Е-планшеты в плотности 105 кл./лунку в 150 мкл полной среды IMDM и инкубировали в стандартных условиях в течение 7–24 ч для прикрепления клеток ко дну планшета. После этого среду заменяли на 150 мкл среды IMDM, содержащей 10% FBS и 0.5, 1, 2.5, 5, 10, 20, 50 мкМ олигонуклеотид OSN4F или 0.5, 1, 2.5, 5, 10, 15, 20 мкМ дуплекс OSN4F/ISN. Клетки инкубировали в течение 24–36 ч в стандартных условиях. Значения клеточных индексов измеряли каждые 30 мин. Дозозависимые кривые выживаемости клеток строили с помощью программного обеспечения MS Excel для временной точки 24 ч после добавления олигонуклеотида/дуплекса к клеткам. Цитотоксичность дуплекса OSN4F/ISN в отношении клеток HEK293T определяли через 36 ч после добавления дуплекса к клеткам вследствие медленной пролиферации клеток HEK293T. Значения IC50 рассчитывали как концентрацию олигонуклеотида/дуплекса, необходимого для снижения клеточного индекса на 50% по сравнению с контрольными клетками, инкубированными в отсутствие олигонуклеотида/дуплекса.
Исследование эффективности трансфекции клеток HEK293T и T98G олигонуклеотидом OSN4F и дуплексом OSN4F/ISN. Клетки HEK293T и T98G высеивали в лунки 24-луночного планшета в концентрации 2 × 105 (HEK293T) или 1.2 × × 105 кл./лунку (T98G) в 500 мкл полной среды IMDM и инкубировали в течение 18 ч для прикрепления клеток. Далее клеточную среду заменяли на 200 мкл среды IMDM без сыворотки и антибиотиков. Олигонуклеотиды (OSNF, OSN1F, OSN4F) или дуплексы (OSN4F/ISN) растворяли в среде Opti-MEM и добавляли к клеткам до финальной концентрации 1 или 5 мкМ для олигонуклеотидов и 5 мкМ для дуплексов (по 50 мкл на лунку). В качестве положительного контроля использовали трансфекцию клеток контрольным олигонуклеотидом OSNF с помощью коммерчески доступного трансфектанта Липофектамина 2000 (Thermo Fisher Scientific, США). Трансфекцию выполняли согласно протоколу фирмы-изготовителя. Клетки инкубировали в течение 4 ч в стандартных условиях. После инкубации клетки снимали с культурального пластика с помощью 2%-ного трипсина (MP Biomedicals, США), ресуспендировали в полной среде IMDM, центрифугировали при 200 g в течение 5 мин, промывали PBS и фиксировали в 2%-ном растворе формальдегида в PBS (10 мин, комнатная температура). Клетки анализировали на проточном цитофлуориметре NovoCyte 3000 (ACEA Biosciences, США). Все экспериментальные точки выполняли в трех повторах для статистического анализа. Эффективность трансфекции характеризовали двумя показателями – процентом флуоресцентных клеток в популяции и средней интенсивностью флуоресценции клеток в образце.
ЗАКЛЮЧЕНИЕ
Авторами данной работы впервые получен новый класс олигонуклеотидных производных – триазиниламидофосфаты. Для синтеза данных соединений на этапе окисления была использована реакция Штаудингера между фосфит-триэфирным производным олигонуклеотида и высокореакционноспособным 2-азидо-4,6-дихлор-1,3,5-триазином. Получаемое фосфазеновое производное – общее соединение-прекурсор для всех представителей класса, получаемых дальнейшей обработкой различными замещенными аминами. Такой подход позволяет гибко варьировать вводимые в состав триазинового остова функциональные группы. Результаты анализа полученных олигонуклеотидов методами офВЭЖХ и масс-спектрометрии указывают на высокую эффективность введения триазиниламидофосфатной модификации предложенным методом.
С помощью фотоаффинной модификации и иммуноферментного анализа в экстрактах определено содержание Кu-антигена и PARP1 – потенциальных акцепторов ДНК-дуплексов. Для дальнейших экспериментов были выбраны клеточные линии с резко различающимся содержанием этих белков, что впоследствии облегчит возможность оценки вклада этих белков в действие созданных модифицированных олигонуклеотидов на клетки.
Проведенные эксперименты показали повышенную устойчивость триазиниламидофосфатных олигонуклеотидов к действию клеточных нуклеаз. Олигонуклеотид, несущий два остатка додецила на триазиновом остове, способен эффективно проникать через клеточную мембрану, не проявляя выраженной цитотоксичности.
Предложенный класс триазиниламидофосфатных олигонуклеотидов, несмотря на определенные ограничения в используемом способе их получения, перспективен в области создания терапевтических препаратов нового поколения, направленных на белковые мишени.
Список литературы
Rinaldi C., Wood M. J. A. // Nat. Rev. Neurol. 2017. V. 14. P. 9–21. https://doi.org/10.1038/nrgastro.2016.169
Juskowiak B. // Anal. Bioanal. Chem. 2010. V. 399. P. 3157–3176. https://doi.org/10.1007/s00216-010-4304-5
Yin W., Rogge M. // Clin. Transl. Sci. 2019. V. 12. P. 98–112. https://doi.org/10.1111/cts.12624
Balwani M., Sardh E., Ventura P., Peiró P.A., Rees D.C., Stölzel U., Bissell D.M., Bonkovsky H.L., Windyga J., Anderson K.E., Parker C., Silver S.M., Keel S.B., Wang J.-D., Stein P.E., Harper P., Vassiliou D., Wang B., Phillips J., Ivanova A., Langendonk J.G., Kauppinen R., Minder E., Horie Y., Penz C., Chen J., Liu S., Ko J.J., Sweetser M.T., Garg P., Vaishnaw A., Kim J.B., Simon A.R., Gouya L. // N. Engl. J. Med. 2020. V. 382. P. 2289–2301. https://doi.org/10.1056/NEJMoa1913147
Heo Y. // Drugs. 2020. V. 80. P. 329–333. https://doi.org/10.1007/s40265-020-01267-2
Keung M.Y.T., Wu Y., Vadgama J.V. // J. Clin. Med. 2019. V. 8. P. 435–459. https://doi.org/10.3390/jcm8040435
Miroshnichenko S.K., Patutina O.A., Burakova E.A., Chelobanov B.P., Fokina A.A., Vlassov V.V., Altman S., Zenkova M.A., Stetsenko D.A. // Proc. Natl. Acad. Sci. USA. 2019. V. 116. P. 1229–1234. https://doi.org/10.1073/pnas.1813376116
Dyudeeva E.S., Kupryushkin M.S., Lomzov A.A., Pyshnaya I.A., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 709–718. https://doi.org/10.1134/S1068162019060153
Blotny G. // Tetrahedron. 2006. V. 62. P. 9507–9522. https://doi.org/10.1016/j.tet.2006.07.039
Bucher G., Siegler F., Jens Wolff J. // Chem. Commun. 1999. V. 20. P. 2113–2114. https://doi.org/10.1039/A905971G
Markov O.V., Filatov A.V., Kupryushkin M.S., Chernikov I.V., Patutina O.A., Strunov A.A., Chernolovskaya E.L., Vlassov V.V., Pyshnyi D.V., Zenkova M.A. // Molecules. 2020. V. 25. P. 1–27. https://doi.org/10.3390/molecules25163663
Garrison A.W., Boozer C.E. // J. Am. Chem. Soc. 1968. V. 90. P. 3486–3494. https://doi.org/10.1021/ja01015a035
Fell V.L., Schild-Poulter C. // Mutat. Res. 2015. V. 763. P. 15–29. https://doi.org/10.1016/j.mrrev.2014.06.002
Khodyreva S.N., Lavrik O.I. // Mol. Biol. (Mosk). 2016. V. 50. P. 655–673. https://doi.org/10.7868/S0026898416040030
Kosova A.A., Kutuzov M.M., Evdokimov A.N., Ilina E.S., Belousova E.A., Romanenko S.A., Trifonov V.A., Khodyreva S.N., Lavrik O.I. // Aging. 2019. V. 11. P. 2852–2873. https://doi.org/10.18632/aging.101959
Menon V., Povirk L.F. // DNA Repair. 2016. V. 43. P. 57–68. https://doi.org/10.1016/j.dnarep.2016.05.011
Yang W. // Q. Rev. Biophys. 2011. V. 44. P. 1–93. https://doi.org/10.1017/S0033583510000181
Walker J.R., Corpina R.A., Goldberg J. // Nature. 2001. V. 412. P. 607–614. https://doi.org/10.1038/35088000
Krasner D.S., Daley J.M., Sung P., Niu H. // J. Biol. Chem. 2015. V. 290. P. 18806–18816. https://doi.org/10.1074/jbc.M115.660191
Blier P.R., Griffith A.J., Craft J., Hardin J.A. // J. Biol. Chem. 1993. V. 268. P. 7594–7601.
Ilina E.S., Khodyreva S.N., Berezhnoy A.E., Larin S.S., Lavrik O.I. // Mutat. Res. 2010. V. 685. P. 90–96. https://doi.org/10.1016/j.mrfmmm.2009.08.008
Biade S., Sobol R.W., Wilson S.H., Matsumoto Y. // J. Biol. Chem. 1998. V. 273. P. 898–902.
Bradford M.M. // Anal. Biochem. 1976. V. 72. P. 248–254. https://doi.org/10.1006/abio.1976.9999
Ilina E.S., Khodyreva S.N., Lavrik O.I. // Biochimie. 2018. V. 150. P. 88–99. https://doi.org/10.1016/j.biochi.2018.04.027
Laemmli U.K. // Nature 1970. V. 227. P. 680–685. https://doi.org/10.1038/227680a0
Мазин А.В., Кузнеделов, Краев А.С., Холодилов Н.Г., Блинов А.Г., Кузьминов А.В., Головин С.Я., Наякшин А.М., Соловьев В.В., Ямщиков В.Ф., Кокоза В.А., Иванов С.В., Потапов В.А., Санарбаев М.К., Дианов Г.Л., Протопопов М.О., Калачиков С.М., Богачев С.С., Чикаев Н.А. // Методы молекулярной генетики и генной инженерии / Под. ред. Салганика Р.И. Новосибирск: Наука. Сибирское отделение, 1990. С. 238–239.
Shevelev G.Y., Kabilov M.R., Lomzov A.A., Dovydenko I.S., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 684–698. https://doi.org/10.1134/S1068162019060359
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия