

Биоорганическая химия, 2022, T. 48, № 1, стр. 3-31
Супрамолекулярные системы на основе макроциклических соединений с протеинами. Перспективы применения
Н. Ш. Лебедева 1, *, О. И. Койфман 1, 2
1 Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, ул. Академическая, 1, Россия
2 Ивановский государственный химико-технологический университет
153000 Иваново, Шереметевский просп., 7, Россия
* E-mail: nsl@isc-ras.ru
Поступила в редакцию 07.12.2020
После доработки 28.12.2020
Принята к публикации 31.12.2020
- EDN: XJYVUR
- DOI: 10.31857/S0132342322010079
Аннотация
Супрамолекулярные комплексы протеинов с макрогетероциклическими соединениями – уникальные структуры, существенно отличающиеся от классических супрамолекул как сложностью организации, так и многофункциональностью. Изучение природных и создание синтетических супрамолекулярных систем позволяет влиять на биохимические процессы и управлять ими, а также открывает новые перспективы практического использования макрогетероцикл-протеиновых супрамолекул. Ежегодно возрастающий интерес к изучению супрамолекулярных систем обусловлен их вариабельностью, разнообразием исходных компонентов, возможностью их химической и биохимической модификации, способностью участвовать в фото- и термоиндуцированных реакциях. Макрогетероциклический компонент супрамолекулярной системы не только влияет на аффинность протеиновой части к другим лигандам, но и может придавать супрамолекуле новые свойства. В обзоре проанализированы и обобщены результаты теоретических и экспериментальных исследований движущих сил самоорганизации молекул, процессов образования супрамолекулярных комплексов. Особое внимание уделено исследованиям супрамолекул протеин–макрогетероцикл, имеющих прикладную направленность для создания искусственных фотосистем, применения в биомедицинских целях в фотодинамической, фототермической терапии, для фотоинактивации бактерий, медицинской диагностики. Обозначены проблемы, сложности в развитии каждого направления и потенциальные пути их решения.
Введение........................................................... 3
Движущие силы самосборки супрамолекулярных структур......................................................... 4
Биологические фотосистемы.......................... 6
Искусственные фотосистемы......................... 8
Супрамолекулярные комплексы протеинов с тетрапиррольными макрогетероциклами в медицине............................................................................13
Фотодинамическая терапия............................ 14
Фототермическая терапия............................. 17
Транспортные системы/системы доставки.... 18
Антимикробная фотодинамическая терапия..20
Медицинская диагностика.............................. 22
Заключение.................................................... 24
Список литературы........................................ 24
ВВЕДЕНИЕ
Супрамолекулярная химия фокусируется на нековалентных специфических взаимодействиях, которые приводят к молекулярному распознаванию и процессам самосборки [1]. Супрамолекулярная химия изначально была построена на принципах биомиметики. Уникальные природные супрамолекулярные системы, как сравнительно простые (гемоглобин, цитохромы, ДНК, липиды и др.), так и сложные многокомпонентные (двухслойная клеточная мембрана, капсид вириона, фотосинтетический аппарат растений и бактерий и др.), вдохновляли исследователей на их изучение, а также на разработку и синтез новых многомолекулярных и надмолекулярных архитектур различной сложности и функциональности. Изначально синтетические супрамолекулярные системы были невелики и состояли из относительно простых строительных блоков. Накопленный опыт и знания в области супрамолекулярной химии в настоящее время позволяют создавать впечатляющие супрамолекулярные структуры. Особое место среди них занимают системы на основе макрогетероциклических соединений с протеинами. Именно на их анализе и перспективах применения сфокусирован данный обзор. Свойства самособирающихся систем можно настраивать, манипулируя энергетическими характеристиками составляющих их молекул, поэтому важно понимать, какие движущие силы управляют процессами самосборки и какова их энергетика.
ДВИЖУЩИЕ СИЛЫ САМОСБОРКИ СУПРАМОЛЕКУЛЯРНЫХ СТРУКТУР
Cпецифические взаимодействия – низкоэнергетические, к ним относятся ван-дер-ваальсовые взаимодействия (ориентационные, дисперсионные, индукционные, <5 кДж/моль), донорно-акцепторные (<200 кДж/моль), π–π-взаимодействия (<50 кДж/моль), катион-π-взаимодействия (5–80 кДж/моль) [2–4], водородные связи (4–120 кДж/моль). В биологических системах они определяют структуру таких биомолекул, как ДНК и белки, и отвечают за процесс молекулярного распознавания. В большинстве случаев нековалентные взаимодействия значительно слабее (на 1–3 порядка), чем ковалентные связи. Их энергия, с одной стороны, достаточна, чтобы удерживать молекулы в составе биосупрамолекулярных структур. C другой стороны, эти системы имеют низкий активационный барьер разрушения [1], что позволяет биосупрамолекулярной структуре перестраиваться, реагировать на определенные внешние сигналы и, тем самым, реализовывать свои биологические функции.
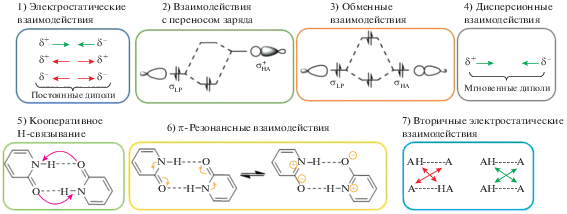
Основные принципы нековалентных взаимодействий были сформулированы более 90 лет назад, например, Полинг дал описание водородных связей в 1930 г. [5]. С тех пор представления о природе водородных связей расширялись и изменялись. Водородные связи зачастую рассматриваются как электростатические взаимодействия между электроотрицательным атомом A и атомом водорода, присоединенным ко второму электроотрицательному атому В [6]. Однако это описание очень схематичное и поверхностное. Оно не объясняет, например, спектральное проявление Н-связей в ЯМР-спектрах (химический сдвиг сигналов 1Н-ЯМР в слабое поле), в ИК-спектрах (изменение колебательной частоты при растяжении связи Н–В), и, самое главное, с точки зрения построения и управления супрамолекулярными структурами, электростатический взгляд на природу Н-связей не объясняет их кооперативный эффект. Водородные связи представляют собой сложное взаимодействие между различными энергетическими компонентами (электростатические взаимодействия, взаимодействия с переносом заряда, обменные взаимодействия, дисперсионные взаимодействия, кооперативные эффекты, π-резонансные взаимодействия и вторичные электростатические взаимодействия) (рис. 1), и их природа до сих пор служит предметом постоянных дискуссий. Более подробно с современными взглядами на природу водородных связей можно ознакомиться в обзорах [7, 8].
По мере накопления данных о взаимодействиях между ароматическими соединениями трансформировались и взгляды на природу π–π-взаимодействия, которое изначально трактовалось как взаимодействие донорно-акцепторного типа, при котором ароматическая система одной молекулы рассматривалась в роли донора π-электронной плотности, а ароматическая система другой – в качестве акцептора [10]. В дальнейшем взгляды на природу π–π-комплексов были пересмотрены, и сейчас они расцениваются как результат двух противоположно направленных сил π–σ-стягивания и π–π-отталкивания [11, 12]. Для π–π-взаимодействий существенна роль поляризации π‑ароматической системы, при которой создается квадрупольный момент с распределением частичных отрицательных и положительных зарядов, зависящих от наличия и природы периферийных заместителей. Именно распределение электронной плотности, наличие электрон-обогащенных и электронодефицитных мест в ароматических молекулах определяет их взаимное расположение в π–π-комплексе [11–14] (рис. 2).
Рис. 2.
Схемы расположения ароматических молекул в π–π-комплексах на примере электрон-обогащенных (бензол и 1,5-диалкоксинафталин) и электронодефицитных молекул (1,4,5,8-нафталинтетракарбоксилатдиимид) с акцентом на места электростатического притяжения или отталкивания [12].

Таким образом, вводя в ароматические соединения электронодонорные или электроноакцепторные заместители и тем самым изменяя распределение электронной плотности в молекулах, можно построить различно организованные супрамолекулярные комплексы. Данный принцип широко применяется для создания различных архитектур и сборок ароматических соединений [15, 16].
Несмотря на огромный прогресс, достигнутый в последние десятилетия как в теоретическом, так и в экспериментальном изучении нековалентных, специфических связей, мы все еще далеки от однозначного понимания нековалентных комплексов [17]. Наглядная демонстрация данного тезиса – работа Černý et al. [18], в которой описана роль H-связей и π-стекинговых взаимодействий в стабилизации биополимеров: ДНК, олигопептидов и белков. Авторы приходят к заключению о доминирующей роли дисперсионных взаимодействий в стабилизации перечисленных биоструктур по сравнению с вкладом в их стабилизацию от Н-связей и π–π-взаимодействий.
Другой важный фактор, определяющий устойчивость супрамолекулярных структур, – количество нековалентных взаимодействий. Самый наглядный пример, отражающий суммарный эффект количества нековалентных взаимодействий, – способность гекконов быстро подниматься по гладким стеклянным вертикальным поверхностям. Оказалось, что на лапке геккона почти пятьсот тысяч ороговевших щетинок-волосков, каждый из которых за счет ван-дер-ваальсовых сил обеспечивает сцепление с любой поверхностью [19], что позволяет удерживать вес животного в вертикальном положении. Принцип аддитивности низкоэнергетических взаимодействий также широко применяется при построении супрамолекулярных систем.
Говоря о движущих силах образования супрамолекулярных комплексов, нельзя не отметить роль растворителя, которая в природных и синтетических супрамолекулярных структурах существенно отличается. В биосистемах растворитель – вода, она определяет гидрофобный эффект. Когда неполярное соединение вносится в воду, происходит нарушение сетки водородных связей и перегруппировка молекул воды вокруг неполярных молекул. Присутствие в воде разных гидрофобных соединений приводит к созданию больших полостей для размещения скоплений неполярных молекул, которые стремятся взаимодействовать друг с другом для уменьшения контакта с водой. Таким образом, для минимизации контакта неполярных молекул с водой и минимизации разрушения структуры воды формируются агрегированные структуры [20]. Гидрофобный эффект играет решающую роль в формировании амфифильных мицеллярных агрегатов [2], сворачивании глобулярных белков, функционировании биомембран [21, 22], формировании фосфолипидных бислоев [23]. С точки зрения термодинамики, растворитель может влиять на образование супрамолекулярной системы не только энтропийно. Например, растворитель существенно влияет и на энергию π–π-взаимодействия, причем в полярных растворителях, как правило, устойчивость π–π-комплексов выше, чем в неполярных [12]. Процессы самосборки очень чувствительны к изменениям растворителя из-за различной термодинамической стабильности промежуточных и конечных комплексов, ассоциатов, образующихся в разных средах [24]. В биосупрамолекулярных структурах растворитель не только участвует в образовании супрамолекулярной системы, но и выступает ее частью, обеспечивая стабильность/лабильность системы, управление и воздействие на супрамолекулы, осуществляет транспортную функцию [25, 26]. Напротив, в синтетических супрамолекулярных системах роль растворителя, как правило, сводится к стадии образования, самосборке системы, влиянии на рост и архитектуру супрамолекулярных систем [25], а затем от растворителя стремятся избавиться [25–27].
Другая отличительная особенность синтетических супрамолекулярных систем, существенно отличающих их от большинства биоструктур, заключается в том, что синтетические системы находятся в состоянии равновесия. Поэтому зачастую синтетические системы также называют равновесными сборками, для них характерна термодинамическая стабильность в течение длительного времени. Синтетические супрамолекулярные системы находятся под термодинамическим контролем, в то время как биологические супрамолекулярные системы контролируются кинетически [28]. Действительно, большинство химических процессов в клетке (ферментативный катализ фосфорилирования [29], трансмембранная ионная накачка [30], синтез белка в рибосомах [1] и др.) невозможны без использования свободной энергии реакции гидролиза АТФ. Mattia et al. [1] предлагают различать супрамолекулярные системы, которые находятся в кинетической ловушке, и системы, которые далеки от равновесия и требуют непрерывной подачи энергии для поддержания стационарного состояния. Первые – кинетически запертые сборки/системы, которые могут существовать длительное время, находятся в локальном энергетическом минимуме, и для перехода в глобальный минимум (более энергетически выгодные структуры) требуются высокие энергии активации. Вторые – системы, требующие постоянной подкачки энергии. Если подача энергии прекращается, система разрушается, переходя в термодинамически минимальное состояние. Непрерывное энергетическое преобразование, которому подвергаются эти структуры, позволяет им иметь интересные, а иногда и непредсказуемые, возникающие эмерджентные свойства и функции, такие как однонаправленное движение, возникновение градиента концентрации и др. Поэтому создание подобных систем, контролируемых энергетически извне, очень перспективно и может позволить исследователям приблизиться к природным совершенным биосупрамолекулярным системам.
В биосупрамолекулярных системах основным источником энергии служит АТФ, а в синтетических супрамолекулярных системах, возможно, подобным источником энергии могут выступать макрогетероциклические соединения порфиринового, хлоринового и фталоцианинового классов. Перечисленные макрогетероциклические соединения имеют ароматическую природу. Их особенность заключается в том, что они могут поглощать энергию света в широком спектральном диапазоне, а благодаря длительному времени жизни в возбужденном триплетном состоянии (1–6 нс) способны передавать эту энергию другим молекулам или генерировать фототок [31].
БИОЛОГИЧЕСКИЕ ФОТОСИСТЕМЫ
В природе соединения порфиринового ряда входят в состав фотосинтетического аппарата высших зеленых растений, большинства водорослей и фотосинтезирующих бактерий, обеспечивающего трансформацию солнечной энергии в химическую, синтез большей части органического вещества, а также кислорода. Преобразование световой энергии в полезную химическую энергию выполняется четырьмя многосубъединичными мембранно-белковыми комплексами (рис. 3). Два комплекса – белок-пигментные суперкомплексы, называемые фотосистема I (PSI) и фотосистема II (PSII). Они функционируют как молекулярные фотовольтаики – испускают электроны при поглощении световой энергии. Каждая фотосистема состоит из основного комплекса и периферической светособирающей антенной системы – I (LHCI) для PSI и II (LHCII) для PSII [32]. PSI и PSII содержат большое количество молекул хлорофилла. Например, в структуре PSI термофильной цианобактерии Synechococcus elongatus содержится 96 молекул хлорофилла [33], а в PSI мезофильной цианобактерии Synechocystis sp. PCC6803 – 591 молекула хлорофилла [34]. Именно хлорофиллы с другими пигментами в PSI и PSII поглощают световую энергию и передают ее в центр фотосинтетической реакции, дополнительно вызывая возбуждение хлорофиллов, известных как P680 для PSII и P700 для PSI, чтобы инициировать перемещение протонов через мембрану. Третий комплекс, цитохром b6 f, опосредует транспорт электронов между двумя фотосистемами и в дальнейшем способствует образованию протон-движущей силы (pmf). Четвертый комплекс, АТФаза F-типа (CF1CF0-АТФ-синтаза), обеспечивает синтез молекулы АТФ. Дополняют эти комплексы молекула пластохинона (PQ) и небольшой водорастворимый медь-связывающий белок, называемый пластоцианином (PC) [35]. Четыре основных комплекса (фотосистемы I и II, цитохром b6 f и АТФ-синтаза) сохраняются от цианобактерий до высших растений.
Рис. 3.
Схематическое изображение бислоя тилакоидной мембраны со встроенными белковыми комплексами, участвующими в транспорте электронов. Пунктирная стрелка – циклический перенос электрона [35].
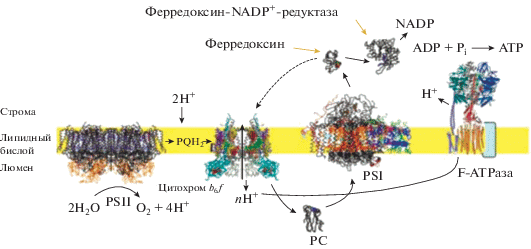
Фотосистема II использует световую энергию для каталитического окисления воды [36, 37]. Этот процесс служит основным источником электронов, которые впоследствии переносятся через мембрану на цитохром b6 f [38–41] молекулами пластохинона (PQ), а затем в фотосистему I [33, 42] через просвет тилакоидов с помощью небольшого водорастворимого белка пластоцианина [43]. Электроны впоследствии переносятся из PSI на окислительно-восстановительный белок ферредоксин в строме [44, 45]. Наконец, ферредоксин окисляется ферредоксин-NADP+-редуктазой, которая восстанавливает NADP+ до NADPН [33, 46]. Одновременно протонный градиент, генерируемый через тилакоиды, управляет образованием молекул АТФ с помощью АТФ-синтазы [46, 47]. Все вышеупомянутые компоненты находятся в сплюснутых пузырьках, называемых тилакоидами, которые также обеспечивают среду для передачи энергии. Самая сложная форма тилакоидных мембран обнаружена в хлоропластах высших растений. У них тилакоиды образуют сложные, сильно взаимосвязанные трехмерные ламеллярные сети, отличающиеся дифференциацией на два различных морфологических домена: цилиндрическая стопка, состоящая из множества плотно прижатых слоев, называемых грана, и неупакованные мембранные области, которые соединяют между собой граны, называемые ламелла стромы [35]. PSI расположена в ламелле стромы тилакоида, тогда как PSII – в многослойном гран-домене [48].
Сложная организация фотосинтетического аппарата обеспечивает не только реализацию основных биохимических функций, но и способность тонкой подстройки системы к изменению внешних условий (уровень и интенсивность света, температура и др.). Уровень регуляции определяется взаимодействиями между антенными системами и фотосистемами [34]. В случае растений было показано, что светособирающая антенна PSI, состоящая из четырех тесно связанных антенных хлорофилл-содержащих белков, связывается с реакционным центром с различной стехиометрией, в зависимости от условий освещения и других факторов окружающей среды [35], тем самым препятствуя фотоокислению и обеспечивая эффективность свето-акклиматизационных реакций [34]. Кроме того, в составе фотосинтетического аппарата есть фоторецепторы (включая две фотосистемы, действующие как светочувствительные датчики) и преобразователи сигналов, которые задействованы в формировании реакции акклиматизации [49, 50]. Даже поверхностное рассмотрение архитектуры и строения фотосинтетического аппарата свидетельствует о сложности его организации и взаимодействия составных частей. Это не удивительно, ведь фотосинтетический аппарат – уникальное творение, созданное природой за несколько миллиардов лет эволюции.
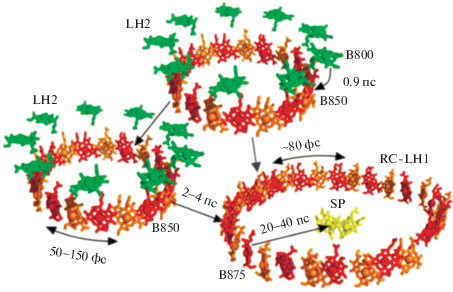
Биологические системы самосборки хромофоров поражают своей упорядоченностью и гармоничностью (рис. 4). В комплексах хромофор–белок точная ориентация пигментов белковой матрицей обеспечивает эффективную ступенчатую передачу энергии к реакционному центру, где и происходит разделение зарядов.
Рис. 4.
Схема процессов передачи энергии светового возбуждения от антенных систем LH2 и LH1 к специальной паре реакционного центра бактериального фотосинтеза [51].
ИСКУССТВЕННЫЕ ФОТОСИСТЕМЫ
Сложная самоорганизация естественных фотосистем обеспечивает эффективный перенос энергии и электронов. Архитектура этих фотосенсибилизирующих молекул определяет фотофизику накопления и переноса энергии, тушение, сочетание с реакционным центром по скорости и химизму. Безусловно, создаваемые искусственные супрамолекулярные системы хоть и основаны на способности соединений порфиринового и хлоринового рядов к поглощению световой энергии и ее трансформации, но организованы значительно проще. Рассмотрим созданные искусственные аналоги светособирающих антенн. Типичная искусственная фотосинтетическая система должна состоять из антенны, содержащей набор хромофоров, которые эффективно поглощают солнечный свет во всем или максимально широком диапазоне солнечного спектра и передают эту энергию реакционному центру. Реакционный центр – это место, где поглощенная энергия возбуждения обеспечивает эффективный фотоиндуцированный перенос электронов к акцептору электронов для создания состояний с разделенными зарядами. Таким образом, реакционный центр можно рассматривать как фотоэлектрическое устройство нанометрового масштаба. Кроме того, реакционный центр может быть соединен с катализатором окисления воды и катализатором восстановления протонов для превращения воды в водород или синтеза заданного вещества (топлива).
Одна из первых проблем, возникающих при конструировании светособирающих антенн, – организация самоассоциатов хромофорных соединений. Причины поиска именно ассоциатов хромофорных соединений очевидны – ни один хромофор не способен эффективно обеспечивать поглощение и передачу световой энергии в широком спектральном диапазоне. Одна из возможностей преодоления этой проблемы – работа с агрегатами хромофоров, которые благодаря экситонному взаимодействию могут иметь гораздо более широкие диапазоны поглощения [52, 53].
Для достижения конечной цели – создания искусственной светособирающей антенны – используются различные приемы сборки хромофорных соединений, среди которых можно выделить следующие: 1) мультихромофорные массивы, 2) кольцеобразные светособирающие антенны, 3) звездообразные светособирающие антенны, 4) антенны для сбора света в форме ветряных мельниц, 4) полиады, 5) дендрические системы.
В последнее время был достигнут значительный прогресс в возможностях синтеза длинных мультихромофорных стержневидных массивов порфиринов, в которых порфирины ковалентно соединены и образуют олигомерные структуры. Чередующаяся ортогональная конформация между соседними порфириновыми единицами в мезосвязанных массивах порфиринов сводит к минимуму возможность π-электронного сопряжения в массиве, несмотря на чрезвычайно короткое расстояние между порфиринами, которое составляет 8.4 Å. Массивы демонстрируют дипольное экситонное взаимодействие, при этом длина делокализации экситона (когерентная длина) распространяется на ~4–5 [54] соседних порфириновых единицы, что практически совпадает со значением, определенным в кольце B850 в LH2 (рис. 4) [55].
В научной литературе широко представлены различные конструкции ассоциатов хромофоров, как ковалентно связанных, так и образованных за счет специфических нековалентных взаимодействий [56].
Перечисленные выше структурные организации хромофоров не нашли широкого применения, т.к. остались нерешенными проблемы переноса и разделения заряда. Для идеальных фотосинтетических искусственных систем необходимые параметры процесса переноса электрона между донором и акцептором – быстрое разделение и относительно медленная рекомбинация зарядов. Фотоиндуцированный одноступенчатый перенос электрона может быть достигнут путем создания простых донорно-акцепторных (D–A) пар. В качестве донора выступает хромофор, он же действует как первичный донор электронов, который передает электрон акцептору (процесс разделения заряда, CS), как показано на рис. 5a. Из-за близости донора и акцептора ион-радикальная пара, образующаяся при фотовозбуждении, быстро рекомбинирует в исходное основное состояние (процесс рекомбинации заряда, CR).
Рис. 5.
Принципиальная схема для одноступенчатого (a), многоступенчатого переноса электронов (б) и переноса энергии в сочетании с процессами переноса электронов (в). CS – процесс разделения заряда, CR – процесс рекомбинации заряда [57].
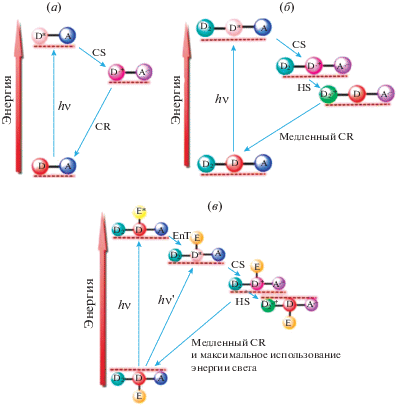
Для предотвращения рекомбинации были разработаны многомодульные системы для генерации долгоживущих состояний с разделенными зарядами [58]. Для реализации многоступенчатого переноса электрона требуются по крайней мере три вида молекул: D2–D–A или D–A–A2, где D и D2 – первичные и вторичные доноры электронов, а A и A2 – первичные и вторичные акцепторы электронов соответственно [59, 60]. Легкое окисление D2 и легкое восстановление A2 – основные требования молекулярного дизайна светособирающей антенны. Фотохимические события при возбуждении D в D2–D–A показаны на рис. 5б, конечный результат которых – состояние с разделенным зарядом ${\text{D}}_{{\text{2}}}^{{\centerdot + }}$–D–${{{\text{A}}}^{{\centerdot - }}}$. На рис. 5в приведена принципиальная схема функционирования антенны, работающей в более широком спектральном диапазоне за счет добавления фотосенсибилизатора (Е).
Потенциальные доноры (D и D2) и акцепторы (А и A2) хорошо известны [61]. Наибольшей популярностью в качестве акцепторов пользуются фуллерены С60/С70, графен и углеродные нанотрубки, т.к. они обладают уникальными электронными характеристиками, обеспечивающими высокую подвижность и локализацию разделенных зарядов [62, 63]. В качестве примера можно привести пентаду [64], состоящую из ферроцена (донор электронов), мезосвязанного тримера порфирина (фотосенсибилизатор) и фуллерена (акцептор электронов). Полученная структура характеризуется чрезвычайно долгоживущим состоянием с разделенными зарядами (0.53 мкс), полученным с высоким квантовым выходом (0.83), что сравнимо с естественной фотосинтетической системой. Недавно были получены супрамолекулярные светособирающие структуры, состоящие из четырех фуллеренов и геометрических структур, построенных из кубических блоков порфиринов с ароматическими спейсерами [65]. Благодаря уникальным особенностям упаковки, которые могут обеспечить быстрое разделение и делокализацию заряда, подобные архитектуры демонстрируют сильные взаимодействия с переносом заряда, сверхдлительное время жизни в состоянии с разделенными зарядами (до 20 нс) и высокую фотопроводимость 3.1 × 10–5 см2 В–1 с–1.
При конструировании полиад в качестве акцепторов могут быть использованы и сами хромофоры [66], при этом, как правило, их самосборка осуществляется за счет аксиальной координации периферийных заместителей одного хромофора (порфирина) на металле-комплексообразователе другого хромофора (фталоцианине). Подробно ключевые аспекты фотохимии подобных полиадных искусственных фотосинтетических систем рассмотрены в многочисленных обзорах [67–69].
Созданные диады и триады могут быть внедрены в мембраны и другие биоструктуры, например, Steinberg-Yfrach et al. [70] разработана фотосинтетическая мембрана (рис. 6), состоящая из липосомного бислоя, триады и фермента АТФ-синтазы. Протонный насос приводится в действие векторным фотоиндуцированным переносом электронов в молекулярной триаде каротин–порфирин–нафтохинон (C–P–Q), которая генерирует частицы ${{{\text{C}}}^{{\centerdot + }}}$–P–${{{\text{Q}}}^{{\centerdot - }}}$ при возбуждении видимым светом. Внутримолекулярный окислительно-восстановительный потенциал, представленный катион-радикалом каротиноида и анион-радикалом нафтохинона, связан с перемещением протона липофильным хиноном (Qs). Восстановление происходит вблизи внешней границы раздела би--слой–вода, когда Qs принимает электрон от анион-радикала нафтохинона с образованием ${\text{Qs}}{{_{{}}^{{\centerdot - }}}^{{}}}$. После протонирования ${\text{Qs}}{{_{{}}^{{\centerdot - }}}^{{}}}$ вблизи внешней границы раздела с водой семихинон (H${{{\text{Q}}}^{\centerdot }}$ ) в результате реакций диффузии или самообмена между молекулами Qs доставляет протон и электрон к месту окислительного потенциала (${{{\text{C}}}^{{\centerdot + }}}$–P–Q), расположенному вблизи внутренней поверхности мембраны. Окисление H${{{\text{Q}}}^{\centerdot }}$ вблизи внутренней водной границы раздела приводит к выбросу протонов во внутрилипосомный объем. При накоплении достаточного количества протонов запускается образование АТФ из аденозиндифосфата (АДФ) и неорганического фосфата (Pi).
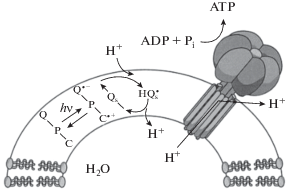
Выше приведены примеры сравнительно простых антенных систем, производительность которых не превышает 5–9%, хотя эффективность неорганических сенсибилизированных красителем солнечных элементов за последние три десятилетия существенно повысилась (с 7.12% в 1991 г. до 12.3% в 2020 г. [71, 72]) и приближается к рентабельному порогу в 15%. Повысить эффективность искусственных светособирающих систем можно за счет изменения архитектуры, аналогичной организации сотовой системы связи. Подобная иерархия обеспечивает увеличение захвата фотонов. Благодаря наличию антенн в биосистемах почти каждый захваченный фотон приводит к полезному разделению зарядов, т.е. квантовая эффективность близка к единице [73]. Перестроение и переключение подобных антенных систем происходит гораздо быстрее и эффективнее. Кроме того, для расширения диапазона поглощения в искусственных антеннах можно использовать несколько разных хромофоров.
Наибольшее соответствие предъявляемым требованиям демонстрируют дендрические архитектуры. Дендримеры – это четко определенные гиперразветвленные трехмерные макромолекулы с регулярным древовидным набором ответвлений [64, 74]. Для звездообразной и конусовидной серий дендримерных антенн эффективность передачи энергии от возбужденного синглетного состояния периферийных порфиринов к сердцевине, как правило, значительно снижается с увеличением номера поколения, причем в случае конусовидных систем это явление проявляется ярче. С другой стороны, уменьшение числа поколений (генераций) отрицательно сказывается на общей фотохимической картине, т.к. приводит к уменьшению числа хромофоров. Среди разнообразных массивов хромофоров дендритные каркасы представляются наиболее подходящими для трехмерного расположения большего количества молекул хромофоров и их эффективного взаимодействия. Дендритные каркасы действуют как энергетические воронки и обеспечивают векторную передачу энергии [63, 67]. Тем не менее существует одна проблема, связанная с дендримерной архитектурой, заключающаяся в том, что векторная передача энергии в большинстве синтезированных архитектур происходит от периферии к ядру дендримера (рис. 7а). Для практического применения захват энергии внутри ядра бесполезен, поскольку доступ других реагентов стерически заблокирован, и кроме нежелательной рекомбинации заряда может происходить только термическая дезактивация. Обратный дендримерный поток энергии, схематично представленный на рис. 7б, где энергия передается от центра к периферии и, в конечном итоге, на искусственный реакционный центр, до сих пор не реализован [73].
Рис. 7.
Дендритные архитектуры: (a) – классический дендример, действующий как энергетическая ловушка; (б) – архитектура, позволяющая фотосенсибилизировать реагент R [73].
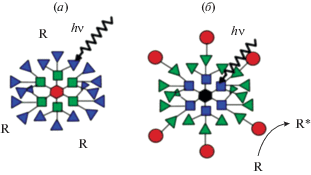
Возможно, в дальнейшем решение проблемы будет найдено при конструировании конусных структур, в которых ядро будет доступно. Стратегия дендритной сборки довольно перспективна, однако получение таких сложных молекул с приемлемым выходом – очень сложной задача, требующая много времени. Поэтому супрамолекулярная система самосборки стала привлекательной альтернативной стратегией для построения хорошо упорядоченных систем с множеством хромофоров на биологических матрицах.
Циклодипептидные и пептидные фрагменты могут быть удобным мотивом для образования супрамолекулярных ансамблей, поскольку они обеспечивают множественные сайты водородных связей с симметрией C2 [75–78]. Димерные структуры, состоящие из двух порфиринов и дипептида, в зависимости от внешних условий и концентрации, образуют различные архитектуры от волокнообразных до тороидальных структур [76, 79], а также рН управляемых архитектур (рис. 8) [80, 81].
Рис. 8.
1 – Супрамолекулярные порфирин-полипептидные системы: (а) – слабое взаимодействие порфирин–порфирин в нейтральных условиях; (б) – взаимодействие J-типа при pH 3.5 [80]; 2 – переключение pH порфирин-пептидных ансамблей. В кислых растворах (pH 3.6) пептид имеет неупорядоченную вторичную структуру, связанные с пептидом порфирины образуют J-агрегаты. В нейтральных растворах (pH 7.6) связывание порфирина с пептидом индуцирует формирование α-спиральной вторичной структуры пептида [81].

Фотокаталитическая система на основе порфиринов, модифицированных пептидами, намного более эффективна, чем система на основе порфирина без пептидного звена. Это позволило создать на основе хромофоров и пептидов искусственные фотосинтетические системы, позволяющие получать, например, L-глутамат [77], кислород [82], аминофенол [78] и другие вещества.
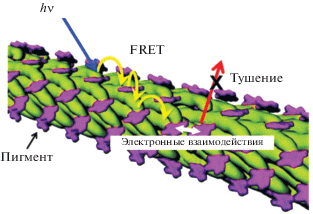
Исследований фотоаккумулятивных и фотокаталитических систем на основе хромофоров и белков проводится значительно меньше. Во-первых, это обусловлено тем, что иммобилизация массива хромофоров должна осуществляться в строго определенном архитектурном паттерне, а природные белки, как правило, нерегулярны. Во-вторых, существенные сложности возникают при сборке хромофор-содержащих белков [83]. В природе сборка белков осуществляется с помощью шаперонов. Как реализовать это в искусственных условиях – вопрос, не имеющий пока четкого ответа. Кроме того, фотоактивность хромофоров и способность к генерации активных форм кислорода может привести к дефектам в белковой матрице и, соответственно, в полученной конечной структуре. Возможный путь преодоления перечисленных трудностей – использование белков с преимущественной β-складчатостью. Особенность белков с преобладанием указанной вторичной структуры состоит в том, что они имеют гидрофобную поверхность и склонны к формированию упорядоченных амилоидоподобных структур. Как недавно было показано [84–86], связывание порфиринов глобулярными α-протеинами приводит к экспонированию гидрофобных участков полипептидной цепи на поверхность глобулы, переходу части разупорядоченных и α-цепей в β-складки и образованию агрегатов различной архитектуры [87, 88]. Доказана иммобилизация массива хромофоров цинк(II)протопорфирина IX на β-листах протеинов [89, 90]. В созданной системе экситоны могут диффундировать вдоль стопок на расстояние >60 нм в оптимальных условиях.
Белковые структуры в естественных светособирающих комплексах играют решающую роль в модуляции уровней энергии отдельных пигментов посредством взаимодействия с остатками белка и соседними пигментами, что, в свою очередь, влияет на путь передачи энергии возбуждения [91]. Поэтому включение синтетической диады в белковую матрицу может быть альтернативным подходом к созданию улучшенной фотохимической системы, что наглядно продемонстрировано в работе Hu et al. [92] на примере искусственных фотосинтетических реакционных центров, сконструированных на поверхности белка (рис. 9). Сравнительный анализ фотофизики диады и диады, помещенной в апомиоглобин, показал, что время жизни в состоянии с разделенными зарядами без белка составляет 1 мкс, а с белком – не менее 2 мс, т.е. многоступенчатый перенос электрона возможен только в случае супрамолекулярной системы, в состав которой включен белок. Подобно естественному фотосинтетическому реакционному центру, включение кофакторов в белковый каркас (апомиоглобин) позволяет пространственно разделить фотогенерированные продукты и тем самым избежать быстрой рекомбинации зарядов.
Рис. 9.
Схематическое изображение восстановления кофакторов (I) и (II) апомиоглобином с получением триад на основе миоглобина: Mb(FeIIIOH2)-Ru2+-BXV4+ и Mb(Zn)-Ru2+-BXV4+ [92].

В связи со сложностями включения хромофоров в природные белки исследователи все чаще обращаются к искусственным белкам [93–95], называемым “макетами” (“maquettes”) [68, 95]. Их достоинства очевидны: они устойчивы, имеют требуемый паттерн, высокую аффинность к хромофорам, что в совокупности обеспечивает эффективную и управляемую самосборку фоточувствительных систем.
Недавние исследования показали, что природные биологические структуры могут служить в качестве шаблонов, управляющих наноразмерной организацией пигментов через химические связи или электростатические взаимодействия. В качестве шаблонов может использоваться ДНК [96, 97], РНК [98, 99] или капсиды вирусов [100]. Например, бактериофаг M13 имеет оболочку – нитевидную структуру длиной ~880 нм и диаметром ~6.5 нм [101], состоящую из ~2700 копий белков α-спиральной оболочки. Оболочка вируса легко подвергается химической модификации благодаря наличию терминальных NH2-групп и легкодоступных на поверхности остатков лизина для обеспечения последующей конъюгации с тетрапиррольными хромофорами. Расположение центров конъюгации (~5400 на вирус), указанных выше терминальных групп и аминокислотных остатков лизина на поверхности вируса обеспечивает экситонное взаимодействие конъюгированных с ними молекул порфиринов (рис. 10) [101].
Рис. 10.
Самосборка пигмента (Zn(II)дейтеропорфирин IX 2,4-бис(этиленгликоль)) на оболочке бактериофага M13 [101].
Подводя итог этого раздела, хочется отметить, что создание искусственных антенн представляет интерес не только для развития альтернативной энергетики, искусственного фотосинтеза, но и может найти применение в биомедицинских целях, например, для мультиплексной маркировки, визуализации в клетках прокариот и млекопитающих, включая линии клеток человека [102]. Однако исследования в данном научном направлении находятся в зачаточном состоянии. Больший успех достигнут в направлении исследования свойств конкретных белков с тетрапиррольными хромофорными соединениями и изучении практического применения этих супрамолекулярных систем.
СУПРАМОЛЕКУЛЯРНЫЕ КОМПЛЕКСЫ ПРОТЕИНОВ С ТЕТРАПИРРОЛЬНЫМИ МАКРОГЕТЕРОЦИКЛАМИ В МЕДИЦИНЕ
Из всех биомолекул белки, вероятно, наиболее интересны, особенно с молекулярной и структурной точек зрения. Белки участвуют практически во всех биологических процессах, и их супрамолекулярная модуляция (например, с помощью небольших молекул) лежит в основе действия большинства лекарств [103]. Как правило, белки работают согласованно с помощью так называемых белок-белковых взаимодействий. Управление белок-белковыми взаимодействиями открывает широкие перспективы для создания новых видов лекарств. Таким образом, белки могут выступать как в роли биологических мишеней (например, в фотодинамической терапии (ФДТ), фотоинактивации бактерий и вирусов (ФДИ, аФДТ)), использоваться для визуализации с помощью порфиринов, хлоринов, фталоцианинов, а также выступать компонентами лекарственных препаратов. Сывороточные белки крови могут использоваться в качестве транспортных систем, обеспечивающих распределение препарата с помощью кровеносной системы и модулирующих связывание порфиринов с другими белками. В данном разделе обобщены достижения последних лет в перечисленных научных направлениях.
Фотодинамическая терапия
Фотодинамическая терапия (ФДТ) – хорошо зарекомендовавший себя неоперабельный метод лечения онкологических заболеваний, различных патологий, в том числе некоторых кожных заболеваний (псориаз, папилломавирусные инфекции, кожный лейшманиоз и др. [104]). В основе метода лежит световое воздействие на фотосенсибилизатор (ФС – порфирин, хлорин, фталоцианин и др.). Полученная ФС световая энергия трансформируется в конечном итоге в химические реакции, приводящие к поражению мишени, гибели клетки. К настоящему времени выделяют четыре типа фотоиндуцированных реакций. В реакциях типа I ФС в возбужденном триплетном состоянии реагирует с биомолекулами путем передачи электрона, в результате чего радикалы и ионы-радикалы вступают в реакцию с молекулярным кислородом, вызывая образование активных форм кислорода (ОН-радикал, супероксидный радикал). В реакциях типа II происходит прямая передача энергии от ФС в триплетном возбужденном состоянии к кислороду в основном триплетном состоянии с образованием синглетного кислорода, который проявляет высокую цитотоксичность [105]. Фоточувствительные молекулы механизма типа III обычно выступают сенсибилизаторами-носителями антиоксидантов, увеличивающими генерацию синглетного кислорода и снижающими концентрацию природных свободных радикалов в клетках-мишенях [106]. Цитотоксический механизм типа IV связан с ФС, которые не способны взаимодействовать с молекулярной мишенью, но при световом возбуждении происходит их внутримолекулярная трансформация (фотоизомерация), способствующая связыванию светочувствительной молекулы с ее молекулярной мишенью [106]. Подавляющее большинство препаратов для ФДТ – это порфирины, хлорины и фталоцианины, обладающие интенсивным поглощением в области 650–850 нм и окисляющие биосубстраты преимущественно по механизму II. Значительный ряд фотосенсибилизаторов был разработан для ФДТ, но только некоторые из них одобрены для эффективного использования: в статьях [107, 108] приводятся структурные формулы лекарственных соединений, торговые называния препаратов для лечения различных нозологических форм.
Существует огромное количество прекрасных обзоров, посвященных ФДТ, фотохимии, фотофизике ФС, вопросам синтеза новых ФС с улучшенными фотоокислительными свойствами, распределению и накоплению в органеллах клеток, фото- и темновой токсичности ФС [105, 109–114], поэтому в данном разделе акцент будет сделан на нерешенных проблемах ФДТ и возможных подходах к их решению с помощью конъюгатов ФС с белками и пептидами. Функционирование последних основано на принципах супрамолекулярой химии, при этом супрамолекула образуется за счет белок-белковых взаимодействий между конъюгатом и экзогенными белками или белковыми частями рецепторов.
Кислород тканей – один из ключевых компонентов для образования 1O2. Известно, что по мере роста опухоли быстро нарушается кровоснабжение, приводя к гипоксии тканей. Таким образом, успешное развитие методов ФДТ требует решения задач, связанных с созданием кислород-независимых подходов. Интенсивно ведется поиск ФС, способных реализовать фотоокисление по типу I, положительные результаты получены для некоторых катионных порфиринов [115] и фосфор-замещенных порфиринов [116].
Проблему кислород-независимых подходов к ФДТ также пытаются решить с использованием других стратегий. Первая стратегия основана на создании носителей, которые одновременно доставляют в пораженную клетку фотосенсибилизатор и O2. Как правило, это загруженные наночастицы [8–10], перфторуглероды [117, 118]. Основные недостатки данного подхода – большой единичный объем препарата и связанная с этим низкая проникающая способность. Кроме того, трудно решаемая проблема – высвобождение O2 в требуемом пораженном участке. Например, перфторуглероды – прекрасные носители O2, но высвобождение загруженного O2 начинается сразу после инъекции в кровоток, потому что O2 высвобождается в основном посредством диффузии, управляемой градиентами концентрации [119, 120]. Вторая стратегия основана на создании конъюгатов фотосенсибилизаторов с каталазой или каталазоподобными материалами. Каталаза – один из важнейших ферментов, который может катализировать разложение внутриклеточной перекиси водорода (H2O2) на O2 и H2O. Достоверно известно, что внутриклеточный уровень H2O2 в опухолях всегда выше. Например, была создана конструкция, включающая каталазу и альгинатдиальдегид, фотосенсибилизатор бенгальский розовый. Полученный конъюгат был загружен в кальцинированнные пористые частицы [121]. Более эффективные конъюгаты сконструированы на основе поли-(D,L-молочной-cо-гликолевой кислоты) – носителя, в который инкапсулировали каталазу и фотосенсибилизатор (метиленовый синий) [122]. Phua et al. разработали наносистему [123], также содержащую каталазу, в которой каталазу сначала конъюгировали с функционализированной β-циклодекстрином гиалуроновой кислотой с образованием наночастиц, а затем наночастицами загружали модифицированный адамантаном хлорин е6. Основной недостаток данной стратегии – проблема с доставкой загруженного в полимер или наночастицы конъюгата, низкая туморотропность, сложность получения конъюгата.
Альтернативная стратегия основана на создании фотосенсибилизаторов, способных участвовать в окислительных процессах по типу I, т.е. связанных с процессами фотоиндуцированного переноса электрона, генерацией радикальных форм. В данном направлении работает несколько научных групп. Например, под руководством Hamblin было показано, что при использовании в качестве фотосенсибилизатора наночастиц (TiO2)n, органических красителей (бенгальский розовый, метиленовый синий, порфирин [124, 125]) в присутствии неорганических солей процессы фотоокисления биосубстратов реализуются по механизму I. Иной стратегии, основанной на создании системы ФДТ, способной переносить кислород, придерживались Wang et al. [126]. Ими был получен новый фотосенсибилизатор-носитель с кислородной самокомпенсирующейся способностью. Он был синтезирован путем конъюгирования гемоглобина (Hb) с полимерными мицеллами, образованными триблок-сополимерами поли(этиленгликоля)–блок-поли(акриловой кислоты)–блок-полистирол (PEG–b-PAA–b-PS) (рис. 11).
Рис. 11.
(а) – Синтез ZnPc-мицелл, загруженных в мицеллы гемоглобина (HbMs); (б) – сравнение фото- и темновой токсичности клеток HeLa по отношению к HbMs, ZnPc-мицеллам и ZnPc-мицеллам с HbMs [126].
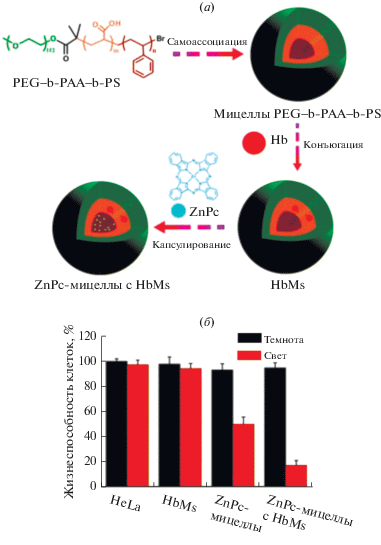
Фталоцианин цинка (ZnPc) был инкапсулирован в конъюгированные с гемоглобином мицеллы в качестве фотосенсибилизатора. Было подтверждено, что полученные мицеллы генерируют больше синглетного кислорода и вызывают большую фотоцитотоксичность для клеток HeLa in vi-tro по сравнению с мицеллами, нагруженными ZnPc без гемоглобина.
Другая проблема ФДТ – это повышение избирательного накопления фотоактивируемых молекул в опухолевой ткани. Ковалентное присоединение порфиринов и родственных макроциклов к пептидам и белкам может решить эту проблему благодаря их усиленному клеточному захвату, благоприятному субклеточному распределению и способности нацеливания белковой части конъюгатов на рецепторы или ферменты, чрезмерно экспрессируемые данной тканью или клеткой. Таким образом, супрамолекула образуется непосредственно в организме за счет белок-белковых взаимодействий между эндогенным конъюгатом и экзогенными белками.
Например, в работе Yu et al. сообщается о конъюгате фталоцианин–пептид (Tyr-His-Trp-Tyr-Gly-Tyr-Thr-Pro-Gln-Asn-Val-Ile) [127], который нацелен на рецептор эпидермального фактора роста (EGFR) и продемонстрировал более высокий клеточный захват клетками эпидермоидной карциномы A431 со сверхэкспрессией EGFR по сравнению с клетками аденокарциномы молочной железы человека MCF7 с низкой экспрессией EGFR. Оказалось, что предварительная обработка клеток A431 указанным выше пептидом значительно ингибировала клеточное поглощение конъюгата, а сам конъюгат проявлял исключительную цитотоксичность, активируемую светом, в отношении клеток A431. Эти данные показывают, что вектор – это пептид, который участвует в белок-белковых взаимодействиях с EGFR. Высокую эффективность в лечении базальноклеточного и плоскоклеточного рака, а также предраковых состояний, таких как солнечный кератоз, демонстрирует конъюгат трикатионного 5-(4-карбоксифенил)-10,15,20-трис(4-метилпиридиния)-4-ил)порфирин трииодида с поли-S-лизином [128]. Циклический пептид RGD, выступающий лигандом для рецептора интегрина αvβ3, был ковалентно конъюгирован с фотосенсибилизатором на основе хлорофилла. Данный конъюгат позволил улучшить визуализацию опухоли и увеличить эффективность ФДТ через 2–4 ч после инъекции, а также продемонстрировал высокую специфичность [129].
Подобных примеров в литературе огромное количество, все это свидетельствует о перспективности подхода и, возможно, скорого внедрения в лечебную практику. В обзорной статье Biscaglia et al. [130] представлены аминокислотные последовательности, обеспечивающие самонаведение на опухолевые клетки, а также пептиды, проникающие в клетки с локализацией в ядре и митохондриях. Потенциально все они могут быть использованы в конъюгатах с ФС. Широко применяется стратегия нацеливания для увеличения сродства ФС к опухолям за счет ковалентной связи порфиринов с аминокислотными конъюгатами. Известно, что порфирины, связанные с аминокислотами, приобретают дополнительные свойства, важные для ФДТ. Среди них водорастворимость [131], способность интеркалировать последовательности ДНК с высокой специфичностью [132–135] и высокое поглощение клетками опухоли [136].
Конъюгаты ФС с антителами (Ab) представляют особый интерес из-за их чрезвычайно высокой селективности и сродства к соответствующим антигенам. Конъюгация ФС с Ab приводит к образованию активных молекул, нацеленных на опухоль и способных связывать антигены или рецепторы, которые сверхэкспрессируются в опухолях. У млекопитающих антитела делятся на пять изотипов (IgA, IgD, IgG, IgE, IgM). На сегодняшний день антитела можно разделить на четыре основные группы: поликлональные, моноклональные, фрагменты антител и нанотела. Наиболее отчетливые различия можно проследить между поли- и моноклональными антителами. Поликлональные антитела относительно недороги, их сравнительно легко получать и хранить, они обладают высокой стабильностью, связываются с несколькими эпитопами и имеют хорошую чувствительность, но они характеризуются разным составом, т.к. выделяются из сыворотки крови иммунизированных животных. Моноклональные антитела более дорогие, обладают высокой специфичностью по отношению к одному эпитопу и чрезвычайно гомогенны [137]. Несколько маркеров опухолевых клеток были исследованы для активного фотодинамического нацеливания, включая рецепторы трансферрина (TR), фолиевой кислоты (FR), фактора роста, рецепторы липопротеинов низкой плотности (LDL), интегрина, инсулина и переносчики глюкозы. Другие потенциальные маркеры опухолевых клеток часто включают гликопротеины, такие как опухолевые маркеры рака легких (например, CEA, CYFRA21-1 и NSE), желудочно-кишечные опухолевые маркеры (например, CEA, CA199, группа CA242 и CA724), маркеры рака молочной железы (CA153), рака яичников (CA125), гепатоцеллюлярной карциномы (AFP), рака простаты (PSA) и хориокарциномы (HCG) [138]. Наиболее распространенные мембранные рецепторы, на которые могут быть нацелены конъюгаты ФС–mAb, описаны в статье Fernandes et al. [139]. Синтетические подходы к получению конъюгатов антитело–фотосенсибилизатор широко представлены в литературе [137, 140–143].
Терапевтический потенциал конъюгата ФС с антителами зависит от его локализации. Антитела, как правило, не могут проходить сквозь клеточную мембрану, поэтому конъюгаты антитело–ФС построены на антителах, нацеленных в основном на рецепторы, доступные на клеточной мембране. Тем не менее известны случаи интернализации конъюгатов ФС–Ab, обладающих, по сведениям Vrouenraets et al. [144], большей фотодинамической активностью, чем конъюгаты, накапливающиеся снаружи на клеточной мембране. Считается, что чуть ли не единственный механизм интернализации ФС–Ab – это эндоцитоз, опосредованный рецепторами. Например, у клеточных линий рака молочной железы MDA-MB-231 и MCF-7 выявлена сверхэкспрессия рецепторов гонадотропин-рилизинг-гормона. Учитывая это, был получен конъюгат рилизинг-гормона гонадотропина с β-карбоксифталоцианином цинка (ZnPc-COOH), который поглощался клетками посредством эндоцитоза и обладал высокой фототоксичностью в отношении вышеупомянутых клеточных линий [145].
По сравнению с традиционной ФДТ, таргетная фотодинамическая терапия (тФДТ) улучшает соотношение между эффективностью и токсичностью при лечении солидных злокачественных новообразований [137] благодаря высокой специфичности связывания конъюгата ФС–Ab аберрантно экспрессирующими рецепторами-мишенями при сохранении прилегающих нормальных тканей.
Буквально несколько лет назад среди медиков, биологов, биохимиков и медицинских химиков было распространено мнение о том, что оптимальные механизмы клеточной гибели после ФДТ – аутофагия и апоптоз [146]. При этих механизмах клеточной гибели ферменты и клеточные осколки не попадают в межклеточную жидкость и не вызывают воспаление (как в случае некроза) здоровых тканей. В последнее время эти взгляды пересматриваются, т.к. эффективный терапевтический метод должен не только на время искоренить болезнь, но и обеспечить защиту от нее в будущем за счет активации иммунной системы. ФДТ может вызывать острое воспаление и привлекать иммунные клетки [147, 148]. Понимание механизма, способствующего развитию иммунитета, поможет в разработке эффективных агентов для иммунотерапии. В качестве доводов, подтверждающих данный тезис, можно привести следующее: фотоактивация конъюгата ФС–Ab приводит к фотоокислению биосубстратов клеток, вызывает клеточную гибель, что активирует иммунный ответ, который в дальнейшем способствует уничтожению раковой опухоли. Однако эти иммунные клетки (Т-клетки), эффективные для борьбы с онкологическим заболеванием, подавляются другими иммунными регуляторными Т-клетками (Treg). Поэтому в настоящее время перспективны разработки конъюгатов и супрамолекулярных систем, вызывающих иммунный ответ, нацеленный на Treg. По мнению ряда исследователей [139, 149, 150], эти работы могут стать основой для иммунотерапии, помогающей иммунной системе пациентов распознавать и уничтожать раковые клетки.
Следует отметить и другие перспективы использования конъюгатов ФС с белками, пептидами и аминокислотами. В первую очередь, это возможность генерирования различных форм активного кислорода, которые позволяют концентрировать окислительное повреждение в целевой области. Хорошо известно, что аминокислотные остатки в белках имеют разную окислительную способность. Выделяют уязвимые аминокислотные остатки, среди которых Cys, His, Met, Tyr и Trp [151–154]. Время жизни активных форм кислорода, генерируемых ФС, невелико, как и расстояние, на которое они могут удалиться от ФС до перехода кислорода в триплетную форму. Поэтому если в составе конъюгата или супрамолекулы ФС–пептид (белок) будут уязвимые аминокислоты, это может способствовать запуску каскадного окисления, приводящего к повреждению мишени [155–157]. Однако изучение механизмов окисления белка, тем более опосредованного эндогенными аминокислотными остатками конъюгатов, – чрезвычайно сложная задача, но, возможно, в обозримом будущем мы увидим успехи и в данном научном направлении.
Фототермическая терапия
Фототермическая терапия (ФТТ) – один из методов неинвазивного лечения онкологических заболеваний (гипертермия рака), основанный на генерации ФС тепла [158]. Известно, что денатурация белков, нарушение клеточной мембраны и абляция опухолевых тканей происходят при температуре 40–43°C и выше [158]. Селективная ФТТ достигается за счет фотоактивации ФС, накопленных на участках опухоли, поглощенная энергия фотонов преобразуется в тепло, достаточное для индукции клеточной гипертермии. Изначально это научное направление развивалось на основе нанохимии неорганических материалов, включая углеродные нанотрубки, наноструктуры золота, поглощающие в ближнем ИК-диапазоне (наностержни, нанооболочки, наноклетки и полые наносферы) [114, 158]. Однако в последнее время стали появляться сообщения об использовании для ФТТ конъюгатов пептид–фотосенсибилизатор [159]. Основные принципы построения супрамолекулярных систем для ФТТ – высокая загрузка фотосенсибилизатором и подавление флуоресценции для обеспечения диссипации энергии по тепловому пути. Lovell et al. [160] удалось создать наноструктуры (пузырьки), образованные из самоорганизующихся липидных бислоев с порфиринами, которые, по сути, представляли собой фототермический преобразователь. После внутривенной инъекции лабораторным животным супрамолекулярные структуры накапливались в опухолях и позволяли выявлять индуцированную лазерным облучением фототермическую абляцию опухоли. Супрамолекулярное конструирование позволило Zou et al. [161] получить наноточки (рис. 12), состоящие из конъюгата дифенилаланин–пептид–порфирин. Супрамолекулярная агрегация обеспечила полное тушение флуоресценции ФС и ингибирование генерации активных форм кислорода, что привело к термическому рассеиванию поглощенной световой энергии с высокой эффективностью преобразования (54.2%). После лазерного облучения средняя температура в течение 10 мин на участках опухоли увеличилась до 58.1°C, что привело к некрозу опухоли.
Рис. 12.
(а) – Получение фототермических наноточек пептид–порфирин (PPP-ND) путем самосборки конъюгата пептид–порфирин (TPP-G-FF); (б) – ТЕМ-изображение PPP-ND [161]; (в) – фотоакустическое изображение во времени после внутривенной инъекции PPP-ND; (г) – ИК-изображения внутривенных PPP-ND, введенных мышам при непрерывном облучении; (д) – профили роста опухоли после ФТТ [161].
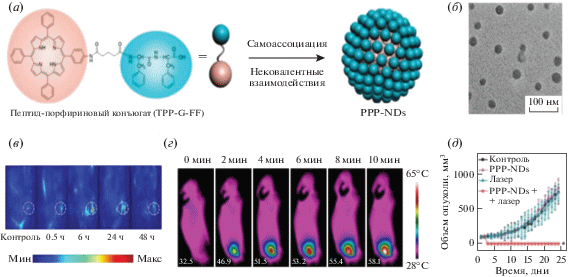
Еще более сложная супрамолекулярная система предложена Li et al. [162]. Она состоит из фталоцианинов, содержащих на периферии гидрофильные триэтиленгликольные остатки. В водных средах происходит самоагрегация с образованием наночастиц, устойчивых в водных средах, однако в присутствии целевых белков происходит частичная дезагрегация, а мономеризованные фталоцианины становятся способными к флуоресценции. Авторы утверждают, что полученные супрамолекулярные структуры обладают ФТТ-эффектом, а управляемый белком триггер флуоресценции обеспечивает высокую эффективность для визуализации опухолевых клеток с высокими отношениями сигнал/фон, а также смену терапии с ФТТ на ФДТ. Безусловно, направление ФТТ будет в дальнейшем развиваться, т.к. демонстрирует потенциальную возможность кислород-независимой терапии солидных опухолей.
Транспортные системы/ системы доставки
Ограниченная растворимость фотосенсибилизаторов в воде и склонность к агрегации часто ухудшают их фотофизические свойства, что приводит к снижению биодоступности соединений, поэтому для фотосенсибилизаторов порфиринового, хлоринового и фталоцианинового рядов, независимо от степени гидрофобности, необходима транспортная система [163]. Транспортная система позволяет не только повысить биодоступность и снизить токсичность ФС, но и обеспечивает дополнительные преимущества, такие как усиление мембранной абсорбции, молекулярная стабилизация и улучшение водорастворимости, а также доступность лекарств в биологических системах [164]. Субклеточная локализация ФС в митохондриях, лизосомах, эндоплазматическом ретикулуме и плазматической мембране (различных органеллах) играет решающую роль в задействованном механизме гибели клеток (апоптотическая, некротическая и связанная с аутофагией гибель клеток) [165]. Фактически конъюгация ФС с этими нетоксичными молекулами показала улучшение их амфифильности, биосовместимости и доступности на поверхности мембран раковых клеток [128, 166].
ФДТ требует использовать наномолярные и микромолярные количества ФС, транспортные системы (пептиды и белки) могут обеспечить перенос таких количеств ФС [167]. Более 20 лет интенсивные усилия направлены на развитие таргетной фотодинамической терапии. За этот период были достигнуты очевидные успехи, но многие проблемы остались не решены, особенно в части доставки ФС с помощью пептидов и белков.
Хотя пептиды можно считать отличными векторами для ФС [168], присутствие нескольких положительно заряженных аминокислот в их последовательности может вызывать сильные неспецифические взаимодействия, что делает их неприменимыми для многих приложений in vivo. Чтобы избежать преждевременного захвата конъюгата, ФС связывают с “активируемыми” пептидами [169]. Конъюгированный пептид можно нейтрализовать другой противоположно заряженной пептидной последовательностью за счет полиэлектролитных взаимодействий или за счет противоположно заряженного пептида, связанного через специфический протеолитически расщепляемый линкер [170]. После накопления конъюгата в опухоли противоположно заряженный сегмент может быть избирательно отщеплен матриксными металлопротеиназами, присутствующими во внеклеточном матриксе опухоли, в результате конъюгат высвобождается. Исследования in vivo на животной модели показали достаточную стабильность активируемого конъюгата в кровотоке и его интенсивное накопление в опухоли, что позволяло визуализировать раковые клетки [171]. Более того, повторные обработки конъюгатом при освещении уменьшали размер и вес солидной опухоли с низкой системной токсичностью для животного [169].
Предлагаются и более сложные подходы к эффективной доставке ФС с нацеливанием в митохондрии пораженных клеток [172]. Например, в работе Tong et al. [172] синтезирован “умный наноноситель”, состоящий из трех функциональных пептидных единиц: 1) гептапептид со специфической способностью связываться с рецептором трансферрина, сверхэкспрессируемый в высокопролиферативных линиях раковых клеток; 2) последовательность, расщепляемая катепсином B (GFLG); 3) последовательность пептида, нацеленного на митохондрии (CGKRK), несущего на N-конце остаток адамантана. Через этот остаток пептид был нековалентно связан с β-циклодекстрином, функционализированным с помощью ФС хлорина е6 (рис. 13). Транспорт ФС в митохондрии пораженных клеток [172] происходит в три стадии: 1) внутриклеточное накопление комплекса, опосредованное рецептором, в клетках, сверхэкспрессирующих рецептор трансферрина; 2) ферментативное расщепление последовательности GFLG катепсином B после интернализации, приводящее к получению другой последовательности, направленной в митохондрии; 3) накопление ФС в митохондриях. Протекание каждой стадии в клетках легко контролировалось с помощью флуоресцентной микроскопии, а при световом облучении была обнаружена дозозависимая фотодинамическая цитотоксичность.

В статье Li et al. [173] описан подход инженерии плазматической мембраны для доставки ФС с использованием контактных клеток (“тро-янский конь”) путем закрепления функционализированных конъюгатов на клеточных носителях. Конъюгаты, фиксирующие мембрану, со-стоят из положительно заряженной тетрааргининовой пептидной последовательности, встраиваемого в мембрану фрагмента на основе пальмитиновой кислоты, а также лизинового линкера, модифицированого камптотецином, протопорфирином IX. Таким образом, порфирин был прикреплен к клетке-носителю с помощью последовательности, богатой аргинином, заканчивающейся остатком пальмитиновой кислоты для встраивания в мембрану. Туморотропные свойства клеток-носителей позволяли накапливать порфирин на участке опухоли, а при освещении только опухолевые клетки, находящиеся в тесном контакте с клетками-носителями, разрушались с помощью ФТД.
Как уже отмечалось выше, при конъюгировании ФС с пептидами и белками сохраняется проблема интернализации ФС. Эндоцитозное поглощение работает не всегда, поэтому проблема проникновения ФС в клетку до сих пор не решена. Но попытки заставить клетку интернализировать конъюгат интенсивно предпринимаются. Заметное улучшение доступности ФС в субклеточных компартментах ожидается от технологии фотохимической интернализации [174–176].
Принципиально иная стратегия транспортной системы для ФС заключается в использовании глобулярных эндогенных белков. При введении в кровоток большинство препаратов связываются с различными белками сыворотки, включая альбумин и липопротеины высокой и низкой плотности, за счет нековалентных взаимодействий. Считается, что транспортировка ФС этими носителями может улучшить его внутриклеточное накопление через рецептор-опосредованный эндоцитоз [177]. Альбумин представляет собой самый распространенный сывороточный белок в крови человека, его концентрация на порядок превышает общую концентрацию всех белков. Увеличение метаболизма и пролиферация раковых клеток вызывают сильный и быстрый обмен сывороточного альбумина в опухолях. Биохимическое основание нацеливания ФС на опухолевые клетки альбуминами – способность модифицированных альбуминов и липопротеинов низкой плотности связываться со скавенджер-рецепторами, сверхэкспрессированными на макрофагах. Эти скавенджер-рецепторы способны связывать широкий спектр различных соединений, включая альбумин, и переносить их в клеточные компартменты. Известно, что изначально макрофаги М1 (активированные макрофаги) [178] препятствуют развитию опухолевых клеток, а макрофаги М2 за счет иммуноредактирования, наоборот, способствуют их развитию [179]. Содержание макрофагов может составлять до 50% от массы клеток при некоторых видах рака [180]. Кроме того, использование альбумина в качестве транспортной системы оправдано повышенной проницаемостью и удержанием макромолекул в опухолевых тканях (эффект EPR) [181, 182].
Альбумин способен связывать ФС порфиринового [157], фталоцианинового [183] и хлоринового рядов, используя различные сайты связывания, а также область между доменами [184]. Локализация ФС в альбуминовой глобуле зависит от природы макрогетероцикла, его периферийных заместителей. Простое решение по выделению ФС в опухолевых клетках из альбуминовой транспортной системы продемонстрировано на примере комплексов бычьего сывороточного альбумина с Fe(III)-протопорфирином [185]. При изменении рН среды происходит выгрузка ФС, а т.к. рН у опухолевых клеток кислый [186], возможно, этот механизм будет работать и in vivo. Некоторые исследования были предприняты для ковалентного связывания различных фотосенсибилизаторов с измененным альбумином для нацеливания на опухолевые клетки с опосредованным участием скавенджер-рецепторов [187].
Следует отметить, что использование альбумина в качестве транспортных белков для ФС не всегда возможно. Как было показано недавно [88], некоторые тетрапиррольные ФС при связывании альбуминовой глобулой инициируют агрегацию белка, что не только обесценивает транспортные свойства сывороточного белка крови, но и может привести к негативным для здоровья последствиям (нарушению осмотического давления, интоксикации, отекам, гипоальбуминемии).
Антимикробная фотодинамическая терапия
Помимо онкологических другие заболевания, такие как бактериальные, грибковые и вирусные инфекции, которые характеризуются общей неконтролируемой пролиферацией клеток и наличием нежелательных клеток микроорганизмов, можно лечить с помощью фотодинамической терапии. В этом случае метод называется антимикробной фотодинамической терапией (аФДТ) или фотодинамической инактивацией (ФДИ) [188–190]. В последние годы наблюдается рост числа исследований in vitro и in vivo, показывающих положительные результаты в отношении аФДТ, применяемой в стоматологии: в периодонтологии, эндодонтии и при инфекциях слизистых оболочек [191]. Нами недавно был опубликован обзор по фотоинактивации вирусов [192], поэтому в данном обзоре в основном будут затронуты вопросы аФДТ. Как показали исследования последних лет, индивидуальные ФС менее эффективны, чем их конъюгаты с белками [193]. Из глобулярных белков в качестве конъюгатов с ФС для аФДТ рассматриваются апомиоглобин, β-лактоглобулин, сывороточные белки крови и др. Перечисленные носители имеют общие характерные черты: водорастворимость и наноразмерность, что обеспечивает биодоступность соединения. Кроме того, указанные белки наделены гидрофобными сайтами связывания, по своей природе биосовместимы и легко выделяются в больших количествах.
По составу клеточной стенки бактерии делятся на две группы: грамположительные (Staphylococcus aureus, Bacillus subtilis, Enterococcus faecalis и др.) и грамотрицательные (Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumonia и др.) [194, 195]. Строение клеточной стенки играет определяющую роль в чувствительности бактерий к ФС (рис. 14), которая существенно ниже у грамотрицательных бактерий.

Высокая восприимчивость видов грамположительных бактерий к ФС по сравнению с грам-отрицательными бактериями объясняется присутствием относительно пористого слоя пептидогликана и липотейхоевой кислоты в их клеточной стенке, что позволяет молекулам ФС диффундировать к целевым сайтам внутри клетки. Напротив, клеточная стенка грамотрицательных бактерий содержит отрицательно заряженный липополисахарид, который препятствует проникновению нейтральных или анионных порфиринов из внешней среды в бактериальную клетку. Именно поэтому подавляющее большинство исследований изначально было связано с изучением катионных ФС, но даже их эффективность существенно повышается за счет конъюгации с пептидами и белками [188]. Это было обнаружено, например, для конъюгатов полипептидов и белков с различными по природе ФC [198–200]. Считается, что лучшая эффективность аФДТ с использованием конъюгатов ФС с белками и пептидами основана на дезорганизующем действии белков или пептидов на клеточную стенку бактерий, что увеличивает ее проницаемость, или на электростатическом взаимодействии, например, катионного пептида конъюгата с отрицательно заряженным липополисахаридом грамотрицательных бактерий [201–204].
В научной литературе представлена масса решений повышения селективности и эффективности аФДТ, среди них – синтез конъюгатов ФС с нацеленными биологически активными соединениями, как, например, недавно полученный конъюгат катионного порфирина с липопептидом полимиксина. Полимиксины известны как вещества, повышающие проницаемость мембраны грамотрицательных бактерий. Конъюгат действительно показал широкий спектр антибактериальной активности при световом облучении и высокую эффективность. Известны примеры конъюгатов ФС с моноклональными антителами или бактериофагами, что позволяет избирательно связываться со специфическими структурами микроорганизма – мишенями. Наиболее доступные и достоверно установленные внеклеточные и внутриклеточные мишени, а также общий обзор возможных целевых локусов бактерий представлены в статьях Liu et al. [196] и Alves et al. [205]. Возможно, в дальнейшем потребуются более сложные нацеливающие конструкции, позволяющие ФС воздействовать на геном патогена, т.к. остальные целевые мишени (липиды, белки патогена) требуют большей дозы ФС для нарушения мембраны или окисления белка [206–208]. Но уже сейчас очень много положительных результатов в борьбе с болезнетворными бактериями, в том числе склонными к образованию биопленок, которые способствуют хронизации и рецидиву инфекций, а также возникновению устойчивости патогенов к антибиотикам [209–212].
Множественная лекарственная устойчивость патогенных бактерий стала серьезной угрозой для здоровья населения. ВОЗ предупреждает, что приближается “постантибиотическая” эра. Уже существуют грамположительные и грамотрицательные супербактерии, такие как Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumo-ni-ae, Acinetobacter baumannii, Pseudomonas aeruginosa и виды Enterobacter, так называемые патогены ESKAPE, способные противостоять антибиотикам почти всех типов/классов. Поэтому поиск новых подходов к борьбе с бактериями с множественной лекарственной устойчивостью становится все более важным. В этом контексте антибактериальная фотодинамическая терапия – самый многообещающий подход к лечению инфекций, вызываемых бактериями с множественной устойчивостью к антибиотикам.
Медицинская диагностика
Известно, что многие тетрапиррольные макрогетероциклические соединения демонстрируют усиленное флуоресцентное излучение в красной или ближней ИК-области, поэтому порфирины, хлорины и другие родственные соединения могут найти применение в качестве контрастных агентов ближнего ИК-диапазона при флуоресцентной визуализации [213, 214], что позволяет создать интегративную систему диагностики и терапии (тераностики), которая представляет собой новый подход к лечению рака. Сочетание такой техники с ФДТ позволяет проводить более ориентированное и эффективное лечение, помимо предоставления информации о распределении фотосенсибилизатора и гибели клеток [215–217]. Очень перспективны для визуализации хлорины и бактериохлорины, подходящие для практического применения, т.к. эти соединения показывают узкие полосы поглощения и излучения в красной или ближней ИК-области с хорошим стоксовым сдвигом (>85 нм) [218–223]. Наряду с высокими квантовыми выходами, высокая туморотропность ФС не менее значима для диагностики. Многие водорастворимые ФС могут селективно накапливаться в опухолевых тканях и вводиться внутривенно [224], но селективность их накопления существенно повышается при использовании конъюгатов ФС с липопротеинами низкой плотности, моноклональными антителами и другими белками. Например, хлорин e6 в составе конъюгата с моноклональными антителами ОС125 накапливается в 3–4 раза эффективнее, чем свободный хлорин е6 [225]. Еще большую актуальность приобретает проблема использования транспортной системы, которая возникает в случае гидрофобных ФС. В клетках рака молочной железы наблюдается сверхэкспрессия рецепторов HER2. В работе Agadjanian et al. [226] сообщалось о визуализации in vivo и терапевтической эффективности коррола, нековалентно связанного с модифицированным герегулином белком (Her Ga), направленным на рецепторы HER2. Этот конъюгат не проявлял токсичности для сердца или других органов. Фотовозбуждение того же конъюгата привело к усилению цитотоксичности, приводящей к апоптозу [226].
Визуализация – необходимый инструмент, поскольку он позволяет проводить наблюдение и анализировать течение заболевания, отслеживать изменения, происходящие с опухолью, а также проверять адекватность лечения. Недавно была создана мультимодельная наноплатформа на основе порфирин-липопротеинов, предназначенная для позитронно-эмиссионной томографии (ПЭТ), флуоресцентной визуализации и ФДТ. Данная система, как сообщается в работе Muhanna et al. [227], демонстрирует большой потенциал для повышения точности определения стадии рака полости рта и, возможно, его лечения. С использованием клинически релевантной модели карциномы полости рта кролика VX-2 была обнаружена не только первичная опухоль, но также метастазы опухоли к региональным лимфатическим узлам с помощью предоперационной визуализации (позитронно-эмиссионной томографии) и интраоперационной флуоресцентной визуализации (рис. 15).
Рис. 15.
Обнаружение основной опухоли (карциномы VX-2) и ее метастаза в лимфатическом узле кролика: (а) – фармакокинетический профиль наночастиц у кроликов с VX-2 (n = 4); (б) – распределение 64Сu-меченных порфирин-липопротеиновых наночастиц (64Сu-PLP) в мышцах (4), опухолях (1) и лимфатических узлах (2) количественно определено с помощью позитронно-эмиссионной томографии (ПЭТ). Поглощение PLP опухолью и лимфатическими узлами было значительно выше, чем поглощение мышцами (n = 4, р < 0.05); (в) – 3D-изображение шеи кролика через 24 ч после инъекции 64Сu-PLP, полученное методом ПЭТ (красная стрелка – опухоль, белая стрелка – лимфатический узел); (г) – распределение 64Сu-PLP в основных органах здоровых кроликов и кроликов с карциномой VX-2, измеренное с помощью γ-счетчика: 1 – опухоль, 2 – лимфатический узел, 3 – слюнная железа, 4 – мышцы, 5 – легкое, 6 – сердце, 7 – печень, 8 – селезенка, 9 – почка; (д) – ex vivo флуоресценция опухоли, лимфатического узла и других основных органов кроликов с карциномой VX-2 после ПЭТ [227].

В последние годы количество исследований биомаркеров для ранней диагностики заболеваний постоянно растет [65]. Создание зондов для визуализации биомаркеров в живых тканях – сложная задача [138]. Например, количественные и качественные изменения коллагена I типа, основного компонента внеклеточного матрикса кожи и других жизненно важных органов, часто связаны с патофизиологией этих органов. Коллагелин-специфический пептид [65], связывающий коллаген, был ковалентно связан с тетрафенилпорфирином в качестве флуоресцентного молекулярного зонда для диагностической визуализации раннего фиброза печени. Хроническое заболевание печени характеризуется накоплением белков внеклеточного матрикса, особенно коллагена, который разрушает архитектуру печени, образуя фиброзный рубец. В работе Chilakamarthi et al. [228] показано, что конъюгат тетрафенилпорфирина с коллагелином специфически связывается с коллагеном, а коллагелин без тетрафенилпорфирина плохо связывается с коллагеном. В условиях ex vivo и in vivo зонд может дифференцировать фиброз печени, что делает его перспективным для использования при лечении хронических заболеваний печени.
Сравнительно новый метод визуализации – фотоакустическая визуализация. Данный метод отличается большей проникающей способностью и высоким пространственным разрешением [229, 230]. В основе метода лежит поглощение света молекулами, создающее термически индуцированный скачок давления, который производит ультразвуковые волны, подлежащие детектированию. Рассмотренные выше ФС для ФТТ обладают всеми необходимыми свойствами для фотоакустической визуализации и в ряде работ демонстрируют отличную работоспособность [231–234].
ЗАКЛЮЧЕНИЕ
Супрамолекулярные системы на основе макроциклических соединений с протеинами находят широкое применение в различных областях науки и техники. Уникальные фотохимические и фотофизические свойства супрамолекулярных систем позволяют применять их при создании светотрансформирующих (светособирающих) систем, искусственного фотосинтеза, в медицине для диагностики и лечения широкого ряда заболеваний, включая онкологию и бактериальные инфекции. Супрамолекулярный подход позволяет не только улучшить уже созданные молекулярные устройства или используемые лекарственные формы, но и демонстрирует развитие новых направлений, например, фототермическую терапию или создание супрамолекулярных систем, участвующих в фотопроцессах по типам I, III и IV, что, возможно, решит проблему кислородной зависимости ФДТ. Искусственная система сбора световой энергии на основе супрамолекул ФС с белками, вероятно, позволит достигнуть столь необходимого пространственно-разделенного переноса экситонов. Еще предстоит решить огромное количество вопросов, но, по мнению авторов, область исследований супрамолекулярных систем обещает стремительно развиваться в ближайшие годы.
Список литературы
Mattia E., Otto S. // Nat. Nanotechnol. 2015. V. 10. P. 111–119. https://doi.org/10.1038/nnano.2014.337
Lombardo D., Kiselev M.A., Magazù S., Calandra P. // Adv. Phys. 2015. V. 2015. https://doi.org/10.1155/2015/151683
Li Q., Han C., Horton S.R., Fuentes-Cabrera M., Sumpter B.G., Lu W., Bernholc J., Maksymovych P., Pan M. // ACS Nano. 2012. V. 6. P. 566–572. https://doi.org/10.1021/nn203952e
Northrop B.H., Zheng Y.-R., Chi K.-W., Stang P.J. // Acc. Chem. Res. 2009. V. 42. P. 1554–1563. https://doi.org/10.1021/ar900077c
Pauling L. // J. Am. Chem. Soc. 1931. V. 53. P. 1367–1400. https://doi.org/10.1021/ja01355a027
McNaught A.D., Wilkinson A. // Compendium of Chemical Terminology. Oxford: Blackwell Science, 1997.
van der Lubbe S.C., Fonseca Guerra C. // Chemistry – an Asian Journal. 2019. V. 14. P. 2760–2769. https://doi.org/10.1002/asia.201900717
Phipps M.J., Fox T., Tautermann C.S., Skylaris C.-K. // Chem. Soc. Rev. 2015. V. 44. P. 3177–3211. https://doi.org/10.1039/C4CS00375F
Van Der Lubbe S.C., Zaccaria F., Sun X., Guerra C.F. // J. Am. Chem. Soc. 2019. V. 141. P. 4878–4885. https://doi.org/10.1021/jacs.8b13358
Petersen J. // Phys. Rev. D. 1977. V. 15. P. 574
Hunter C.A., Sanders J.K. // J. Am. Chem. Soc. 1990. V. 112. P. 5525–5534. https://doi.org/10.1021/ja00170a016
Martinez C.R., Iverson B.L. // Chemical Science. 2012. V. 3. P. 2191–2201. https://doi.org/10.1039/C2SC20045G
Wheeler S.E. // J. Am. Chem. Soc. 2011. V. 133. P. 10262–10274. https://doi.org/10.1021/ja202932e
Ninković D.B., Blagojević Filipović J.P., Hall M.B., Brothers E.N., Zarić S.D. // ACS Cent. Sci. 2020. V. 6. P. 420–425. https://doi.org/10.1021/acscentsci.0c00005
Lanigan N., Assoud A., Zhang W., Wang X. // Chem. Eur. J. 2019. V. 25. P. 12221–12227. https://doi.org/10.1002/chem.201903138
Percec V., Glodde M., Bera T., Miura Y., Shiyanovskaya I., Singer K., Balagurusamy V., Heiney P., Schnell I., Rapp A. // Nature. 2002. V. 419. P. 384–387. https://doi.org/10.1038/nature01072
Hobza P., Zahradník R., Müller-Dethlefs K. // Collect. Czech. Chem. Commun. 2006. V. 71. P. 443–531. https://doi.org/10.1135/cccc20060443
Černý J., Hobza P. // PCCP. 2007. V. 9. P. 5291–5303. https://doi.org/10.1039/B704781A
Autumn K., Liang Y.A., Hsieh S.T., Zesch W., Chan W.P., Kenny T.W., Fearing R., Full R.J. // Nature. 2000. V. 405. P. 681–685. https://doi.org/10.1038/35015073
van der Vegt N.F., Nayar D. // J. Phys. Chem. B. 2017. V. 121. P. 9986–9998. https://doi.org/10.1021/acs.jpcb.7b06453
Fenimore P.W., Frauenfelder H., Magazu S., McMahon B.H., Mezei F., Migliardo F., Young R.D., Stroe I. // Chem. Phys. 2013. V. 424. P. 2–6. https://doi.org/10.1016/j.chemphys.2013.06.023
Meyer E.A., Castellano R.K., Diederich F. // Angew. Chem. Int. Ed. 2003. V. 42. P. 1210–1250. https://doi.org/10.1002/anie.200390319
Steed J.W., Atwood J.L. // Supramolecular Chemistry. John Wiley & Sons, 2013.
Kuehl C.J., Huang S.D., Stang P.J. // J. Am. Chem. Soc. 2001. V. 123. P. 9634–9641. https://doi.org/10.1021/ja0114355
Li C.-P., Du M. // Chem. Commun. 2011. V. 47. P. 5958–5972. https://doi.org/10.1039/C1CC10935A
Lebedeva N. SH., Mal’kova E.A., V’yurgun A.I. // Rev. J. Chem. 2012. V. 2. P. 20–50. https://doi.org/10.1134/S2079978011040029
Safont-Sempere M.M., Fernández G., Würthner F. // Chem. Rev. 2011. V. 111. P. 5784–5814. https://doi.org/10.1021/cr100357h
Pascal R., Pross A., Sutherland J.D. // Open Biology. 2013. V. 3. P. 130156. https://doi.org/10.1098/rsob.130156
Zhao Y., She N., Ma Y., Wang C., Cao Z. // ACS Catal. 2018. V. 8. P. 4143–4159. https://doi.org/10.1021/acscatal.8b00006
Zhang Z., Li P., Kong X.-Y., Xie G., Qian Y., Wang Z., Tian Y., Wen L., Jiang L. // J. Am. Chem. Soc. 2018. V. 140. P. 1083–1090. https://doi.org/10.1021/jacs.7b11472
Stromberg J.R., Marton A., Kee H.L., Kirmaier C., Diers J.R., Muthiah C., Taniguchi M., Lindsey J.S., Bocian D.F., Meyer G.J. // J. Phys. Chem. C. 2007. V. 111. P. 15464–15478. https://doi.org/10.1021/jp0749928
Pan X., Cao P., Su X., Liu Z., Li M. // Biochim. Biophys. Acta Bioenerg. 2020. V. 1861. P. 148038. https://doi.org/10.1016/j.bbabio.2019.06.010
Gao J., Wang H., Yuan Q., Feng Y. // Front. Plant Sci. 2018. V. 9. P. 357. https://doi.org/10.3389/fpls.2018.00357
Toporik H., Li J., Williams D., Chiu P.-L., Mazor Y. // Nat. Struct. Mol. Biol. 2019. V. 26. P. 443–449. https://doi.org/10.1038/s41594-019-0228-8
Nevo R., Charuvi D., Tsabari O., Reich Z. // Plant J. 2012. V. 70. P. 157–176. https://doi.org/10.1111/j.1365-313X.2011.04876.x
Ferreira K.N., Iverson T.M., Maghlaoui K., Barber J., Iwata S. // Science. 2004. V. 303. P. 1831–1838. https://doi.org/10.1126/science.1093087
Lubitz W., Chrysina M., Cox N. // Photosynth. Res. 2019. P. 1–21. https://doi.org/10.1007/s11120-019-00648-3
Cramer W.A., Hasan S.S. // In: Cytochrome Comple-xes: Evolution, Structures, Energy Transduction, and Signaling, vol. 41 / Eds. Cramer W., Kallas T. Dordrecht: Springer, 2016. P. 177–207. https://doi.org/10.1007/978-94-017-7481-9_9
Cramer W.A. // Photosynth. Res. 2019. V. 139. P. 53–65. https://doi.org/10.1007/s11120-018-0585-x
Malone L.A., Qian P., Mayneord G.E., Hitchcock A., Farmer D.A., Thompson R.F., Swainsbury D.J., Ranson N.A., Hunter C.N., Johnson M.P. // Nature. 2019. V. 575. P. 535–539. https://doi.org/10.1038/s41586-019-1746-6
Dumas L., Chazaux M., Peltier G., Johnson X., Alric J. // Photosynth. Res. 2016. V. 129. P. 307–320. https://doi.org/10.1007/s11120-016-0298-y
Mazor Y., Borovikova A., Caspy I., Nelson N. // Nat. Plants. 2017. V. 3. P. 1–9. https://doi.org/10.1038/nplants.2017.14
Höhner R., Pribil M., Herbstová M., Lopez L.S., Kunz H.-H., Li M., Wood M., Svoboda V., Puthiyavee-til S., Leister D. // Proc. Natl. Acad. Sci. USA. 2020. V. 117. P. 15354–15362. https://doi.org/10.1073/pnas.2005832117
Fukuyama K. // Photosynth. Res. 2004. V. 81. P. 289–301. https://doi.org/10.1023/B:PRES.0000036882.19322.0a
Cashman D.J., Zhu T., Simmerman R.F., Scott C., Bruce B.D., Baudry J. // J. Mol. Recognit. 2014. V. 27. P. 597–608. https://doi.org/10.1002/jmr.2384
Guo H., Suzuki T., Rubinstein J.L. // Elife. 2019. V. 8. P. e43128. https://doi.org/10.7554/eLife.43128.001
Hahn A., Vonck J., Mills D.J., Meier T., Kühlbrandt W. // Science. 2018. V. 360. P. eaat4318. https://doi.org/10.1126/science.aat4318
Dekker J.P., Boekema E.J. // Biochim. Biophys. Acta Bioenerg. 2005. V. 1706. P. 12–39. https://doi.org/10.1016/j.bbabio.2004.09.009
Anderson J.M., Chow W.S., Park Y.-I. // Photosynth. Res. 1995. V. 46. P. 129–139. https://doi.org/10.1007/BF00020423
Pascal A.A., Liu Z., Broess K., van Oort B., van Ame-rongen H., Wang C., Horton P., Robert B., Chang W., Ruban A. // Nature. 2005. V. 436. P. 134–137. https://doi.org/10.1038/nature03795
Squire R., March N., Rubio A., Ingles J., Cunningham W. // arXiv preprint arXiv:1608.06913. 2016. https://arxiv.org/abs/1608.06913
Frischmann P.D., Mahata K., Würthner F. // Chem. Soc. Rev. 2013. V. 42. P. 1847–1870. https://doi.org/10.1039/C2CS35223K
Lebedeva N.S., Gubarev Y.A., Koifman O.I. // Mendeleev Commun. 2015. V. 25. P. 307–309. https://doi.org/10.1016/j.mencom.2015.07.027
Aratani N., Osuka A., Kim Y.H., Jeong D.H., Kim D. // Angew. Chem. Int. Ed. 2000. V. 39. P. 1458–1462. https://doi.org/10.1002/(SICI)1521-3773(20000417)-39:8<1458::AID-ANIE1458>3.0.CO;2-E
Kim Y.H., Jeong D.H., Kim D., Jeoung S.C., Cho H.S., Kim S.K., Aratani N., Osuka A. // J. Am. Chem. Soc. 2001. V. 123. P. 76–86. https://doi.org/10.1021/ja0009976
Takahashi R., Kobuke Y. // J. Am. Chem. Soc. 2003. V. 125. P. 2372–2373. https://doi.org/10.1021/ja028325y
D’Souza F. // Coord. Chem. Rev. 2016. V. 322. P. 104–141. https://doi.org/10.1016/j.ccr.2016.05.012
Natali M., Scandola F. // In: Applied Photochemistry. Lecture Notes in Chemistry, vol. 92 / Еds. Bergami-ni G., Silvi S. Springer, Cham, 2016. P. 1–66. https://doi.org/10.1007/978-3-319-31671-0_1
Lee S.H., Matula A.J., Hu G., Troiano J.L., Karpovich C.J., Crabtree R.H., Batista V.S., Brudvig G.W. // ACS Appl. Mater. Interfaces. 2019. V. 11. P. 8000–8008. https://doi.org/10.1021/acsami.8b20996
Wróbel D., Graja A. // Coord. Chem. Rev. 2011. V. 255. P. 2555–2577. https://doi.org/10.1016/j.ccr.2010.12.026
Shukla J., Singh V.P., Mukhopadhyay P. // Chem-istryOpen. 2020. V. 9. P. 304–324. https://doi.org/10.1002/open.201900339
Yu X., Wang B., Kim Y., Park J., Ghosh S., Dhara B., Mukhopadhyay R.D., Koo J., Kim I., Kim S. // J. Am. Chem. Soc. 2020. V. 142. P. 12596–12601. https://doi.org/10.1021/jacs.0c05339
Panda M.K., Ladomenou K., Coutsolelos A.G. // Coord. Chem. Rev. 2012. V. 256. P. 2601–2627. https://doi.org/10.1016/j.ccr.2012.04.041
Imahori H. // J. Phys. Chem. B. 2004. V. 108. P. 6130–6143. https://doi.org/10.1021/jp038036b
Muzard J., Sarda-Mantel L., Loyau S., Meulemans A., Louedec L., Bantsimba-Malanda C., Hervatin F., Marchal-Somme J., Michel J.B., Le Guludec D. // PLoS One. 2009. V. 4. P. e5585. https://doi.org/10.1371/journal.pone.0005585
Lourenço L.M., Hausmann A., Schubert C., Neves M.G., Cavaleiro J.A., Torres T., Guldi D.M., Tomé J.P. // ChemPlusChem. 2015. V. 80. P. 832. https://doi.org/10.1002/cplu.201500005
El-Khouly M.E., El-Mohsnawy E., Fukuzumi S. // J. Photochem. Photobiol. C. 2017. V. 31. P. 36–83. https://doi.org/10.1016/j.jphotochemrev.2017.02.001
Otsuki J. // J. Mater. Chem. 2018. V. 6. P. 6710–6753. https://doi.org/10.1039/C7TA11274B
Beletskaya I., Tyurin V.S., Tsivadze A.Y., Guilard R., Stern C. // Chem. Rev. 2009. V. 109. P. 1659–1713. https://doi.org/10.1021/cr800247a
Steinberg-Yfrach G., Rigaud J.-L., Durantini E.N., Moore A.L., Gust D., Moore T.A. // Nature. 1998. V. 392. P. 479–482. https://doi.org/10.1038/33116
O’Regan B., Grätzel M. // Nature. 1991. V. 353. P. 737–740. https://doi.org/10.1038/353737a0
Zeng K., Tong Z., Ma L., Zhu W.-H., Wu W., Xie Y. // Energy & Environmental Science. 2020. https://doi.org/10.1039/C9EE04200H
Balaban T.S. // Acc. Chem. Res. 2005. V. 38. P. 612–623. https://doi.org/10.1021/ar040211z
Choi M.S., Yamazaki T., Yamazaki I., Aida T. // Angew. Chem. Int. Ed. 2004. V. 43. P. 150–158. https://doi.org/10.1002/anie.200301665
Jang W.-D., Aida T. // Macromolecules. 2003. V. 36. P. 8461–8469. https://doi.org/10.1021/ma034221b
Kim D., Heo J., Ham S., Yoo H., Lee C.-H., Yoon H., Ryu D., Kim D., Jang W.-D. // Chem. Commun. 2011. V. 47. P. 2405–2407. https://doi.org/10.1039/C0CC04654J
Kim S., Kim J.H., Lee J.S., Park C.B. // Small. 2015. V. 11. P. 3623–3640. https://doi.org/10.1002/smll.201500169
Zou Q., Zhang L., Yan X., Wang A., Ma G., Li J., Möhwald H., Mann S. // Angew. Chem. 2014. V. 126. P. 2398–2402. https://doi.org/10.1002/ange.201308792
Tao K., Xue B., Frere S., Slutsky I., Cao Y., Wang W., Gazit E. // Chem. Mater. 2017. V. 29. P. 4454–4460. https://doi.org/10.1021/acs.chemmater.7b00966
Pepe-Mooney B.J., Kokona B., Fairman R. // Biomacromolecules. 2011. V. 12. P. 4196–4203. https://doi.org/10.1021/bm201354m
Kuciauskas D., Kiskis J., Caputo G.A., Gulbinas V. // J. Phys. Chem. B. 2010. V. 114. P. 16029–16035. https://doi.org/10.1021/jp108685n
Kim J.H., Nam D.H., Lee Y.W., Nam Y.S., Park C.B. // Small. 2014. V. 10. P. 1272–1277. https://doi.org/10.1002/smll.201302627
Zhao L., Zou H., Zhang H., Sun H., Wang T., Pan T., Li X., Bai Y., Qiao S., Luo Q. // ACS Nano. 2017. V. 11. P. 938–945. https://doi.org/10.1021/acsnano.6b07527
Lebedeva N.S., Gubarev Y.A., Vyugin A.I., Koifman O.I. // J. Lumin. 2015. V. 166. P. 71–76. https://doi.org/10.1016/j.jlumin.2015.04.041
Lebedeva N.S., Yurina E.S., Guseinov S.S., Gubarev Y.A., Syrbu S.A. // Dyes and Pigments. 2019. V. 162. P. 266–271. https://doi.org/10.1016/j.dyepig.2018.10.034
Lebedeva N.S., Gubarev Y.A., Koifman O.I. // J. Incl. Phenom. Macrocycl. Chem. 2017. V. 88. P. 191–198. https://doi.org/10.1007/s10847-017-0718-4
Lebedeva N.S., Yurina E.S., Gubarev Y.A., Syrbu S.A., Koifman O.I. // J. Porphyrins Phthalocyanines. 2020. V. 25. P. 145–152. https://doi.org/10.1142/S1088424621500061
Lebedeva N.S., Yurina E.S., Gubarev Y.A., Koifman O.I. // J. Incl. Phenom. Macrocycl. Chem. 2019. P. 1–8. https://doi.org/10.1007/s10847-019-00947-1
Fry H.C., Garcia J.M., Medina M.J., Ricoy U.M., Gosztola D.J., Nikiforov M.P., Palmer L.C., Stupp S.I. // J. Am. Chem. Soc. 2012. V. 134. P. 14646–14649. https://doi.org/10.1021/ja304674d
Solomon L.A., Sykes M.E., Wu Y.A., Schaller R.D., Wiederrecht G.P., Fry H.C. // ACS Nano. 2017. V. 11. P. 9112–9118. https://doi.org/10.1021/acsnano.7b03867
Raszewski G., Saenger W., Renger T. // Biophys. J. 2005. V. 88. P. 986–998. https://doi.org/10.1529/biophysj.104.050294
Hu Y.-Z., Tsukiji S., Shinkai S., Oishi S., Hamachi I. // J. Am. Chem. Soc. 2000. V. 122. P. 241–253. https://doi.org/10.1021/ja991406i
Anderson J.R., Armstrong C.T., Kodali G., Lichtenstein B.R., Watkins D.W., Mancini J.A., Boyle A.L., Farid T.A., Crump M.P., Moser C.C. // Chemical Science. 2014. V. 5. P. 507–514. https://doi.org/10.1039/C3SC52019F
Cohen-Ofri I., van Gastel M., Grzyb J., Brandis A., Pinkas I., Lubitz W., Noy D. // J. Am. Chem. Soc. 2011. V. 133. P. 9526–9535. https://doi.org/10.1021/ja202054m
Kodali G., Mancini J.A., Solomon L.A., Episova T.V., Roach N., Hobbs C.J., Wagner P., Mass O.A., Aravindu K., Barnsley J.E. // Chemical Science. 2017. V. 8. P. 316–324. https://doi.org/10.1039/C6SC02417C
Woller J.G., Hannestad J.K., Albinsson B. // J. Am. Chem. Soc. 2013. V. 135. P. 2759–2768. https://doi.org/10.1021/ja311828v
Lebedeva N.S., Yurina E.S., Gubarev Y.A., Syrbu S.A. // Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2018. V. 199. P. 235–241. https://doi.org/10.1016/j.saa.2018.03.066
Vilona D., Di Lorenzo R., Carraro M., Licini G., Trainotti L., Bonchio M. // Journal of Materials Che-mistry B. 2015. V. 3. P. 6718–6730. https://doi.org/10.1039/C5TB00924C
Aljabri M.D., Jadhav R.W., Al Kobaisi M., Jones L.A., Bhosale S.V., Bhosale S.V. // ACS Omega. 2019. V. 4. P. 11408–11413. https://doi.org/10.1021/acsomega.9b00909
Endo M., Fujitsuka M., Majima T. // Chem. Eur. J. 2007. V. 13. P. 8660–8666. https://doi.org/10.1002/chem.200700895
Nam Y.S., Shin T., Park H., Magyar A.P., Choi K., Fantner G., Nelson K.A., Belcher A.M. // J. Am. Chem. Soc. 2010. V. 132. P. 1462–1463. https://doi.org/10.1021/ja908812b
Hou Y.-N., Ding W.-L., Jiang S.-P., Miao D., Tan Z.-Z., Hu J.-L., Scheer H., Zhao K.-H. // Biochim. Biophys. Acta Mol. Cell Res. 2019. V. 1866. P. 277–284. https://doi.org/10.1016/j.bbamcr.2018.11.008
Uhlenheuer D.A., Petkau K., Brunsveld L. // Chem. Soc. Rev. 2010. V. 39. P. 2817–2826. https://doi.org/10.1039/B820283B
De Annunzio S.R., Costa N.C.S., Mezzina R.D., Gra-minha M.A., Fontana C.R. // Int. J. Mol. Sci. 2019. V. 20. P. 3861. https://doi.org/10.3390/ijms20163861
Abrahamse H., Hamblin M.R. // Biochem. J. 2016. V. 473. P. 347–364. https://doi.org/10.1042/BJ20150942
Scherer K.M., Bisby R.H., Botchway S.W., Parker A.W. // Anti-Cancer Agents Med. Chem. 2017. V. 17. P. 171–189. https://doi.org/10.2174/1871520616666160513131723
Ormond A.B., Freeman H.S. // Materials. 2013. V. 6. P. 817–840. https://doi.org/10.3390/ma6030817
de Oliveira K.T., Momo P.B., de Assis F.F., Ferreira M.A.B., Brocksom T.J. // Current Organic Synthesis. 2014. V. 11. P. 42–58. https://doi.org/10.2174/15701794113106660085
Zhang J., Jiang C., Longo J.P.F., Azevedo R.B., Zhang H., Muehlmann L.A. // Acta Pharmaceutica Sinica B. 2018. V. 8. P. 137–146. https://doi.org/10.1016/j.apsb.2017.09.003
Chilakamarthi U., Giribabu L. // The Chemical Record. 2017. V. 17. P. 775–802. https://doi.org/10.1002/tcr.201600121
Tian J., Huang B., Nawaz M.H., Zhang W. // Coord. Chem. Rev. 2020. V. 420. P. 213410. https://doi.org/10.1016/j.ccr.2020.213410
Rak J., Pouckova P., Benes J., Vetvicka D. // Anticancer Res. 2019. V. 39. P. 3323–3339. https://doi.org/10.21873/anticanres.13475
Iqbal Z., Chen J., Chen Z., Huang M. // Curr. Drug Metab. 2015. V. 16. P. 816–832. https://doi.org/10.2174/1389200217666151120165404
Rizwan M., Rasheed T., Raza A., Bilal M., Yahya R., Yar M., Iqbal H.M. // J. Drug Deliv. Sci. Technol. 2019. V. 51. P. 70–82. https://doi.org/10.1016/j.jddst.2019.02.014
Lebedeva N.S., Yurina E.S., Gubarev Y.A., Lyubimtsev A.V., Syrbu S.A. // J. Photochem. Photobiol. A: Chem. 2018. V. 353. P. 299–305.https://doi.org/10.1016/j.jphotochem.2017.11.037
Hirakawa K. // Photodynamic Therapy – from Basic Science to Clinical Research. IntechOpen, 2020. https://doi.org/10.5772/intechopen.77705
Day R.A., Estabrook D.A., Logan J.K., Sletten E.M. // Chem. Commun. 2017. V. 53. P. 13043–13046. https://doi.org/10.1039/C7CC07038A
Hu D., Zhong L., Wang M., Li H., Qu Y., Liu Q., Han R., Yuan L., Shi K., Peng J. // Adv. Funct. Mater. 2019. V. 29. P. 1806199. https://doi.org/10.1002/adfm.201806199
Riess J.G. // Artificial Cells, Blood Substitutes, and Biotechnology. 2005. V. 33. P. 47–63. https://doi.org/10.1081/BIO-200046659
Song X., Feng L., Liang C., Yang K., Liu Z. // Nano Lett. 2016. V. 16. P. 6145–6153. https://doi.org/10.1021/acs.nanolett.6b02365
Zhao J., Fei J., Du C., Cui W., Ma H., Li J. // Chem. Commun. 2013. V. 49. P. 10733–10735. https://doi.org/10.1039/C3CC46969G
Chen H., Tian J., He W., Guo Z. // J. Am. Chem. Soc. 2015. V. 137. P. 1539–1547. https://doi.org/10.1021/ja511420n
Phua S.Z.F., Yang G., Lim W.Q., Verma A., Chen H., Thanabalu T., Zhao Y. // ACS Nano. 2019. V. 13. P. 4742–4751. https://doi.org/10.1021/acsnano.9b01087
Hamblin M.R. // Expert Review of Anti-Infective Therapy. 2017. V. 15. P. 1059–1069. https://doi.org/10.1080/14787210.2017.1397512
Wen X., Zhang X., Szewczyk G., El-Hussein A., Huang Y.-Y., Sarna T., Hamblin M.R. // Antimicrob. Agents Chemother. 2017. V. 61. P. e00467-17. https://doi.org/10.1128/AAC.00467-17
Wang S., Yuan F., Chen K., Chen G., Tu K., Wang H., Wang L.-Q. // Biomacromolecules. 2015. V. 16. P. 2693–2700. https://doi.org/10.1021/acs.biomac.5b00571
Yu L., Wang Q., Wong R.C.-H., Zhao S., Ng D.K., Lo P.-C. // Dyes and Pigments. 2019. V. 163. P. 197–203. https://doi.org/10.1016/j.dyepig.2018.11.055
Gomes A.T., Neves M.G., Cavaleiro J.A. // Anais da Academia Brasileira de Ciências. 2018. V. 90. P. 993–1026. https://doi.org/10.1590/0001-3765201820170811
Srivatsan A., Ethirajan M., Pandey S.K., Dubey S., Zheng X., Liu T.-H., Shibata M., Missert J., Morgan J., Pandey R.K. // Mol. Pharm. 2011. V. 8. P. 1186–1197. https://doi.org/10.1021/mp200018y
Biscaglia F., Gobbo M. // Peptide Science. 2018. V. 110. P. e24038. https://doi.org/10.1002/pep2.24038
Schneider R., Tirand L., Frochot C., Vanderesse R., Tho-mas N., Gravier J., Guillemin F., Barberi-Heyob M. // Anti-Cancer Agents Med. Chem. 2006. V. 6. P. 469–488. https://doi.org/10.2174/187152006778226503
Biron E., Voyer N. // Chem. Commun. 2005. P. 4652–4654. https://doi.org/10.1039/B508380J
Urbanska K., Pawlicki M. // J. Org. Chem. 2020. V. 85. P. 8196–8202. https://doi.org/10.1021/acs.joc.0c00335
Pathak P., Yao W., Hook K.D., Vik R., Winnerdy F.R., Brown J.Q., Gibb B.C., Pursell Z.F., Phan A.T., Jaya-wickramarajah J. // J. Am. Chem. Soc. 2019. V. 141. P. 12582–12591. https://doi.org/10.1021/jacs.9b03250
Biron E., Voyer N. // Org. Biomol. Chem. 2008. V. 6. P. 2507–2515. https://doi.org/10.1039/b803281e
Bourre L., Giuntini F., Eggleston I.M., Wilson M., MacRobert A.J. // Mol. Cancer Ther. 2008. V. 7. P. 1720–1729. https://doi.org/10.1158/1535-7163.MCT-08-0092
Sandland J., Boyle R.W. // Bioconj. Chem. 2019. V. 30. P. 975–993. https://doi.org/10.1021/acs.bioconjchem.9b00055
Lin L., Xiong L., Wen Y., Lei S., Deng X., Liu Z., Chen W., Miao X. // J. Biomed. Nanotechnol. 2015. V. 11. P. 531–554. https://doi.org/10.1166/jbn.2015.2090
Fernandes S.R., Fernandes R., Sarmento B., Pereira P.M., Tomé J.P. // Org. Biomol. Chem. 2019. V. 17. P. 2579–2593. https://doi.org/10.1039/C8OB02902D
Hudson R., Carcenac M., Smith K., Madden L., Clarke O., Pelegrin A., Greenman J., Boyle R. // Br. J. Cancer. 2005. V. 92. P. 1442–1449. https://doi.org/10.1038/sj.bjc.6602517
Shi Q., Tao Z., Yang H., Fan Q., Wei D., Wan L., Lu X. // Drug Deliv. 2017. V. 24. P. 1818–1830. https://doi.org/10.1080/10717544.2017.1407011
Sato K., Watanabe R., Hanaoka H., Harada T., Nakajima T., Kim I., Paik C.H., Choyke P.L., Kobayashi H. // Mol. Oncol. 2014. V. 8. P. 620–632. https://doi.org/10.1016/j.molonc.2014.01.006
Sato K., Nagaya T., Choyke P.L., Kobayashi H. // The-ranostics. 2015. V. 5. P. 698. https://doi.org/10.7150/thno.11559
Vrouenraets M.B., Visser G.W., Loup C., Meunier B., Stigter M., Oppelaar H., Stewart F.A., Snow G.B., van Dongen G.A. // Int. J. Cancer. 2000. V. 88. P. 108–114. https://doi.org/10.1002/1097-0215(20001001)88:1<108:: AID-IJC17>3.0.CO;2-H
Xu P., Chen J., Chen Z., Zhou S., Hu P., Chen X., Huang M. // PLoS One. 2012. V. 7. P. e37051. https://doi.org/10.1371/journal.pone.0037051
Tesniere A., Panaretakis T., Kepp O., Apetoh L., Ghi-ringhelli F., Zitvogel L., Kroemer G. // Cell Death & Differentiation. 2008. V. 15. P. 3–12. https://doi.org/10.1038/sj.cdd.4402269
Kepp O., Tesniere A., Schlemmer F., Michaud M., Senovilla L., Zitvogel L., Kroemer G. // Apoptosis. 2009. V. 14. P. 364–375. https://doi.org/10.1007/s10495-008-0303-9
Castano A.P., Mroz P., Wu M.X., Hamblin M.R. // Proc. Natl. Acad. Sci. USA. 2008. V. 105. P. 5495–5500. https://doi.org/10.1073/pnas.0709256105
Habermeyer B., Guilard R. // Photochem. Photobiol. Sci. 2018. V. 17. P. 1675–1690. https://doi.org/10.1039/c8pp00222c
Duan W., Smith K., Savoie H., Greenman J., Boyle R.W. // Org. Biomol. Chem. 2005. V. 3. P. 2384–2386. https://doi.org/10.1039/B504334D
Klotz L.-O., Kröncke K.-D., Sies H. // Photochem. Photobiol. Sci. 2003. V. 2. P. 88–94. https://doi.org/10.1039/B210750C
Liu M., Zhang Z., Cheetham J., Ren D., Zhou Z.S. // Anal. Chem. 2014. V. 86. P. 4940–4948. https://doi.org/10.1021/ac500334k
Giménez R.E., Vargová V., Rey V., Turbay M.B.E., Abatedaga I., Vieyra F.E.M., Zanini V.I.P., Ortiz J.H.M., Katz N.E., Ostatná V. // Free Radical Biol. Med. 2016. V. 94. P. 99–109. https://doi.org/10.1016/j.freeradbiomed.2016.02.014
Gracanin M., Hawkins C.L., Pattison D.I., Davies M.J. // Free Radical Biol. Med. 2009. V. 47. P. 92–102. https://doi.org/10.1016/j.freeradbiomed.2009.04.015
Silvester J.A., Timmins G.S., Davies M.J. // Free Radical Biol. Med. 1998. V. 24. P. 754–766. https://doi.org/10.1016/S0891-5849(97)00327-4
Kollar J., Machacek M., Halaskova M., Lenco J., Kucera R., Demuth J., Rohlickova M., Hasonova K., Miletin M., Novakova V. // J. Med. Chem. 2020. V. 63. P. 7616–7632. https://doi.org/10.1021/acs.jmedchem.0c00481
Sannikova N.E., Timofeev I.O., Chubarov A.S., Lebedeva N.S., Semeikin A.S., Kirilyuk I.A., Tsentalovich Y.P., Fedin M.V., Bagryanskaya E.G., Krumkacheva O.A. // J. Photochem. Photobiol. B: Biol. 2020. V. 211. P. 112008. https://doi.org/10.1016/j.jphotobiol.2020.112008
Jiang B.-P., Hu L.-F., Shen X.-C., Ji S.-C., Shi Z., Liu C.-J., Zhang L., Liang H. // ACS Appl. Mater. Interfaces. 2014. V. 6. P. 18008–18017. https://doi.org/10.1021/am504860c
Li S., Zou Q., Xing R., Govindaraju T., Fakhrullin R., Yan X. // Theranostics. 2019. V. 9. P. 3249. https://doi.org/10.7150/thno.31814
Lovell J.F., Jin C.S., Huynh E., Jin H., Kim C., Rubinstein J.L., Chan W.C., Cao W., Wang L.V., Zheng G. // Nature Materials. 2011. V. 10. P. 324–332. https://doi.org/10.1038/nmat2986
Zou Q., Abbas M., Zhao L., Li S., Shen G., Yan X. // J. Am. Chem. Soc. 2017. V. 139. P. 1921–1927. https://doi.org/10.1021/jacs.6b11382
Li X., Kim C.-Y., Lee S., Lee D., Chung H.-M., Kim G., Heo S.-H., Kim C., Hong K.-S., Yoon J. // J. Am. Chem. Soc. 2017. V. 139. P. 10880–10886. https://doi.org/10.1021/jacs.7b05916
Zhou Y., Liang X., Dai Z. // Nanoscale. 2016. V. 8. P. 12394–12405. https://doi.org/10.1039/C5NR07849K
Peng L., Liu S., Feng A., Yuan J. // Mol. Pharm. 2017. V. 14. P. 2475–2486. https://doi.org/10.1021/acs.molpharmaceut.7b00160
Mazzaglia A., Angelini N., Darcy R., Donohue R., Lombardo D., Micali N., Sciortino M.T., Villari V., Scolaro L.M. // Chem. Eur. J. 2003. V. 9. P. 5762–5769. https://doi.org/10.1002/chem.200304861
Sortino S., Mazzaglia A., Scolaro L.M., Merlo F.M., Valveri V., Sciortino M.T. // Biomaterials. 2006. V. 27. P. 4256–4265. https://doi.org/10.1016/j.biomaterials.2006.03.035
Bryden F., Savoie H., Rosca E.V., Boyle R.W. // Dalton Trans. 2015. V. 44. P. 4925–4932. https://doi.org/10.1039/C4DT02949F
Eggleston I.M., Yaghini E., Dondi R., Tewari K., Edler K.J., Loizidou M., MacRobert A.J. // ECS Meeting Abstracts, 2020. https://iopscience.iop.org/article/10.1149/MA2020-0112947mtgabs/meta
Li S.-Y., Cheng H., Qiu W.-X., Liu L.-H., Chen S., Hu Y., Xie B.-R., Li B., Zhang X.-Z. // ACS Appl. Mater. Interfaces. 2015. V. 7. P. 28319–28329. https://doi.org/10.1021/acsami.5b08637
Jiang T., Olson E.S., Nguyen Q.T., Roy M., Jennings P.A., Tsien R.Y. // Proc. Natl. Acad. Sci. USA. 2004. V. 101. P. 17867–17872. https://doi.org/10.1073/pnas.0408191101
Peltek O.O., Muslimov A.R., Zyuzin M.V., Timin A.S. // J. Nanobiotechnology. 2019. V. 17. P. 90. https://doi.org/10.1186/s12951-019-0524-9
Tong H., Du J., Li H., Jin Q., Wang Y., Ji J. // Chem. Commun. 2016. V. 52. P. 11935–11938. https://doi.org/10.1039/C6CC06439F
Li S.Y., Qiu W.X., Cheng H., Gao F., Cao F.Y., Zhang X.Z. // Adv. Funct. Mater. 2017. V. 27. P. 1604916. https://doi.org/10.1002/adfm.201604916
Zhu K., Liu G., Hu J., Liu S. // Biomacromolecules. 2017. V. 18. P. 2571–2582. https://doi.org/10.1021/acs.biomac.7b00693
Xu X., Li Y., Liang Q., Song Z., Li F., He H., Wang J., Zhu L., Lin Z., Yin L. // ACS Appl. Mater. Interfaces. 2018. V. 10. P. 256–266. https://doi.org/10.1021/acsami.7b15896
Han K., Lei Q., Wang S.B., Hu J.J., Qiu W.X., Zhu J.Y., Yin W.N., Luo X., Zhang X.Z. // Adv. Funct. Mater. 2015. V. 25. P. 2961–2971. https://doi.org/10.1002/adfm.201500590
Sharman W.M., van Lier J.E., Allen C.M. // Adv. Drug Del. Rev. 2004. V. 56. P. 53–76. https://doi.org/10.1016/j.addr.2003.08.015
Ma J., Liu L., Che G., Yu N., Dai F., You Z. // BMC Cancer. 2010. V. 10. P. 112. https://doi.org/10.1186/1471-2407-10-112
Hanahan D., Weinberg R.A. // Cell. 2011. V. 144. P. 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Colegio O.R., Chu N.-Q., Szabo A.L., Chu T., Rhebergen A.M., Jairam V., Cyrus N., Brokowski C.E., Eisenbarth S.C., Phillips G.M. // Nature. 2014. V. 513. P. 559–563. https://doi.org/10.1038/nature13490
Kratz F. // J. Controlled Release. 2008. V. 132. P. 171–183. https://doi.org/10.1016/j.jconrel.2008.05.010
Maeda H., Wu J., Sawa T., Matsumura Y., Hori K. // J. Controlled Release. 2000. V. 65. P. 271–284. https://doi.org/10.1016/S0168-3659(99)00248-5
Li X., Jeong K., Lee Y., Guo T., Lee D., Park J., Kwon N., Na J.-H., Hong S.K., Cha S.-S. // Theranostics. 2019. V. 9. P. 6412. https://doi.org/10.7150/thno.35210
Lebedeva N., Popova T., Kozbial M., Wszelaka-Rylik M., Gubarev Y., Gierycz P. // J. Porphyrins Phthalocyanines. 2011. V. 15. P. 223–229. https://doi.org/10.1142/S1088424611003185
Lebedeva N.S., Yurina E.S., Gubarev Y.A., Koif-man O.I. // Mendeleev Commun. 2017. V. 27. P. 47–49. https://doi.org/10.1016/j.mencom.2017.01.014
Sadeghi M., Ordway B., Rafiei I., Borad P., Fang B., Koomen J.L., Zhang C., Yoder S., Johnson J., Damaghi M. // Front. Oncol. 2020. V. 10. P. 304. https://doi.org/10.3389/fonc.2020.00304
Pereira P.M., Carvalho J.J., Silva S., Cavaleiro J.A., Schneider R.J., Fernandes R., Tomé J.P. // Org. Biomol. Chem. 2014. V. 12. P. 1804–1811. https://doi.org/10.1039/C3OB42082E
Dai T., Huang Y.-Y., Hamblin M.R. // Photodiagn. Photodyn. Ther. 2009. V. 6. P. 170–188. https://doi.org/10.1016/j.pdpdt.2009.10.008
Hamblin M.R., Hasan T. // Photochem. Photobiol. Sci. 2004. V. 3. P. 436–450. https://doi.org/10.1039/B311900A
Huang L., Dai T., Hamblin M.R. // In: Photodynamic Therapy. Methods in Molecular Biology (Methods and Protocols), vol. 635 / Eds. Gomer C. Humana Press, Totowa, NJ, 2010. P. 155–173. https://doi.org/10.1007/978-1-60761-697-9_12
Carrera E., Dias H., Corbi S., Marcantonio R., Bernardi A., Bagnato V., Hamblin M., Rastelli A.N.S. // Laser P-hysics. 2016. V. 26. P. 123001. https://doi.org/10.1088/1054-660X/26/12/123001
Lebedeva N., Gubarev Y., Koifman M., Koifman O. // Molecules. 2020. V. 25. P. 4368. https://doi.org/10.3390/molecules25194368
Hally C., Delcanale P., Nonell S., Viappiani C., Abbruzzetti S. // Transl. Biophotonics. 2020. V. 2. P. e201900031. https://doi.org/10.1002/tbio.201900031
Hugenholtz P. // Genome Biology. 2002. V. 3. P. reviews0003.1. https://doi.org/10.1186/gb-2002-3-2-reviews0003
Khan H.A., Ahmad A., Mehboob R. // Asian Pac. J. Trop. Biomed. 2015. V. 5. P. 509–514. https://doi.org/10.1016/j.apjtb.2015.05.001
Liu Y., Qin R., Zaat S.A., Breukink E., Heger M. // Transl. Res. 2015. V. 1. P. 140. https://doi.org/10.18053/jctres.201503.002
Banfi S., Caruso E., Buccafurni L., Battini V., Zazzaron S., Barbieri P., Orlandi V. // J. Photochem. Photobiol. B: Biol. 2006. V. 85. P. 28–38. https://doi.org/10.1016/j.jphotobiol.2006.04.003
Tomé J.P., Neves M.G., Tomé A.C., Cavaleiro J.A., Soncin M., Magaraggia M., Ferro S., Jori G. // J. Med. Chem. 2004. V. 47. P. 6649–6652. https://doi.org/10.1021/jm040802v
Zhao Y., Ying J.-W., Sun Q., Ke M.-R., Zheng B.-Y., Huang J.-D. // Dyes and Pigments. 2020. V. 172. P. 107834. https://doi.org/10.1016/j.dyepig.2019.107834
Reinhardt A., Neundorf I. // Int. J. Mol. Sci. 2016. V. 17. P. 701. https://doi.org/10.3390/ijms17050701
Malik Z., Ladan H., Nitzan Y. // J. Photochem. Photobiol. B: Biol. 1992. V. 14. P. 262–266. https://doi.org/10.1016/1011-1344(92)85104-3
Rovaldi C., Pievsky A., Sole N., Friden P., Rothstein D., Spacciapoli P. // Antimicrob. Agents Chemother. 2000. V. 44. P. 3364–3367. https://doi.org/10.1128/AAC.44.12.3364-3367.2000
Hamblin M.R., O’Donnell D.A., Murthy N., Rajagopa-lan K., Michaud N., Sherwood M.E., Hasan T. // J. Antimicrob. Chemother. 2002. V. 49. P. 941–951. https://doi.org/10.1093/jac/dkf053
Beyene B.B., Wassie G.A. // BMC Chemistry. 2020. V. 14. P. 1–8. https://doi.org/10.1186/s13065-020-00701-6
Alves E., Faustino M.A., Neves M.G., Cunha A., Tome J., Almeida A. // Future Med. Chem. 2014. V. 6. P. 141–164. https://doi.org/10.4155/fmc.13.211
Pereira M., Faustino M., Tomé J., Neves M., Tome A., Cavaleiro J., Cunha A., Almeida A. // Photochem. Photobiol. Sci. 2014. V. 13. P. 680–690. https://doi.org/10.1039/c3pp50408e
Laganowsky A., Reading E., Allison T.M., Ulmschneider M.B., Degiacomi M.T., Baldwin A.J., Robinson C.V. // Nature. 2014. V. 510. P. 172–175. https://doi.org/10.1038/nature13419
Dosselli R., Millioni R., Puricelli L., Tessari P., Arrigoni G., Franchin C., Segalla A., Teardo E., Reddi E. // Journal of Proteomics. 2012. V. 77. P. 329–343. https://doi.org/10.1016/j.jprot.2012.09.007
Liu X., Chang Q., Ferrer-Espada R., Leanse L.G., Goh X.S., Wang X., Gelfand J.A., Dai T. // Photochem. Photobiol. 2020. V. 96. P. 611–617. https://doi.org/10.1111/php.13241
Gonçalves P.J., Bezzerra F.C., Teles A.V., Menezes L.B., Alves K.M., Alonso L., Alonso A., Andrade M.A., Borissevitch I.E., Souza G.R. // J. Photochem. Photobiol. A: Chem. 2020. V. 391. P. 112375. https://doi.org/10.1016/j.jphotochem.2020.112375
Rossi G.G., Guterres K.B., da Silveira C.H., Moreira K.S., Burgo T.A.L., Iglesias B.A., de Campos M.M.A. // Microb. Pathog. 2020. V. 148. P. 104455. https://doi.org/10.1016/j.micpath.2020.104455
Guterres K.B., Rossi G.G., Moreira K.S., Burgo T.A.L., Iglesias B.A. // BioMetals. 2020. V. 33. P. 269–282. https://doi.org/10.1007/s10534-020-00251-3
Luo S., Zhang E., Su Y., Cheng T., Shi C. // Biomaterials. 2011. V. 32. P. 7127–7138. https://doi.org/10.1016/j.biomaterials.2011.06.024
Lee D.-E., Koo H., Sun I.-C., Ryu J.H., Kim K., Kwon I.C. // Chem. Soc. Rev. 2012. V. 41. P. 2656–2672. https://doi.org/10.1039/C2CS15261D
Kondo M., Mitsui T., Ito S., Kondo Y., Ishigure S., Dewa T., Yamashita K., Nakamura J., Oura R., Nango M. // J. Colloid Interface Sci. 2007. V. 310. P. 686–689. https://doi.org/10.1016/j.jcis.2007.02.029
Rahimi R., Tehrani A.A., Fard M.A., Sadegh B.M.M., Khavasi H.R. // Catal. Commun. 2009. V. 11. P. 232–235. https://doi.org/10.1016/j.catcom.2009.09.022
Teiten M.-H., Even P., Burgos P., Frochot C., Aubert S., Carré M.-C., Bolotine L., Merlin J.-L., Guillemin F., Viriot M.-L. // Comptes Rendus Biologies. 2002. V. 325. P. 487–493. https://doi.org/10.1016/S1631-0691(02)01446-4
Kee H.L., Nothdurft R., Muthiah C., Diers J.R., Fan D., Ptaszek M., Bocian D.F., Lindsey J.S., Culver J.P., Holten D. // Photochem. Photobiol. 2008. V. 84. P. 1061–1072. https://doi.org/10.1111/j.1751-1097.2008.00409.x
Kee H.L., Diers J.R., Ptaszek M., Muthiah C., Fan D., Lindsey J.S., Bocian D.F., Holten D. // Photochem. Photobiol. 2009. V. 85. P. 909–920. https://doi.org/10.1111/j.1751-1097.2008.00532.x
Lee S.J., Koo H., Jeong H., Huh M.S., Choi Y., Jeong S.Y., Byun Y., Choi K., Kim K., Kwon I.C. // J. Controlled Release. 2011. V. 152. P. 21–29. https://doi.org/10.1016/j.jconrel.2011.03.027
Otvagin V.F., Kuzmina N.S., Krylova L.V., Volovetsky A.B., Nyuchev A.V., Gavryushin A.E., Meshkov I.N., Gorbunova Y.G., Romanenko Y.V., Koifman O.I. // J. Med. Chem. 2019. V. 62. P. 11182–11193. https://doi.org/10.1021/acs.jmedchem.9b01294
Otvagin V.F., Nyuchev A.V., Kuzmina N.S., Grishin I.D., Gavryushin A.E., Romanenko Y.V., Koifman O.I., Belykh D.V., Peskova N.N., Shilyagina N.Y. // Eur. J. Med. Chem. 2018. V. 144. P. 740–750. https://doi.org/10.1016/j.ejmech.2017.12.062
Kuzmina N.S., Otvagin V.F., Krylova L.V., Nyuchev A.V., Romanenko Y.V., Koifman O.I., Balalaeva I.V., Fedorov A.Y. // Mendeleev Commun. 2020. V. 30. P. 159–161. https://doi.org/10.1016/j.mencom.2020.03.009
Machinskaya E.A., Ivanova-Radkevich V.I. // Biome-dical Photonics. 2013. V. 2. P. 28–32. https://www.pdt-journal.com/jour/article/view/24
Соболев А., Розенкранц А., Гилязова Д. // Биофизика. 2004. Т. 49. С. 351–379.
Agadjanian H., Ma J., Rentsendorj A., Valluripalli V., Hwang J.Y., Mahammed A., Farkas D.L., Gray H.B., Gross Z., Medina-Kauwe L.K. // Proc. Natl. Acad. Sci. USA. 2009. V. 106. P. 6105–6110. https://doi.org/10.1073/pnas.0901531106
Muhanna N., Cui L., Chan H., Burgess L., Jin C.S., MacDonald T.D., Huynh E., Wang F., Chen J., Irish J.C. // Clin. Cancer. Res. 2016. V. 22. P. 961–970. https://doi.org/10.1158/1078-0432.CCR-15-1235
Chilakamarthi U., Kandhadi J., Gunda S., Thatipalli A.R., Jerald M.K., Lingamallu G., Reddy R.C., Chaudhuri A., Pande G. // Exp. Cell Res. 2014. V. 327. P. 91–101. https://doi.org/10.1016/j.yexcr.2014.05.005
Wang L.V., Hu S. // Science. 2012. V. 335. P. 1458–1462. https://doi.org/10.1126/science.1216210
Pu K., Shuhendler A.J., Jokerst J.V., Mei J., Gambhir S.S., Bao Z., Rao J. // Nat. Nanotechnol. 2014. V. 9. P. 233–239. https://doi.org/10.1038/nnano.2013.302
Merkes J.M., Zhu L., Bahukhandi S.B., Rueping M., Kiessling F., Banala S. // Int. J. Mol. Sci. 2020. V. 21. P. 3082. https://doi.org/10.3390/ijms21093082
Hester S.C., Kuriakose M., Nguyen C.D., Mallidi S. // Photochem. Photobiol. 2020. V. 96. P. 260–279. https://doi.org/10.1111/php.13217
Liu Y., Bhattarai P., Dai Z., Chen X. // Chem. Soc. Rev. 2019. V. 48. P. 2053–2108. https://doi.org/10.1039/C8CS00618K
Wang S., Lin J., Wang T., Chen X., Huang P. // Theranostics. 2016. V. 6. P. 2394. https://doi.org/10.7150/thno.16715
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия