

Биоорганическая химия, 2022, T. 48, № 1, стр. 115-126
Изменение метаболизма сфингомиелина и церамида в структурах головного и спинного мозга трансгенных мышей линии FUS (FUS(1-359)), моделирующих боковой амиотрофический склероз
М. А. Шупик 1, У. А. Гутнер 1, А. А. Устюгов 2, А. П. Резвых 3, С. Ю. Фуников 3, О. А. Малошицкая 4, С. А. Соколов 4, А. Т. Лебедев 4, А. В. Алесенко 1, *, **
1 Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, ул. Косыгина, 4, Россия
2 Институт физиологически активных веществ РАН
142432 Черноголовка, Северный проезд, 1, Россия
3 Институт молекулярной биологии им. В.А. Энгельгардта РАН
119991 Москва, ул. Вавилова, 32, Россия
4 Московский государственный университет имени М.В. Ломоносова
Москва, Ленинские горы, 3, Россия
* E-mail: mariashupik@gmail.com
** E-mail: alicealessenko@gmail.com
Поступила в редакцию 11.03.2021
После доработки 22.03.2021
Принята к публикации 26.03.2021
- EDN: ZDMFVO
- DOI: 10.31857/S0132342322010134
Аннотация
Обнаружены изменения экспрессии генов ферментов метаболизма сфинголипидов у трансгенных мышей линии FUS (FUS(1-359)), моделирующих боковой амиотрофический склероз (БАС). Проанализированы профили молекулярных видов сфингомиелинов (СМ) и церамидов (ЦЕР) в структурах головного мозга и в спинном мозге. В процессе развития БАС изменения в спектре молекулярных видов ЦЕР отмечены только в спинном мозге, где уровни практически всех ЦЕР снижаются по отношению к контролю через 2 и 3 месяца течения заболевания. Через 4 месяца развития патологии наблюдается повышение экспрессии гена кислой церамидазы, в результате чего может образовываться сфингозин, обладающий выраженными проапоптотическими свойствами. Анализ экспрессии генов ферментов обмена ЦЕР и галактозилцерамида (ГалЦер) указывает на возможность участия ГалЦЕР в качестве источника для поддержания уровня ЦЕР на терминальной стадии БАС. Таким образом, метаболизм сфинголипидов представляет собой перспективную область исследования процессов, связанных с БАС, как в контексте поиска потенциальных диагностических маркеров и эффективных лекарственных средств, так и для объяснения патогенеза БАС.
ВВЕДЕНИЕ
Боковой амиотрофический склероз (БАС) – неизлечимое мультисистемное нейродегенеративное заболевание, поражающее главным образом верхние и нижние двигательные нейроны центральной нервной системы. Потеря этих групп нейронов приводит к неуклонно нарастающей мышечной слабости и атрофии, а в конечном итоге – к гибели пациента в течение 3–5 лет после постановки диагноза, в основном вследствие паралича дыхательных путей. Около 90% всех случаев БАС имеют неизвестную этиологию и классифицируются как спорадические, остальные 10% – наследственные формы БАС [1]. Число больных БАС в мире неуклонно растет из-за старения населения (характерный возраст начала заболевания – 50–75 лет) и к 2040 г. предположительно составит ~400 тыс. человек [2].
Генетический фон БАС исключительно разнообразен. С БАС ассоциированы ~30 генов [3]. Наиболее распространены мутации в генах C9orf72, SOD1, TDP-43 и FUS. В европейских популяциях чаще встречаются мутации C9orf72 (наследственная форма БАС – 33.7% и спорадическая – 5.1%), затем SOD1 (14.8 и 1.2% соответственно), TDP-43 (4.2 и 0.8%) и FUS (2.8 и 0.3%), а в азиатских – SOD1 (30.0 и 1.5%), затем FUS (6.4 и 0.9%), C9orf72 (2.3 и 0.3%) и TDP-43 (1.5 и 0.2%) [4].
Мутации генов проявляются в многообразных патологических механизмах БАС, которые выражаются в нарушении гомеостаза белков, метаболизма липидов и РНК, репарации ДНК, функций митохондрий и эндоплазматического ретикулума, нуклеоцитоплазматического, эндосомального и везикулярного транспорта, аутофагии, а также эксайтотоксичности, нейровоспалительных реакциях и др. [5–10]. Успехи в изучении генетических основ БАС привели к созданию многочисленных моделей этого заболевания [11]. Наиболее изучены SOD-мутантные модели, т.к. мутации в гене SOD были открыты первыми, как причинные для БАС, еще в 1993 г. Последние 10 лет наблюдается неуклонный интерес к созданию и изучению моделей БАС, экспрессирующих аберрантные формы РНК-связывающих белков, в первую очередь TDP-43 и FUS, потому что нарушение экспрессии, дисфункция и агрегация этих белков – основная отличительная черта БАС [12–16].
И наследственные, и спорадические формы БАС характеризуются дислипидемией, изменением обмена липидов следующих классов: фосфолипидов [17], триглицеридов, жирных кислот [18], а также сфинголипидов (СФЛ), которые необходимы для развития и поддержания функциональной целостности нервной ткани [19–22]. Класс СФЛ чрезвычайно разнообразен и включает сотни молекул, которые служат как основными компонентами мембран, так и ключевыми регуляторами ряда важнейших клеточных процессов: пролиферации и дифференцировки, адгезии и миграции, межклеточного взаимодействия, вне- и внутриклеточной передачи сигналов, воспаления и старения, мито- и аутофагии, апоптоза, некроптоза и др. [23–27]. Особую роль в метаболизме СФЛ играет церамид (ЦЕР) [28], выступающий гидрофобной основой для других подклассов СФЛ: сфингомиелинов (СМ), цереброзидов, ганглиозидов, сульфатидов и др. Образование и деградация ЦЕР строго регулируется несколькими десятками ферментов, каждый из которых обладает уникальными свойствами и разнообразным распределением в тканях и органеллах [19, 28, 29] (рис. 1).

Согласно большинству данных литературы, в том числе нашим ранее полученным данным, повышение уровня ЦЕР в основном рассматривается как ключевое цитотоксическое и/или проапоптотическое событие [30]. О возможности изменений спектра ЦЕР и СМ при БАС впервые сообщили Cutler et al. в 2002 г., которые продемонстрировали повышенное содержание двух молекулярных видов ЦЕР и одного молекулярного вида СМ в поясничном отделе спинного мозга больных спорадической формой БАС и у трансгенных мышей линии SOD1G93A – модели семейной формы БАС [31]. Затем Dodge et al. показали, что уровень ЦЕР и СМ и активность ферментов их метаболизма у мышей SOD1G93A может определяться стадией развития БАС [20].
Участие CФЛ в патогенезе БАС в настоящее время изучается только несколькими группами исследователей и только на моделях мышей, несущих мутации в гене SOD [20, 31–33]. В наших экспериментах использовалась принципиально другая модель, в которой ключевые признаки патогенеза БАС человека имитировались мутациями в гене FUS (так называемая FUSопатия [34]).
Цель данного исследования – определение изменений метаболизма сфингомиелина и церамида в структурах головного мозга (гиппокампе, коре и мозжечке) и в спинном мозге трансгенных мышей линии FUS(1-359), моделирующих боковой амиотрофический склероз.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Уровни СФЛ особенно высоки в нервной системе [35, 36], и изменения сфинголипидного профиля и активности ферментов метаболизма ЦЕР и СМ проявляются в патогенезе многих нейродегенеративных заболеваний: при болезнях Паркинсона [36–38], Хантингтона [35], Альцгеймера [36, 37, 39], рассеянном склерозе [26] и др. [28]. Не исключение и БАС. Дисрегуляция метаболизма СФЛ, включая СМ и ЦЕР, показана с использованием наиболее изученных моделей БАС на мышах – линиях SOD1G86R [33] и SOD1G93A [20, 31]. Мы в своей работе использовали трансгенных мышей линии FUS(1-359), моделирующих характеристики БАС человека [34]. Данный вид FUSопатии приводит к повреждению аксонов мотонейронов, нейровоспалению и снижению численности мотонейронов и периферических нервных волокон. Клиническая картина характеризуется относительно ранней манифестацией и быстрым летальным исходом заболевания. Аберрантный метаболизм ЦЕР может приводить к гибели нейронов в результате окислительного стресса и апоптоза, как у животных моделей, так и у пациентов, страдающих нейродегенеративными заболеваниями [38–41]. Структуры головного мозга мышей (гиппокамп, кора и мозжечок) и спинной мозг были выделены на разных стадиях развития БАС на сроках 2, 3 и 4 месяца у трансгенных мышей линии FUS(1‑359) и у контрольных животных соответствующего возраста.
Анализ экспрессии генов метаболизма ЦЕР и СМ в спинном мозге мышей при развитии БАС. В спинном мозге трансгенных мышей линии FUS(1-359), моделирующих характеристики БАС человека, и в контрольной группе методом РНК-секвенирования нами был изучен уровень экспрессии генов ферментов, участвующих в метаболизме ЦЕР: кислой сфингомиелиназы (a-SMase) – Smpd1, нейтральной сфингомиелиназы (nSMase) – Smpd3, сфингомиелинсинтазы-1 (SMS1) – Sgms1, сфингомиелинсинтазы-2 (SMS2) – Sgms2, кислой церамидазы (aC-Dase) – Asah1, нейтральной церамидазы (nC-Dase) – Asah2, галактозилцерамидазы (GALC) – Galc, галактозилцерамидсинтазы (GalCS) – Ugt8a и церамидсинтаз (CerS) – Lass1–6. На рис. 2–4 представлены данные по уровню экспрессии генов наиболее значимых изоформ изученных ферментов.
Рис. 2.
Экспрессия генов нейтральной (Smpd3) (а) и кислой (б) сфингомиелиназ (Smpd1) у мышей линии FUS(1-359) и мышей контрольной группы. Стандартное отклонение обозначено верхними и нижними границами усов графика, интерквартильный размах – верхней и нижней границами ящика, черными точками обозначены “выбросы”.

Рис. 3.
Экспрессия генов сфингомиелинсинтазы 1 (Sgms1) (а), сфингомиелинсинтазы 2 (Sgms2) (б) и кислой церамидазы (Asah1) (в) у мышей линии FUS(1-359) и мышей контрольной группы. Стандартное отклонение обозначено верхними и нижними границами усов графика, интерквартильный размах – верхней и нижней границами ящика, черными точками обозначены “выбросы”. *** Статистически достоверное различие с контролем, p < 0.01.

Рис. 4.
Экспрессия генов галактозилцерамидазы (Galc) (а) и галактозилтрансферазы (Ugt8a) (б) у мышей линии FUS(1‑359) и мышей контрольной группы. Стандартное отклонение обозначено верхними и нижними границами усов графика, интерквартильный размах – верхней и нижней границами ящика, черными точками обозначены “выбросы”. * Cтатистически достоверное различие с контролем, p < 0.05.

Анализ экспрессии генов SMase (кодируют ферменты, генерирующие ЦЕР из СМ) показывает, что уровень экспрессии гена Smpd3, кодирующего nSMase, практически не меняется в ходе развития БАС у мышей, а экспрессия гена Smpd1, кодирующего aSMase, через 2 мес. была ниже, чем в контроле (рис. 2). Через 3 и 4 мес. достоверных отличий от контроля не обнаружено. Из представленных данных следует, что, вероятно, в ходе развития модели БАС интенсивная генерация ЦЕР из СМ не осуществляется. Пониженная экспрессия гена фермента кислой изоформы SMase, активность которой в первую очередь связывают с реализацией цитотоксического действия ЦЕР, может указывать на запуск защитных механизмов в ЦНС трансгенных животных на досимптоматической стадии БАС.
Изменение экспрессии генов Sgms1 и Sgms2, которые кодируют ферменты SMS1 и SMS2, синтезирующие СМ из ЦЕР, идет разнонаправленно. Уровень экспрессии гена изоформы SMS1, локализованной в аппарате Гольджи, повышается через 4 мес. развития БАС по сравнению с показателями нормы, а уровень экспрессии SMS2, расположенной на плазматической мембране, напротив, понижается (рис. 3а и 3б), что может указывать на специфическую роль отдельных пулов СМ и ЦЕР в ходе развития БАС.
При анализе экспрессии генов, кодирующих различные изоформы CerS, которые отвечают за синтез ЦЕР de novo, мы не обнаружили отличий в уровнях экспрессии по сравнению с соответствующими контрольными уровнями (данные не представлены).
Наиболее интересные результаты получены для генов ферментов Asah1 (образует сфингозин из ЦЕР), GALC (ЦЕР из ГалЦЕР) и Ugt8a (Гал-ЦЕР из ЦЕР). Повышение экспрессии гена Asah1 через 4 мес. развития FUS-опосредованной протеинопатии (рис. 3в) указывает на то, что ЦЕР под действием Asah1 может превращаться в сфингозин на терминальной стадии БАС, когда интенсивно гибнут клетки спинного мозга от апоптоза, индуцируемого сфингозином. Это предположение находит подтверждение в нашей недавней работе, в которой был проведен масс-cпектрометрический анализ сфингоидных оснований на разных стадиях развития FUSопатии и обнаружено повышенное в несколько раз по сравнению с показателями контрольных животных содержание сфингозина в тканях спинного мозга [42]. Кроме того, активация экспрессии гена Galc, кодирующего GALC (фермент, ответственный за образование ЦЕР из ГалЦЕР), на этом же сроке (рис. 4 а) дает основание полагать, что в процессе развития БАС происходит генерация дополнительных количеств ЦЕР в результате ферментативной деградации ГалЦЕР этим ферментом, т.е. активируется так называемый “путь спасения” для образования ЦЕР. Важно отметить, что при этом есть указания на ингибирование обратного процесса, а именно образование ГалЦЕР из ЦЕР синтазой ГалЦЕР, поскольку экспрессия гена Ugt8a снижается (рис. 4 б). Таким образом, можно предположить, что сфингозин на поздней симптоматической стадии БАС, скорее всего, образуется из ЦЕР, в том числе возникающего при гидролизе ГалЦЕР.
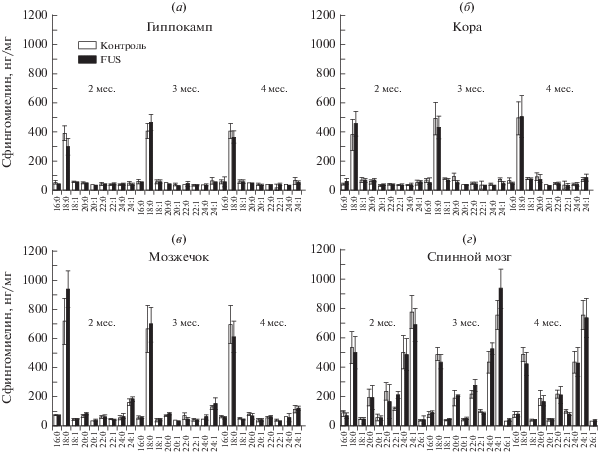
Анализ спектра молекулярных видов сфингомиелинов в гиппокампе, коре головного мозга, мозжечке и в спинном мозге в процессе развития БАС. Мы изучили уровни суммарных СМ и наиболее распространенных молекулярных видов СМ d18:1 с длиной жирной кислоты от С16 до С24 (d18:1/16:0, d18:1/18:0, d18:1/18:1, d18:1/20:0, d18:1/20:1, d18:1/22:0, d18:1/22:1, d18:1/24:0, d18:1/24:1) в гиппокампе, коре головного мозга, мозжечке и спинном мозге трансгенных мышей линии FUS(1-359), моделирующих характеристики БАС человека, и мышей контрольной группы методом высокоэффективной жидкостной хроматографии c тандемной масс-спектрометрией (ВЭЖХ/МС). Анализ суммарных СМ показал их практическую неизменность во всех изученных структурах ЦНС в процессе развития протеинопатии по сравнению с соответствующими показателями контроля (рис. 5). Общее содержание СМ практически одинаково в гиппокампе и коре головного мозга, несколько выше в мозжечке, а в спинном мозге – в несколько раз выше, чем в структурах головного мозга (рис. 5). Данное наблюдение согласуется с имеющимися представлениями о том, что миелиновые оболочки (основным компонентом которых выступает СМ) спинного мозга значительнее обогащены липидами, чем ткани головного мозга.
Рис. 5.
Общее содержание сфингомиелина в структурах ЦНС мышей линии FUS(1-359) и мышей контрольной группы. Приведены средние значения и стандартная ошибка среднего.

Среди отдельных молекулярных видов СМ в гиппокампе, коре головного мозга и мозжечке преобладает СМ d18:1/18:0 (рис. 6а–6в), что согласуется с имеющимися данными литературы о сфинголипидном составе структур головного мозга. Спинной мозг характеризуется более разнообразным в количественном выражении спектром СМ. Преобладает СМ d18:1/24:1, далее в равных соотношениях представлены СМ d18:1/18:0 и d18:1/24:0, затем d18:1/20:0 и d18:1/22:0, d18:1/16:0 и d18:1/22:1 (рис. 6г).
Рис. 6.
Содержание молекулярных видов сфингомиелина в структурах ЦНС мышей линии FUS(1-359) и мышей контрольной группы: (а) – гиппокамп, (б) – кора головного мозга, (в) – мозжечок, (г) – спинной мозг. Приведены средние значения и стандартная ошибка среднего.
Изменение содержания молекулярных видов церамидов в гиппокампе, коре головного мозга, мозжечке и в спинном мозге в процессе развития БАС. В наших экспериментах показано, что у мышей линии FUS(1-359) отсутствуют достоверные изменения суммарного уровня всех изученных нами молекулярных видов ЦЕР в отделах головного мозга (гиппокампе, коре и мозжечке) по сравнению с соответствующим значениями у мышей контрольной группы (рис. 7). Однако в спинном мозге наблюдается понижение уровня суммарных ЦЕР через 2 мес. после индукции БАС по сравнению с показателями контроля (рис. 7). Подобно СМ, мы исследовали изменения в уровнях молекулярных видов ЦЕР с аналогичными жирными кислотами в их составе: d18:1/16:0, d18:1/18:0, d18:1/18:1, d18:1/20:0, d18:1/20:1, d18:1/22:0, d18:1/22:1, d18:1/24:0, d18:1/24:1 (рис. 8а–8г).
Рис. 7.
Общее содержание церамида в структурах ЦНС мышей линии FUS(1-359) и мышей контрольной группы. Приведены средние значения и стандартная ошибка среднего. * Cтатистически достоверное различие с контролем, p < 0.05.

Рис. 8.
Содержание молекулярных видов ЦЕР в структурах ЦНС мышей линии FUS(1-359) и мышей контрольной группы: (а) – гиппокамп, (б) – кора головного мозга, (в) – мозжечок, (г) – спинной мозг. Приведены средние значения и стандартная ошибка среднего.
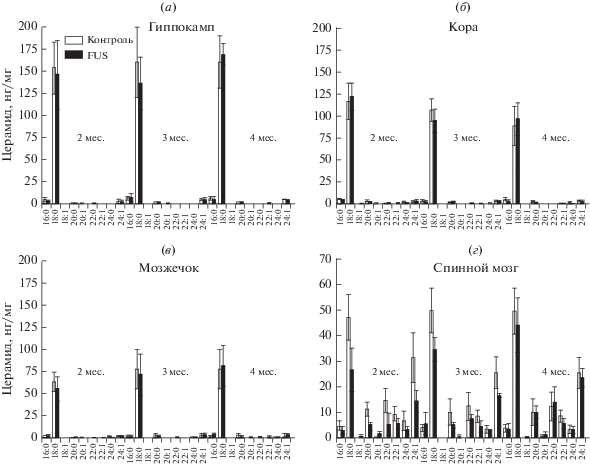
В структурах головного мозга наблюдается преобладание ЦЕР d18:1/18:0 – основного молекулярного вида, характерного для нейронов. В спинном мозге, где более выражено содержание миелина, преобладает также ЦЕР d18:1/24:1 и выше процентное содержание остальных молекулярных видов ЦЕР. В процессе развития заболевания изменения в спектре молекулярных видов ЦЕР отмечены только в спинном мозге, где уровень практически всех ЦЕР снижается по отношению к контролю через 2 и 3 мес. течения заболевания. На четвертый месяц, так же как и для суммарных ЦЕР на этот срок, их уровень соответствует контрольному (рис. 8г).
Подобное поведение ЦЕР может определять его превращение в сфингозин путем гидролиза ЦЕР Asah1. Сфингозин обладает более выраженными проапоптотическими свойствами по сравнению с ЦЕР, его появление в клетках ЦНС способствует более активной их гибели.
Обсуждение результатов. Эксперименты по определению уровня экспрессии генов ферментов, катализирующих как образование, так и расщепление СМ, не показали существенных изменений, что согласуется с неизменным содержанием СМ и спектра его индивидуальных молекулярных видов на всех изученных нами сроках БАС. Также при FUSопатии не отмечено изменений уровня мРНК ни одной из изоформ CerS (Lass1–6). Ранее мы показали неизменность экспрессии гена еще одного фермента, вовлеченного в процесс синтез ЦЕР de novo, – 3-кетодигидросфингозинредуктазы – у FUS-трансгенных мышей на всех изученных сроках [43]. Полученные нами данные указывают на то, что синтез ЦЕР при БАС происходит не за счет образования de novo, а по катаболическим путям. К аналогичному заключению пришли Dodge et al., продемонстрировавшие это на модели БАС SOD1G93A у мышей [20]. Образование ЦЕР может происходить в результате повышения экспрессии гена фермента GALC, генерирующего ЦЕР из Гал-ЦЕР, т.к. изученные нами образцы характеризуются повышенной экспрессией гена Galc. На этом фоне ингибируется обратный процесс, а именно образование ГалЦЕР из ЦЕР синтазой ГалЦЕР, поскольку экспрессия гена Ugt8a снижается. Предположение о роли ГалЦЕР в качестве источника для поддержания уровня ЦЕР согласуется и с некоторыми результатами работы Dodge et al. в части, проведенной на образцах серого вещества спинного мозга пациентов с БАС, у которых обнаруживается повышение активности ферментов, генерирующих ЦЕР из гексозилцерамидов, GALC и GBA2 [20].
Также нами обнаружено увеличение экспрессии гена Asah1, кодирующего фермент, который превращает ЦЕР в еще более активный проапоптотический агент – сфингозин. В своей недавней работе, проведенной на той же модели мышей линии FUS(1-359), мы исследовали аспекты метаболизма сфингоидных оснований при БАС и показали, что активация экспрессии генов ферментов обмена сфингозина и повышенное содержание этого проапоптотического метаболита сфингомиелинового цикла обнаруживаются на терминальной стадии FUSопатии в спинном мозге мышей [43]. Таким образом, можно предположить, что ЦЕР, образующийся вследствие ферментативной деградации ГалЦЕР, не накапливается при патологии БАС у мышей, а превращается в сфингозин, который приводит к гибели клеток спинного мозга.
Один из аспектов передачи сигналов ЦЕР в клетках – регуляция гибели клеток посредством формирования каналов ЦЕР во внешних мембранах митохондрий [44]. Различие метаболических профилей митохондрий в спинном и головном мозге может объяснять различия в характере изменений сфинголипидного профиля в головном и спинном мозге. Различие метаболических профилей митохондрий в спинном и головном мозге проявляется в том, что митохондрии в спинном мозге производят больше АФК и имеют более выраженные окислительные повреждения по сравнению с митохондриями головного мозга. Эти тканеспецифические отличия могут быть причиной того, что определенные области ЦНС особенно уязвимы, а также могут играть важную роль в специфических возрастных и нейродегенеративных заболеваниях, таких как БАС [45]. Более того, митохондриальная функция, по-видимому, неодинакова не только в клетках разных типов тканей, но даже в разных областях клеток одного типа, например, в синаптических и несинаптических митохондриях [46], а с возрастом эти различия увеличиваются еще больше [45, 47]. Также в спинном мозге значительно выше повреждения митохондриальной ДНК, чем в головном мозге [48]. Это может усиливать характерные для БАС нарушения в работе митохондрий из-за повышения вероятности мутаций белков, кодируемых митохондриальной ДНК и, как следствие, их дисфункции [45].
Изменения уровня СФЛ могут различаться в зависимости от области отдела мозга. Так, при болезни Паркинсона показано, что в передней части поясной извилины коры головного мозга содержание СМ понижено, а в затылочной коре изменений нет. Кроме этого, в поясной коре уровни СМ С23:0, С24:1 и С26:1 снижены, а концентрации С18:1 и С20:0 увеличены [49]. Таким образом, наблюдаемое нами отсутствие принципиальных изменений в исследованных структурах головного мозга может отражать как принципиальное отличие в их метаболизме от спинного мозга, так и необходимость более детального разделения тканей для выявления изменений, которые могут быть характерны для более локальных участков и нивелируются при анализе липидов, выделенных из более крупных образцов. При анализе данных следует также принимать во внимание тот факт, что сфинголипидный состав как отдельных органов, так и биологических жидкостей различен, и он отличается у разных организмов [20]. С учетом роли СФЛ в патогенезе БАС ведется поиск новых препаратов, влияющих на метаболизм СФЛ. В настоящее время клинические испытания безопасности фазы IIа для лечения БАС проходит единственный препарат из класса СФЛ – Fingolimod [50], эффективность которого доказана у мышей SOD1G93A на симптоматической стадии БАС при диагностируемой двигательной дисфункции [32].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Модель БАС. В качестве модели БАС использовали линию трансгенных мышей FUS(1-359) (Биоресурсная коллекция Института физиологически активных веществ РАН, Россия), проявляющих характеристики БАС. В нервной системе животных экспрессируется трансгенная последовательность, кодирующая аберрантную форму белка FUS человека с удаленным сигналом ядерной локализации под нейрон-специфичным промотором Thy1. Линию стабильно поддерживали в гемизиготном состоянии согласно ранее описанному протоколу с переводом животных на генетический фон CD1 [34]. Гетерозиготных животных определяли методом конвенционной ПЦР с использованием праймеров (5′-TCTTTGTGCAAGGCCTGGGT-3′ и 3′-AGAAGCAAGACCTCTGCAGAG-5′) при следующих условиях: 2 мин предварительной денатурации при 94°C, далее 30 циклов: 94°C – 15 с, 58°C – 30 с, 72°C – 40 с, детектируя фрагмент размером 255 п.н. [34]. У трансгенных мышей накапливаются белковые агрегаты в цитоплазме нервных клеток. Данный вид FUS-протеинопатии приводит к повреждению аксонов мотонейронов, нейровоспалительным реакциям и снижению числа тел мотонейронов. Такая клиническая картина характеризуется относительно ранней манифестацией заболевания (2.5–4.5 мес.) и быстрым летальным исходом (в течение нескольких дней с момента манифестации). Изменения метаболического и стрессового ответа предшествуют началу открытого моторного заболевания БАС [51]. Поэтому у модельных мышей выделяли структуры головного мозга (гиппокамп, кору, мозжечок) и спинной мозг в 2, 3 и 4 мес. жизни, что соответствует досимптомной, ранней симптоматической и симптоматической стадиям БАС. Для одной экспериментальной точки использовали семь мышей-самцов. В контрольную группу входили не содержащие трансгенную кассету однопометные особи того же возраста, что и модельные мыши.
Анализ экспрессии генов. Анализ экспрессии генов ферментов метаболизма сфинголипидов в спинном мозге мышей проводили методом РНК-секвенирования. Библиотеки мРНК для секвенирования получали с помощью набора для выделения полиА(+)-фракции мРНК и набора для создания библиотек NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Bi-olabs, США) по методике, предложенной производителем [52]. Cеквенирование полиА(+)-обогащенной фракции РНК проводили на платформе Illumina NextSeq 500 (Illumina, США) в Центре коллективного пользования “Геном” Института молекулярной биологии им. В.А. Энгельгардта РАН.
Обработку данных проводили при помощи программного обеспечения PPLine [53]. Она включала триммирование коротких прочтений по качеству и длине при помощи программы Trimmomatic [54], выравнивание прочтений на референсный геном версии GRCm38 программой STAR [55] и подсчет прочтений, выравненных на гены, программой htseq-count [56].
Статистическую обработку данных и вычисление дифференциальной экспрессии генов производили при помощи пакета edgeR [57], используемого в среде статистической обработки данных языка R [58]. В ходе парного сравнения экспериментальных групп для определения достоверности дифференциальной экспреcсии использовали стандартный F-тест Фишера с применением метода Беньямини–Хохберга для внесения поправки на множественное тестирование [59]. Для визуализации полученных данных применяли пакет ggplot2 [60].
Анализ сфинголипидов. Липиды отделов головного (кора, мозжечок и гиппокам) и спинного мозга экстрагировали по методу Bligh и Dyer [61]. Церамиды и сфингомиелины анализировали методом хромато-масс-спектрометрии [62]. Выделенные липиды упаривали под током азота и растворяли в смеси хлороформ/метанол в соотношении 2 : 1 (v/v). Анализ липидов проводили методом ВЭЖХ/МС на приборе TSQ Endura (Thermo Scientific, США), снабженном ВЭЖХ-колонкой EclipsePlusC8 (Agilent, США), в режиме мониторинга множественных реакции (ММР) при давлении в ячейке соударений 1.5 мТорр, разрешение на Q1 и Q3 1.2 Да. Для ЦЕР проводили фрагментацию исходных протонированных и дегидратированных молекул при энергии 25 В до иона с m/z 264.4, dwell time 25 мс. Для СМ проводили фрагментацию исходных протонированных молекул при энергии 25 В до иона с m/z 184.1, dwell time 25 мс. Параметры источника ионизации: температура нагревателя 300°С, температура капилляра 340°С, поток sheath gas 45 arb, поток aux gas 13 arb, поток sweep gas 1 arb. Для построения калибровочных зависимостей использовали СМ и ЦЕР d18:1/16:0, d18:1/18:0, d18:1/18:1, d18:1/20:0, d18:1/22:0, d18:1/24:0 и d18:1/24 (Avanti, США). Для каждого липида вычисляли отношение площади под хроматографическим пиком этой молекулы к площади хроматографического пика молекулы стандарта.
ЗАКЛЮЧЕНИЕ
Впервые охарактеризован профиль молекулярных видов СМ и ЦЕР, а также уровни экспрессии генов ферментов их метаболизма в ЦНС трансгенных мышей линии FUS(1-359), моделирующих характеристики БАС на разных сроках развития FUS-протеинопатии. Показано, что изменения содержания ЦЕР и экспрессии генов ферментов его обмена происходят в спинном мозге животных на всех стадиях БАС. Таким образом, метаболизм СФЛ представляет собой перспективную область исследования процессов, связанных с БАС, как в контексте поиска потенциальных диагностических маркеров и эффективных лекарственных средств, так и для объяснения патогенеза БАС.
Список литературы
Talbott E.O., Malek A.M., Lacomis D. // Handb. Clin. Neurol. 2016. V. 138. P. 225–238. https://doi.org/10.1016/B978-0-12-802973-2.00013-6
Arthur K.C., Calvo A., Price T.R., Geiger J.T., Chiò A., Traynor B.J. // Nat. Commun. 2016. V. 7. P. 12408. https://doi.org/10.1038/ncomms12408
Chia R., Chiò A., Traynor B.J. // Lancet Neurol. 2018. V. 17. P. 94–102. https://doi.org/10.1016/S1474-4422(17)30401-5
Zou Z.Y., Zhou Z.R., Che C.H., Liu C.Y., He R.L., Huang H.P.J. // Neurol. Neurosurg. Psychiatry. 2017. V. 88. P. 540–549. https://doi.org/10.1136/jnnp-2016-315018
Shi P., Gal J., Kwinter D.M., Liu X., Zhu H. // Biochim. Biophys. Acta. 2010. V. 1802. P. 45–51. https://doi.org/10.1016/j.bbadis.2009.08.012
Peters O.M., Ghasemi M., Brown R.H., Jr. // J. Clin. Invest. 2015. V. 125. P. 1767–1779. https://doi.org/10.1172/JCI71601
Van Damme P., Dewil M., Robberecht W., Van Den Bosch L. // Neurodegener Dis. 2005. V. 2. P. 147–159. https://doi.org/10.1159/000089620
Chitnis T., Weiner H.L. // J. Clin. Invest. 2017. V. 127. P. 3577–3587. https://doi.org/10.1172/JCI90609
Hardiman O., Al-Chalabi A., Chio A., Corr E.M., Logroscino G., Robberecht W., Shaw P.J., Simmons Z., van den Berg L.H. // Nat. Rev. Dis. Primers. 2017. V. 3. P. 17085. https://doi.org/10.1038/nrdp.2017.85
van Damme P., Robberecht W., Van Den Bosch L. // Dis. Model Mech. 2017. V. 10. P. 537–549. https://doi.org/10.1242/dmm.029058
Van Den Bosch L. // J. Biomed. Biotechnol. 2011. V. 2011. P. 348765. https://doi.org/10.1155/2011/348765
Naumann M., Pal A., Goswami A., Lojewski X., Japtok J., Vehlow A., Naujock M., Günther R., Jin M., Stanslowsky N., Reinhardt P., Sterneckert J., Frickenhaus M., Pan-Montojo F., Storkebaum E., Poser I., Freischmidt A., Weishaupt J.H., Holzmann K., Troost D., Ludolph A.C., Boeckers T.M., Liebau S., Petri S., Cordes N., Hyman A.A., Wegner F., Grill S.W., Weis J., Storch A., Hermann A. // Nat. Commun. 2018. V. 9. P. 335. https://doi.org/10.1038/s41467-017-02299-1
Vance C., Rogelj B., Hortobágyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K.L., Tripathi V., Al-Saraj S., Al-Chalabi A., Leigh P.N., Blair I.P., Nicholson G., de Belleroche J., Gallo J.M., Miller C.C., Shaw C.E. // Science. 2009. V. 323. P. 1208–1211. https://doi.org/10.1126/science.1165942
Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., Valdmanis P., Rouleau G.A., Hosler B.A., Cortelli P., de Jong P.J., Yoshinaga Y., Haines J.L., Pericak-Vance M.A., Yan J., Ticozzi N., Siddique T., McKenna-Yasek D., Sapp P.C., Horvitz H.R., Landers J.E., Brown R.H., Jr. // Science. 2009. V. 323. P. 1205–1208. https://doi.org/10.1126/science.1166066
Conlon E.G., Manley J.L. // Genes Dev. 2017. V. 31. P. 1509–1528. https://doi.org/10.1101/gad.304055.117
Скворцова В.И., Мальцев А.В., Григорьев В.В., Овчинников Р.К., Роман А.Ю., Ефимова А.Д., Ковражкина Е.А. // Мол. биология. 2017. Т. 51. С. 387–399. https://doi.org/10.7868/S0026898417020094
Arima H., Omura T., Hayasaka T., Masaki N., Hanada M., Xu D., Banno T., Kobayashi K., Takeuchi H., Kadomatsu K., Matsuyama Y., Setou M. // Neuroscience. 2015. V. 297. P. 127–136. https://doi.org/10.1016/j.neuroscience.2015.03.060
Fitzgerald K.C., O’Reilly É.J., Falcone G.J., Mc-Cullough M.L., Park Y., Kolonel L.N., Ascherio A. // JAMA Neurol. 2014. V. 71. P. 1102–1110. https://doi.org/10.1001/jamaneurol.2014.1214
Blasco H., Veyrat-Durebex C., Bocca C., Patin F., Vourc’h P., Kouassi Nzoughet J., Lenaers G., Andres C.R., Simard G., Corcia P., Reynier P. // Sci. Rep. 2017. V. 7. P. 17652. https://doi.org/10.1038/s41598-017-17389-9
Dodge J.C., Treleaven C.M., Pacheco J., Cooper S., Bao C., Abraham M., Cromwell M., Sardi S.P., Chuang W.L., Sidman R.L., Cheng S.H., Shihabuddin L.S. // Proc. Nat. Acad. Sci. USA. 2015. V. 112. P. 8100–8105. https://doi.org/10.1042/bst0320144
van Echten-Deckert G., Herget T. // Biochim. Biophys. Acta. 2006. V. 1758. P. 1978–1994. https://doi.org/10.1016/j.bbamem.2006.06.009
Piccinini M., Scandroglio F., Prioni S., Buccinnà B., Loberto N., Aureli M., Chigorno V., Lupino E., DeMarco G., Lomartire A., Rinaudo M.T., Sonnino S., Prinetti A. // Mol. Neurobiol. 2010. V. 41. P. 314–340. https://doi.org/10.1007/s12035-009-8096-6
Hannun Y.A., Obeid L.M. // Nat. Rev. Mol. Cell Biol. 2008. V. 9. P. 139–150. https://doi.org/10.1038/nrm2329
Hannun Y.A., Obeid L.M. // Nat. Rev. Mol. Cell Biol. 2018. V. 19. P. 175–191. https://doi.org/10.1038/nrm.2017.107
Nganga R., Oleinik N., Ogretmen B. // Adv. Cancer Res. 2018. V. 140. P. 1–25. https://doi.org/10.1016/bs.acr.2018.04.007
Chaurasia B., Summers S.A. // Annu. Rev. Physiol. 2021. V. 83. P. 303–330. https://doi.org/.org/10.1146/annurev-physiol-031620-093815
Zhang X., Matsuda M., Yaegashi N., Nabe T., Kitatani K. // Cells. 2020. V. 9. P. 627. https://doi.org/10.3390/cells9030627
Mullen T.D., Hannun Y.A., Obeid L.M. // Biochem. J. 2012. V. 441. P. 789–802. https://doi.org/10.1042/bj20111626
Zelnik I.D., Ventura A.E., Kim J.L., Silva L.C., Futerman A.H. // Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2020. V. 1865. P. 158489. https://doi.org/10.1016/j.bbalip.2019.06.015
Futerman A.H., Hannun Y.A. // EMBO Rep. 2004. V. 5. P. 777–782. https://doi.org/10.1038/sj.embor.7400208
Cutler R.G., Pedersen W.A., Camandola S., Rothstein J.D., Mattson M.P. // Ann. Neurol. 2002. V. 52. P. 448–457. https://doi.org/10.1002/ana.10312
Potenza R.L., De Simone R., Armida M., Mazziotti V., Pèzzola A., Popoli P., Minghetti L. // Neurotherapeutics. 2016. V. 13. P. 918–927. https://doi.org/10.1007/s13311-016-0462-2
Henriques A., Croixmarie V., Priestman D.A., Rosenbohm A., Dirrig-Grosch S., D’Ambra E., Huebecker M., Hussain G., Boursier-Neyret C., Echaniz-Laguna A., Ludolph A.C., Platt F.M., Walther B., Spedding M., Loeffler J.P., Gonzalez De Aguilar J.L. // Hum. Mol. Genet. 2015. V. 24. P. 7390–7405. https://doi.org/10.1093/hmg/ddv439
Дейкин А.В., Ковражкина Е.А., Овчинников Р.К., Броновицкий Е.В., Разинская О.Д., Смирнов А.П., Ермолкевич Т.Г., Еляков А.Б., Попов А.Н., Федоров Е.Н., Лыткина О.А., Кухарский М.С., Тарасова Т.В., Шелковникова Т.А., Устюгов А.А., Нинкина Н.Н., Гольдман И.Л., Садчиков Е.Р., Бачурин С.О., Скворцо-ва В.И. // Журнал неврологии и психиатрии им. С.С. Корсакова. 2014. Т. 114. С. 63–70.
Giusto N.M., Roque M.E., Ilincheta De Boschero M.G. // Li-pids. 1992. V. 27. P. 835–839. https://doi.org/10.1007/BF02535859
Di Pardo A., Basit A., Armirotti A., Amico E., Castaldo S., Pepe G., Marracino F., Buttari F., Digilio A.F., Maglione V. // Front Neurosci. 2017. V. 11. P. 698. https://doi.org/10.3389/fnins.2017.00698
Lin G., Wang L., Marcogliese P.C., Bellen H.J. // Trends Endocrinol. Metab. 2019. V. 30. P. 106–117. https://doi.org/10.1016/j.tem.2018.11.003
Czubowicz K., Jęśko H., Wencel P., Lukiw W.J., Strosznajder R.P. // Mol. Neurobiol. 2019. V. 56. P. 5436–5455. https://doi.org/10.1007/s12035-018-1448-3
Brugg B., Michel P.P., Agid Y., Ruberg M. // J. Neurochem. 1996. V. 66. P. 733–739. https://doi.org/10.1046/j.1471-4159.1996.66020733.x
France-Lanord V., Brugg B., Michel P.P., Agid Y., Ruberg M. // J. Neurochem. 1997. V. 69. P. 1612–1621. https://doi.org/10.1046/j.1471-4159.1997.69041612.x
Bras J., Singleton A., Cookson M.R., Hardy J. // FEBS J. 2008. V. 275. P. 5767–5773. https://doi.org/10.1111/j.1742-4658.2008.06709.x
Гутнер У.А., Шупик М.А., Малошицкая О.А., Соколов С.А., Резвых А.П., Фуников С.Ю., Лебедев А.Т., Устюгов А.А., Алесенко А.В. // Биохимия. 2019. Т. 84. С. 1437–1449. https://doi.org/10.1134/S0320972519100063
Filippov V., Song M.A., Zhang K., Vinters H.V., Tung S., Kirsch W.M., Yang J., Duerksen-Hughes P.J. // J. Alzheimer’s Dis. 2012. V. 29. P. 537–547. https://doi.org/10.3233/JAD-2011-111202
Siskind L.J., Kolesnick R.N., Colombini M. // J. Biol. Chem. 2002. V. 277. P. 26796–267803. https://doi.org/10.1074/jbc.M200754200
Yonutas H.M., Pandya J.D., Sullivan P.G. // J. Bio-e-nerg. Biomembr. 2015. V. 47. P. 149–154. https://doi.org/10.1007/s10863-014-9593-5
Brown M.R., Sullivan P.G., Geddes J.W. // J. Biol. Chem. 2006. V. 281. P. 11658–11668. https://doi.org/10.1074/jbc.M510303200
Brown M.R., Geddes J.W., Sullivan P.G. // J. Bioenerg. Biomembr. 2004. V. 36. P. 401–406. https://doi.org/10.1023/B:JOBB.0000041775.10388.23
Sullivan P.G., Rabchevsky A.G., Keller J.N., Lovell M., Sodhi A., Hart R.P., Scheff S.W. // J. Comp. Neurol. 2004. V. 474. P. 524–534. https://doi.org/10.1002/cne.20130
Bienias K., Fiedorowicz A., Sadowska A., Prokopiuk S., Car H. // Pharmacol. Rep. 2016. V. 668. P. 570–581. https://doi.org/10.1016/j.pharep.2015.12.008
Berry J.D., Paganoni S., Atassi N., Macklin E.A., Goyal N., Rivner M., Simpson E., Appel S., Grasso D.L., Mejia N.I., Mateen F., Gill A., Vieira F., Tassinari V., Perrin S. // Muscle Nerve. 2017. V. 56. P. 1077–1084. https://doi.org/10.1002/mus.25733
Pharaoh G., Sataranatarajan K., Street K., Hill S., Gregston J., Ahn B., Kinter C., Kinter M., Van Remmen H. // Front. Neurosci. 2019. V. 13. P. 487. https://doi.org/10.3389/fnins.2019.00487
Funikov S.Y., Rezvykh A.P., Mazin P.V., Morozov A.V., Maltsev A.V., Chicheva M.M., Vikhareva E.A., Evgen’ev M.B., Ustyugov A.A. // Neurogenetics. 2018. V. 19. P. 189–204. https://doi.org/10.1007/s10048-018-0553-9
Krasnov G.S., Dmitriev A.A., Kudryavtseva A.V., Shargunov A.V., Karpov D.S., Uroshlev L.A., Melnikova N.V., Blinov V.M., Poverennaya E.V., Archacov A.I., Lisitsa A.V., Ponomarenko E.A. // J. Proteome Res. 2015. V. 14. P. 3729–3737. https://doi.org/10.1021/acs.jproteome.5b00490
Bolger A.M., Lohse M., Usadel B. // Bioinformatics. 2014. V. 30. P. 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Dobin A., Davis C.A., Schlesinger F., Drenkow J., Za-leski C., Jha S., Batut P., Chaisson M., Gingeras T.R. // Bioinformatics. 2013. V. 29. P. 15–21. https://doi.org/10.1093/bioinfor-matics/bts635
Anders S., Pyl P.T., Huber W. // Bioinformatics. 2015. V. 31. P. 166–169. https://doi.org/10.1101/002824
Robinson M.D., McCarthy D.J., Smyth G.K. // Bioinformatics. 2010. V. 26. P. 139–140. https://doi.org/10.1093/bioinformatics/btp616
RC Team // R Language Definition, R Foundation for Statistical Computing, 2000. Vienna, Austria.
Thissen D., Steinberg L., Kuang D. // J. Educ. Behav. Stat. 2002. V. 27. P. 77–83. https://doi.org/10.3102/10769986027001077
Wickham H. // Wiley Interdisciplinary Reviews. Comput. Statistics. 2011. V. 3. P. 180–185. https://doi.org/10.1002/wics.147
Bligh T.G., Dyer W.J. // Can. J. Biochem. Physiol. 1959. V. 37. P. 911–917. https://doi.org/10.1139/o59-099
Sullards M.C. // Methods Enzymol. 2000. V. 312. P. 32–45. https://doi.org/10.1016/S0076-6879(00)12898-8
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия