

Биоорганическая химия, 2021, T. 47, № 3, стр. 391-399
Региоселективный синтез, структура и хемосенсибилизирующая противоопухолевая активность циклической гидроксамовой кислоты на основе DL-валина
И. В. Выстороп 1, Г. В. Шилов 1, А. В. Черняк 1, Е. Н. Климанова 1, Т. Е. Сашенкова 1, С. Г. Клочков 2, М. Е. Неганова 2, *, Ю. Р. Александрова 2, У. Ю. Аллаярова 1, Д. В. Мищенко 1, 3
1 ФГБУН “Институт проблем химической физики” РАН
142432 Черноголовка, просп. академика Семенова, 1, Россия
2 ФГБУН “Институт физиологически активных веществ” РАН
142432 Черноголовка, Северный проезд, 1, Россия
3 Научно-образовательный центр “Медицинская химия” Московского государственного областного
университета в г. Черноголовка
141014 Мытищи, ул. Веры Волошиной, 24, Россия
* E-mail: neganova83@mail.ru
Поступила в редакцию 10.07.2020
После доработки 22.07.2020
Принята к публикации 26.07.2020
Аннотация
Реакция DL-валингидроксамовой кислоты с триацетонамином проходит как N,N'-региоселективная циклоконденсация с образованием (±)-1-гидрокси-3-изопропил-7,7,9,9-тетраметил-1,4,8-триазаспиро[4,5]декан-2-она. Исследование антиметастатической и противоопухолевой активности полученной гидроксамовой кислоты in vivo методом комбинированной терапии с цитостатиком алкилирующего типа на модели экспериментальной перевиваемой меланомы B16 мышей показало способность данного соединения повышать чувствительность опухоли к действию известного противоопухолевого препарата циклофосфамида, примененного в субтерапевтической дозе. Хемосенсибилизирующая активность гидроксамовой кислоты в комбинации с циклофосфамидом привела к повышению противоопухолевого действия цитостатика почти в 2 раза, а также заметному снижению количества метастазов, что выражалось в увеличении индекса ингибирования метастазирования до 74%.
ВВЕДЕНИЕ
Онкологические заболевания – глобальная проблема современного общества, затрагивающая миллионы пациентов [1, 2]. На сегодняшний день злокачественные новообразования занимают второе место среди причин смертности, уступая лидирующую позицию лишь сердечно-сосудистым патологиям [3]. Так, согласно данным Всемирной организации здравоохранения, ежегодный темп прироста онкозаболеваний составляет ~1.5%, при этом в 2018 г. было зарегистрировано более 9.6 млн смертельных случаев от онкопатологий.
Используемые в настоящее время противоопухолевые агенты характеризуются низким терапевтическим индексом и, как следствие, высоким токсическим действием не только на неопластические клетки, но и на здоровые клетки организма, что может быть связано с разнообразием патогенеза и этиологии опухолей, их развитием, симптоматикой и феноменом резистентности. Поэтому не вызывает сомнения актуальность поиска высокоэффективных малотоксичных онкопрепаратов с целью получения лекарственных средств нового поколения с различными механизмами действия.
Одним из перспективных классов химических соединений с подтвержденным противоопухолевым потенциалом выступают гидроксамовые кислоты (ГК), которые обладают широким спектром биологической активности [4–6]. На сегодняшний день FDA одобрено использование трех представителей данного класса для лечения кожной и периферической Т-клеточной лимфомы и множественной миеломы [7]: вориностат (SAHA, Золинза) [8], панобиностат (Фаридак) [9] и белиностат (Белеодаг) [10], что наглядно подтверждает перспективность поиска антинеопластических агентов среди гидроксамовых кислот. Анализ современных тенденций в исследованиях противоопухолевых свойств соединений этого класса при терапии онкологических заболеваний показывает, что антинеопластический потенциал гидроксамовых кислот может реализовываться по нескольким направлениям: 1) использование монотерапии, нацеленной на эффективное ингибирование гистоновых деацетилаз (HDACs) [5, 11]; 2) создание на основе ГК полифункциональных препаратов с разными фармакофорными фрагментами, воздействующими одновременно на несколько различных звеньев патологии [12, 13]; 3) применение ГК в качестве хемосенсибилизаторов при комбинированной терапии с цитостатиками [14].
Ранее было показано, что реакции глицингидроксамовой (GlyГК) и ряда других DL-α-аминогидроксамовых кислот с ацетоном в избытке кетона осуществляются как региоселективные циклоконденсации N,N'-типа с образованием 2,2-диметил-3-гидроксиимидазолидин-4-она (Iа) и его 5-замещенных производных (Ib–e) соответственно с умеренными выходами 55–83% [15–17].
Аналогичным способом образуются спироциклические ГК (II) и (III) на основе реакции GlyГК и DL-аланингидроксамовой кислоты (DL-AlaГК) соответственно с триацетонамином (ТАА) в этаноле с высокими выходами (70–80%) [18].
Исследование хемосенсибилизирующей противолейкозной активности циклических ГК (II) и (III) в комбинации с цитостатиками алкилирующего типа на модели лейкоза Р388 показало способность кислот (II) и (III) усиливать действие цитостатиков, что привело к увеличению продолжительности жизни (УПЖ) животных экспериментальных групп на 30–50% [18].
В настоящей работе изучена возможность осуществления региоселективной реакции циклоконденсации пространственно затрудненной гидрофобной DL-валингидроксамовой кислоты (DL-ValГК) с ТАА с целью получения гомолога кислот (II) и (III). Для изучения взаимозависимости “структура–активность” в ряду производных 1-гидрокси-1,4,8-триазаспиро[4,5]декан-2-она проанализированы структурные особенности полученной спироциклической ГК (IV), а также исследованы ее спектральные данные, хемосенсибилизирующая и антиметастатическая активности в терапии экспериментальной перевиваемой меланомы B16 мышей in vivo с цитостатиком алкилирующего типа – циклофосфамидом.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез и спектральные данные гидроксамовой кислоты (IV). Исследование реакции DL-ValГК с ТАА (схема 1 ) в мягких условиях (соотношение реагентов 1 : 1.3, кипячение в этаноле в течение 2 ч), аналогичных проведению реакции GlyГК и DL-AlaГК с ТАА, показало, что хотя реакция и приводит к образованию спироциклической гидроксамовой кислоты (IV), но исходная кислота DL-ValГК вступает в реакцию не полностью (<40%). Попытка изменения условий реакции путем увеличения продолжительности проведения реакции до 5 ч и применения 2-кратного избытка кетона приводит к почти полному расходованию DL-ValГК (до 95%). Однако реакция осложняется образованием олигомерных продуктов, что снижает выход целевого продукта (IV) (<45%).

Схема 1 . Синтез спироциклической гидроксамовой кислоты (IV).
Вероятно, причиной пониженной реакционной способности DL-ValГК служит увеличение пространственной затрудненности и понижение растворимости DL-ValГК по сравнению с GlyГК и DL-АlaГК. В условиях накопления в реакционной смеси побочного продукта – воды и отсутствия выпадения в виде осадка кислоты (IV) из реакционной среды это способствует смещению равновесия реакции в обратном направлении.
Поэтому были предложены более эффективные способы проведения реакции DL-ValГК с ТАА: 1) поэтапный способ – с добавлением новых порций ТАА к непрореагировавшей DL-ValГК (3‒4 этапа); 2) способ дробной отгонки реакционной воды в виде тройной азеотропной смеси EtOH–бензол–Н2О с периодическим добавлением в реакционную среду абсолютированного бензола. В обоих случаях реакция проводится в мягких условиях и позволяет достичь приемлемого выхода продукта (IV) (64–70%).
Строение кислоты (IV) доказано методами масс-спектроскопии высокого разрешения, ИК-, 1D- (1H,13C (с подавлением или без подавления 1Н-13С ССВ)) и 2D-спектроскопии ЯМР (COSY 1H-1H и HSQC 1Н-13С) в растворах CDCl3 и DMSO-d6.
Уширенные полосы в области 2700–2200 см–1 (νmax = 2616 и 2490 см–1) в ИК-спектрах (KBr) и положительные тест-реакции с FeCl3 в MeOH подтверждают наличие ОН-группы в кислоте (IV). Это исключает образование циклического гидроксамата (–CO–NH–OR) посредством возможной альтернативной циклоконденсации N,O-типа.
Согласно данным спектров 1Н-ЯМР и 13С‑ЯМР, кислота (IV) образует кристаллосольваты двух видов: 1) с MeCN и Н2О в мольном соотношении 2 : 1 : 1 соответственно (т. пл. 152–153°С (из MeCN), (IVa)); 2) с диоксаном в эквимолярном соотношении (т. пл. 158–159°C (из диоксана), (IVb)), аналогично гомологам (II) (с MeCN в соотношении 1 : 1) и (III) (с диоксаном в соотношении 2 : 1) [18].
Спектры 1Н-ЯМР обеих форм (IVa, b) в CDCl3 в концентрации 10 мкМ почти идентичны и носят отличия, обусловленные присутствием сигналов сольватных растворителей δН 2.03 (MeCN) или δН 3.73 м.д. (диоксан).
Отнесение сигналов углеродов кислоты (IV) проведено анализом спектров HSQC 1Н-13С форм (IVa) в DMSO-d6 и (IVb) в CDCl3 (рис. 1). Отсутствие кросс-пиков сигналов углеродов групп NCN, β-СМе2 и С=О с сигналами каких-либо протонов в спектрах HSQC 1Н-13С форм (IVa, b) подтверждает апротонный характер этих углеродов.
Рис. 1.
2,2-Диметил-3-гидроксиимидазолидин-4-он (Iа) и его 5-замещенные производные (Ib–e), спироциклические гидроксамовые кислоты (II) и (III).

Химические сдвиги сигнала углерода NCN в спектрах 13С-ЯМР (δС 77.5 (DMSO-d6, (IVa), 78.3 м.д. (CDCl3, (IVb)), отвечающие тетраэдрическому характеру атома углерода, подтверждают спироциклическую структуру кислоты (IV), аналогично данным спектров для кислот (II) (δС 79.6, DMSO-d6) и (III) (80.3, CD3OD) [18]. С другой стороны, отсутствие сигналов тригонального углерода группы C=N в спектрах 13С-ЯМР (δС > 150 м.д.) [19] и наличие сигнала протона NH-группы имидазолидинового цикла в спектрах 1Н-ЯМР (δН 2.32 (DMSO-d6, с 0.4 М, (IVa), 1.55 (CDCl3, с 0.05 М, (IVa, b), 1.48 м.д. (CDCl3, 0.25 M, (IVb)), аналогично данным спектров для гомологов кислот (II) (δН 3.10) и (III) (2.61 м.д., DMSO-d6) [18], исключает структуру соответствующей ациклической азометиновой формы кислоты (IV) (HONH(C=O)CH(CHMe2)N=R, где R – тетраметилпиперидиновый цикл.
Отсутствие сигналов азометиновой формы в спектрах 1Н- и 13С-ЯМР указывает на отсутствие медленной в шкале времени ЯМР кольчато-цепной таутомерии ГК (IV) в CDCl3 и DMSO-d6.
Отнесение сигнала протона NH-группы имидазолидинового цикла кислоты (IV) основано на сопоставлении данных спектров COSY 1H-1H (наблюдаются кросс-пики и вицинальные КССВ протонов NH- и СНСО-групп) и HSQC 1Н-13С (отсутствуют кросс-пики сигнала протона NH-группы с сигналом какого-либо углерода) в растворах CDCl3 и DMSO-d6. В спектре 1Н-ЯМР кислоты (IV) в DMSO-d6 наблюдается дезэкранирующий эффект для NH-группы по сравнению со спектром в CDCl3.
Сигналы протонов метильных групп кислоты (IV) в спектрах 1Н-ЯМР в CDCl3 и DMSO-d6 отнесены в соответствии со следующей зависимостью их химических сдвигов: δН (МеА < MeB < MeC < MeD). При этом наблюдается обратная последовательность величин химических сдвигов протонов (δН (МеА, МеВ, 1.0–1.2) < δH (MeC, MeD, 1.3–1.5 м.д.)) и углеродов Ме-групп (δС (МеА, МеВ, ~35) > δC (MeC, MeD, ~29 м.д.)) пиперидинового цикла.
Известно, что сигналы 13С аксиальных Ме-групп резонируют в более сильном поле по сравнению с сигналами экваториальных Ме-групп циклогексана (в конформации “кресло” на ~4.5 м.д.) [20]. Это позволяет предположить для групп МеС, МеD и МеА, МеВ кислоты (IV), соответственно, а- и е-ориентации при пиперидиновом цикле.
Отнесение сигналов углеродов α1- и α2-метиленовых групп пиперидинового цикла кислоты (IV) в растворах CDCl3 и DMSO-d6 основано на следующей зависимости химических сдвигов этих сигналов: δα2-С (~45 м.д.) > δα1-С (~40 м.д.), аналогично сигналам кислоты (III) (δα2-С ~ 45.3 м.д., δα1-С ~ ~ 40.8 м.д.) в CD3OD [18]. Для ахиральной кислоты (II) наблюдается изохронность сигналов углеродов метиленовых групп (δα1-С, δα2-С 41.3 м.д., DMSO-d6) [18].
Углерод α1-СН2-группы пиперидинового цикла, вероятно, экранируется неподеленной электронной парой (НЭП) атома N1. Углероды метиленовых групп С6 и С7 находятся, соответственно, в транс- и цис-положениях к псевдо-а-ориентации H1N, а значит, соответственно, в цис- и транс-положениях к псевдо-е НЭП при атоме N1, т.е. атомы псевдо-а-С6 и псевдо-е-С7 соответствуют, вероятно, углеродам α1-СН2- и α2-СН2-групп.
Сигналы протонов АВ-систем α1- и α2-метиленовых групп пиперидинового цикла отнесены сопоставлением данных спектров COSY 1H-1H и HSQC 1Н-13С. Наблюдается существенно большая разница относительного химического сдвига α2-СН2-группы (∆δАВ 0.53–0.58 (CDCl3), 0.17–0.20 м.д. (DMSO-d6)) по сравнению с α1‑СН2-группой (∆δАВ 0.16–0.2 (CDCl3), 0.05 м.д. (DMSO-d6)) кислоты (IV). Аналогичное соотношение ∆δАВ отмечается и для гомологов (II) (0.05) и (III) (0.25 и 0.07 м.д.) в DMSO-d6 [18]. В растворе CDCl3 кислоты (IV) это происходит в основном за счет дезэкранирования слабопольного протона α2-НВ по сравнению с протоном α1-НВ (∆δ (α1,α2-НВ) 0.28 м.д.). В то же время сильнопольные протоны α2-НА и α1-НВ метиленовых групп показывают меньшее различие химических сдвигов их сигналов (∆δ (α1,α2-НА) 0.09 м.д.). В растворе DMSO-d6 наблюдается изохронность сигналов протонов α1-НА и α2-НА метиленовых групп кислоты (IV).
Сравнение величин ∆δАВ метиленовых групп кислоты (IV) указывает на большую конформационную подвижность α1-СН2-группы в процессе интерконверсии пиперидинового цикла, которая осуществляется, вероятно, по механизму осциллирующего псевдовращения [19].
Интересно, что конформационная подвижность пиперидинового цикла в кислоте (IV), как и в кислотах (II) и (III) [18], выше в биполярном апротонном растворителе DMSO-d6, склонном к образованию ассоциатов с молекулами растворенного в нем вещества, по сравнению с малополярным CDCl3.
В спектрах 1Н-ЯМР обеих форм (IVa, b) в CDCl3 наблюдается дальняя КССВ 4JН,Н 1.6 Гц (с 0.25 М), 2.0 Гц (с 0.05 М) между сильнопольными протонами α1-НА и α2-НА метиленовых групп. Это возможно при экваториальной ориентации этих протонов в конформации “кресло” пиперидинового цикла, поскольку реализуется транс-W-конфигурация разделяющих их связей, тогда как для слабопольных протонов α1-НВ и α2-НВ этих групп аксиальной ориентации реализуется цис-конфигурация разделяющих их связей, при которой не должно наблюдаться заметной 4JН,Н.
Приписывание сильнопольным (НА) и слабопольным (НВ) протонам α-метиленовых групп пиперидинового цикла кислоты (IV) в спектрах 1Н-ЯМР в CDCl3, соответственно, экваториальной и аксиальной ориентаций подтверждается анализом спектров 1Н-ЯМР в CDCl3 и DMSO-d6 аналогичных циклических ГК [20], имеющих незамещенные α- и β-метиленовые группы пиперидинового цикла. Согласно характеру вицинальных взаимодействий протонов этих групп, для слабопольных протонов α-НВ и β-НВ, в отличие от сильнопольных протонов α-НА и β-НА, наблюдается большая КССВ (3J (α-НВ, β-НВ) ≥ 10 Гц), что возможно только для аксиальных протонов транс-ориентации в преимущественной конформации “кресло” пиперидинового цикла [21, 22].
Наблюдаемые в спектрах 1Н-ЯМР (IVa, b) КССВ протона группы СНС=О с протонами СНМе2 (3J (CHCO, CHMe2) 4.8–4.9 (CDCl3), 6.3–6.5 Гц (DMSO-d6)) и NH-групп (3J (CHСО, NH) 10.2–10.3 (c 0.05 M, CDCl3), 9.9 (c 0.25 M, CDCl3), 10.7 Гц (DMSO-d6)) свидетельствует, соответственно, о взаимной цис- и транс-ориентации этих протонов, т.е. конформационной однородности имидазолидинового цикла в растворе сильно различающихся по полярности растворителей.
Острая токсичность гидроксамовой кислоты (IV) in vivo. При изучении острой токсичности на мышах-гибридах линии BDF1 показано, что при однократном внутрибрюшинном введении соединения (IV) гибель животных отмечалась на 1-е и 2-е сутки наблюдения после инъекции раствора гидроксамовой кислоты в дозе 500–900 мг/кг. У выживших животных каких-либо клинических проявлений интоксикации и изменений в поведенческих реакциях не наблюдалось до конца опыта. На рис. 2 представлена кривая нарастания токсичности гидроксамовой кислоты (IV) по мере увеличения дозы, исходя из которой была определена величина LD50 = 730.4 ± 32.4 мг/кг, а также были рассчитаны токсические дозы: LD16 = = 639.7; LD84 = 833.9 и LD100 = 885.7 мг/кг.
Рис. 2.
Кривая зависимости “доза–эффект” для определения параметров острой токсичности после однократного внутрибрюшинного введения раствора гидроксамовой кислоты (IV) мышам-гибридам линии BDF1.
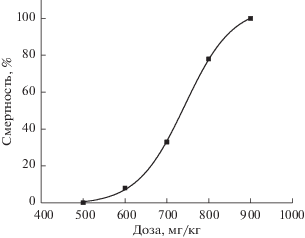
Результаты исследования острой токсичности позволяют отнести соединение (IV) к III классу токсичности, т.е. данная гидроксамовая кислота – умеренно токсичное соединение в соответствии с ГОСТ 12.1.007–76.
Хемосенсибилизирующая противоопухолевая активность кислоты (IV) in vivo. Для оценки хемосенсибилизирующего противоопухолевого и антиметастатического действия гидроксамовой кислоты (IV) на модели экспериментальной перевиваемой меланомы B16 на самцах мышей-гибридов линии BDF1 для внутрибрюшинного введения была взята одна третья часть от величины LD50, которая составила 233 мг/кг. При монотерапии гидроксамовой кислотой и цитостатиком было выявлено достоверное увеличение индекса ингибирования метастазирования (ИИМ) – до 12.82 и 38.46% соответственно. При применении комбинированной химиотерапии циклофосфамидом с исследуемым соединением (IV) наблюдалось двукратное увеличение данного показателя по сравнению с группой циклофосфамид (до 74.36%) и появление эффекта усиления противоопухолевого действия известного цитостатика, что может свидетельствовать о проявлении данной ГК адъювантной способности. Терапия меланомы B16 низкотоксичной кислотой (IV) в комбинации с циклофосфамидом позволила достичь торможения роста опухоли животных экспериментальных групп до 31.56%, что более чем в 1.5 раза выше данного показателя при монотерапии циклофосфамидом (табл. 1). Ранее нами также было показано усиление противолейкозного действия циклофосфамида в комбинированной терапии лимфолейкоза Р388, которое выражалось в увеличении выживаемости животных до 38% и увеличении средней продолжительности жизни невылеченных животных почти в 2 раза по сравнению с монотерапией данным цитостатиком [23].
Таблица 1.
Хемосенсибилизирующий эффект кислоты (IV) на модели экспериментальной перевиваемой меланомы B16 мышей
| Препарат | Разовая доза, мг/кг |
Режим введения, сут | Среднее количество метастаз | Индекс ингибирования метастазиро-вания, % | Средний вес опухоли, г | Торможение роста опухоли, % |
|---|---|---|---|---|---|---|
| Циклофосфамид | 20 | 2, 7 | 6.00 ± 1.39* | 38.46 | 7.95 ± 1.34 | 20.26 |
| (IV) | 233 | 2–9 | 8.50 ± 1.70 | 12.82 | 9.71 ± 0.59 | 2.62 |
| Циклофосфамид + + (IV) | 20 + 233 | 2, 7 + 2–9 | 2.50 ± 1.11*** | 74.36 | 6.83 ± 1.22** | 31.56 |
| Контроль | – | – | 9.75 ± 2.05 | – | 9.97 ± 1.34 | – |
Введение разветвленной изопропильной группы при имидазолидиновом цикле приводит к значительному повышению хемосенсибилизирующей способности кислоты валинового ряда (IV) по сравнению с ее гомологами глицинового (II) и аланинового ряда (III), активность которых была описана нами ранее [21]. Наибольшее отличие в хемосенсибилизирующей активности наблюдается при совместном действии кислоты (IV) и циклофосфамида (ИИМ ~74%, УПЖ >280% [23]).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез гидроксамовой кислоты (IV). ИК-спектры образцов соединений, запрессованных в таблетки с KBr, регистрировали на спектрометре Specord-82M (Analytik Jena, Германия) в области 400–4000 см–1. Масс-спектры высокого разрешения снимали на квадрупольном времяпролетном масс-спектрометре Sciex QStar (SCIEX, США) с ортогональным вводом ионов и на масс-спектрометре Thermo Fisher Exactive (Thermo Fisher Scientific, США) с источником ионизации электрораспылением и масс-анализатором Orbitrap. Для ионизации были взяты растворы исходных веществ в ацетонитриле с концентрацией ~10 мкМ. При расчете молекулярных масс использовали следующие величины атомных масс: Н – 1.007825, О – 15.994915, С – 12.000000, N – 14.003074. Спектры ЯМР регистрировали на спектрометре Bruker Avance III-500 (Bruker, Германия) с частотой 500.20 МГц (1H) и 125.775 МГц (13C). Химические сдвиги ядер 1Н и 13С определяли относительно остаточных сигналов растворителя (δ, м.д., 2.50 и 39.52 для DMSO-d6, 7.26 и 77.16 для CDCl3 соответственно). Температуры плавления определяли на микронагревательном столике Boetius RNMK (Boetius, Германия). Элементный анализ (С, Н, N) выполняли на микроанализаторе Vario MicroCube Elementar (Elementar, Германия). DL-ValГК ((R,S)-2-амино-3-метилбутирогидроксамовая кислота, (R,S)-N-гидроксиамид-2-амино-3-метилмасляной кислоты) синтезировали по известной методике [24].
1-Гидрокси-3-изопропил-7,7,9,9-тетраметил-1,4,8-триазаспиро[4,5]декан-2-он (IV). Масс-спектр (ESI), m/z: 270.2192 (M + H)+. C14H27N3O2. Вычислено: 270.2176 (M + H)+.
Гидроксамовая кислота (IVa). Cпектр 1Н-ЯМР (CDCl3, с 0.05 М, δ, м.д., J, Гц): 0.96 (д, 3Н, СНМеА, 3J 6.9), 1.06 (д, 3Н, СНМеВ, 3J 6.9), 1.20 (с, 3Н, β-МеА), 1.23 (с, 3Н, β-МеВ), 1.44 (с, 3Н, β-МеС), 1.46 (с, 3Н, β-МеD), 1.46 (д.д, 1Н, α2-НА, 2J 13.9, 4Jα1-HA 2.0), 1.55 (д.д, 2Н, NH + α1-HA, J 13.6, J 2.0), 1.71 (д, 1Н, α1-НВ, 2J 13.6), 1.99 (д, 1Н, α2-НВ, 2J 13.9), 2.03 (с, 1.5 Н, MeCN), 2.10 (д. гепт, 1Н, СНМе2, 3JМе 6.9, 3JСНСО 4.8), 3.33 (д.д, 1Н, СНСО, 3JNH 10.2, 3JСНМе2 4.8), 4.96 (уш. с, 2Н, 0.5Н2О + + NOH).
Спектр 1Н-ЯМР (DMSO-d6, с 0.4 М, δ, м.д., J, Гц): 0.93 (д, 3Н, СНМеА, 3J 6.6), 1.02 (д, 3Н, СНМеВ, 3J 6.7), 1.05 (с, 3Н, β-МеА), 1.06 (с, 3Н, β-МеВ), 1.34 (с, 3Н, β-МеС), 1.35 (с, 3Н, β-МеD), 1.42 (уш. д, 2Н, α1‑НА + α2-НА, 2J 13.3), 1.47 (д, 1Н, α1-НВ, 2J 13.3), 1.62 (д.д, 1Н, α2-HВ, 2J 13.5, J 2.4), 1.90 (д. гепт, 1Н, СНМе2, 3JМе 6.9, 3JСНСО 6.5), 2.08 (с, 1.5Н, 0.5 MeCN), 2.32 (уш. д, 1Н, NH, 3JCHCO 10.7), 3.07 (д.д, 1Н, СНСО, 3JNH 10.7, 3JСНМе2 6.3), 3.71 (уш. с, Н2О + NOH).
Спектр 13С-ЯМР (DMSO-d6, с 0.4 М, δ, м.д., J, Гц): 1.42 (к, 0.5С, MeCN, 1J 135.8), 18.67 (к.м, 1С, СНМеА, 1J 125.8), 18.95 (к.м, 1С, СНМеВ, 1J 125.8), 29.15 (к.м, 1С, β-Ме, 1J 125.8), 29.27 (д.м, 1С, СНМе2, 1J 127.0), 29.34 (к.м, 1С, β-Ме, 1J 125.8), 35.25 (к.м, 1С, β-Ме, 1J 125.8). 35.35 (к.м, 1С, β-Ме, 1J 125.8), 39.69 (т, 1С, α1-СН2, 1J 127.0), 44.19 (т, 1С, α2-СН2, 1J 127.0), 50.11 (м, 1С, β-СМе2), 50.20 (м, 1С, β‑СМе2), 59.88 (д, 1С, СНСО, 1J 139.6), 77.45 (м, 1С, NCN), 118.06 (к, 0.5С, MeCN, 2J 10.1), 171.27 (д.д, 1С, С=О, J 3.8, J 2.5).
Гидроксамовая кислота (IVb). Спектр 1Н-ЯМР (CDCl3, с 0.05 М, δ, м.д., 3J, Гц): 0.97 (д, 3Н, СНМеА, 3J 6.9), 1.07 (д, 3Н, СНМеВ, 3J 6.9), 1.20 (с, 3Н, β‑МеА), 1.23 (с, 3Н, β-МеВ), 1.44 (с, 3Н, β-МеС), 1.46 (с, 3Н, β-МеD), 1.47 (д.д, 1Н, α2-НА, 2J 13.9, 4Jα2-HA 2.0), 1.55 (д.д, 2Н, NH + α1-HA, J 13.4, J 2.0), 1.69 (д, 1Н, α1-НВ, 2J 13.7), 1.97 (д, 1Н, α2-НВ, 2J 14.0), 2.12 (д. гепт, 1Н, СНМе2, 3JМе 6.9, 3JСНСО 4.9), 3.35 (д.д, 1Н, СНСО, 3JNH 10.3, 3JСНМе2 4.9), 3.73 (с, 8Н, диоксан).
Спектр 1Н-ЯМР (CDCl3, с 0.25 М, δ, м.д., 3J, Гц): 0.88 (д, 3Н, СНМеА, 3J 6.9), 0.99 (д, 3Н, СНМеВ, 3J 6.9), 1.15 (с, 3Н, β-МеА), 1.17 (с, 3Н, β-МеВ), 1.38 (д.д, 1Н, α2-НА, 2J 13.9, 4Jα2-HA 1.6), 1.39 (с, 3Н, β-МеС), 1.41 (с, 3Н, β-МеD), 1.48 (д.д, 2Н, NH + α1-HA, J 13.5, J 1.6), 1.68 (д, 1Н, α1-НВ, 2J 13.5), 1.96 (д, 1Н, α2-НВ, 2J 13.9), 2.04 (д. гепт, 1Н, СНМе2, 3JМе 6.9, 3JСНСО 4.8), 3.24 (д.д, 1Н, СНСО, 3JNH 9.9, 3JСНМе2 4.8), 3.65 (с, 8Н, диоксан), 6.06 (уш.с, 1Н, NOH).
Спектр 13С-ЯМР (CDCl3, с 0.25 М, δ, м.д., 3J, Гц): 17.66 (1С, СНМеА), 18.88 (1С, СНМеВ), 28.46 (1С, β-МеD), 28.74 (1C, β-МеC), 28.94 (1C, СНМе2), 35.04 (1C, β-МеB), 35.23 (1C, β-МеA), 40.68 (1C, α1‑CH2), 45.63 (1C, α2-CH2), 51.15 (2C, β-CМе2), 60.71 (1C, CHCO), 67.07 (4C, диоксан), 78.34 (1C, NCN), 170.91 (1C, C=O).
Острая токсичность гидроксамовой кислоты (IV). Острую токсичность (LD50) определяли на мышах-гибридах BDF1, полученных из УНУ Питомник и виварий ФГБУН “Институт проблем химической физики” РАН, при однократном внутрибрюшинном введении водных растворов кислоты (IV) по методу, описанному в работе [25]. Исследования острой токсичности соединения (IV) проводили в соответствии с ГОСТ ISO 10993-11-2011. В качестве тест-объекта использовали самцов мышей-гибридов линии BDF1 весом 21–24 г (n = 30). Животных содержали в соответствии с методическими рекомендациями по содержанию лабораторных животных в вивариях со свободным доступом к воде и пище. В каждую опытную группу рандомизированно распределяли по шесть особей с использованием в качестве основного критерия массу тела так, чтобы различия в данном параметре между особями не превышали 20%. Соединения вводили животным однократно внутрибрюшинно в виде водного раствора в диапазоне доз 500–900 мг/кг массы тела животного. Для учета гибели и оценки общего клинического состояния мышей наблюдения за животными проводили непрерывно в течение первых 30 мин после введения вещества, затем с интервалом 1 ч в течение 4 ч и далее ежедневно один раз в день. Общий срок наблюдения составил 14 сут. Для определения токсических доз строили кривую нарастания токсичности по мере увеличения дозы. Расчет доз LD16, LD50, LD84 и LD100 проводили с использованием метода пробит-анализа.
Хемосенсибилизирующая активность гидроксамовой кислоты (IV). Исследование хемосенсибилизирующей активности гидроксамовой кислоты (IV) проводили на модели экспериментальной перевиваемой меланомы B16 на самцах мышей-гибридов линии BDF1 весом 22–24 г (n = 32), полученных из УНУ Питомник и виварий ФГБУН “Институт проблем химической физики” РАН. Мышей содержали в клетках со свободным доступом к воде и пище. Опухоли трансплантировали подкожно (инокулум измельченной опухолевой ткани в изотоническом растворе NaCl 1 : 5 в объеме 0.2 мл). Противоопухолевый цитостатик циклофосфамид (в субтерапевтической дозе) и соединение (IV) вводили в виде водных растворов внутрибрюшинно. Дозы и режимы введения (сутки после трансплантации опухоли) указаны в табл. 1. Отдельная группа животных-опухоленосителей, получавшая вместо инъекций препаратов растворитель, служила контролем. Каждая группа содержала восемь мышей. Наблюдение за животными проводили ежедневно в течение 24 сут, после чего мышей выводили из эксперимента путем дислокации шейных позвонков, извлекали внутренние органы, проводили взвешивание опухоли и подсчет метастазов в легких. Антиметастатическую активность соединений определяли по изменению индекса ингибирования метастазирования (ИИМ) по формуле:
ЗАКЛЮЧЕНИЕ
Было показано, что реакция DL-валингидроксамовой кислоты с триацетонамином проходит как N,N'-региоселективная циклоконденсация с образованием (±)-1-гидрокси-3-изопропил-7,7,9,9-тетраметил-1,4,8-триазаспиро[4,5]декан-2-она. В ходе исследования антиметастатической и противоопухолевой активности синтезированной циклической гидроксамовой кислоты (IV) была обнаружена ее способность повышать чувствительность экспериментальной перевиваемой меланомы В16 мышей к действию циклофосфамида (известного цитостатика алкилирующего действия, использованного в субтерапевтической дозе), проявляя тем самым хемосенсибилизирующие свойства. Было показано значительное снижение количества метастазов, а также торможение роста опухоли у животных экспериментальной группы. Полученные результаты свидетельствуют о перспективности синтеза циклических гидроксамовых кислот на основе DL-валина в качестве потенциальных противоопухолевых агентов.
Список литературы
Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. // CA Cancer J. Clin. 2015. V. 65. P. 87–108. https://doi.org/10.3322/caac.21262
Torre L.A., Siegel R.L., Jemal A. // Adv. Exp. Med. Biol. 2016. V. 893. P. 1–19. https://doi.org/10.1007/978-3-319-24223-1_1
Harding M.C., Sloan C.D., Merrill R.M., Harding T.M., Thacker B.J., Thacker E.L. // Prev. Chronic Dis. 2018. V. 15. P. 180151. https://doi.org/10.5888/pcd15.180151external icon
Pontiki E., Hadjipavlou-Litina D. // Med. Res. Rev. 2012. V. 32. P. 1–165. https://doi.org/10.1002/med.20200
Manal M., Chandrasekar M.J.N., Gomathi Priya J., Nanjan M.J. // Bioorg. Chem. 2016. V. 67. P. 18–42. https://doi.org/10.1016/j.bioorg.2016.05.005
Вартанян А.А., Хоченков Д.А., Хоченкова Ю.А., Мачкова Ю.С., Хачатрян Д.С., Колотаев А.В., Балаев А.Н., Охманович К.А., Осипов В.Н. // Биоорг. химия. 2020. Т. 46. С. 207–219. [Vartanyan A.A., Khochenkov D.A., Khochenkova Yu.A., Markova Yu.S., Khachatryan D.S., Korotaev A.V., Balaev A.N., Okhmanovich K.A., Osipov V.N. // Bioorg. Chem. 2020. V. 46. P. 252–263.] https://doi.org/10.1134/S106816202002017X
Shah R.R. // Drug Saf. 2019. V. 42. P. 235–245.
Duvi M., Talpur R., Ni X., Zhang Ch., Hazarika P., Kelly C., Chiao J.H., Reilly J.F., Ricker J.L., Richon V.M., Frankel S.R. // Blood. 2007. V. 109. P. 31–39. https://doi.org/10.1182/blood-2006-06-025999
Atadja P. // Cancer Lett. 2009. V. 280. P. 233–241. https://doi.org/10.1016/j.canlet.2009.02.019
Sawas A., Radeski D., O’Connor O.A. // Ther. Adv. Hematol. 2015. V. 6. P. 202–208. https://doi.org/10.1177/2040620715592567
Qin H.-T., Li H.-Q., Liu F. // Expert Opin. Ther. Pat. 2016. V. 27. P. 1–15. https://doi.org/10.1080/13543776.2017.1276565
Rajulu G.G., Naik H.S.B., Viswanadhan A., Thiruvengadam J., Rajesh K., Ganesh S., Jagadheshan H., Kesavan P.K. // Chem. Pharm. Bull. 2014. V. 62. P. 168–175. https://doi.org/10.1248/cpb.c13-00797
Zhang X., Zhang J., Tong L., Luo Yu., Su M., Zang Y., Li J., Lu W., Chen Y. // Bioorg. Med. Chem. 2013. V. 21. P. 3240–3244. https://doi.org/10.1016/j.bmc.2013.03.049
Rikiishi H., Shinohara F., Sato T., Sato Y., Suzuki M., Echigo S. // Int. J. Oncol. 2007. V. 30. P. 1181–1188.
Vystorop I.V., Lyssenko K.A., Kostyanovsky R.G. // Mendeleev Commun. 2002. V. 12. P. 85–87. https://doi.org/10.1070/MC2002v012n03ABEH001603
Vystorop I.V., Aliev Z.G., Andreeva N.Yu., Atovmyan L.O., Fedorov B.S. // Russ. Chem. Bull., 2000. V. 49. P. 182–183. https://doi.org/10.1070/MC2002v012n03ABEH001603
Vystorop I.V., Nelyubina Yu.V., Voznesensky V.N., Sun W.-H., Lodygina V.P., Lyssenko K.A., Kostyanovsky R.G. // Mendeleev Commun. 2010. V. 20. P. 106–108. https://doi.org/10.1016/j.mencom.2010.03.014
Vystorop I.V., Konovalova N.P., Nelyubina Yu.V., Varfolomeev V.N., Fedorov B.S., Sashenkova T.E., Berseneva E.N., Lyssenko K.A., Kostyanovsky R.G. // Russ. Chem. Bull. 2010. V. 59. P. 127–135. https://doi.org/10.1007/s11172-010-0055-x
Hogradi M. // Akademiai Kiado, Budapest, chapter 1.2.3, 1981.
Williams D.H., Fleming I. // Stereochemistry. Basic Concept and Applications / Akademiai Kiado, Budapest, chapter 1.2.3, 1981.
Vystorop I.V., Konovalova N.P., Nelyubina Yu.V., Chernyak A.V., Sashenkova T.E., Klimanova E.N., Utienyshev A.N., Fedorov B.S., Shilov G.V., Kostyanovsky R.G. // Russ. Chem. Bull. 2013. V. 62. P. 1272–1281. https://doi.org/10.1007/s11172-013-0176-0
Vystorop I.V., Konovalova N.P., Sashenkova T.E., Berseneva E.N., Chernyak A.V., Fedorov B.S., Kostyanovsky R.G. // Mendeleev Commun. 2011. V. 21. P. 239–241. https://doi.org/10.1016/j.mencom.2011.09.002
Mishchenko D.V., Neganova M.E., Klimanova E. N., Sashenkova T. E., Klochkov S.G., Shevtsova E.F., Vystorop I.V., Tarasov V.V., Chubarev V.N., Samsonova A.N., Ashraf G.Md., Barreto G., Yarla N.S., Aliev G. // Curr. Cancer Drug Targets. 2018. V. 18. P. 365–371. https://doi.org/10.2174/1568009617666170623104030
Gunningham K.G., Newbold G.T., Spring F.S., Stark J. // Chem. Soc. 1949. P. 2091.
Руководство по проведению доклинических исследований лекарственных средств. Часть первая / Под ред. Миронова А.Н. М.: Гриф и К, 2012. 944 с.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия