

Биоорганическая химия, 2022, T. 48, № 4, стр. 381-414
АТР-зависимые Lon-протеазы в системе контроля качества клеточных белков
А. М. Куджаев 1, А. Г. Андрианова 1, А. Е. Гущина 2, И. В. Смирнов 1, Т. В. Ротанова 1, *
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 Protein Structure Section, Center for Structural Biology, National Cancer Institute
MD 21702 Frederick, USA
* E-mail: tatyana.rotanova@ibch.ru
Поступила в редакцию 09.11.2021
После доработки 19.11.2021
Принята к публикации 24.11.2021
- EDN: VQROGR
- DOI: 10.31857/S0132342322040133
Аннотация
Система контроля качества белков (СКК) играет ведущую роль в поддержании сохранности клеточного протеома во всех природных царствах. В обзоре обобщены сведения о строении и структурных особенностях молекулярных шаперонов и энергозависимых протеаз, формирующих СКК, показана важная роль белков, принадлежащих к ААА+-суперсемейству, дана развернутая характеристика АТР-зависимых Lon-протеаз как особого семейства в СКК, обсуждены различия между ферментами отдельных подсемейств Lon-протеаз, приведены современные данные о структуре и уникальных механизмах функционирования ААА+-белков – представителей СКК.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ......................................................381
Система контроля качества белков в клетках бактерий и эукариот............................382
Молекулярные шапероны системы контроля качества белков..................................................382
Внутриклеточные АТР-зависимые протеазы и мультисубъединичные протеолитические комплексы.............................................................387
Lon-протеазы как особое семейство в системе контроля качества клеточных белков......................395
Общая характеристика семейства Lon. Подсемейства Lon-протеаз...............................395
LonA-протеазы – уникальный подкласс ААА+-белков.......................................................396
Cовременные представления о структурных особенностях и механизмах функциони- рования LonA-протеаз и других AAA+-белков системы контроля качества...............................401
Спиральная организация гексамеров ААА+-белков – базовое условие транслокации белковых мишеней/субстратов...........................401
Крио-ЭM-структуры EcLon и других LonA-протеаз.....................................................403
ЗАКЛЮЧЕНИЕ................................................408
СПИСОК ЛИТЕРАТУРЫ................................409
ВВЕДЕНИЕ
Поддержание гомеостаза клеточных белков имеет первостепенное значение для роста и выживания клеток. Существование внутриклеточных белков в нативном состоянии – необходимое условие для выполнения клеткой самых разнообразных функций. Однако вследствие возможных ошибок в транскрипции и трансляции, мутаций генома и различных стрессовых воздействий в клетках накапливаются поврежденные и мутантные белки, а также белки с нарушенной структурой, которые не только утрачивают функции, свойственные нативным белкам, но и могут формировать токсичные для клетки неупорядоченные агрегаты. Известно также, что до 30% вновь синтезированных белков никогда не достигают нативного состояния, что создает серьезную угрозу функционированию и жизнеспособности клеток. Кроме того, нарушение сворачивания (фолдинга) клеточных белков и их агрегация – основные причины развития как возрастных дегенеративных заболеваний, так и различных других патологий.
Для преодоления нарушений протеостаза в клетках сформировались мощные сети системы контроля качества (СКК) белков, которая играет ведущую роль в поддержании сохранности клеточного протеома во всех природных царствах. Семейство АТР-зависимых Lon-протеаз – один из ключевых участников СКК.
СИСТЕМА КОНТРОЛЯ КАЧЕСТВА БЕЛКОВ В КЛЕТКАХ БАКТЕРИЙ И ЭУКАРИОТ
Система контроля качества внутриклеточных белков состоит из молекулярных шаперонов и энергозависимых протеаз (рис. 1) [1–3]. Компоненты СКК контролируют сворачивание вновь синтезированных полипептидов и их сборку в функциональные комплексы, обеспечивают распознавание и высокоэффективное связывание белков с нарушенной структурой и их последующий рефолдинг, а также осуществляют выборочное удаление из клетки избыточных и поврежденных белков путем их деградации энергозависимыми протеазами (в бактериях, а также в митохондриях и хлоропластах эукариот) или мультикомпонентной убиквитин-протеасомной системой (в цитозоле эукариот) [4–7].
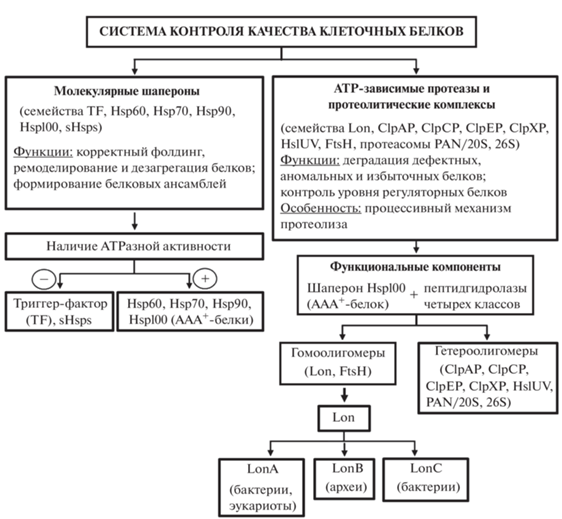
Молекулярные шапероны системы контроля качества белков
Сообщество молекулярных шаперонов СКК [2, 8, 9] наряду с триггер-фактором (TF, 48 кДа), связанным с рибосомой и участвующим в котрансляционном сворачивании новосинтезированных белков [10], включает целый ряд семейств белков теплового шока (Heat shock proteins, Hsps), различающихся молекулярными массами (от 12–43 кДа у sHsps (small Hsps, малые белки теплового шока) до 100 кДа у белков семейства Hsp100) (рис. 1) [2, 9, 11–15]. При этом белки Hsp60, Hsp70, Hsp90 и Hsp100 (табл. 1) обладают АТРазной активностью, что определяет динамический характер их существования и обеспечивает высокую конформационную гибкость. Условно молекулярные шапероны разделяются на группы фолдеров, холдеров и анфолдеров [16]. Фолдеры (семейства Hsp60, Hsp70 и Hsp90) участвуют в сворачивании белковых мишеней, предотвращая нежелательные взаимодействия между белками, еще не достигшими нативного состояния; холдеры (sHsps) удерживают частично фолдированные мишени, не влияя на их конформацию; анфолдеры (Hsp100), обладающие транслоказной активностью, разворачивают белковые субстраты [9, 16]. Для большинства семейств шаперонов получены данные рентгеноструктурного анализа их представителей [17].
Таблица 1.
Системы АТР-зависимых шаперонов СКК (по данным работы Saibil [13])
| Шапероны | Кофакторы | Основные функции |
|---|---|---|
| Система Hsp60 (шаперонины) | ||
| GroEL (Escherichia coli), HSP60/CPN60, CCT
(млекопитающие), thermosome (археи) |
GroES, HSP10/CPN10, prefoldin | Фолдинг и предотвращение агрегации белков |
| Система Hsp70 | ||
| DnaK (E. coli), HSP70, Ssa, Ssb (Saccharomyces cerevisiae), BiP/GRP78 (млекопитающие, растения) |
DnaJ, GrpE, NEF, HSP40, Sis1, Hdj1, Hsp110, Grp170, Sil1 | Разворачивание, дезагрегация и фолдинг белков, стабилизация полипептидов, регуляция ответа на тепловой шок, отбор субстратов для деградации |
| Система Hsp90 | ||
| HptG (E. coli), GRP94, TRAP1 (эукариоты), Hsc82, Hsp82 (дрожжи) |
HOP, p50, AHA1, AIP, FKPB52, p23, UNC45 | Связывание, стабилизация и созревание стероидных рецепторов и протеинкиназ, регуляция отбора и превращений субстратов, сборка миозина |
| Система Hsp100 (ААА+-белки) | ||
| ClpA, ClpB, ClpC, ClpE, ClpX, HslU (бактерии, эукариоты), PAN (археи), p97, Hsp104, Hsp101, Hsp78, RPT1–RPT6 (эукариоты) | DnaK/HSP70, ClpS | Разворачивание и ремоделирование белков, термотолерантность, солюбилизация белковых агрегатов |
Высококонсервативное семейство шаперонов Hsp70, участвующее в большом количестве разнообразных биологических процессов, сформировано, главным образом, каноническими белками DnaK у бактерий и подобными им белками Hsp70 у эукариот [12, 18]. Сверх того, эукариоты содержат также ряд неканонических шаперонов Hsp70, таких как белки Grp78, Grp170 и Hsp110 [19]. Представители семейства локализованы как в цитозоле, так и в различных органеллах (митохондрии, эндоплазматический ретикулум, хлоропласты) [12, 18].
В физиологических условиях шапероны Hsp70 способствуют фолдингу белковых мишеней, а при стрессе они предотвращают агрегацию развернутых белков и, кроме того, совместно с шаперонами семейства Hsp100 проводят солюбилизацию и рефолдинг агрегированных белков [20]. В структуре Hsp70 различают три домена: N-концевой АТРазный, центральный субстрат-связывающий и вариабельный С-концевой, который участвует как в связывании мишеней, так и во взаимодействии с различными партнерскими белками и модулировании шапероновой функции [9, 18].
Функционирование Hsp70 основано на изменении сродства шаперона к белковым мишеням в зависимости от природы связанного нуклеотида – АТР-форма Hsp70 проявляет пониженную по сравнению с ADP-формой аффинность к белкам. Регуляторами АТРазной и шапероновой активностей Hsp70 служат кошапероны группы Hsp40/J-домен-содержащих белков и факторы нуклеотидного обмена (NEF) (табл. 1) [9, 21]. Связывание белков-мишеней с шапероном опосредуется предварительным образованием комплексов субстрат–кошаперон с участием С-концевого домена Hsp40, после чего комплекс доставляется к Hsp70. Взаимодействие АТРазного домена шаперона с N-концевым J-доменом Hsp40 стимулирует гидролиз связанного АТР, а действие фактора NEF ускоряет последующие стадии высвобождения нуклеотида (в ADP-форме) и связывания новой молекулы АТР, сопровождающиеся диссоциацией белкового субстрата.
В недавних исследованиях установлено, что и эукариотические шапероны Hsp70, и бактериальные DnaK существуют не только в мономерной форме, но и как димеры, тримеры и более высокие олигомеры. На равновесие в таких смесях влияет присутствие нуклеотида и его природа (ATP или ADP) [22].
Шапероны Hsp60 или шаперонины оказались первым идентифицированным семейством шаперонов, для которого была выявлена важная роль в биогенезе клеточных белков и которое широко распространено в бактериях и в митохондриях, пластидах и цитоплазме эукариот (табл. 1) [12, 18]. Функциональными структурами шаперонинов служат тетрадекамеры, образованные двумя гептамерными кольцами, в каждом из которых сформирована центральная полость. Эти структуры способны в своих противоположных полостях попеременно связывать и инкапсулировать белки, подлежащие рефолдингу [9].
Наиболее изученный представитель семейства Hsp60 – шаперонин GroEL из Escherichia coli, функционирующий в комплексе с кошаперонином GroES – белком теплового шока семейства Hsp10 (табл. 1) [9, 12, 18, 23, 24]. Мономеры GroEL построены из трех доменов: апикального (Ap), шарнирного промежуточного (Hinge) и С-концевого экваториального (Eq), несущего АТРазный центр [23–25]. Кольцевые гептамеры GroEL, состыкованные своими Eq-доменами, образуют зеркально симметричные тороиды с двумя изолированными гидрофобными полостями, способными в АТР-связанном состоянии размещать развернутые полипептиды размером до 60 кДа [9, 12]. Кошаперонины GroES, которые также образуют циклические куполообразные гептамеры, поочередно прикрывают торцы тороида GroEL, взаимодействуя с его входными отверстиями, сформированными Ар-доменами. Эти превращения в сопряжении с гидролизом АТР вызывают значительные конформационные изменения шаперонина, которые приводят к увеличению размеров и гидрофилизации полости, содержащей инкапсулированный субстрат, а это способствует фолдингу мишени, последующему ее высвобождению вместе с ADP и кошаперонином из комплекса с GroEL и началу нового цикла рефолдинга белкового субстрата [9, 12].
Следует отметить, что при фолдинге крупных субстратов, в том числе мультидоменных белков-мишеней, инкапсулирование которых в полость комплекса GroEL/GroES невозможно, реализуется альтернативный механизм, согласно которому связывание определенного фрагмента белка-мишени и кошаперонина GroES происходит на противоположных кольцах тетрадекамера GroEL [9, 26].
Шапероны Hsp90 присутствуют в высоких концентрациях в бактериях и эукариотах и не обнаруживаются в археях. У бактерий обычно выявляется единственный Hsp90-шаперон, в то время как эукариоты содержат по два шаперона в цитозоле, и, кроме того, белки Hsp90 локализованы в митохондриях, эндоплазматическом ретикулуме и в хлоропластах [27–29]. Эукариотические шапероны Hsp90 функционируют в составе комплексных систем, включающих как другие шапероны (в частности, Hsp70), так и сеть разнообразных кошаперонов (табл. 1) [9, 12, 27–30]. Семейство Hsp90 вовлечено в процессы созревания ключевых сигнальных белков, а также участвует в сборке и разборке различных белковых комплексов. Белками-клиентами для Hsp90 служат различные протеинкиназы, факторы транскрипции, рецепторы стероидных гормонов и др. Интересно, что бактериальные шапероны Hsp90 не имеют собственных кошаперонов, и их ремоделирующая функция обеспечивается исключительно путем взаимодействия с системой шаперона Hsp70 [31].
По структурной организации Hsp90 – это гомодимеры, протомеры которых включают N-концевой нуклеотид-связывающий (N) и центральный (М) домены, формирующие АТРазный модуль, а также С-концевой домен, ответственный за димеризацию шаперона [28, 29]. Частично развернутые клиентские белки зачастую доставляются к Hsp90 посредством системы шаперона Hsp70 и связываются с М- и С-доменами Hsp90. Регуляция функциональной активности Hsp90-белков эукариот осуществляется с помощью набора кошаперонов, которые в определенном порядке связываются с разными доменами шаперона, включая специфический С-концевой мотив MEEVD, отсутствующий у бактериальных Hsp90. Апо-форма Hsp90, представляющая собой “раскрытый” V-образный димер, образует сложный комплекс с белком-клиентом и необходимыми кошаперонами. Взаимодействие этого комплекса с АТР приводит к возникновению контактов между N-доменами с образованием “закрытой” формы шаперона и к ремоделированию связанной на его поверхности белковой мишени. В результате гидролиза АТР происходит диссоциация N-доменов с образованием ADP-связанного “полураскрытого” V-димера, из которого высвобождаются белок-клиент, ADP и неорганический фосфат, а Hsp90 вновь принимает открытую конформацию [28, 29]. Следует отметить, что для достижения нативного состояния освобожденный субстрат обычно нуждается в дополнительных стадиях рефолдинга [12, 28].
В то время как рассмотренные выше семейства Hsp60, Hsp70 и Hsp90 составляют группу шаперонов-фолдеров, малые белки теплового шока (sHsps) выполняют роль холдеров в системе контроля качества клеточных белков. Шапероны sHsps образуют прочные долгоживущие комплексы с широким набором клеточных белков в их ненативных конформациях и тем самым предотвращают агрегацию субстратов [32–35]. Белки sHsps представляют собой наиболее распространенное, но низкоконсервативное семейство шаперонов. Их общим отличительным признаком служит наличие в центральной части последовательности домена размером ~100 а.о. со структурой α-кристаллина хрусталика глаза млекопитающих, который фланкирован неструктурированными N- и С-концевыми фрагментами протяженностью 24–247 а.о. и <20 а.о. соответственно [9, 12, 32–36].
Характерная особенность sHsp-шаперонов – полидисперсность их четвертичных структур, что отражается в формировании ансамблей из крупных олигомеров с изменяющимся числом субъединиц. В частности, в клетках человека олигомеры могут включать 12–32 субъединицы sHsp-шаперона [36], а в клетках растений – до 40 [37]. Такие олигомеры часто имеют консервативную структурную организацию в виде сферического или дискообразного комплекса с пронизывающим его центральным каналом [33–36].
В противоположность другим семействам шаперонов, каждый олигомер sHsp способен удерживать по несколько молекул белковых мишеней, высвобождение которых из комплексов с sHsps происходит в кооперации с АТР-зависимыми шаперонами. На этом основании sHsp-белки рассматриваются в качестве “резервуаров” ненативных белков, сохраняющих их для последующего рефолдинга с помощью других семейств шаперонов, в частности системы шаперона Hsp70 [9, 12, 33–35].
В то же время следует заметить, что несмотря на значительный прогресс в понимании механизмов функционирования sHsp-шаперонов, достигнутый за последнее время, вопросы их специфичности, связанные с разнообразием последовательностей и структур шаперонов, остаются малоизученными и требуют дальнейшего исследования.
Семейство Hsp100/Clp, вовлеченное во множество важнейших клеточных процессов (в том числе в мембранный транспорт, регуляцию цитоскелета, биогенез органелл, инициацию репликации ДНК и активацию транскрипции), занимает особое место в системе контроля качества клеточных белков. Шапероны Hsp100 представляют группу “анфолдеров”, которые, с одной стороны, осуществляют разворачивание белковых мишеней перед последующими стадиями фолдинга, а с другой – служат АТРазными компонентами всех протеаз и мультисубъединичных комплексов СКК, деградирующих белки-субстраты. Эти шапероны принадлежат к широко распространенному суперсемейству AAA+-белков (АТРаз, ассоциированных с различными клеточными активностями) (рис. 1), которые функционируют как кольцевые гексамерные структуры, использующие энергию гидролиза АТР для ремоделирования своих белковых мишеней [38–41]. Hsp100-семейство в бактериях представлено белками ClpA, ClpB, ClpC, ClpE, ClpX, ClpY (HslU) и некоторыми другими (в частности, АТРазными составляющими Lon- и FtsH-протеаз) (табл. 1) [2, 9, 12, 41]. У эукариот подобные белки, относящиеся к семействам Hsp104, Hsp101 и Hsp78, встречаются в митохондриях клеток млекопитающих, а также в растениях и дрожжах [12, 42, 43].
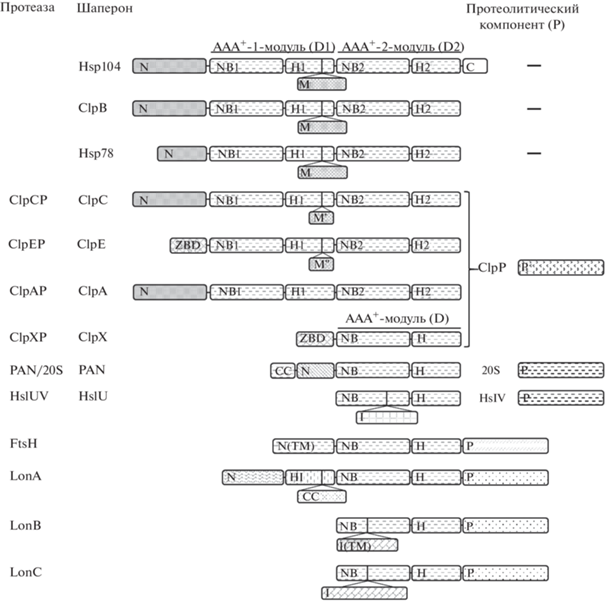
По количеству АТРазных составляющих представители семейства Hsp100 делятся на два класса: к классу I относятся белки, включающие два так называемых ААА+-модуля (D1 и D2, рис. 2), а к классу II – белки, содержащие только один такой модуль D (рис. 2) [9, 41]. Кроме АТРазных модулей ААА+-белки содержат также структурно независимые вспомогательные домены (экстрадомены), которые либо расположены перед ААА+-модулями (N-концевые (N) и Zn-связывающие (ZBD) домены), либо локализованы внутри ААА+-модулей (инсерционные (I) домены) (рис. 2) [9, 41]. Представленные на рис. 2 белки Hsp104, ClpB и Hsp78 функционируют исключительно как шапероны, в то время как белки ClpA, ClpC, ClpE, ClpX и HslU в комплексах с протеолитическими составляющими (белками ClpP или HslV, см. ниже) осуществляют селективную деградацию внутриклеточных белков. Шапероны Hsp104 отличаются от других представителей семейства наличием небольшого С-концевого домена (рис. 2), участвующего в формировании функционально активной структуры шаперона и во взаимодействии с белками-субстратами [44, 45].
Рис. 2.
Доменная организация молекулярных шаперонов семейства Hsp100 и АТР-зависимых протеаз системы контроля качества белков. N – N-концевые домены; ZBD – цинк-связывающие домены; NB, NB1 и NB2 – нуклеотид-связывающие домены; М, M' и M'' – вставочные (middle) домены; I – инсерционные домены; Н, Н1 и Н2 – α-спирализованные домены; С – С-концевой домен; Р – протеазные субъединицы/домены; CC – coiled-coil-область; ТМ – трансмембранные фрагменты.
Ремоделирование белков-мишеней шаперонами Hsp100/Clp включает ряд последовательных стадий. Отбор субстратов происходит путем взаимодействия шаперонов с неструктурированными участками мишеней, содержащими специфические мотивы узнавания (дегроны), причем в некоторых случаях связывание субстратов опосредуется участием дополнительных адаптерных белков [41]. Связывание нуклеотидов способствует стабилизации бочкообразной гексамерной структуры шаперона, а разворачивание мишеней осуществляется путем их “принудительного” перемещения (транслокационная активность шаперона) через центральный канал гексамера Hsp100/Clp за счет энергии гидролиза АТР. На следующей стадии развернутые белки высвобождаются из комплексов с шаперонами благодаря конформационным изменениям последних, обусловленным преобразованием АТР в ADP, и подвергаются рефолдингу – либо спонтанному, либо с участием молекулярных шаперонов других семейств, что определяется специфичностью субстрата [44–47].
Наиболее изученные представители семейства Hsp100 – бактериальные шапероны ClpB и белок Hsp104 из дрожжей, которые вместе с менее исследованным шапероном Hsp78 отличаются от шаперонов других семейств СКК проявлением дезагрегационной функции наряду с транслокационной и анфолдазной [48]. Это свойство шаперонов ClpB/Hsp104, относящихся к ААА+-белкам класса I, обусловлено наличием в их первом ААА+-модуле (D1) вставочного пропеллерообразного домена (“серединный” или middle (M) domain) с конформацией coiled-coil (СС) (рис. 2) [49]. Следует отметить, что шапероны-дезагрегазы семейства Hsp100 обнаруживаются в бактериях, дрожжах и растениях и не встречаются в клетках теплокровных животных [50, 51].
Дезагрегирующая функция ClpB и Hsp104 реализуется совместно с белками системы шаперона Hsp70/DnaK (табл. 1) [52], роль которых заключается в изменении физических свойств белковых агрегатов, что приводит к высвобождению отдельных полипептидов из агрегированных субстратов и перемещению их к ААА+-шаперону. Подвижный М-домен, локализующийся на внешней стороне гексамерного кольца ClpB, регулирует как АТРазную активность самого шаперона, так и DnaK-зависимое связывание мишени с аксиальным каналом ClpB [53]. Далее происходят транслокация и разворачивание полипептида с последующим высвобождением его в среду для рефолдинга [47].
Внутриклеточные АТР-зависимые протеазы и мультисубъединичные протеолитические комплексы
Все АТР-зависимые протеазы и протеолитические комплексы СКК относятся к суперсемейству ААА+-белков (рис. 1 и 2) и выступают критическими регуляторами состава клеточного протеома [2, 54–59]. Показано, что энергозависимый протеолиз отвечает за деградацию более чем 90% белков внутри клетки [55]. Этот процесс исключительно важен для устранения дефектных, неправильно свернутых или агрегированных белков, накопленных в результате стрессовых ситуаций, а также для регуляции внутриклеточного протеолиза при обычных условиях путем контроля концентрации большинства короткоживущих регуляторных белков [54–56, 59].
В клетках прокариот и в органеллах эукариот бифункциональные энергозависимые протеазы представлены пятью семействами гетероолигомерных ферментов (ClpAP, ClpСP, ClpEP, ClpXP, HslUV (ClpYQ)) и двумя семействами гомоолигомерных ферментов (FtsH и Lon – подсемейства LonA и LonC) [56, 57] (рис. 2). В археях аналогичные функции выполняются Lon-протеазами (подсемейство LonВ) и мультисубъединичной архейной протеасомой (PAN/20S), которая представляет собой комплекс протеасомоактивирующей нуклеотидазы (proteasome-activating nucleotidase, PAN) с протеолитическим компонентом – 20S-протеасомой [58]. В цитоплазме и ядрах эукариот функционируют 26S-протеасомы – мультисубъединичные ансамбли, которые состоят из 20S-протеасом (протеолитические коры) и регуляторных 19S-комплексов, включающих ААА+-АТРазы [59, 60]. При этом все гетероолигомерные протеазы и протеолитические комплексы СКК характеризуются общей архитектурой: их бочкообразные гепта- или гексамеры, образованные двухярусными кольцами пептидгидролаз, активные сайты которых изолированы внутри центральной деградационной камеры, формируют комплексы с кольцевыми гексамерами ААА+-АТРаз регуляторных компонентов, которые проявляют анфолдазную активность, направленную на разворачивание белков-мишеней для последующей их транслокации в деградационную камеру [55–57].
АТРазные составляющие протеаз СКК, преобразующие энергию АТР в механическую работу, выполняют важнейшую роль в деградации белковых субстратов. Весь процесс превращения белка-субстрата можно разделить на отдельные этапы, при этом распознавание белковой мишени, ее разворачивание и транслокация в деградационную камеру осуществляются АТРазным компонентом ААА+-протеазы, а собственно гидролиз белка-субстрата – пептидазным.
Этап узнавания и связывания мишени, как правило, не зависит от АТР и может быть либо прямым, либо “косвенным”, т.е. опосредованным адаптерными белками. Специфическими мотивами узнавания часто служат упомянутые выше дегроны – пептидные фрагменты, локализованные вблизи (или внутри) неструктурированных N- или С-концевых областей белка, или даже N-концевые аминокислоты [54, 56, 57]. Известны также дегроны, представляющие собой “прикрепленные” пептидные последовательности (тэги). Один из ярких примеров тэга – фрагмент SsrA-tag – AAXXXXXALAA (где X – любая аминокислота), который узнается протеазами ClpAP, ClpXP, FtsH, Lon и архейной протеасомой [54, 56].
В отличие от АТР-зависимых протеаз бактерий и протеасом архей, функционирование 26S-протеасом эукариот обычно опосредовано предварительной селективной модификацией белков-мишеней, подлежащих деградации, путем ковалентного присоединения специфической метки – цепочки из нескольких молекул убиквитина (белок из 76 а.о.) [59–61]. Вследствие этого дегроны мишеней 26S-протеасом представлены двумя частями: универсальным полиубиквитиновым тэгом, обеспечивающим связывание мишени с регуляторным компонентом каталитического комплекса, и неструктурированным фрагментом, который служит областью инициации протеолиза [60, 61]. Вместе с тем показано, что возможен также убиквитин-независимый путь деградации белков 26S-протеасомой. Его реализации способствует ряд так называемых прямых протеасомных сигналов, к которым относятся либо специфические фрагменты последовательности, либо посттрансляционные модификации мишеней, либо их конъюгация с химическими веществами, приводящая к связыванию с протеасомой по принципу заряд-зарядовых взаимодействий [62].
АТР-зависимые протеазы отличаются от классических протеолитических ферментов следующими уникальными характеристиками: 1) высокой селективностью взаимодействия с белковыми мишенями при отсутствии выраженной специфичности по отношению к аминокислотам, образующим расщепляемые связи; 2) наличием транслоказной активности; 3) сопряжением протеолитической активности с гидролизом АТР; 4) процессивным механизмом деградации белков-субстратов (c образованием 10–15-членных пептидных продуктов без высвобождения высокомолекулярных интермедиатов); 5) мультисубъединичной (гомо- или гетероолигомерной) организацией [54, 56, 57].
ААА+-модули – характеристические компоненты АТР-зависимых протеаз. Выступая представителями суперсемейства ААА+-белков, АТР-зависимые протеазы содержат в своей структуре как высококонсервативные ААА+-модули, состоящие из 200–250 а.о., так и дополнительные N‑концевые (N) или инсерционные (I) экстрадомены (рис. 2). АТРазные модули формируются большими нуклеотид-связывающими (NB или α/β) и малыми α-спирализованными (Н или α) доменами. Любая АТР-зависимая протеаза содержит либо один ААА+-модуль, либо два топологически подобных ААА+-модуля, включающих наборы различных консенсусных элементов (рис. 2 и 3) [39, 56, 63–66].
Рис. 3.
Вторичная структура и консенсусные элементы ААА+-модулей шаперонов Hsp100 и АТР-зависимых протеаз системы контроля качества белков (по данным работ Puchades et al. [39], Chang et al. [65] и Miller et al. [66]). Прямоугольники – α-спирали, стрелки – β-тяжи, NB-домен – нуклеотид-связывающий домен, Н-домен – α-спирализованный домен, показаны мотивы Уолкера А и В (WA и WB), петли pore loop-1 (GYVG), pore loop-2 и RKH, мотив межсубъединичного сигналинга (ISS), область вторичной гомологии (SRH), остатки сенсор-1 (s1), сенсор-2 (s2) и “аргининовый палец” (Rf). Обозначены сайты включения экстрадоменов в протеазах LonB, LonC (ILons) и HslUV (IHslUV).
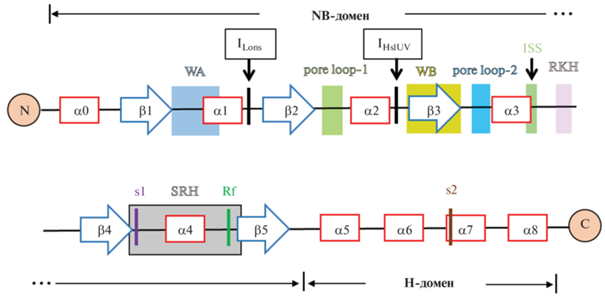
В NB-доменах ААА+-модулей локализованы консервативные мотивы Уолкера А и В. Мотив А (WA, называемый также Р-петлей, Р-loop), играющий важную роль как в связывании нуклеотида и координации иона металла, так и в формировании гексамерного АТРазного кольца, представлен последовательностью GX2GZGK[T/S], где X и Z – любые аминокислоты, но хотя бы один из Х-остатков – это остаток пролина. Мотив В (WB) представляет собой гексапептид Φ4DE, где Φ – остатки гидрофобных аминокислот. Наряду с координацией иона металла, мотив WB принимает участие в гидролизе АТР, при этом консервативный остаток глутамата активирует молекулу воды для нуклеофильной атаки на γ-фосфат нуклеотида. Кроме мотивов Уолкера, ААА+-модули содержат ряд других консервативных участков, включая мотив межсубъединичного сигналинга (inter-subunit signaling, ISS), область вторичной гомологии (second region of homology, SRH), а также остатки sensor-1 (s1), sensor-2 (s2) и “аргининовый палец” (Arg-finger, Rf) (рис. 3) [39, 56, 63–68].
Мотив ISS, представленный α-спиралью из нескольких консервативных остатков (рис. 3), имеет большое значение для координации связывания и гидролиза АТР в соседних субъединицах кольцевого ААА+-гексамера. Взаимодействие С‑концевой аспарагиновой (или глутаминовой) кислоты ISS-мотива с Rf-остатком собственной субъединицы, который воспринимает статус нуклеотида, связанного в соседней субъединице, запускает цикл гидролиза АТР, необходимый для транслокации субстрата [65].
Остаток Rf локализован в SRH-области (фрагмент полипептидной цепи из 15–20 а.о.), расположенной в С-концевой части NB-домена (рис. 3) [56, 64–68]. К этой же области относится остаток s1 (обычно аспарагин, реже – серин, треонин или гистидин), также взаимодействующий с γ-фосфатом молекулы ATP соседней субъединицы и координирующий взаимодействие атакующей молекулы воды с остатками WB-мотива.
Совокупность консенсусных мотивов SRH-области критически важна для передачи между субъединицами ААА+-комплекса сигналов конформационных изменений, сопровождающих акт гидролиза нуклеотида. Вследствие своей функциональной значимости и в силу отсутствия SRH-фрагмента в других нуклеозидтрифосфатазах, эта область служит отличительной характеристикой для белков ААА+-суперсемейства [64–66].
Кроме рассмотренных консенсусных элементов, NB-домены ААА+-белков содержат специфические фрагменты последовательности, которые формируют петли трех типов, обращенные внутрь аксиального (осевого) канала гексамера белка [69]. Это так называемые петли pore loop-1 (или GYVG), pore loop-2 и RKH, которые участвуют в связывании, разворачивании и перемещении молекулы белка-мишени внутри полости канала вплоть до протеолитической камеры (в случае ААА+-протеаз) (рис. 3). Петля RKH локализуется у входа в осевой канал, наиболее консервативная петля pore loop-1 – в его центральной части, а петля pore loop-2 – на выходе из канала. При этом ведущую роль в разворачивании белков-субстратов и обеспечении транслоказной активности ААА+-протеаз играют петли pore loop-1.
В малом Н-домене ААА+-модуля, сформированном четырьмя α-спиралями, локализован мотив sensor-2 (рис. 3). Он содержит консервативный остаток аргинина (иногда – лизина) (s2), который взаимодействует с α-фосфатом АТР и опосредует конформационные изменения, сопряженные с циклом связывания/гидролиза нуклеотида, а также участвует в межсубъединичных взаимодействиях [64, 66]. Следует упомянуть, что в противоположность членам AAA+-суперсемейства у представителей классических AAA-АТРаз в положении s2 обычно содержится остаток аланина [64, 66].
Особенности доменной и структурной организации АТР-зависимых протеаз и мультисубъединичных комплексов. Известно, что подавляющее большинство ААА+-белков взаимодействует с функциональными белковыми партнерами, образуя комплексные гетероолигомерные структуры, АТРазные составляющие которых – самостоятельные субъединицы [64]. В редких случаях АТРазный и функциональный компоненты ААА+-белка локализованы в единой полипептидной цепи [56, 64, 70]. С другой стороны, как упоминалось выше, суперсемейство ААА+-белков делится на классы I и II [41]. В первом случае ААА+-белки содержат два ААА+-модуля (D1 и D2), разделенных вставками разной длины, а во втором – единственный ААА+-модуль (D), подобный D2-модулю белков класса I [41]. В этом отношении сообщество АТР-зависимых протеаз системы контроля качества белков объединяет представителей обоих классов ААА+-белков: к классу I относятся АТРазные компоненты ClpAP/СР/ЕР-протеаз, а к классу II – протеаз ClpXP, HslUV, FtsH, Lon и комплексов архейных PAN/20S-протеасом и эукариотических 26S-протеасом [56, 70]. Доменное строение протеаз СКК показано на рис. 2, а их структурная организация охарактеризована в табл. 2.
Таблица 2.
Характеристика АТР-зависимых протеаз и протеолитических комплексов, осуществляющих селективный внутриклеточный протеолиз
| Протеаза/ комплекс | ФК в четв. структуре* | Функциональные компоненты (ФК) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| АТРазные компоненты | пептидгидролазные компоненты | ||||||||||
| название | n** | ААА+-модули | экстра-домен | название | n** | остатки каталитических центров | MEROPS | ||||
| количест-во*** | MD | Clan | Family | ||||||||
| ААА+-протеазы I класса | |||||||||||
| ClpCP | Su | ClpC | 6/12 | 2 | M' | N | ClpP | 14 | Ser, His, Asp | SK | S14 |
| ClpEP | Su | ClpE | 6/12 | 2 | M'' | N | ClpP | 14 | Ser, His, Asp | SK | S14 |
| ClpAP | Su | ClpA | 6/12 | 2 | – | N | ClpP | 14 | Ser, His, Asp | SK | S14 |
| ААА+-протеазы и протеолитические комплексы II класса | |||||||||||
| ClpXP | Su | ClpX | 6/12 | 1 | – | N | ClpP | 14 | Ser, His, Asp | SK | S14 |
| PAN/20S | Su | PAN | 6/12 | 1 | – | N | β-Su в 20S | 14 | Thr | PB | T1B |
| 26S-протеасома | Su | Rpt(1–6) в 19S | 6/12 | 1 | – | N | β-Su(1,2,5) в 20S | 6 из 14 | Thr | PB | T1B |
| HslUV | Su | HslU | 6/12 | 1 | – | I | HslV | 12 | Thr | PB | T1B |
| FtsH | Dom | FtsH | 6 | 1 | – | N | FtsH | 6 | His, Glu, His, Asp | MA | M41 |
| LonA | Dom | LonA | 6 | 1 | – | N | LonA | 6 | Ser, Lys | SJ | S16 |
| LonB | Dom | LonB | 6 | 1 | – | I | LonB | 6 | Ser, Lys | SJ | S16 |
| LonС | Dom | LonC | 6 | 1 | – | I | LonC | 6 | Ser, Lys | SJ | S16 |
Примечание: Su – субъединица; Dom – домен; MD – наличие инсерционного М-домена в D1-модуле; 20S – 20S-протеасомы (протеолитические коры архейной (PAN/20S) и эукариотической (26S) протеасом); PAN – Proteasome Activating Nucleotidase (регуляторная составляющая PAN/20S-протеасомы); 19S – регуляторный комплекс 26S-протеасомы; Rpt – Regulatory particle triphosphatase (ААА+-АТРаза). * Функциональные компоненты в четвертичной структуре протеазы или протеолитического комплекса. ** Количество субъединиц/доменов в ФК. *** Количество ААА+-модулей в субъединице.
АТРазные и протеазные составляющие семейств Lon и FtsH локализованы в единой полипептидной цепи, и эти ферменты функционируют как гомоолигомеры. Однако большинство ААА+-протеаз – это гетероолигомерные комплексы, в которых АТРазный и протеазный компоненты представлены индивидуальными субъединицами (рис. 2).
Особенность протеаз ClpCP и ClpEP – наличие в α-спирализованных доменах Н1 их D1-модулей вставочных “серединных” (middle) доменов (M' и M''), обладающих СС-конформацией, но различающихся своими размерами. Показано, что эти домены участвуют во взаимодействии протеаз со специфическими адаптерными белками, которые опосредуют связывание и деградацию белковых мишеней [70, 71]. Подобные им вставочные домены (М), также имеющие СС-конформацию, обнаружены в шаперонах ClpB, Hsp104 и Hsp78 (рис. 2, см. подраздел “Молекулярные шапероны СКК”) [49, 72]. Однако М-домены более чем вдвое превышают M'- и M''-домены по размеру и выполняют другие функции – они обеспечивают дезагрегазную активность шаперонов [72].
Разнообразие протеазных компонентов АТР-зависимых протеаз и протеасом, вовлеченных в селективный АТР-зависимый протеолиз, отражено в табл. 2, из которой видно, что внутриклеточная деградация белков осуществляется ферментами, представляющими четыре разных клана в классификации пептидгидролаз MEROPS: каталитический центр протеолитической субъединицы ClpР, общей для гетероолигомерных протеаз ClpАР, ClpСР, ClpЕР и ClpХР, представлен классической триадой Ser–His–Asp, у протеаз HslUV, а также в β-субъединицах 20S-протеасом архей и эукариот каталитически активны N-концевые остатки треонина, мембраносвязанная протеаза FtsH – Zn-зависимая металлопротеаза, а в активных центрах Lon-протеаз функционирует каталитическая диада Ser–Lys [73].
Функционально активными формами бактериальных гетероолигомерных АТР-зависимых протеаз (ClpАР, ClpСР, ClpЕР, ClpХР и HslUV) выступают бочкообразные комплексы из состыкованных колец, в которых два центральных пептидазных кольца (гептамерных в случае ClpP или гексамерных в случае HslV) с каталитическими центрами, обращенными внутрь аксиальной полости (деградационная камера), фланкированы с одной или с обеих сторон гексамерными кольцами регуляторных АТРазных субъединиц (ClpА, ClpС, ClpЕ, ClpХ или HslU), выполняющих функции узнавания, разворачивания и транслокации субстрата [56, 57, 70, 74]. Показано, что ассоциация АТРаз ClpA, ClpC, ClpЕ и ClpХ с пептидазой ClpP происходит с участием специальных “стыковочных петель”, локализованных в ААА+-модулях шаперонов между остатками sensor-1 NB-доменов и Н-доменами [75]. Гомоолигомерные протеазы Lon и FtsH собираются в бочкообразные структуры из шести идентичных субъединиц.
Аналогично бактериальным ААА+-протеазам формируются более сложные мультисубъединичные комплексы протеасом архей (PAN/20S) и эукариот (26S), у которых протеолитические компоненты (20S) представляют тетраярусные гептамерные кольца из внутренних каталитических β-субъединиц и внешних регуляторных α-субъединиц, одинаковых в 20S-комплексах архей (структурная формула α7/β7β7/α7) и различающихся в 20S-комплексах эукариот (α1–7/β1–7β1–7/α1–7) [76].
Регуляторные комплексы протеасом (PAN и 19S), обладающие АТРазной активностью, содержат 6 субъединиц у архей и 19 субъединиц у эукариот, при этом основой обеих структур служат гексамерные кольца ААА+-белков (соответственно, PAN и Rpt(1–6) – Regulatory particle triphosphatases, табл. 2) [58, 60]. Эти регуляторные комплексы находятся в непосредственном взаимодействии с гептамерами α-субъединиц протеолитических коров протеасом.
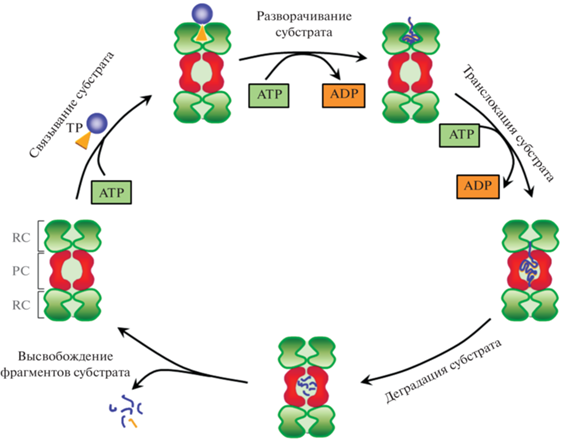
На рис. 4 представлен общий принцип функционирования АТР-зависимых протеаз и мультисубъединичных протеолитических комплексов. Распознавание и связывание белка-мишени (ТР – target protein) осуществляется регуляторными ААА+-компонентами (RC – regulatory complexes), локализованными во внешних кольцах активного комплекса фермента. На следующих этапах происходит разворачивание молекулы субстрата и транслокация ее в область протеолитических центров. В деградационной камере, расположенной внутри ферментного комплекса, осуществляется процессивный гидролиз субстрата с последующим высвобождением продуктов. Образовавшиеся олигопептидные фрагменты могут в дальнейшем гидролизоваться АТР-независимыми протеазами до коротких пептидов и, наконец, клеточными аминопептидазами до свободных аминокислот.
Рис. 4.
Схематическое представление механизма ATP-зависимой деградации белков в клетках бактерий, архей и эукариот (по данным работ Gottesman S. [54] и Bittner et al. [122]). RC – регуляторный АТРазный компонент (Clp(A, С, Е, Х), HslU, PAN или 19S-комплекс); PC – протеолитический компонент (ClpP, HslV или 20S-протеасома); TP – белок-мишень.
Основные характеристики некаталитических экстрадоменов АТР-зависимых протеаз. Специфические для каждого семейства АТР-зависимых протеаз и протеасом экстрадомены их АТРазных составляющих в большинстве случаев представлены вариабельными “не АТРазными” N-концевыми (N) доменами, предшествующими ААА+-модулям, и реже – вставочными (I) доменами, локализованными внутри NB-доменов между мотивами WA и WB (рис. 2) [41, 56, 67]. Обычно экстрадомены служат местами связывания субстратов [56]. Они выполняют функцию первичного распознавания субстрата-мишени, а также играют важную роль в его разворачивании и/или контролируют доступ мишеней к связывающим центрам, локализованным внутри ААА+-модулей. Экстрадомены протеаз LonB и FtsH включают также трансмембранные фрагменты, обеспечивающие взаимодействие ферментов с клеточными мембранами.
Вовлечение в селективный отбор субстратов белков-адаптеров, способных к специфическому взаимодействию как с мишенями, так и с ААА+-партнерскими белками, зачастую приводит к модулированию функциональных свойств последних [41, 77]. В качестве примера в табл. 3 представлены адаптерные белки, функционирующие в комплексе с ААА+-протеазами в клетках E. coli и Bacillus subtilis. При этом следует заметить, что обычно LonA-протеазам не требуется никаких адаптеров, и обнаруженный в 2015 г. адаптерный белок SmiA используется ферментом из B. subtilis исключительно для деградации активатора биосинтеза жгутиков SwrA [78]. Для протеаз FtsH и ClpEP из этих же организмов до настоящего времени адаптеров не обнаружено [77].
Большинство белковых адаптеров связываются с N-концевыми экстрадоменами ААА+-белков, что обеспечивает оптимальное взаиморасположение ААА+-олигомера и “заякоренной” через этот белковый линкер мишени. Обнаружено также, что некоторые адаптерные белки могут влиять на активность ААА+-партнеров. При этом сами белки-адаптеры также могут подвергаться регуляции (например, путем взаимодействия с небольшими белками-антиадаптерами и/или посредством фосфорилирования) [77].
N-экстрадомены Clp-шаперонов – АТРазных компонентов ряда ААА+-протеаз. Шапероны Clp-семейств системы контроля качества характеризуются N-концевыми экстрадоменами двух типов. Прежде всего, это α-спирализованные N-домены молекулярных шаперонов ClpВ и Hsp104 и родственных им АТРазных субъединиц (ClpA и ClpС) гетероолигомерных протеаз ClpAР и ClpСР (рис. 2), которые проявляют высокую гомологию на уровне своих первичных и вторичных структур [79, 80]. Эти домены содержат по восемь α-спиралей, формирующих тандемы из двух структурных повторов, каждый из которых включает четыре α‑спирали и имеет размер ~70 а.о. Установлено, что N-домены ClpA, ClpВ и ClpС выполняют роль регуляторов специфичности своих шаперонов и обеспечивают их взаимодействие с широким набором субстратов-мишеней [80, 81]. Наряду с этим показано, что N-домен ClpA имеет также исключительное значение для образования активного комплекса с ClpР-пептидазой [82].
Для шаперона ClpA выявлен единственный адаптерный белок ClpS (табл. 3), связывание которого с N-доменом приводит к ингибированию деградации известных субстратов ClpAP – SsrA-меченых белков. В то же время комплекс ClpAP/ClpS обнаруживает способность к распознаванию и расщеплению некоторых белков, агрегированных в результате действия теплового шока. Таким образом, взаимодействие ClpS c экстрадоменом модифицирует специфичность ClpA и перенаправляет активное функционирование шаперона на деградацию агрегированных белков [83].
Для ClpC из B. subtilis известен целый ряд адаптерных белков (MecA, YpbH, McsB и др.) (табл. 3). Связывание N-домена ClpC с белком-адаптером MecA играет важную роль при деградации большинства субстратов протеолитическим комплексом ClpСP [70]. Показано, что в узнавании и связывании адаптерных белков ClpC-шапероном кроме N-концевого экстрадомена участвует также локализованный в ААА+-модуле D1 (рис. 2) вставочный M'-домен (58 а.о). При этом детали участия адаптерных белков в доставке субстратов к комплексу ClpCP до сих пор остаются неизвестными [71, 77, 80, 84].
Рентгеноструктурные исследования показали, что взаимодействия экстрадоменов ClpA и ClpC с их адаптерами ClpS и MecA носят сходный характер. Оба адаптера используют подобные α-спирали для взаимодействия cо структурно подобными областями N-доменов шаперонов, при этом в комплексах шаперон–адаптер формируются одинаковые наборы водородных связей [84].
Вместе с тем ClpC кардинально отличается от других шаперонов семейства Clp/Hsp100, поскольку все виды его активности, включая распознавание и связывание белков-субстратов, гексамеризацию, шапероновую активность, образование функционального комплекса ClpCP и деградацию белковых мишеней, опосредованы обязательным взаимодействием шаперона с адаптерными белками. Выявлен регуляторный механизм, в соответствии с которым в отсутствие белков-субстратов происходит инактивация комплекса ClpCP за счет деградации связанного белка-адаптера [80, 81].
Несмотря на высокое подобие шаперона ClpВ и регуляторных шаперонов АТР-зависимых протеаз ClpA и ClpC, адаптерные белки для ClpB до настоящего времени не обнаружены [77]. N-экстрадомены ClpB участвуют во взаимодействии с рядом белковых мишеней, однако их роль в основной функции шаперона – ClpB-опосредованной дезагрегации белков – все еще до конца не изучена. Показано, что ряд вариантов ClpB, несущих точечные мутации в экстрадомене, проявляет пониженную дезагрегационную активность, однако некоторые делеционные формы ClpB из разных источников с полностью удаленным N‑доменом сохраняют активность, соизмеримую с активностью полноразмерного белка [16]. Кроме того, существование в Mycoplasma sp. гомологов ClpB, лишенных N-домена, подтверждает необязательность наличия N-доменов в ClpB-шаперонах для дезагрегации белков [16]. При этом однозначно установлено, что непосредственное участие в реализации дезагрегационной функции ClpB принимает характеристический для белков ClpB/Hsp104 М-домен, внедренный в ААА+-1-модуль шаперона (рис. 2) [85, 86].
Для белков семейства Hsp104 показано, что их N-экстрадомены помимо взаимодействия с белками-мишенями вовлечены также в процессы олигомеризации самих шаперонов [87].
Другой тип N-концевых экстрадоменов Clp-шаперонов представляют гомологичные Zn-связывающие домены (ZBD) протеаз ClpXP и ClpEP, которые относятся к разным классам ААА+-белков (рис. 2). При этом можно отметить, что шаперон ClpE проявляет также сходство с ClpС, поскольку в своем D1-модуле он содержит вставочный M''-домен (53 а.о), более чем на 60% подобный M'-домену ClpС (рис. 2). ZBD-экстрадомены ClpX и ClpE имеют размеры ~60 а.о. и включают по четыре остатка цистеина, координирующих атом Zn [88].
Показано, что ZBD-домен ClpX E. coli отвечает за узнавание некоторых субстратов (в частности, белков λO и MuA) и, кроме того, связывает адаптерный SspB-белок (табл. 3), направляющий субстраты к осевому каналу ClpX для разворачивания [88]. О белке ClpE известно, что он участвует в процессе дезагрегации и в последующей деградации агрегированных белков, а его N-концевой экстрадомен необходим для проявления базовой АТРазной активности шаперона in vitro [70, 89].
На примере ClpX установлено, что индивидуальные ZBD-экстрадомены образуют стабильные димеры, в то время как в кольцевых гексамерах полноразмерных шаперонов шесть ZBD-доменов формируют тримеры димеров, локализованные на поверхности гексамерных колец AAA+-модулей ClpX [88].
N-концевые экстрадомены AAA+-АТРаз протеасом. ААА+-регуляторные комплексы, активирующие протеолитические 20S-коры протеасом, в случае архей сформированы гомогексамерами протеасомоактивирующей нуклеотидазы PAN, а в случае эукариот представлены 19S-ансамблями, состоящими из 19 субъединиц, шесть неидентичных ААА+-белков которых (Rpt(1–6), табл. 2) образуют кольцевые гексамерные “основания”, непосредственно взаимодействующие с 20S-корами [90–92]. Топологически подобные белки PAN и Rpt, как и другие ААА+-АТРазы, включают N‑концевые экстрадомены (PAN-N и Rpt-N соответственно), которые участвуют в формировании гексамеров, в узнавании и связывании белковых мишеней и совместно с собственными ААА+-модулями превращают химическую энергию в механическую работу.
Экстрадомены PAN-N и Rpt-N начинаются с α-спиральных сегментов, за которыми следуют глобулярные β-структурированные С-концевые участки, формирующие олигосахарид-связывающий (OB) фолд [91, 92]. Архитектурно экстрадомены PAN-N и Rpt-N представляют собой тримеры димеров, в которых на гексамерных кольцах ОВ-фрагментов располагаются три CC-участка, образованные парами N-концевых α-спиральных сегментов соседствующих субъединиц [91, 92]. Эти структуры через короткие линкеры соединяются с гексамерными кольцами ААА+-модулей PAN и Rpt, имеющими каноническую укладку ААА+-АТРаз [91].
В функционально активных протеасомах, включающих и регуляторные, и протеолитические компоненты, кольца ААА+-модулей ассоциируются с протеолитическим кором 20S и управляют ATP-зависимым разворачиванием белка-субстрата, тогда как дистальные комплексы экстрадоменов образуют входные отверстия каналов транслокации субстрата [93].
Инсерционные экстрадомены HslUV-протеаз. Характерная особенность АТРазного компонента гетероолигомерной протеазы HslUV – отсутствие N-концевого домена. Вставочные α-спирализованные экстрадомены (I-домены) HslU размером ~130 а.о., формирующие в кольцевом олигомере воронкообразную полость, расположены в NB-домене шаперона вслед за спиралью α2 и в непосредственной близости от мотива WB (рис. 3) [94, 95]. Гексамер, образованный I-доменами, обеспечивает отбор полипептидных субстратов для деградации, способствует транслокации субстратов в протеолитическую камеру HslUV-комплекса, а также участвует в гидролизе АТР и деградации белковых мишеней [94, 95].
N-экстрадомены FtsH-протеаз. FtsH – единственная мембраносвязанная ААА+-протеаза бактерий. Функционально активной формой фермента, нацеленного на деградацию мембраносвязанных белков, служит кольцевой гомогексамер [96]. N-экстрадомены FtsH-протеаз содержат трансмембранные сегменты, сформированные двумя α-спиралями [96, 97]. Показано, что N-домен влияет на олигомеризацию и стабильность FtsH [98]. Белковые адаптеры для FtsH-протеаз неизвестны, однако существует целый набор белковых модуляторов и ассоциированных факторов (HflK, HflC, HflD, MgtR, SpoVM), которые участвуют в функционировании ферментов [90]. Аналоги FtsH обнаружены в митохондриях и хлоропластах эукариот. Установлено высокое подобие N-экстрадоменов бактериальных и эукариотических FtsH-протеаз [97]. Обнаружена также особая роль FtsH как единственной ААА+-протеазы, имеющей критическое значение для выживаемости E. coli [99].
Три типа экстрадоменов протеаз семейства Lon. Lon-протеазы – это единственное семейство АТР-зависимых протеаз, которое в системе контроля качества клеточных белков представлено тремя подсемействами (LonА, LonВ и LonC, см. далее подраздел “Общая характеристика семейства Lon-протеаз. Подсемейства Lon”), имеющими сходные ААА+-модули и протеазные домены, но кардинально различающимися строением и локализацией своих экстрадоменов (рис. 2 и 5) [100]. Согласно современной концепции, предстоящая ААА+-модулю пролонгированная N‑концевая область LonА-протеаз в действительности сформирована двумя доменами [101] – собственно N-концевым β-структурированным экстрадоменом (N) [102, 103] и следующим за ним “вставочным” α-спирализованным доменом [101, 104]. При этом вставочный домен включает олигопептидный фрагмент (~100 а.о.) с СС-конформацией и носит название HI(CC) (helical inserted with CC-fragment – инсерционный спирализованный с CC-фрагментом) домен [101, 105].
Рис. 5.
Доменная организация и консенсусные элементы Lon-протеаз различных подсемейств. S* и K* – каталитически активные остатки протеолитического центра, Ф – остаток гидрофобной аминокислоты, Х – остаток любой аминокислоты, РА и РВ – протеазные домены А-типа (коралловый) и В-типа (бирюзовый), АА, АВ и АВ* – ААА+-модули соответственно А-типа (голубой), В-типа и В*-типа “вырожденный” (синие), NB – нуклеотид-связывающие домены, Н – α-спирализованные домены. Экстрадомены ферментов включают: N-домен (фиолетовый) и HI(CC) – вставочный α-спирализованный домен (зеленый) с участком coiled-coil (СС) (красный) у протеаз LonA, I – вставочный домен с трансмембранным (ТМ) сегментом (серый) у LonB и I* – вставочный α-спирализованный домен (заштрихован) у LonC; консервативные фрагменты показаны красным цветом; замены аминокислот в консервативных фрагментах выделены синим цветом.
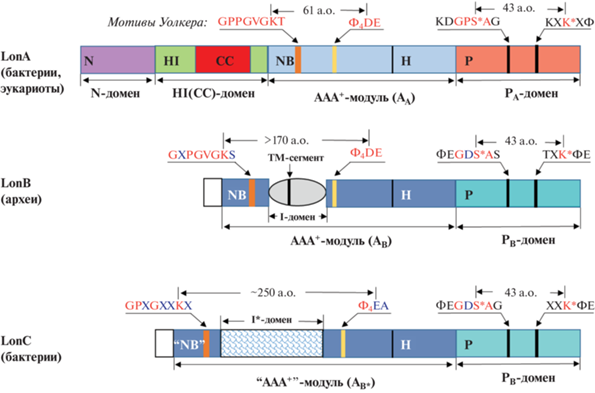
Ферменты подсемейств LonВ и LonC, лишенные N-экстрадоменов, содержат разные по размеру инсерционные экстрадомены (I и I*), включающие, соответственно, 124–140 и 185–191 а.о., которые внедрены, как и у HslUV, внутрь NB-доменов их собственных ААА+-модулей (рис. 5) [73, 100, 106–108]. Однако, в отличие от HslUV, I-домены Lon-протеаз локализованы вблизи мотива WA, непосредственно после спиралей α1 (рис. 3). Особенностью экстрадоменов LonB-протеаз служит наличие включенных в их последовательности трансмембранных сегментов (ТМ).
Сходство и различия экстрадоменов ААА+-белков СКК. Приведенные выше данные по экстрадоменам ААА+-белков – представителей системы контроля качества клеточного протеома – показывают, что эти некаталитические фрагменты ААА+-протеаз и протеолитических комплексов разнообразны по структуре: они могут быть полностью α-спирализованными (как N-домены ClpA, ClpB и ClpС, а также I-домены HslU и LonC), могут иметь α/β-структуру (ZBD-домены ClpX и ClpE), могут представлять β-структурный фолд (ОВ) с включением N-концевых CC-фрагментов (N-домены ААА+-АТРаз регуляторных комплексов эукариотических и архейных протеасом), и, наконец, их последовательности могут содержать спиральные фрагменты, обеспечивающие взаимодействие с клеточными мембранами (N-концевой домен FtsH и I-домен LonB-протеаз). В настоящее время для большинства экстрадоменов ААА+-белков СКК получены рентгеноструктурные данные (в том числе для ClpA E. coli PDB: 1K6K; ClpB E. coli PDB: 1KHY; ClpB Thermus thermofilus PDB: 1QVR; ClpC B. subtilis PDB: 2Y1Q; ClpX E. coli PDB: 2DS7; Hsp104 S. cerevisiae PDB: 5U2U; Hsp104 Candida albicans PDB: 5U2L; HslU E. coli PDB: 1YYF; HslU Haemophilus influenzae PDB: 1G41; FtsH E. coli PDB: 4V0B; FtsH Thermotoga maritima PDB: 4M8A и др.) и выявлена их роль в связывании белков-мишеней и/или белковых адаптеров.
Практически все экстрадомены ААА+-белков СКК объединены одним общим свойством – их размеры не превышают 150–190 а.о. В этом отношении протеазы подсемейства LonA занимают особое место, поскольку только у них некаталитическая N-концевая область включает от 300 до более чем 400 аминокислот и состоит из двух структурно независимых доменов – N и HI(CC) (рис. 2) [100, 101]. При этом кристаллическая структура определена только для N-домена [102–104], а пространственная укладка инсерционного HI(CC)-домена остается невыясненной до сих пор. До последнего времени участие каждого из этих доменов в функционировании полноразмерного фермента также не было охарактеризовано в полной мере.
Вместе с тем LonА-протеазы играют ключевую роль в функционировании системы контроля качества в бактериях и эукариотах. Так, известно, что в клетках E. coli именно LonА-протеаза обеспечивает >50% протеолиза аномальных белков [109], а у эукариот LonА-протеазы – ведущие ферменты, расщепляющие окисленные белки митохондриального матрикса. Установлено, что многие патологии (в частности, нейродегенеративные нарушения и развитие ряда онкологических заболеваний) сопровождаются изменениями содержания LonА-протеазы в клетках [110–113]. Сведения об участии LonА-протеаз в патологических процессах и о возможности их применения в медицинских целях широко представлены в современной литературе (обзоры [114–118]). Существенная биологическая роль LonА-протеаз определяет важность их структурно-функционального исследования.
Lon-ПРОТЕАЗЫ КАК ОСОБОЕ СЕМЕЙСТВО В СИСТЕМЕ КОНТРОЛЯ КАЧЕСТВА КЛЕТОЧНЫХ БЕЛКОВ
Общая характеристика семейства Lon. Подсемейства Lon-протеаз
Согласно современной классификации MEROPS [119], семейство Lon-протеаз S16 клана SJ формируется, главным образом, подсемействами А, В и С (табл. 2). Lon-протеаза из E. coli (EcLon), относящаяся к наиболее представительному подсемейству LonA, исторически была первой экспериментально обнаруженной АТР-зависимой пептидгидролазой (1981 г.) [120], и до настоящего времени она остается основной моделью при изучении ферментов семейства Lon.
К 2004 г. семейство Lon-протеаз насчитывало уже более 100 ферментов из различных источников. Сравнительный анализ их первичных структур позволил выделить в общем пуле два подсемейства – крупное LonA (80 ферментов) и менее репрезентативное LonB [100] (рис. 5). На текущий момент в базе данных MEROPS [119] насчитывается ~2300 LonA-протеаз и >200 LonB-ферментов. Наряду с рассмотренными выше различиями в экстрадоменах, протеазы LonA и LonB характеризуются также разными типами протеолитических центров: окружение каталитически активных остатков в подсемействах представлено фрагментами KDGPS*AG и KXK*XФ (у LonA) или ΦEGDS*AS и TXK*ФЕ (у LonB), где S* и K* – каталитические остатки серина и лизина, в обоих случаях отстоящие друг от друга на 43 а.о., Х – любая, а Ф – гидрофобная аминокислота (жирным шрифтом выделены консервативные остатки, подчеркнуты различные по природе остатки) (рис. 5). Помимо этого, присутствуют отличия и в аминокислотных сегментах AAA+-модулей, формирующих мотивы Уолкера и их окружение: так, мотивы WA представлены пептидами GPPGVGKT (LonA) и GХPGVGKS (LonB), а подобные мотивы WB (Ф4DE) фланкированы различающимися пептидами: у LonA это NP и IDK, а у LonB – KG и INT [100].
Как упоминалось выше, LonA-протеазы преимущественно обнаруживаются в прокариотах (цитозольные ферменты) и эукариотах (ферменты митохондрий, хлоропластов и пероксисом). Между тем, несколько протеаз LonA-типа идентифицировано также у отдельных представителей архей (в том числе Methanosarcina sp., Methanobacterium formicicum и Methanolobus psychrophilus) [119].
Что касается протеаз LonB, то они представлены в основном в архебактериях. Однако в течение последнего десятилетия протеазы с LonB-типом протеолитического центра (окружение каталитических остатков: ΦEGDS*AG и XXK*ΦE) обнаружены и у ряда термофильных бактерий (в частности, Marinithermus hydrothermalis, Meiothermus taiwanensis, T. thermophilus и др.). В своих АТРазных компонентах эти ферменты также проявляют подобие с LonB-протеазами. Однако они не способны к гидролизу АТР из-за вырожденности АТРазных центров вследствие замен существенных консервативных остатков в мотивах Уолкера и их окружении: GPХGXXKX (WA) и GGΦ4EAXXΦ (WB), а также на участках sensor-1 и sensor-2. Еще одной отличительной характеристикой новой группы ферментов служит наличие дополнительного по сравнению с LonB-протеазами α-спирализованного фрагмента размером 90–91 а.о. в экстрадомене, локализованном внутри потенциального ААА+-модуля (рис. 5) [100, 107, 121]. Несмотря на то что такие ферменты могут только связывать, но не гидролизовать АТР, они тоже проявляют способность к селективной деградации развернутых белковых субстратов, но по АТР-независимому механизму [121]. К настоящему времени именно это сообщество ферментов оформилось в немногочисленное (в пределах 10 представителей) третье подсемейство Lon-протеаз – LonС (рис. 5) [119].
Современные исследования протеаз семейства Lon направлены, прежде всего, на получение данных о пространственных структурах этих ферментов и механизмах их функционирования. При этом семейство Lon-протеаз достаточно полно охарактеризовано в части АТРазных и пептидгидролазных свойств представителей всех трех подсемейств [108, 122–127]. Вместе с тем вследствие особенностей организации некаталитической области структурно-функциональная характеристика самого крупного подсемейства LonА в целом остается недостаточно изученной, и для ряда научных групп она служит программой продолжающихся исследований.
LonA-протеазы – уникальный подкласс ААА+-белков
Индивидуальные субъединицы гомоолигомерных LonA-протеаз образованы пятью доменами (рис. 5). Функциональными выступают протеазный С-концевой домен (Р) и центральные нуклеотид-связывающий (NB) и α-спирализованный (H) домены, которые формируют АТРазный ААА+-модуль. В неординарной для ААА+-белков СКК некаталитической N-концевой области ферментов локализованы два домена: N-концевой (N, включающий 110–130 а.о. у бактериальных ферментов и 165–220 а.о. у эукариотических) и инсерционный (HI(CC), 178–186 а.о. у ферментов из любых источников).
Поскольку пространственные структуры полноразмерных LonA-протеаз до самого последнего времени не были известны, структурные сведения о представителях подсемейства ограничивались преимущественно 3D-данными для ряда фрагментов ферментов из различных источников: E. coli, B. subtilis, Brevibacillus thermoruber, M. taiwanensis, Mycobacterium avium и мозга человека (EcLon, BsLon, BtLon, MtLon, MaLon и HumLon соответственно). Эти данные полностью согласуются с предсказанными вторичными структурами LonA-протеаз и дают представление о трехмерных структурах четырех из пяти их доменов – N, NB, H и P [102–105, 128–135].
Результаты рентгеноструктурного анализа показали, что структуры доменов NB и Н, составляющих ААА+-модули LonA-протеаз, топологически подобны соответствующим фрагментам классических ААА+-белков (в частности, единственным D-модулям протеаз ClpХР и HslUV, а также D2-модулям шаперонов ClpB/Hsp104 и регуляторных компонентов АТР-зависимых протеаз ClpАР, ClpСР и ClpЕР) [63, 67, 122].
Отличительными именно для LonA-протеаз оказались структуры их N-концевых и протеазных доменов. Установлено, что β-структурированные N-домены бактериальных LonA-протеаз, состоящие из семи β-тяжей и двух α-спиральных фрагментов, обнаруживают выраженное сходство с N-доменом гипотетического белка BPP1347 из Bordetella parapertussis [102], который относится к группе РНК-связывающих белков, обладающих PUA-подобной архитектурой [136]. N-домены эукариотических LonA-протеаз проявляют высокое подобие с N-доменами бактериальных ферментов и при этом в большинстве своем содержат крупные вставочные фрагменты размером до 100 а.о. в шпильке между шестым и седьмым β‑тяжами [105].
Характеристический фолд Р-доменов, который отличает LonA-протеазы от других серин-лизиновых пептидгидролаз (в частности, от ферментов семейств S24 и S26 клана SF) [128], в 2004 г. послужил основой для выведения всего семейства Lon (S16) в индивидуальный структурный клан SJ в базе данных MEROPS [119].
И лишь упаковка индивидуального HI(CC)-домена, образованного восемью α-спиралями (α3–α10 в субъединице LonА), четыре из которых (α6–α9) по предсказанию могут формировать CC-участок в протомере фермента [101], до сих пор не определена. Вместе с тем известны кристаллические структуры некоторых укороченных бактериальных LonA-протеаз, которые наряду с одним или несколькими доменами фермента включают либо пять N-концевых спиралей HI(CC)-домена (форма EcLon (1–245) [104]), либо три его С-концевые спирали (формы EcLon (235–584), BsLon (240–774) и MtLon (242–793) [103, 105, 131]). Рассмотрение всех известных структурных фрагментов LonA-протеаз, полученных с помощью рентгеновской кристаллографии, показывает, что на настоящем этапе только для EcLon существует набор 3D-структур тех участков фермента, которые в совокупности покрывают его полную аминокислотную последовательность (рис. 6) [104, 105, 128, 129]. Это позволяет считать EcLon-протеазу не только базовым ферментом, но и структурной моделью для общего пула LonA-протеаз.
Рис. 6.
Кристаллические структуры фрагментов EcLon-протеазы: (1–245) (а), (235–584) (б) и (585–784) (в) (по данным работ Li et al. [104], Rotanova et al. [105] и Botos et al. [128]). (a) – N-домен (M1–Y116) показан оливковым цветом, спирали HI(CC)-домена показаны желтым (α3), голубым (α4), оранжевым (α5) и фиолетовым (α6 и α7, СС-участок) цветами; (б) – спирали HI(CC)-домена показаны фиолетовым (α8, α9, СС-участок) и темно-синим (α10) цветами, NB‑домен – зеленым, спирали H-домена окрашены так же, как спирали α(3–5) и α10 HI(CC)-домена; (в) – спирали Р-домена показаны синим цветом, β-тяжи – красным.

Различие функций доменов, формирующих N‑концевую область LonA-протеаз. Однозначное подтверждение двухдоменной организации N‑концевой области LonA-протеаз было получено экспериментально на примере EcLon-протеазы путем сравнительного исследования функционирования фермента и его модифицированных форм. При этом были использованы рекомбинантная форма EcLon, ее укороченные модификации, не содержащие либо консервативной части N‑домена (Lon-d106) [137], либо 172 N-концевых остатков вплоть до начала СС-участка (Lon-d172) [138], делеционные формы, лишенные полного HI(CC)-домена или его СС-участка (Lon-dHI(CC) и Lon-d(CC) соответственно [139, 140]), точечные мутанты с заменами потенциально важных остатков, локализованных в разных участках HI(CC)-домена (Lon-R164A, Lon-R192A и Lon-Y294A) [141], и тройной мутант LonEKR, несущий замены трех заряженных поверхностных остатков (E34, K35 и R38) в N-домене фермента [142] (рис. 7).
Рис. 7.
Модифицированные и мутантные формы EcLon-протеазы, использованные для изучения вклада некаталитической N-концевой области в функционирование фермента. Консервативные элементы: WА и WB – мотивы Уолкера, s1 и s2 – сенсорные остатки, Rf – остаток “аргининовый палец”, Ser679 и Lys722 – каталитические остатки. Показаны гексагистидиновые тэги (Н6-тэги) на С-конце EcLon и ее различных форм (в форме Lon-d172 Н6-тэг локализован на N-конце белка).
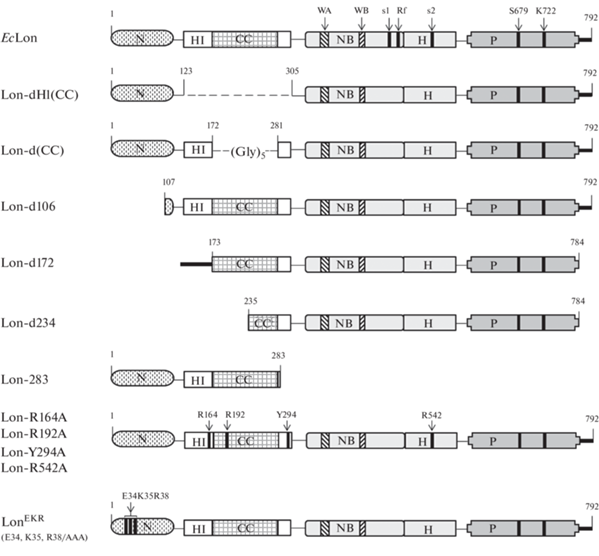
Изучение АТРазной, пептидазной и протеолитической активностей, а также аутолитического расщепления EcLon-протеазы и ее модифицированных форм, проведенное как в базовых условиях, так и при влиянии эффекторов (белковые субстраты в случае гидролиза АТР и нуклеотиды или их комплексы с ионами Mg2+ в случае других типов активности) показало, что оба домена N-концевой области важны для функционирования индивидуальных каталитических центров LonА-протеаз – АТРазного и пептидазного, а также для осуществления аллостерических взаимодействий между ними. При этом ключевую роль в реализации уникального процессивного механизма гидролиза белкового субстрата играет инсерционный HI(CC)-домен, который вовлечен в формирование функционально активной гексамерной структуры фермента и во взаимодействия ЕсLon-протеазы с нуклеотидами и белковыми субстратами. В отношении N-домена сделано заключение о том, что его основная функция – обеспечение конформационной стабильности ЕсLon-протеазы в классических условиях сопряжения протеолиза с гидролизом АТР.
Структурное сходство между LonA-протеазами и ClpB-шаперонами. Для получения представления о структуре инсерционного HI(CC)-домена в составе протомера фермента были предприняты попытки совмещения фрагментов EcLon-протеазы, представленных на рис. 6. Однако такая задача оказалась невыполнимой вследствие чрезвычайной гибкости региона, формирующего HI(CC)-домен. Тем не менее путем сравнительного анализа кристаллических структур форм EcLon, содержащих фрагменты HI(CC)-домена (рис. 6а, 6б), и АТРазных модулей ClpВ-шаперонов (рис. 8а) было показано, что LonA-протеазы могут представлять собой новый необычный подкласс AAA+-белков [105].
Рис. 8.
Кристаллическая структура шаперона TtClpB (а) и сопоставление фрагмента TtClpB (Е331–A854) (б) с фрагментами EcLon-протеазы (E124–D245) (в) и (A247–F584) (г), потенциально формирующими HI(CC)-домен (по данным работ Lee et al. [49] и Rotanova et al. [105]). (a) – Мономер TtClpB: N-домен показан золотистым цветом, NB1-домен – светло-зеленым, NB2-домен – темно-зеленым, вставочный М-домен – фиолетовым цветом; (б) – TtClpB, лишенный N- и NB1-доменов (Е331–A854); (в) – фрагмент HI(CC)-домена EcLon-протеазы (E124–D245); (г) – фрагмент EcLon(А235–F584), включающий С-концевую часть HI(CC)-домена и ААА+-модуль (домены NB и Н). Спирали доменов H1, H2 (TtClpB), HI(CC) и Н (EcLon) окрашены как на рис. 6. Серым фоном объединены фрагменты EcLon, составляющие HI(CC)-домен.
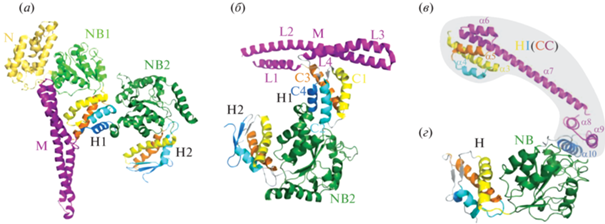
Основанием для такого анализа послужило обнаруженное сходство [100, 101, 105] первичных и вторичных структур HI(CC)-доменов LonA-протеаз и Н1-доменов D1-модулей шаперонов ClpB с внедренными в них пропеллерообразными М-доменами, обладающими СС-структурой (Н1(М)-фрагмент на рис. 2). Общий специфический признак сопоставляемых фрагментов – наличие “длинных” α-спиралей (55 а.о. у LonA-протеаз и 58 а.о. у ClpB-шаперонов, рис. 6а и 8а соответственно), степень подобия которых превышает 50% при общей степени сходства СС-сегментов ~36%.
Было проведено сопоставление α-спирализованной составляющей D1-модуля ClpB-шаперона T. thermophilus (TtClpB, домены Н1 и М на рис. 8б) и фрагментов EcLon, потенциально формирующих HI(CC)-домен фермента (рис. 8в и 8г – участки, объединенные серым фоном). Структурная эквивалентнность “длинных” спиралей (L2 и α7), совпадение общего количества спиралей в CC-сегментах (L1–L4 и α6–α9) и возможное сходство их топологического расположения с учетом различий в размерах малых спиралей (рис. 8б–8г) позволили выдвинуть гипотезу о том, что архитектура вставочного HI(CC)-домена LonА-протеаз может быть подобной архитектуре Н1(М)-фрагмента ClpB-шаперонов [100, 101, 105].
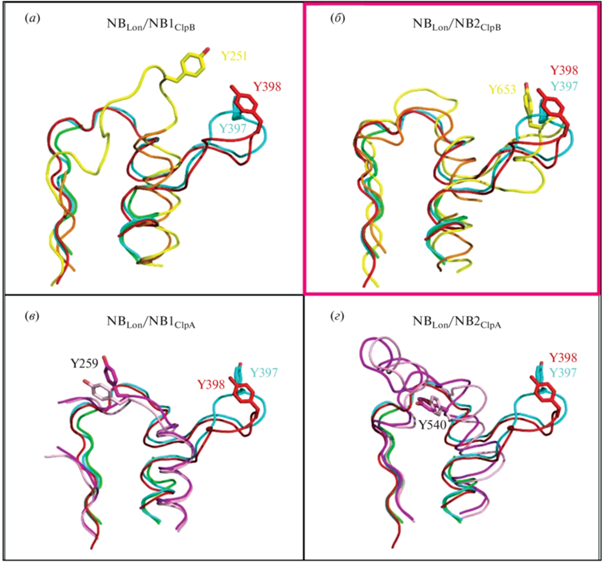
Данные в поддержку этого предположения были получены при сравнительном изучении расположения ключевой консервативной петли pore loop-1 (GYVG) ААА+-модулей в структурах LonA-протеаз и Clp-шаперонов, проведенном путем наложения соответствующих структурных фрагментов NB-доменов LonА-протеаз на фрагменты NB1- и NB2-доменов шаперонов ClpВ, а также топологически подобных им шаперонов ClpА, у которых отсутствует вставочный М-домен. При этом было установлено, что ориентация GYVG-петли в LonА-протеазах кардинально отличается от ее положения в NB1-доменах ClpВ и в обоих NB-доменах ClpА (рис. 9а, 9в и 9г), но однозначно и безусловно совпадает с ее расположением в NB2-доменах ClpB (рис. 9б) [105]. Полученные результаты позволили сделать заключение о том, что и предстоящие NB-доменам α-спирализованные HI(CC)-домены LonА-протеаз, и Н1(М)-фрагменты ClpB-шаперонов также могут проявлять определенное топологическое сходство.
Рис. 9.
Сопоставление фрагментов, включающих аксиальные петли pore loop-1 (GYVG), в кристаллических структурах NB-доменов LonА-протеаз и NB1(NB2)-доменов шаперонов ClpA и ClpB (по данным работы Rotanova et al. [105]). Цвета фрагментов последовательностей: EcLon – красный, BsLon – ярко-зеленый, MtLon – голубой, EcClpB – желтый, TtClpB – оранжевый, EcClpА-1 – розовый и EcClpA-2 – пурпурный.
Совокупные данные рентгеноструктурного анализа представляют достаточно обоснованные аргументы в пользу справедливости гипотезы о том, что инсерционный HI(CC)-домен можно рассматривать в качестве α-спирализованного компонента гипотетического добавочного ААА+-модуля, который мог локализоваться в LonА-протеазах подобно D1-модулю шаперонов ClpB (рис. 2), однако утратил свой нуклеотид-связывающий домен. На этом основании Rotanova et al. [105] предлагают признать протеазы подсемейства LonA особым, ранее неохарактеризованным подклассом AAA+-белков.
СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О СТРУКТУРНЫХ ОСОБЕННОСТЯХ И МЕХАНИЗМАХ ФУНКЦИОНИРОВАНИЯ LonA-ПРОТЕАЗ И ДРУГИХ ААА+-БЕЛКОВ СИСТЕМЫ КОНТРОЛЯ КАЧЕСТВА
В самое последнее время наметился значительный прогресс в определении трехмерных структур и выявлении особенностей фундаментальных механизмов функционирования ААА+-белков – этих динамических молекулярных машин, которые обеспечивают структурную базу для осуществления транслокации белковых субстратов. Основой достигнутых успехов служит широкое использование криоэлектронной микроскопии (крио-ЭМ) высокого разрешения, позволяющей визуализировать как конформационные превращения собственно ААА+-АТРаз, так и отдельные этапы фермент-субстратных взаимодействий у ААА+-протеаз системы контроля качества белков, включающие распознавание и связывание субстратов, их транслокацию и последующую деградацию. Новейшие достижения в этой области и перспективные направления новых исследований были представлены и подробно освещены на Втором cимпозиуме по ААА+-белкам (Keystone, 2020 [143]).
Спиральная организация гексамеров ААА+-белков – базовое условие транслокации белковых мишеней/субстратов
Представление об общей архитектуре ААА+-белков как циклических гексамеров было получено в результате их рентгеновской кристаллографии. При этом в ряде случаев ААА+-белки обнаруживались в виде открытых гексамерных колец или спиралевидных комплексов, что свидетельствовало о возможности больших конформационных изменений у этих ферментов [144]. Вместе с тем вопросы о взаимодействии ААА+-белков со своими субстратами и о путях сопряжения АТРазной и транслокационной функций долгое время оставались неизученными.
Прорывным исследованием в этом отношении оказалась работа 2013 г. [145], в которой при помощи крио-ЭМ впервые было показано, что взаиморасположение шести субъединиц (Rpt1–Rpt6) регуляторного 19S-комплекса дрожжевой 26S-протеасомы, связанной с субстратом, представляет тип правосторонней “винтовой лестницы”. Это открытие было подтверждено в последующие годы решением крио-ЭМ-структур более чем 50 ААА+-белков с различной функциональностью [39, 44, 144, 146–151]. В принятой на настоящий момент модели комплекса ААА+-белок*S (где S – белковый субстрат) АТРазные субъединицы (или протомеры) “обволакивают” со всех сторон белковую мишень, локализованную в вертикальном центральном канале ААА+-белка (рис. 10) [144]. При этом пять субъединиц (A–E), связанных с АТР, но имеющих различные конформации [144, 146, 152], непосредственно контактируют с белком-субстратом посредством петель pore loop-1 (GYVG), обращенных внутрь канала. В то же самое время шестая субъединица F (“стыковочная”, “seam”), пребывающая сначала в ADP-связанном, а затем в аро-состоянии, оказывается свободной от субстрата и располагается между верхней (А) и нижней (Е) субъединицами (рис. 10). Установлено, что соседние субъединицы, контактирующие с субстратом, разделены расстоянием в 6 Å (дистанция в 2 а.о. в последовательности субстрата) и повернуты друг относительно друга на 60° (отражение симметричности кольцевого гексамера). Особо следует отметить, что в отсутствие субстратов ААА+-белки могут принимать множество различных конформаций (формируя в том числе плоские симметричные кольца), что определяется различием их экстрадоменов и/или включением в некоторых случаях дополнительных вставочных элементов структуры [39, 144, 147].
Рис. 10.
Схема механизма разворачивания и последовательной транслокации полипептидного субстрата в ААА+-белках (по данным работ Puchades et al. [39] и Gates et al. [144]). Показано взаиморасположение протомеров (A–F) в гексамере ААА+-белка и их нуклеотидное состояние; отмечены сайты связывания ААА+-модулей в последовательности белка-субстрата; “seam” – стыковочная субъединица.
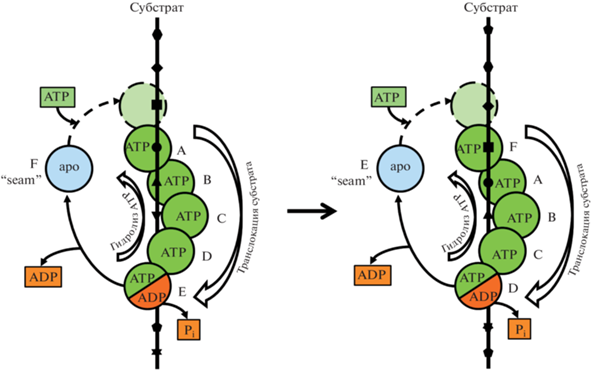
Механизм поэтапной транслокации белка-мишени, регулируемой гидролизом АТР, подтвержден для целого ряда ААА+-белков [39, 144, 146, 147], причем установлено, что ключевую роль в процессе транслокации играют крупномасштабные конформационные перестройки внутри ААА+-модулей, происходящие с участием междоменных линкеров. Аллостерическое влияние нуклеотидного состояния АТРазных субъединиц ААА+-гексамера приводит к различному расположению ключевых остатков тирозина петель GYVG в аксиальном канале, что обеспечивает прочный захват субстрата и реализацию последовательного кругового цикла гидролиза АТР (“around-the-ring ATP hydrolysis cycle”), первый этап которого происходит в нижней субъединице E. Образование ADP и высвобождение фосфата дестабилизируют междоменные взаимодействия внутри этой субъединицы (в частности, за счет увеличения угла поворота между NB- и H-доменами ААА+-модуля) и, кроме того, ослабляют ее взаимодействия с другими субъединицами (вследствие нарушения связи ISS-мотива и остатка “аргининовый палец” с нуклеотидом соседней субъединицы). Все это приводит к отсоединению субъединицы E как от субстрата, так и от смежной субъединицы D и ее передвижению к вершине спирали (рис. 10). Одновременно АТР-содержащие субъединицы (A–D), связанные с субстратом, продвигаются вниз по спирали в виде единой жесткой конструкции (rigid body), обеспечивая тем самым разворачивание и нисходящую транслокацию субстрата внутри центрального канала. Вместе с тем в отделенной на предыдущей стадии стыковочной seam-субъединице F происходит обмен ADP на АТР, и она снова становится способной к установлению стабильного взаимодействия с соседней субъединицей (A) и к связыванию с белковой мишенью в сайте, расположенном на 2 а.о. выше, чем сайт связывания субъединицы A (рис. 10). Таким образом, эта модель предполагает регулярное пошаговое перемещение ААА+-гексамера вдоль субстрата. При этом в каждом цикле транслокации гидролизуется одна молекула АТР, и в любой момент времени четыре субъединицы, связанные с АТР, остаются в контакте с субстратом, предотвращая его случайное “скольжение” в канале или высвобождение.
Для большей наглядности Puchades et al. [39] проводят аналогию между представленным “каскадным” механизмом транслокации и подтягиванием саней с помощью привязанной к ним веревки, когда согласованно работают шесть рук: они поочередно дотягиваются до веревки, схватывают ее и тянут (механизм “hand-over-hand” – “из руки в руку”). Руки попеременно отпускают веревку внизу и хватают ее сверху, при этом веревка все время остается плотно зажатой четырьмя “центральными” руками, и каждый цикл сопровождается тяговым усилием. Совокупность подобных движений в ААА+-белке обеспечивает постоянный захват и удерживание субстрата, а также его ступенчатую транслокацию, которая сочетается с последовательными круговыми циклами гидролиза АТР, протекающего в субъединицах гексамерного кольца шаперона в направлении против часовой стрелки (рис. 10).
Описанный выше основной механизм транслокации субстрата в направлении по часовой стрелке, называемый SC/2R (Sequential Clockwise/2-Residue step), зачастую рассматривается как универсальный для ААА+-белков, выполняющих разные функции [144, 146, 149]. Тем не менее существует ряд примеров функционирования белков по альтернативным механизмам. Так, транслокация белкового субстрата шапероном ClpX происходит по модели SC/6R, когда каждый цикл гидролиза АТР сопряжен с перемещением ААА+-гексамера сразу на 6 а.о. [153]. Кроме того, в работе Fei et al. [153] обсуждаются также модели транслокации, реализация которых приводит к изменению не только диапазона перемещения АТРазных субъединиц (вплоть до 30 а.о. и более), но и направления их передвижения (против часовой стрелки). Помимо этого, для некоторых ААА+-шаперонов обнаружено, что цикл транслокации субстрата может быть связан с перемещением не одной, а двух стыковочных АТРазных субъединиц [151].
Крио-ЭM-структуры EcLon и других LonA-протеаз
Проводимое в самые последние годы в ряде лабораторий изучение полноразмерных LonА-протеаз методом крио-ЭМ позволило получить новые данные о строении бактериальных ферментов из E. coli (EcLon) [154], M. taiwanensis (MtLon) [155] и Yersinia pestis (YpLon) [156], а также эукариотической LonА-протеазы человека (HumLon) [157]. Обнаружено, что подобно описанным выше индивидуальным ААА+-шаперонам АТРазные модули протомеров LonА-протеаз, выступающие центральными фрагментами единой полипептидной цепи этих сложных ферментов, которые фланкированы N-концевыми областями и С-концевыми протеазными доменами, также формируют активные гексамерные комплексы с архитектурой “винтовой лестницы” (табл. 4).
Таблица 4.
Характеристика структур LonА-протеаз, полученных методом крио-ЭМ
| Lon-протеаза, источник, а.о., активный центр |
Фрагмент с решенной структурой (ХХX–YYY), мутации |
Структурные данные | Характеристики LonA-протеазы | Ссылки | |||
|---|---|---|---|---|---|---|---|
| PDB ID, разре- шение |
условия определения | AAA+-модуль (ХХX–YYY), конфигурация, (число seam-субъединиц) |
протеазный домен (ХХX–YYY), конформация активного центра | ||||
| белковый субстрат; сайты связывания (ароматический остаток в петле) |
лиганд | ||||||
| EcLon, Escherichia coli, (1–784), S679/K722 |
(247–775), S679A |
6U5Z, 3.50 Å |
Отсутствует | ADP | (326–590), открытая левосторонняя “винтовая лестница”, (0) |
(591–775), аутоингибированный активный центр |
[154] |
| YpLon, Yersinia pestis, (1–784), S679/K722 |
(253–775) | 6V11, 3.80 Å |
Отсутствует | ADP | (326–590), открытая левосторонняя “винтовая лестница”, (0) |
(591–775), аутоингибированный активный центр |
[156] |
| (253–775), E424Q | 6ON2, 3.00 Å |
Y2853 (18 кДа), 7 видимых а.о.; pore loop-1 (Y398) в протомерах A–С |
ATP, ADP, Mg | Закрытая правосторонняя “винтовая лестница”, (2) | Функционально активный центр | ||
| MtLon, Meiothermus taiwanensis, (1–793), S678/K721 |
(245–780) | 6WQH, 3.60 Å |
Ig2 (25 кДа), 11 видимых а.о.; pore loop-1 (Y397) в протомерах A–D, pore loop-2 (W431) в протомерах A–D |
ATP-γ-S, ADP, Bortezomib |
(321–594), закрытая правосторонняя “винтовая лестница”, (2) |
(595–780), функционально активный центр |
[155] |
| HumLon, Homo sapiens, (115–959), S855/K898 |
(421–947) | 7KSL, 3.50 Å |
Отсутствует | ADP | (416–762), открытая левосторонняя “винтовая лестница”, (0) |
(763–947), аутоингибированный активный центр |
[157] |
| (416–947) | 7KSM, 3.2 Å |
Эндогенный субстрат неизвестной структуры,
12 видимых а.о.; pore loop-1 (Y565) в протомерах A–E, pore loop-2 (Y599) в протомерах A, B. Субстрат связан только с ААА+-гексамером |
ATP, ADP, Mg | Закрытая правосторонняя “винтовая лестница”, (1) | Аутоингибированный активный центр | ||
| (415–947) | 7KRZ, 3.2 Å |
Эндогенный субстрат неизвестной структуры,
12 видимых а.о.; pore loop-1 (Y565) в протомерах A–E. Субстрат связан с ААА+-гексамером и с Р-доменом |
ATP, ADP, Mg, Bortezomib | Закрытая правосторонняя “винтовая лестница”, (1) | Функционально активный центр | ||
| HumLon, Homo sapiens, (68–959), S855/K898 |
(410–949), G106P, R563W, K594M |
7P0B, 4.11 Å |
Отсутствует | ADP | (416–762), открытая левосторонняя “винтовая лестница”, (0) |
(763–949), аутоингибированный активный центр |
[160] |
| 7P09, 2.70 Å |
TFAM (25 кДа) (фрагмент), 11 видимых а.о.; pore loop-1 (Y565) в протомерах A–D, pore loop-2 (Y599) в протомерах A–C и E. Субстрат связан только с ААА+-гексамером |
ATP, ADP, Mg | Закрытая правосторонняя “винтовая лестница”, (1) | Функционально активный центр | |||
| (410–949), G106P, R563W, K594M, K898A |
7P0M, 1.75 Å |
TFAM (25 кДа) (фрагмент), 11 видимых а.о.; pore loop-1 (Y565) в протомерах A–F. Субстрат связан с ААА+-гексамером и с Р-доменом |
ATP, ADP, Mg | Закрытая правосторонняя “винтовая лестница”, (1) | Функционально активный центр с мутацией K898A. Субстрат связан с S855 |
||
Примечание: Y2853 – белок-регулятор сенсорной трансдукции из Y. pestis; Ig2 – домены 5 и 6 гелеобразующего фактора АВР-120 из Dictyostelium discoideum; TFAM – транскрипционный фактор А митохондрий; ATP-γ-S – аденозин-5'-[γ-тио]трифосфат; Bortezomib – N-[(1R)-1-(дигидроксиборил)-3-метилбутил]-N-(пиразин-2-илкарбонил)-L-фенилаланинамид.
Вместе с тем следует подчеркнуть, что долгое время даже с помощью крио-ЭМ не удавалось выявить структуру N-концевых областей полноразмерных LonA-протеаз, включающих N-домен и большую часть вставочного α-спирализованного HI(CC)-домена, вследствие высокой гибкости этого региона в протомерах ферментов (табл. 4). При этом использование электронной микроскопии низкого разрешения (ЭМ) для изучения укороченной HumLon-протеазы, лишенной N-домена, показало, что фрагменты HI(CC)-доменов противостоящих субъединиц фермента объединяются в димерные пары (A–D, B–E и C–F), из которых складывается тример димеров, формирующий вход в аксиальный канал, образованный ААА+-модулями фермента [158]. Эти данные успешно подтверждены вновь полученными с помощью крио-ЭМ структурами полноразмерных бактериальной LonA-протеазы из Thermus thermophilus (TtLonA) [159] и HumLon-протеазы [160, 161]. Есть основания полагать, что выявленная организация N-концевых областей LonA-протеаз топологически подобна структурам входных отверстий в аксиальные каналы, сформированным N-концевыми экстрадоменами других шаперонов-транслоказ (ClpX, ClpB, ААА+-АТРазы эукариотических и архейных протеасом [88, 91, 92, 148]) (см. подраздел “Основные характеристики некаталитических экстрадоменов АТР-зависимых протеаз”). Можно ожидать, что углубленный анализ новых структур даст возможность получить также ответы на вопросы о позиции и структурной роли уникальных N-доменов LonA-протеаз, служащих структурными аналогами PUA-доменов (см. подраздел “Различие функций доменов, формирующих N-концевую область LonA-протеаз”), но пока остаются недостаточно охарактеризованными.
На примере полноразмерной EcLon-протеазы методом ЭМ было также обнаружено сосуществование двух олигомерных форм фермента – гексамерной и додекамерной, где додекамеры образованы в результате непосредственного взаимодействия N-концевых областей гексамеров EcLon по типу “голова к голове” [154, 162, 163]. Детали структуры, механизм функционирования и биологическую значимость додекамерной формы LonA-протеаз еще предстоит выяснить.
Участники LonA-подсемейства, исследованные методом крио-ЭМ, характеризуются высоким сходством своих первичных структур (рис. 11). Так, EcLon и YpLon содержат 91.1% консервативных остатков, а общая степень подобия этих ферментов составляет 98.5%. Степень подобия между EcLon и MtLon несколько ниже – 77.9% (при 50.1% консервативных остатков), практически те же цифры характеризуют пару EcLon/TtLon (77.2 и 50.3%), а подобие между EcLon и HumLon составляет 70.7% (34.7% консервативных остатков). Консервативны для всех четырех ферментов 27.2% а.о. (общая степень подобия 55.4%).
Рис. 11.
Сопоставление первичных и вторичных структур LonA-протеаз, для которых получены структурные данные методом крио-ЭМ. Источники LonA-протеаз: Ec – Escherichia coli (MER0000485), Yp – Yersinia pestis (MER0015528), Mt – Meiothermus taiwanensis (MER1037479), Tt – Thermus thermophilus (MER0006064), Hum – Homo sapiens (MER0000495). Наложение первичных структур проведено с помощью программы https://www.ebi.ac.uk/Tools/msa/clustalo/. Элементы вторичных структур ферментов представлены согласно рентгеноструктурным данным (жирный шрифт, красный цвет – α-спирали, розовый – 310-спирали, синий – β-тяжи, курсив – фрагменты, не видимые в кристаллических структурах). В последовательностях Yp, Mt и Hum обычным шрифтом показаны фрагменты, не видимые в крио-ЭМ-структурах; для Tt рентгеноструктурные данные отсутствуют. Показаны консенсусные фрагменты: мотивы Уолкера (WA и WB), петли pore loop-1 и pore loop-2 с ключевыми остатками Tyr и (Met, Trp, Tyr) соответственно, ISS-фрагмент и RKH-петля, остатки сенсор-1 и сенсор-2 (s1 и s2) и “аргининовый палец” (Rf), каталитические остатки серина (S*) и лизина (K*), важный для активности остаток треонина (Т#).
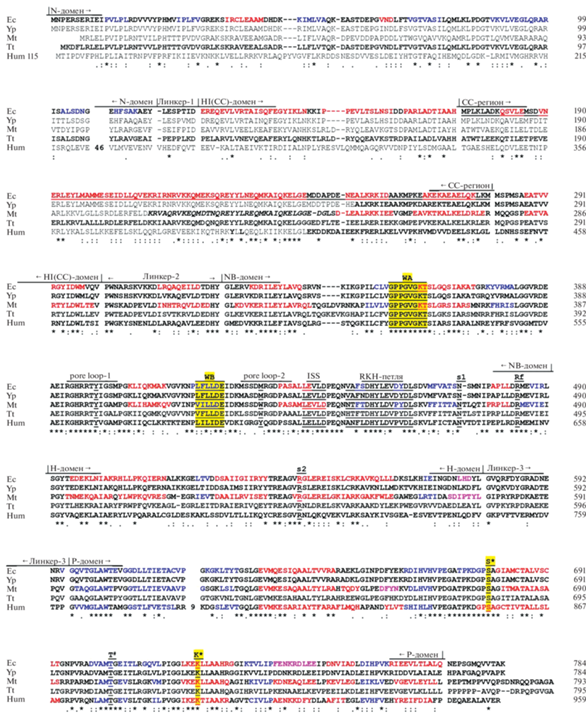
Наименьший уровень сходства обнаруживают N-домены (39.3%), близки по степени подобия HI(CC)- и Н-домены (50.8 и 52.8% соответственно), высокое сходство проявляют нуклеотид-связывающие и протеолитические домены (79.4 и 66.1%). Несмотря на это, в элементах вторичной структуры Р-доменов EcLon, YpLon и HumLon обнаруживаются заметные отличия (в частности, в области каталитического остатка серина, рис. 11), что, однако, практически не нарушает их общей топологии.
Установлено (табл. 4), что в отсутствие транслоцируемого белкового субстрата спиральные гексамеры ААА+-модулей LonА-протеаз, включающие связанный ADP, принимают открытую левостороннюю конфигурацию [154–157, 160]. При этом HumLon олигомеризуется с меньшим шагом спирали, чем ее бактериальные ортологи (EcLon и YpLon) [157]. Той же левосторонней конфигурации соответствует пространственная организация протеазных доменов ферментов, причем их каталитические центры, экспонированные во внешнюю среду вследствие образования обширной полости между верхним и нижним протомерами, пребывают в неактивных конформациях (состояние фермента – “выключено”, “OFF”) [154–157, 160]. Проявляемое “аутоингибирование” активных центров протеаз позволяет предотвратить вероятную в этих условиях беспорядочную деградацию белковых молекул клеточного протеома.
В присутствии белка-субстрата гексамеры LonА-протеаз, содержащие также молекулы ADP, АТР (или его медленно гидролизующегося аналога АТР-γ-S) и ионы Mg2+, подвергаются реорганизации в активное состояние, отвечающее за транслокацию субстрата. Эти изменения приводят к формированию вокруг транслоцируемых полипептидов закрытых спиральных правосторонних ААА+-конформеров и к перестройке протеазных доменов с образованием симметричных планарных структур с активными каталитическими центрами (состояние ферментов – “включено”, “ON”) [154–157, 160]. При этом в структурах ферментов их ААА+-кольца оказываются наклоненными по отношению к центральной оси 6-го порядка колец Р-доменов [160]. Применение борорганического ингибитора бортезомиба приводит к “закреплению” активной конформации протеазных центров [155, 157]. Показано, что конформация активного фермента сохраняется также при введении в HumLon мутаций как по мотиву Уолкера WB, так и по каталитическому остатку лизина [157, 160]. Следует отметить, что важную роль при переходе протеазных доменов от спиральной организации к плоскому состоянию играют линкерные фрагменты, соединяющие ААА+-модули с Р-доменами (рис. 11). На примере YpLon-протеазы отмечено, что левосторонняя спираль гексамера фермента (“OFF”) значительно круче ее правостороннего конформера (“ON”) [156].
Shin et al. [156] полагают, что переход гексамера LonА-протеазы из состояния “OFF” в состояние “ON” инициируется связыванием АТР верхним протомером и взаимодействием его петли pore loop-1 с белковой мишенью, что обеспечивает энергию, необходимую для реализации начального этапа локального разворачивания субстрата, за которым следует процесс последовательного гидролиза АТР, сопровождающийся пошаговой транслокацией развернутого полипептида в протеазную камеру для деградации.
Описанные к настоящему времени структуры функционально активных LonА-протеаз кроме сходства общей архитектуры, определяемой конфигурацией ААА+-модуля, проявляют ряд заметных отличий, обусловленных, главным образом, связыванием белков-субстратов. Так, установлено, что АТРазные кольца бактериальных ферментов (MtLon и YpLon) состоят из четырех нисходящих по спирали ААА+-модулей, взаимодействующих с субстратом, и двух восходящих стыковочных субъединиц [155, 156], в то время как у эукариотической HumLon-протеазы в связывании с субстратом участвуют пять ААА+-модулей, и только одна субъединица выполняет функцию стыковочной [157]. При этом в работе Gesé et al. [160] описана АТР-связанная стыковочная субъединица HumLon, что противоречит нуклеотидному статусу seam-субъединиц, соответствующему основному механизму транслокации (“аро” или комплекс с ADP) (рис. 10). Авторы считают, что обнаруженное состояние отражает промежуточную стадию, которая непосредственно следует за обменом нуклеотидов в стыковочном протомере и предшествует его повторному вовлечению в связывание субстрата.
Еще одно отличие связано с тем, что у некоторых LonA-протеаз во взаимодействии с субстратом кроме основных петель pore loop-1, включающих критически важный остаток Tyr, могут участвовать также петли pore loop-2 при условии, что в них содержится остаток ароматической аминокислоты, способный к связыванию с основной цепью белка-субстрата (ферменты MtLon и HumLon в табл. 4). Дополнительные взаимодействия с петлями pore loop-2 позволяют визуализировать более протяженные фрагменты последовательностей связанных субстратов (7 а.о. в структуре YpLon и 11–12 а.о. в структурах MtLon или HumLon) [155–157, 160].
Наиболее впечатляющим различием служит тот факт, что для активации протеазных доменов у бактериальных ферментов достаточно “захвата” полипептидного субстрата только ААА+-модулями, в то время как для эукариотической HumLon-протеазы дополнительным требованием выступает взаимодействие субстрата также и с протеазными активными центрами (табл. 4) [155–157, 160]. Важная характеристика HumLon – наличие АТР-зависимой шапероновой активности, причем способность к сворачиванию ряда митохондриальных белковых субстратов сохраняется и у протеолитически неактивного фермента [157]. Авторы полагают, что шапероновая и протеолитическая функции – это независимо регулируемые функции HumLon-протеазы, а возможность дифференцированного взаимодействия субстратов с ААА+-модулями и Р-доменами служит проявлением функциональной пластичности, необходимой для регулирования разнообразных ролей этого фермента в митохондриях [157].
ЗАКЛЮЧЕНИЕ
Охарактеризована система контроля качества белков (СКК), играющая ключевую роль в поддержании сохранности клеточного протеома во всех природных царствах, изложены основные сведения о строении и структурных особенностях участников СКК – молекулярных шаперонов, АТР-зависимых протеаз и мультисубъединичных комплексов, а также обобщены современные данные о структуре, функциональных свойствах и механизме действия Lon-протеаз, представляющих одно из важнейших семейств СКК. Значительное внимание уделено характеристике ферментов подсемейства LonA, которые, как показано, можно отнести к особому подклассу ААА+-белков.
Отдельный раздел посвящен применению метода криоэлектронной микроскопии к изучению представителей СКК. Отмечено, что с помощью этого метода определены структуры целого ряда протеаз и шаперонов ААА+-суперсемейства, обладающих транслоказными свойствами, а также выявлены как общие этапы, так и различающиеся стадии в уникальных механизмах их функционирования. Несомненным успехом явилось также определение крио-ЭМ-структур полноразмерных HumLon- и TtLon-протеаз, что позволило смоделировать N-концевые области этих молекул, строение которых ранее не было известно. Однако вследствие низкого разрешения и недостаточного качества карт электронной плотности детали структур на атомном уровне в этих областях ферментов остаются нераскрытыми. При этом очевидно, что несмотря на схожесть общей ориентации характеристических длинных спиралей в HI(CC)-доменах бактериального и эукариотического ферментов, их фактическая архитектура значительно различается. В этой связи остаются актуальными дальнейшие углубленные исследования молекулярных принципов и степени консервативности механизмов действия ААА+-белков СКК (в том числе Lon-протеаз), поскольку они вовлечены в протекание таких процессов, как окислительный стресс, нейродегенеративные нарушения, проявление вирулентности и канцерогенез, что дает основание рассматривать их в качестве перспективных диагностических биомаркеров и терапевтических мишеней при лечении ряда патологий.
Список литературы
Balchin D., Hayer-Hartl M., Hartl F.U. // Science. 2016. V. 353. P. aac4354. https://doi.org/10.1126/science.aac4354
Gottesman S., Wickner S., Maurizi M.R. // Genes Dev. 1997. V. 11. P. 815–823. https://doi.org/10.1101/gad.11.7.815
Kovacs D., Szabo B., Pancsa R., Tompa P. // Arch. Biochem. Biophys. 2013. V. 531. P. 80–89. https://doi.org/10.1016/j.abb.2012.09.010
Jeng W., Lee S., Sung N., Lee J., Tsai F.T.F. // F1000Res. 2015. V. 4. P. 1448. https://doi.org/10.12688/f1000research.7214.1
Schubert U., Anton L.C., Gibbs J., Norbury C.C., Yewdell J.W., Bennink J.R. // Nature. 2000. V. 404. P. 770–774. https://doi.org/10.1038/35008096
Clausen L., Abildgaard A.B., Gersing S.K., Stein A., Lindorff-Larsen K., Hartmann-Petersen R. // Adv. Protein Chem. Struct. Biol. 2019. V. 114. P. 61–83. https://doi.org/10.1016/bs.apcsb.2018.09.002
Mogk A., Huber D., Bukau B. // Cold Spring Harb. Perspect. Biol. 2011. V. 3. https://doi.org/10.1101/cshperspect.a004366
Finka A., Mattoo R.U.H., Goloubinoff P. // Annu. Rev. Biochem. 2016. V. 85. P. 715–742. https://doi.org/10.1146/annurev-biochem-060815-014124
Мельников Э.Э., Ротанова Т.В. // Биоорг. химия. 2010. Т. 36. С. 5–14. [Melnikov E.E., Rotanova T.V. // Russ. J. Bioorg. Chem. 2010. V. 36. P. 1–10.] https://doi.org/10.1134/S1068162010010012
Hoffmann A., Bukau B., Kramer G. // Biochim. Biophys. Acta. 2010. V. 1803. P. 650–661. https://doi.org/10.1016/j.bbamcr.2010.01.017
Jee H. // J. Exerc. Rehabil. 2016. V. 12. P. 255–259. https://doi.org/10.12965/jer.1632642.321
Richter K., Haslbeck M., Buchner J. // Mol. Cell. 2010. V. 40. P. 253–266. https://doi.org/10.1016/j.molcel.2010.10.006
Saibil H. // Nat. Rev. Mol. Cell Biol. 2013. V. 14. P. 630–642. https://doi.org/10.1038/nrm3658
Dahiya V., Buchner J. // Adv. Protein Chem. Struct. Biol. 2019. V. 114. P. 1–60. https://doi.org/10.1016/bs.apcsb.2018.10.001
Mattoo R.U.H., Goloubinoff P. // Cell. Mol. Life Sci. 2014. V. 71. P. 3311–3325. https://doi.org/10.1007/s00018-014-1627-y
Weibezahn J., Bukau B., Mogk A. // Microbial. Cell Fact. 2004. V. 3. P. 1. https://doi.org/10.1186/1475-2859-3-1
Bascos N.A.D., Landry S.J. // Int. J. Mol. Sci. 2019. V. 20. P. 6195. https://doi.org/10.3390/ijms20246195
Voos W. // Biochim. Biophys. Acta. 2013. V. 1833. P. 388–399. https://doi.org/10.1016/j.bbamcr.2012.06.005
Chakafana G., Zininga T., Shonhai A. // Biomolecules. 2019. V. 9. P. 543. https://doi.org/10.3390/biom9100543
Rosenzweig R., Nillegoda N.B., Mayer M.P., Bukau B. // Nat. Rev. Mol. Cell Biol. 2019. V. 20. P. 665–680. https://doi.org/10.1038/s41580-019-0133-3
Kampinga H.H., Craig E.A. // Nat. Rev. Mol. Cell Biol. 2010. V. 11. P. 579–592. https://doi.org/10.1038/nrm2941
Takakuwa J.E., Nitika, Knighton L.E., Truman A.W. // Front. Mol. Biosci. 2019. V. 6. P. 81. https://doi.org/10.3389/fmolb.2019.00081
Walter S. // Cell. Mol. Life Sci. 2002. V. 59. P. 1589–1597. https://doi.org/10.1007/pl00012485
Bar-Lavan Y., Shemesh N., Ben-Zvi A. // Essays Biochem. 2016. V. 60. P. 237–253. https://doi.org/10.1042/EBC20160004
Taguchi H. // J. Mol. Biol. 2015. V. 427. P. 2912–2918. https://doi.org/10.1016/j.jmb.2015.04.007
Chaudhuri T.K., Verma V.K., Maheshwari A. // Prog. Biophys. Mol. Biol. 2009. V. 99. P. 42–50. https://doi.org/10.1016/j.pbiomolbio.2008.10.007
Prodromou C. // Biochem. J. 2016. V. 473. P. 2439–2452. https://doi.org/10.1042/BCJ20160005
Li J., Buchner J. // Biomed. J. 2013. V. 36. P. 106–117. https://doi.org/10.4103/2319-4170.113230
Pearl L.H. // Biopolymers. 2016. V. 105. P. 594–607. https://doi.org/10.1002/bip.22835
Luengo T.M., Mayer M.P., Rüdiger S.G.D. // Trends Cell Biol. 2019. V. 29. P. 164–177. https://doi.org/10.1016/j.tcb.2018.10.004
Genest O., Wickner S., Doyle S.M. // J. Biol. Chem. 2019. V. 294. P. 2109–2120. https://doi.org/10.1074/jbc.REV118.002806
Haslbeck M., Vierling E. // J. Mol. Biol. 2015. V. 427. P. 1537–1548. https://doi.org/10.1016/j.jmb.2015.02.002
Mogk A., Ruger-Herreros C., Bukau B. // Annu. Rev. Microbiol. 2019. V. 73. P. 89–110. https://doi.org/10.1146/annurev-micro-020518-115515
Obuchowski I., Liberek K. // Cell Stress Chaperones. 2020. V. 25. P. 593–600. https://doi.org/10.1007/s12192-020-01094-0
Haslbeck M., Weinkauf S., Buchner J. // J. Biol. Chem. 2019. V. 294. P. 2121–2132. https://doi.org/10.1074/jbc.REV118.002809
Riedl M., Strauch A., Catici D.A.M., Haslbeck M. // Int. J. Mol. Sci. 2020. V. 21. P. 5448. https://doi.org/10.3390/ijms21155448
Waters E.R., Vierling E. // New Phytol. 2020. V. 227. P. 24–37. https://doi.org/10.1111/nph.16536
Neuwald A.F., Aravind L., Spouge J.L., Koonin E.V. // Genome Res. 1999. V. 9. P. 27–43. https://doi.org/10.1101/gr.9.1.27
Puchades C., Sandate C.R., Lander G.C. // Nat. Rev. Mol. Cell Biol. 2020. V. 21. P. 43–58. https://doi.org/10.1038/s41580-019-0183-6
Ogura T., Wilkinson A.J. // Genes Cells. 2001. V. 6. P. 575–597. https://doi.org/10.1046/j.1365-2443.2001.00447.x
Dougan D.A., Mogk A., Zeth K., Turgay K., Bukau B. // FEBS Lett. 2002. V. 529. P. 6–10. https://doi.org/10.1016/s0014-5793(02)03179-4
Voos W., Jaworek W., Wilkening A., Bruderek M. // Essays Biochem. 2016. V. 60. P. 213–225. https://doi.org/10.1042/EBC20160009
Abrahão J., Mokry D.Z., Ramos C.H.I. // Front. Mol. Biosci. 2017. V. 4. P. 60. https://doi.org/10.3389/fmolb.2017.00060
Yokom A.L., Gates S.N., Jackrel M.E., Mack K.L., Su M., Shorter J., Southworth D.R. // Nat. Struct. Mol. Biol. 2016. V. 23. P. 830–837. https://doi.org/10.1038/nsmb.3277
Barends T.R., Werbeck N.D., Reinstein J. // Curr. Opin. Struct. Biol. 2010. V. 20. P. 46–53. https://doi.org/10.1016/j.sbi.2009.12.014
Miller J.M., Lin J., Li T., Lucius A.L. // J. Mol. Biol. 2013. V. 425. P. 2795–2812. https://doi.org/10.1016/j.jmb.2013.04.019
Doyle S.M., Wickner S. // Trends Biochem. Sci. 2009. V. 34. P. 40–48. https://doi.org/10.1016/j.tibs.2008.09.010
Zolkiewski M., Zhang T., Nagy M. // Arch. Biochem. Biophys. 2012. V. 520. P. 1–6. https://doi.org/10.1016/j.abb.2012.01.012
Lee S., Sowa M.E., Watanabe Y.H., Sigler P.B., Chiu W., Yoshida M., Tsai F.T. // Cell. 2003. V. 115. P. 229–240. https://doi.org/10.1016/s0092-8674(03)00807-9
Schramm F.D., Schroeder K., Jonas K. // FEMS Microbiol. Rev. 2020. V. 44. P. 54–72. https://doi.org/10.1093/femsre/fuz026
Voos W. // Res. Microbiol. 2009. V. 160. P. 718–725. https://doi.org/10.1016/j.resmic.2009.08.003
Mogk A., Kummer E., Bukau B. // Front. Mol. Biosci. 2015. V. 2. P. 22. https://doi.org/10.3389/fmolb.2015.00022
Watanabe Y.H., Nakazaki Y., Suno R., Yoshida M. // Biochem. J. 2009. V. 421. P. 71–77. https://doi.org/10.1042/BJ20082238
Gottesman S. // Annu. Rev. Cell Dev. Biol. 2003. V. 19. P. 565–587. https://doi.org/10.1146/annurev.cellbio.19.110701.153228
Koodathingal P., Jaffe N.E., Kraut D.A., Prakash S., Fishbain S., Herman C., Matouschek A. // J. Biol. Chem. 2009. V. 284. P. 18674–18684. https://doi.org/10.1074/jbc.M900783200
Sauer R.T., Baker T.A. // Annu. Rev. Biochem. 2011. V. 80. P. 587–612. https://doi.org/10.1146/annurev-biochem-060408-172623
Mahmoud S.A., Chien P. // Annu. Rev. Biochem. 2018. V. 87. P. 677–696. https://doi.org/10.1146/annurev-biochem-062917-012848
Maupin-Furlow J.A. // Subcell Biochem. 2013. V. 66. P. 297–327. https://doi.org/10.1007/978-94-007-5940-4_11
Ciechanover A. // Cell. 1994. V. 79. P. 13–21. https://doi.org/10.1016/0092-8674(94)90396-4
Bard J.A.M., Goodall E.A., Greene E.R., Jonsson E., Dong K.C., Martin A. // Annu. Rev. Biochem. 2018. V. 87. P. 697–724. https://doi.org/10.1146/annurev-biochem-062917-011931
Ciechanover A., Stanhill A. // Biochim. Biophys. Acta. 2014. V. 1843. P. 86–96. https://doi.org/10.1016/j.bbamcr.2013.07.007
Kudriaeva A., Kuzina E.S., Zubenko O., Smirnov I.V., Belogurov A., Jr. // FASEB J. 2019. V. 33. P. 6852–6866. https://doi.org/10.1096/fj.201802237R
Iyer L.M., Leipe D.D., Koonin E.V., Aravind L. // J. Struct. Biol. 2004. V. 146. P. 11–31. https://doi.org/10.1016/j.jsb.2003.10.010
Lupas A.N., Martin J. // Curr. Opin. Struct. Biol. 2002. V. 12. P. 746–753. https://doi.org/10.1016/s0959-440x(02)00388-3
Chang C.W., Lee S., Tsai F.T.F. // Front. Mol. Biosci. 2017. V. 4. P. 27. https://doi.org/10.3389/fmolb.2017.00027
Miller J.M., Enemark E.J. // Archaea. 2016. V. 2016. P. 9294307. https://doi.org/10.1155/2016/9294307
Wendler P., Ciniawsky S., Kock M., Kube S. // Biochim. Biophys. Acta. 2012. V. 1823. P. 2–14. https://doi.org/10.1016/j.bbamcr.2011.06.014
Ogura T., Whiteheart S.W., Wilkinson A.J. // J. Struct. Biol. 2004. V. 146. P. 106–112. https://doi.org/10.1016/j.jsb.2003.11.008
Martin A., Baker T.A., Sauer R.T. // Mol. Cell. 2008. V. 29. P. 441–450. https://doi.org/10.1016/j.molcel.2008.02.002
Gur E., Ottofueling R., Dougan D.A. // Subcell. Biochem. 2013. V. 66. P. 3–33. https://doi.org/10.1007/978-94-007-5940-4_1
Kirstein J., Schlothauer T., Dougan D.A., Lilie H. Tischendorf G., Mogk A., Bukau B., Turgay K. // EMBO J. 2006. V. 25. P. 1481–1491. https://doi.org/10.1038/sj.emboj.7601042
Mogk A., Bukau B. // Curr. Biol. 2004. V. 14. P. 78–80. https://doi.org/10.1016/j.cub.2003.12.051
Ротанова Т.В., Мельников Э.Э. // Биомед. химия. 2008. Т. 54. С. 512–530. [Rotanova T.V., Melnikov E.E. // Biochem. Moscow Suppl. Ser. B. Biomed. Chem. 2008. V. 2. P. 245–257.] https://doi.org/10.1134/S1990750808030049
Sauer R.T., Bolon D.N., Burton B.M., Burton R.E., Flynn J.M., Grant R.A., Hersch G.L., Joshi S.A., Kenniston J.A., Levchenko I., Neher S.B., Oakes E.S., Siddiqui S.M., Wah D.A., Baker T.A. // Cell. 2004. V. 119. P. 9–18. https://doi.org/10.1016/j.cell.2004.09.020
Kim Y.I., Levchenko I., Fraczkowska K., Woodruff R.V., Sauer R.T., Baker T.A. // Nat. Struct. Biol. 2001. V. 8. P. 230–233. https://doi.org/10.1038/84967
Olivares A.O., Baker T.A., Sauer R.T. // Nat. Rev. Microbiol. 2016. V. 14. P. 33–44. https://doi.org/10.1038/nrmicro.2015.4
Kirstein J., Moliere N., Dougan D.A., Turgay K. // Nat. Rev. Microbiol. 2009. V. 7. P. 589–599. https://doi.org/10.1038/nrmicro2185
Mukherjee S., Bree A.C., Liu J, Patrick J.E., Chien P., Kearns D.B. // Proc. Natl. Acad. Sci. USA. 2015. V. 112. P. 250–255. https://doi.org/10.1073/pnas.1417419112
Lo J.H., Baker T.A., Sauer R.T. // Protein Sci. 2001. V. 10. P. 551–559. https://doi.org/10.1110/ps.41401
Kojetin D.J., McLaughlin P.D., Thompson R.J., Dubnau D., Prepiak P., Rance M., Cavanagh J. // J. Mol. Biol. 2009. V. 387. P. 639–652. https://doi.org/10.1016/j.jmb.2009.01.046
Elsholz A.K.W., Birk M.S., Charpentier E., Turgay K. // Front. Mol. Biosci. 2017. V. 4. P. 44. https://doi.org/10.3389/fmolb.2017.00044
Hinnerwisch J., Reid B.G., Fenton W.A., Horwich A.L. // J. Biol. Chem. 2005. V. 280. P. 40838–40844. https://doi.org/10.1074/jbc.M507879200
Dougan D.A., Reid B.G., Horwich A.L., Bukau B. // Mol. Cell. 2002. V. 9. P. 673–683. https://doi.org/10.1016/s1097-2765(02)00485-9
Wang F., Mei Z., Qi Y., Yan C., Qi H., Wang J., Shi Y. // Nature. 2011. V. 471. P. 331–335. https://doi.org/10.1038/nature09780
Mogk A., Schlieker C., Strub C., Rist W., Weibezahn J., Bukau B. // J. Biol. Chem. 2003. V. 278. P. 17615–17624. https://doi.org/10.1074/jbc.M209686200
Cashikar A.G., Schirmer E.C., Hattendorf D.A., Glover J.R., Ramakrishnan M.S., Ware D.M., Lindquist S.L. // Mol. Cell. 2002. V. 9. P. 751–760. https://doi.org/10.1016/S1097-2765(02)00499-9
Wendler P., Shorter J., Snead D., Plisson C., Clare D.K., Lindquist S., Saibil H.R. // Mol. Cell. 2009. V. 34. P. 81–92. https://doi.org/10.1016/j.molcel.2009.02.026
Donaldson L.W., Wojtyra U., Houry W.A. // J. Biol. Chem. 2003. V. 278. P. 48991–48996. https://doi.org/10.1074/jbc.M307826200
Miethke M., Hecker M., Gerth U. // J. Bacteriol. 2006. V. 188. P. 4610–4619. https://doi.org/10.1128/JB.00287-06
Tomko R.J., Jr., Hochstrasser M. // Cell Biochem. Biophys. 2011. V. 60. P. 13–20. https://doi.org/10.1007/s12013-011-9178-4
Zhang F., Hu M., Tian G., Zhang P., Finley D., Jeffrey P.D., Shi Y. // Mol. Cell. 2009. V. 34. P. 473–484. https://doi.org/10.1016/j.molcel.2009.04.021
Djuranovic S., Hartmann M.D., Habeck M., Ursinus A., Zwickl P., Martin J., Lupas A.N., Zeth K. // Mol. Cell. 2009. V. 34. P. 580–590. https://doi.org/10.1016/j.molcel.2009.04.030
Zhang F., Wu Z., Zhang P., Tian G., Finley D., Shi Y. // Mol. Cell. 2009. V. 34. P. 485–496. https://doi.org/10.1016/j.molcel.2009.04.022
Sousa M.C., Trame C.B., Tsuruta H., Wilbanks S.M., Reddy V.S., McKay D.B. // Cell. 2000. V. 103. P. 633–643. https://doi.org/10.1016/S0092-8674(00)00166-5
Sundar S., Baker T.A., Sauer R.T. // Protein Sci. 2012. V. 21. P. 188–198. https://doi.org/10.1002/pro.2001
Scharfenberg F., Serek-Heuberger J., Coles M., Hartmann M.D., Habeck M., Martin J., Lupas A.N., Alva V. // J. Mol. Biol. 2015. V. 427. P. 910–923. https://doi.org/10.1016/j.jmb.2014.12.024
Ramelot T.A., Yang Y., Sahu I.D., Lee H.W., Xiao R., Lorigan G.A., Montelione G.T., Kennedy M.A. // FEBS Lett. 2013. V. 587. P. 3522–3528. https://doi.org/10.1016/j.febslet.2013.09.009
An J.Y., Sharif H., Kang G.B., Park K.J., Lee J.G., Lee S., Jin M.S., Song J.J., Wang J., Eom S.H. // Biochem. Biophys. Res. Commun. 2018. V. 495. P. 1201–1207. https://doi.org/10.1016/j.bbrc.2017.11.158
Langklotz S., Baumann U., Narberhaus F. // Biochim. Biophys. Acta. 2012. V. 1823. P. 40–48. https://doi.org/10.1016/j.bbamcr.2011.08.015
Rotanova T.V., Melnikov E.E., Khalatova A.G., Makhovskaya O.V., Botos I., Wlodawer A., Gustchina A. // Eur. J. Biochem. 2004. V. 271. P. 4865–4871. https://doi.org/10.1111/j.1432-1033.2004.04452.x
Ротанова Т.В., Дергоусова Н.И., Морозкин А.Д. // Биоорг. химия. 2013. Т. 39. С. 303–319. [Rotanova T.V., Dergousova N.I., Morozkin A.D. // Russ. J. Bioorg. Chem. 2013. V. 39. P. 268–282.] https://doi.org/10.1134/S1068162013030114
Li M., Rasulova F., Melnikov E.E., Rotanova T.V., Gustchina A., Maurizi M.R., Wlodawer A. // Protein Sci. 2005. V. 14. P. 2895–2900. https://doi.org/10.1110/ps.051736805
Duman R.E., Löwe J. // J. Mol. Biol. 2010. V. 401. P. 653–670. https://doi.org/10.1016/j.jmb.2010.06.030
Li M., Gustchina A., Rasulova F.S., Melnikov E.E., Maurizi M.R., Rotanova T.V., Dauter Z., Wlodawer A. // Acta Cryst. 2010. V. 66. P. 865–873. https://doi.org/10.1107/S090744491001955
Rotanova T.V., Andrianova A.G., Kudzhaev A.M., Li M., Botos I., Wlodawer A., Gustchina A. // FEBS Open Bio. 2019. V. 9. P. 1536–1551. https://doi.org/10.1002/2211-5463.12691
Маховская О.В., Козлов С., Ботос И., Степнов А.А., Андрианова А.Г., Гущина А.Е., Влодавер А., Мельников Э.Э., Ротанова Т.В. // Биоорг. химия. 2007. Т. 33. С. 657–660. [Makhovskaya O.V., Kozlov S., Botos I., Stepnov A.A., Andrianova A.G., Gustchina A.E., Wlodawer A., Melnikov E.E., Rotanova T.V. // Russ. J. Bioorg. Chem. 2007. V. 33. P. 610–613.] https://doi.org/10.1134/S1068162007060131
Liao J.H., Kuo C.I., Huang Y.Y., Lin Y.C., Lin Y.C., Yang C.Y., Wu W.L., Chang W.H., Liaw Y.C., Lin L.H., Chang C.I., Wu S.H., Liao J.H., Kuo C.I., Huang Y.Y., Lin Y.C., Lin Y.C. // PLoS One. 2012. V. 7. e40226. https://doi.org/10.1371/journal.pone.0040226
Li J.K., Liao J.H., Li H., Kuo C.I., Huang K.F., Yang L.W., Wu S.H., Chang C.I. // Acta Cryst. 2013. V. 69. P. 1789–1797. https://doi.org/10.1107/S090744491301500X
Gur E., Sauer R.T. // Genes Dev. 2008. V. 22. P. 2267–2277. https://doi.org/10.1101/gad.1670908
Ngo J.K., Pomatto L.C.D., Davies K.J.A. // Redox Biol. 2013. V. 1. P. 258–264. https://doi.org/10.1016/j.redox.2013.01.015
Cheng C.W., Kuo C.Y., Fan C.C., Fang W.C., Jiang S.S., Lo Y.K., Wang T.Y., Kao M.C., Lee A.Y.L. // Cell Death Dis. 2013. V. 4. e681. https://doi.org/10.1038/cddis.2013.204
Liu Y., Lan L., Huang K., Wang R., Xu C., Shi Y., Wu X., Wu Z., Zhang J., Chen L., Wang L., Yu X., Zhu H., Lu B. // Oncotarget. 2014. V. 5. P. 11209–11224. https://doi.org/10.18632/oncotarget.2026
Bulteau A.L., Bayot A. // Biochim. Biophys. Acta. 2011. V. 1807. P. 595–601. https://doi.org/10.1016/j.bbabio.2010.12.011
Voos W., Pollecker K. // Biomolecules. 2020. V. 10. P. 253. https://doi.org/10.3390/biom10020253
Lu B. // Adv. Exp. Med. Biol. 2017. V. 1038. P. 8173–8182. https://doi.org/10.1007/978-981-10-6674-0_12
Pinti M., Gibellini L., Liu Y., Xu S., Lu, B., Cossarizza A. // Cell. Mol. Life Sci. 2015. V. 72. P. 4807–4824. https://doi.org/10.1007/s00018-015-2039-3
Quiros P.M., Espanol Y., Acin-Perez R., Rodriguez F., Barcena C., Watanabe K., Calvo E., Loureiro M., Fernandez-Garcia M.S., Fueyo A., Vazquez J., Enriquez J.A., Lopez-Otin C. // Cell Rep. 2014. V. 8. P. 542–556. https://doi.org/10.1016/j.celrep.2014.06.018
Bota D.A., Davies K.J.A. // Free Rad. Biol. Med. 2016. V. 100. P. 188–198. https://doi.org/10.1016/j.freeradbiomed.2016.06.031
Rawlings N.D., Barrett A.J., Thomas P.D., Huang X., Bateman A., Finn R.D. // Nucl. Acids Res. 2018. V. 46. P. 624–632. https://doi.org/10.1093/nar/gkx1134
Swamy K.S., Goldberg A.L. // Nature. 1981. V. 292. P. 652–654. https://doi.org/10.1038/292652a0
Liao J.H., Ihara K., Kuo C.I., Huang K.F., Wakatsuki S., Wu S.H., Chang C.I. // Acta Cryst. 2013. V. 69. P. 1395–1402. https://doi.org/10.1107/S0907444913008214
Bittner L.M., Arends J., Narberhaus F. // Biopolymers. 2016. V. 105. P. 505–517. https://doi.org/10.1002/bip.22831
Goldberg A.L., Waxman L. // J. Biol. Chem. 1985. V. 260. P. 12029–12034. https://doi.org/10.1016/S0021-9258(17)38980-9
Patterson-Ward J., Tedesco J., Hudak J., Fishovitz J., Becker J., Frase H., McNamara K., Lee I. // Biochim. Biophys. Acta. 2009. V. 1794. P. 1355–1363. https://doi.org/10.1016/j.bbapap.2009.02.015
Vineyard D., Patterson-Ward J., Berdis A.J., Lee I. // Biochemistry. 2005. V. 44. P. 1671–1682. https://doi.org/10.1021/bi048618z
Besche H., Tamura N., Tamura T., Zwickl P. // FEBS Lett. 2004. V. 574. P. 161–166. https://doi.org/10.1016/j.febslet.2004.08.021
Botos I., Melnikov E.E., Cherry S., Kozlov S., Makhovskaya O.V., Tropea J.E., Gusthchina A., Rotanova T.V., Wlodawer A. // J. Mol. Biol. 2005. V. 351. P. 144–157. https://doi.org/10.1016/j.jmb.2005.06.008
Botos I., Melnikov E.E., Cherry S., Tropea J., Khalatova A.G., Rasulova F.S., Dauter Z., Maurizi M.R., Rotanova T.V., Wlodawer A., Gustchina A. // J. Biol. Chem. 2004. V. 279. P. 8140–8148. https://doi.org/10.1074/jbc.M312243200
Botos I., Melnikov E.E., Cherry S., Khalatova A.G., Rasulova F.S., Tropea J.E., Maurizi M.R., Rotanova T.V., Gustchina A., Wlodawer A. // J. Struct. Biol. 2004. V. 146. P. 113–122. https://doi.org/10.1016/j.jsb.2003.09.003
Lee A.Y., Chen Y.D., Chang Y.Y., Lin Y.C., Chang C.F., Huang S.J., Wu S.H., Hsu C.H. // Acta Cryst. 2014. V. 70. P. 218–230. https://doi.org/10.1107/S139900471302631X
Lin C.C., Su S.C., Su M.Y., Liang P.H., Feng C.C., Wu S.H., Chang C.I. // Structure. 2016. V. 24. P. 667–675. https://doi.org/10.1016/j.str.2016.03.001
Tzeng S.R., Tseng Y.C., Lin C.C., Hsu C.Y., Huang S.J., Kuo Y.T., Chang C.I. // eLife. 2021. V. 10. P. e64056. https://doi.org/10.7554/eLife.64056
Su S.C., Lin C.C., Tai H.C., Chang M.Y., Ho M.R., Babu C.S., Liao J.H., Wu S.H., Chang Y.C., Lim C., Chang C.I. // Structure. 2016. V. 24. P. 676–686. https://doi.org/10.1016/j.str.2016.03.003
Chen X., Zhang S., Bi F., Guo C., Feng L., Wang H., Yao H., Lin D. // Protein Sci. 2019. V. 28. P. 1720–1726. https://doi.org/10.1002/pro.3687
Garcia-Nafria J., Ondrovicova G., Blagova E., Levdikov V.M., Bauer J.A., Suzuki C.K., Kutejova E., Wilkinson A.J., Wilson K.S. // Protein Sci. 2010. V. 19. P. 987–999. https://doi.org/10.1002/pro.376
Bertonati C., Punta M., Fischer M., Yachdav G., Forouhar F., Zhou W., Kuzin A.P., Seetharaman J., Abashidze M., Ramelot T.A., Kennedy M.A., Cort J.R., Belachew A., Hunt J.F., Tong L., Montelione G.T., Rost B. // Proteins. 2009. V. 75. P. 760–773. https://doi.org/10.1002/prot.22287
Куджаев А.М., Дубовцева Е.С., Серова О.В., Андрианова А.Г., Ротанова Т.В. // Биоорг. химия. 2016. Т. 42. С. 421–430. [Kudzhaev A.M., Dubovtseva E.S., Serova O.V., Andrianova A.G., Rotanova T.V. // Russ. J. Bioorg. Chem. 2016. V. 42. P. 381–388.] https://doi.org/10.1134/S1068162016040142
Андрианова А.Г., Куджаев А.М., Дубовцева Е.С., Ротанова Т.В. // Биоорг. химия. 2017. Т. 43. С. 357–366. [Andrianova A.G., Kudzhaev A.M., Dubovtseva E.S., Rotanova T.V. // Russ. J. Bioorg. Chem. 2017. V. 43. P. 368–376.] https://doi.org/10.1134/S1068162017040021
Куджаев А.М., Андрианова А.Г., Дубовцева Е.С., Серова О.В., Ротанова Т.В. // Acta Naturae. 2017. Т. 9. С. 79–86. [Kudzhaev A.M., Andrianova A.G., Dubovtseva E.S., Serova O.V., Rotanova T.V. // Acta Naturae. 2017. V. 9. P. 75–81.] https://doi.org/10.32607/20758251-2017-9-2-75-81
Куджаев А.М., Дубовцева Е.С., Серова О.В., Андрианова А.Г., Ротанова Т.В. // Биоорг. химия. 2018. Т. 44. С. 522–532. [Kudzhaev A.M., Dubovtseva E.S., Serova O.V., Andrianova A.G., Rotanova T.V. // Russ. J. Bioorg. Chem. 2018. V. 44. P. 518–527.] https://doi.org/10.1134/S0132342318050081
Куджаев А.М., Андрианова А.Г., Серова О.В., Архипова В.А., Дубовцева Е.С., Ротанова Т.В. // Биоорг. химия. 2015. Т. 41. С. 579–586. [Kudzhaev A.M., Andrianova A.G., Serova O.V., Arkhipova V.A., Dubovtseva E.S., Rotanova T.V. // Russ. J. Bioorg. Chem. 2015. V. 41. P. 518–524.] https://doi.org/10.1134/S1068162015050076
Андрианова А.Г., Куджаев А.М., Абрикосова В.А., Гущина А.Е., Смирнов И.В., Ротанова Т.В. // Acta Naturae. 2020. Т. 12. С. 86–97. [Andrianova A.G., Kudzhaev A.M., Abrikosova V.A., Gustchina A., Smirnov I.V., Rotanova T.V. // Acta Naturae. 2020. V. 12. P. 102–113.] https://doi.org/10.32607/actanaturae.11197
Glynn S.E., Kardon J.L., Mueller-Cajar O., Cho C. // Nat. Struct. Mol. Biol. 2020. V. 27. P. 515–518. https://doi.org/10.1038/s41594-020-0444-2
Gates S.N., Martin A. // Protein Sci. 2019. V. 29. P. 407–419. https://doi.org/10.1002/pro.3743
Matyskiela M.E., Lander G.C., Martin A. // Nat. Struct. Mol. Biol. 2013. V. 20. P. 781–788. https://doi.org/10.1038/nsmb.2616
Puchades C., Rampello A.J., Shin M., Giuliano C.J., Wiseman R.L., Glynn S.E., Lander G.C. // Science. 2017. V. 358. P. eaao0464. https://doi.org/10.1126/science.aao0464
Uchihashi T., Watanabe Y.H., Nakazaki Y., Yamasaki T., Watanabe H., Maruno T., Ishii K., Uchiyama S., Song C.H., Murata K., Iino R., Ando T. // Nat. Commun. 2018. V. 9. P. 2147. https://doi.org/10.1038/s41467-018-04587-w
Rizo A.N., Lin J.B., Gates S.N., Tse E., Bart S.M., Castellano L.M., DiMaio F., Shorter J., South-worth D.R. // Nat. Commun. 2019. V. 10. P. 2393. https://doi.org/10.1038/s41467-019-10150-y
Majumder P., Rudack T., Beck F., Danev R., Pfeifer G., Nagy I., Baumeister W. // Proc. Natl. Acad. Sci. USA. 2019. V. 116. P. 534–539. https://doi.org/10.1073/pnas.1817752116
Lee S., Roh S.H., Lee J., Sung N., Liu J., Tsai F.T.F. // Cell Rep. 2019. V. 26. P. 29–36. https://doi.org/10.1016/j.celrep.2018.12.037
Zhang S., Mao Y. // Biomolecules. 2020. V. 10. P. 629. https://doi.org/10.3390/biom10040629
de la Pena A.H., Goodall E.A., Gates S.N., Lander G.C., Martin A. // Science. 2018. V. 362. P. eaav0725. https://doi.org/10.1126/science.aav0725
Fei X., Bell T.A., Jenni S., Stinson B.M., Baker T.A., Harrison S.C., Sauer R.T. // eLife. 2020. V. 9. P. e52774. https://doi.org/10.7554/eLife.52774
Botos I., Lountos G.T., Wu W., Cherry S., Ghirlando R., Kudzhaev A.M., Rotanova T.V., de Val N., Tropea J., Gustchina A., Wlodawer A. // Curr. Res. Struct. Biol. 2019. V. 1. P. 13–20. https://doi.org/10.1016/j.crstbi.2019.10.001
Li S., Hsieh K.Y., Su S.C., Pintilie G.D., Zhang K., Chang C.I. // J. Biol. Chem. 2021. V. 297. P. 101239. https://doi.org/10.1016/j.jbc.2021.101239
Shin M., Puchades C., Asmita A., Puri N., Adjei E., Wiseman R.L., Karzai A.W., Lander G.C. // Sci. Adv. 2020. V. 6. P. eaba8404. https://doi.org/10.1126/sciadv.aba8404
Shin M., Watson E.R., Song A.S., Mindrebo J.T., Novick S.J., Griffin P.R., Wiseman R.L., Lander G.C. // Nat. Commun. 2021. V. 12. P. 3239. https://doi.org/10.1038/s41467-021-23495-0
Kereiche S., Kovacik L., Bednar J., Pevala V., Kunova N., Ondrovicova G., Bauer J., Ambro L., Bellova J., Kutejova E., Raska I. // Sci. Rep. 2016. V. 6. P. 33631. https://doi.org/10.1038/srep33631
Coscia F., Löwe J. // FEBS Lett. 2021. V. 595. P. 2691–2700. https://doi.org/10.1002/1873-3468.14199
Gesé G.V., Shahzad S., Pardo-Hernández C., Wramstedt A., Falkenberg M., Hällberg B.M. // bioRxiv. 2021. https://doi.org/10.1101/2021.1106.1109.447696
Mohammed I., Schmitz K.A., Schenck N., Topitsch A., Maier T., Abrahams J.P. // bioRxiv. 2021. https://doi.org/10.1101/2021.1107.1128.454137
Vieux E.F., Wohlever M.L., Chen J.Z., Sauer R.T., Baker T.A. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 2002–2008. https://doi.org/10.1073/pnas.1307066110
Brown B.L., Vieux E.F., Kalastavadi T., Kim S.R., Chen J.Z., Baker T.A. // Protein Sci. 2018. V. 28. P. 1239–1251. https://doi.org/10.1002/pro.3553
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия