

Биоорганическая химия, 2022, T. 48, № 5, стр. 520-530
Роль линкерных гистонов в канцерогенезе
А. В. Любителев 1, М. П. Кирпичников 1, 2, В. М. Студитский 1, 3, *, **
1 Московский государственный университет имени М.В. Ломоносова, кафедра биоинженерии биологического факультета
119234 Москва, Ленинские горы, 1, стр. 12, Россия
2 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН”
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
3 Fox Chase Cancer Center
19111 Pennsylvania, Philadelphia, Cottman avenue 333, USA
* E-mail: varanus-salvator@yandex.ru
** E-mail: vasily.studitsky@fccc.edu
Поступила в редакцию 27.08.2020
После доработки 05.09.2020
Принята к публикации 10.09.2020
- EDN: LZABKT
- DOI: 10.31857/S0132342321010140
Аннотация
Линкерные гистоны представляют собой ДНК-связывающие архитектурные белки хроматина, участвующие в формировании наднуклеосомных уровней упаковки хроматина. У млекопитающих известно 11 вариантов этих белков, однако функциональное значение такого разнообразия на настоящий момент до конца не определено. Помимо основной структурной функции линкерные гистоны участвуют в регуляторных взаимодействиях, таких как глобальное нарушение регуляции генов в процессе канцерогенеза. В свою очередь, эти изменения при канцерогенезе могут воздействовать как на линкерные гистоны через мутации кодирующих их генов или изменения в экспрессии этих генов, так и на регуляторные системы клетки, вызывающие перераспределение вариантов линкерных гистонов в интерфазном хроматине, нарушающие их посттрансляционные модификации или формирующие с ними функционально активные комплексы. В некоторых случаях изменения в метаболизме линкерных гистонов могут являться возможной причиной злокачественной трансформации, а также выступать в роли возможных прогностических маркеров. Обсуждаются возможные механизмы таких изменений в канцерогенезе, позволяющие также лучше понять функции вариантов линкерных гистонов и доменов этих белков в нормальных клетках.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ.......................................................520
СТРОЕНИЕ И ВАРИАНТЫ ЛИНКЕРНЫХ ГИСТОНОВ.............................521
ИЗМЕНЕНИЕ ЭКСПРЕССИИ ЛИНКЕРНЫХ ГИСТОНОВ В РАКОВЫХ КЛЕТКАХ..............522
МУТАЦИИ ЛИНКЕРНЫХ ГИСТОНОВ В РАКОВЫХ КЛЕТКАХ...................................523
ВАРИАНТЫ ЛИНКЕРНЫХ ГИСТОНОВ КАК ПРОГНОСТИЧЕСКИЕ МАРКЕРЫ.......526
ЗАКЛЮЧЕНИЕ.................................................527
СПИСОК ЛИТЕРАТУРЫ.................................527
ВВЕДЕНИЕ
Генетический аппарат клетки имеет тонко регулируемую иерархическую организацию, основной структурной единицей которой является нуклеосома. Нуклеосома представляет собой октамер гистонов (Н2А/Н2В/Н3/Н4)2 [1], вокруг которого обернута ДНК длиной приблизительно 147 п.н. [2], формирующих нуклеосомное ядро. Гистоны нуклеосомного ядра являются важнейшими компонентами систем эпигенетической регуляции транскрипционной и репликационной активности, их посттрансляционные модификации отмечают домены активного и неактивного хроматина. Нарушения эпигенетической регуляции отмечены многими авторами в качестве одного из важнейших механизмов канцерогенеза [3, 4]. Еще один, линкерный, гистон Н1 связывается в области входа ДНК в нуклеосомную частицу. Функции гистона Н1, или линкерного гистона, по ряду причин гораздо менее изучены, чем функции гистонов нуклеосомного ядра, что существенно ограничивает понимание эпигенетической регуляции и механизмов канцерогенеза, в которых этот белок может принимать участие. Вместе с тем установлено, что в клетках по меньшей мере некоторых раковых опухолей экспрессия и распределение в интерфазном хроматине различных вариантов линкерных гистонов подвергается значительным изменениям [5, 6]. В свете сведений о том, что варианты линкерного гистона могут принимать участие в регуляции плюрипотентности клеточных линий и раннего эмбрионального развития [7, 8], изучение роли линкерного гистона в канцерогенезе может расширить наше понимание как механизмов злокачественной трансформации, так и биологических функций различных вариантов гистона Н1.
СТРОЕНИЕ И ВАРИАНТЫ ЛИНКЕРНЫХ ГИСТОНОВ
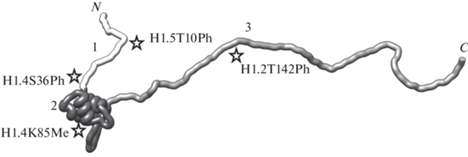
По своей структуре гистон Н1 существенно отличается от гистонов нуклеосомного ядра. Линкерные гистоны не демонстрируют характерной для Н2А, Н2В, Н3 и Н4 гистоновой укладки (α‑спираль, ограниченная двумя более короткими α-спиралями по краям); вместо этого их глобулярная часть свернута в мотив “спираль с крылом”, представляющий собой укладку типа “спираль-поворот-спираль” с небольшой β-шпилькой ближе к С-концу и характерной для разнообразных ДНК-связывающих белков (рис. 1). Важно отметить, что поверхность глобулярного домена заряжена практически целиком положительно [9], в то время как в составе других белков этот мотив обычно несет ярко выраженный дипольный момент [10]. Протяженный С-концевой фрагмент линкерных гистонов насчитывает около 100 а.о. и несет сильный суммарный положительный заряд за счет остатков лизина, аланина и пролина [11] (рис. 1). В водном растворе этот фрагмент не формирует вторичной структуры, однако в присутствии нейтральных детергентов [12] и при связывании с ДНК [12–14] способен формировать вторичную структуру. Данные экспериментов по регистрации spFRET от комплексов гистона Н1 с ДНК и нуклеосомами свидетельствуют о том, что в составе таких комплексов С-концевой фрагмент способен формировать компактную глобулоподобную структуру [15, 16]. За счет высокого суммарного положительного заряда именно этот участок молекул линкерных гистонов, как полагают, отвечает за компактизацию ДНК. N-концевой фрагмент линкерных гистонов значительно короче, чем С-концевой (рис. 1), и вносит значительно меньший вклад в связывание Н1 с нуклеосомой [17].
Рис. 1.
Схема строения молекулы гистона Н1: 1 – N-концевой хвост, 2 – глобулярный домен, 3 – С-концевой хвост. Указаны приблизительные положения сайтов посттрансляционных модификаций различных вариантов линкерных гистонов.
У человека, как и у других млекопитающих, насчитывается 11 различных вариантов линкерного гистона [18], семь из которых экспрессируется в соматических клетках (Н1.1–Н1.5, Н1.0 и Н1.х), тогда как остальные четыре – в половых клетках и их предшественниках (H1t, H1T2, HILS1 и H1oo) [19]. Те из линкерных гистонов, что характерны для соматических клеток, делятся на два вида: экспрессирующиеся в течение S-периода (Н1.1–Н1.5) и экспрессирующиеся конститутивно (Н1.0 и Н1.х); последние называют также замещающими вариантами линкерных гистонов, поскольку в хроматине терминально дифференцированных клеток остальные варианты Н1 заменяются именно на них [18, 19]. В последние годы получены сведения о том, что глобулярные домены определенных вариантов линкерных гистонов способны формировать с нуклеосомой два различающихся по своей геометрии вида комплексов, один из которых характеризуется размещением глобулы линкерного гистона на оси симметрии частицы, а другой – несколько в стороне от нее [20, 21]. Молекулярно-динамические расчеты, вместе с тем, показывают, что в том или ином хроматиновом контексте один и тот же вариант гистона Н1 может формировать оба типа комплексов [22]. Варианты линкерных гистонов различаются по сродству к хроматину [23, 24] и способности его конденсировать [23, 25], а также по скорости обмена после связывания с ним [26, 27]. Несмотря на существующие между этими вариантами различия в аминокислотной последовательности, соотнести их с конкретными особенностями функций каждого из них оказалось затруднительно, поскольку эти варианты, судя по всему, по меньшей мере частично взаимозаменяемы [28]. Так, было установлено, что эмбрионы мышей, нокаутных по генам Н1.0 [29], Н1t [30] или Н1.1 [31], без отклонений развивались y животных, не отличающихся от мышей дикого типа. Никаких существенных отклонений не было обнаружено и при двойном нокауте по гену Н1.0 и гену одного из трех вариантов Н1, экспрессирующихся в S‑периоде: Н1.2, Н1.3 или Н1.4 [28]. Только тройной нокдаун по Н1.2, Н1.3 и Н1.4 привел к гибели эмбрионов мышей в процессе развития [32]. В то же время имеются указания на специфические функции различных соматических вариантов Н1 в клетке, поскольку селективное ингибирование их экспрессии в клетках рака молочной железы приводило к различным последствиям для клеточного метаболизма [33]. Такое противоречие может быть обусловлено тем, что компенсаторные механизмы, обеспечивающие выживание мышей, нокаутных по нескольким вариантам линкерных гистонов, могут не работать в клетках, подвергшихся злокачественной трансформации, либо должны быть запущены на ранних стадиях эмбриогенеза.
ИЗМЕНЕНИЕ ЭКСПРЕССИИ ЛИНКЕРНЫХ ГИСТОНОВ В РАКОВЫХ КЛЕТКАХ
Известно, что в ряде случаев дерегуляция экспрессии генома клетки, ведущая к злокачественной трансформации, сопровождается общей декомпактизацией клеточного хроматина.
На основании данных высокочувствительного секвенирования мРНК линкерных гистонов из Атласа раковых геномов (The Cancer Genome Atlas) было установлено, что транскрипция Н1 часто демонстрирует отличия от нормы, с преобладанием вариантов, экспрессирующихся во время клеточного цикла. Вместе с тем выявляются и специфические изменения в транскрипции этих генов, характерные только для опухолей определенных типов [6].
Анализ транскриптом клеток аденом и аденокарцином яичников при помощи количественной ПЦР позволил выявить ряд различий между профилями экспрессии различных вариантов линкерных гистонов в клетках злокачественных и доброкачественных опухолей [34]. Так, в клетках аденокарцином было выявлено заметное снижение содержания мРНК гистонов Н1.0, Н1.1, Н1.4 и Н1.х, тогда как содержание мРНК Н1.3 было существенно повышено. Выявлено было также общее снижение количества мРНК всех вариантов линкерных гистонов в злокачественных клетках примерно на 40%, произошедшее в основном за счет снижения количества мРНК гистона Н1.0, наиболее характерного для высокодифференцированных клеток. Такие изменения могут быть причиной глобальной декомпактизации хроматина клеток злокачественных опухолей (рис. 2а). Применение выявленных различий в качестве критерия злокачественности позволило верно определить тип опухолевых клеток для 32 из 33 проанализированных образцов. Вместе с тем прямо связать изменения в содержании мРНК с изменениями на уровне белков в случае линкерных гистонов затруднительно, поскольку регуляция их содержания в клетке осуществляется также на уровне процессинга мРНК [35] и скорости деградации белка [36, 37]. Примечательно, что более поздняя работа выявила значительное снижение пролиферационной активности культуры клеток рака яичника OVCAR-3 при повышении экспрессии гистона Н1.3 [38]. Было установлено, что Н1.3 является селективным ингибитором экспрессии небелкового канцерогена – РНК Н19. Эти данные указывают на то, что одни и те же изменения в экспрессии генов линкерных гистонов могут приводить в разных типах раковых клеток к различным эффектам.
Рис. 2.
Возможные механизмы канцерогенеза с участием линкерных гистонов. (а) – Декомпактизация хроматина в результате падения экспрессии линкерных гистонов; (б) – перераспределение вариантов линкерных гистонов внутри хроматина под воздействием повреждений регуляторных систем клетки. (в) – нарушение связывания линкерных гистонов с хроматином в результате мутации; (г) – нарушение взаимодействия регуляторных белков с линкерными гистонами по механизму приобретения или утраты функции.

Различия в экспрессии генов Н1 возможны, судя по всему, не только между раковыми клетками и клетками, не претерпевшими злокачественной трансформации, но и между различными клетками гетерогенных раковых опухолей. Так, для клеток культуры генно-инженерных фибробластов с канцерогенными свойствами [39] было продемонстрировано существенное снижение содержания Н1.0 в клетках со стволовыми свойствами по сравнению с дифференцированными клетками [5]. Похожая ситуация была выявлена и в культурах клеток глиобластомы и рака груди, где высокодифференцированные опухоли демонстрировали более высокое содержание гистона Н1.0, чем низкодифференцированные. Выявлены были также различия в метилировании CpG-островков ДНК гена Н1.0: клетки, проявлявшие свойства стволовых, демонстрировали существенно повышенный уровень метилирования области вокруг промотора гена Н1.0. Образцы опухолевых клеток естественного происхождения также демонстрируют обратную зависимость между уровнем метилирования ДНК области промотора гена Н1.0 и содержанием кодируемого этим геном белка. При этом повышение уровня экспрессии Н1.0 генно-инженерными методами в клетках со стволовыми свойствами существенно снижало их пролиферативный и опухолеформирующий потенциал, тогда как снижение экспрессии Н1.0 имело обратный эффект. Снижение экспрессии гена Н1.0 приводило к воспроизводимым, хотя чаще всего и незначительным, изменениям экспрессии более 800 генов, располагающихся преимущественно в АТ-богатых участках хромосом. При этом в клетках с низким содержанием Н1.0, предпочтительно связывающегося с GC-богатыми областями ДНК [40], наблюдалось снижение количества связанного Н1.0 именно в АТ-богатых областях, характерных для сайтов старта транскрипции генов, отвечающих за стволовые свойства, и обладающих высокой механической жесткостью, снижающей стабильность формирующихся на таких участках нуклеосом [41].
Функциональная гетерогенность раковых опухолей приобретает все большее клиническое значение [42], в силу чего данные о роли различных вариантов Н1, и особенно Н1.0 как маркера высокодифференцированных клеток, в формировании этой гетерогенности также могут быть важны для формирования стратегий терапии злокачественных образований.
МУТАЦИИ ЛИНКЕРНЫХ ГИСТОНОВ В РАКОВЫХ КЛЕТКАХ
Было установлено, что мутации гистонов нуклеосомного ядра являются одним из важнейших механизмов злокачественной трансформации клеток [43]. Наилучшим образом изучена роль мутации Н3К37М, препятствующей работе эпигенетических регуляторных механизмов клетки [44]. Систематические мутации гистона Н1 поддаются изучению значительно хуже, поскольку функции различных его вариантов и роль посттрансляционных модификаций таких вариантов в эпигенетической регуляции гораздо менее изучены. Тем не менее повторяющиеся мутации генов линкерных гистонов были обнаружены в образцах клеток фолликулярной лимфомы [45, 46], хронической лимфоцитарной лейкемии [47], диффузной крупноклеточной В-лимфомы [48] и раковых опухолей толстой кишки [49].
В клетках фолликулярной лимфомы мутации в генах Н1 были выявлены для 27% образцов, в шести из которых были обнаружены множественные мутации; большую их часть составляли соматические миссенс-мутации [45]. Высокая частота мутаций генов Н1 в клетках фолликулярной лимфомы была подтверждена независимым исследованием [46]. Вместе с тем установить точные механизмы участия обнаруженных мутаций в канцерогенезе на настоящий момент затруднительно. В пользу участия этих мутаций в злокачественной трансформации говорит, в частности, тот факт, что они были предпочтительно локализованы в участках генов, кодирующих глобулярный и С‑концевой домены гистонов Н1 [45], которые отвечают за связывание с ДНК. Высокое отношение несинонимичных замен и инсерций/делеций к синонимичным (5.1 : 1), как и тот факт, что мутации генов Н1 и других генов с достоверной ролью в канцерогенезе являются взаимоисключающими [45], также говорит в пользу физиологической значимости обнаруженных изменений.
Участие Н1 в большом количестве внутриклеточных процессов позволяет рассматривать в качестве потенциальных механизмов действия подобных мутаций как потерю функций, так и приобретение. Наиболее очевидным возможным объяснением могла бы быть декомпактизация ДНК вследствие снижения прочности связывания с ней мутантного Н1 (рис. 2б). В то же время, поскольку обнаруженные мутации редко затрагивали больше одного гена в диплоидном хромосомном наборе, а даже полная делеция одного или нескольких вариантов Н1 не вызывала заметных физиологических изменений [28, 32], такое объяснение нельзя считать в полной мере удовлетворительным. В правильности такого объяснения заставляет усомниться и то, что значительная часть обнаруженных мутаций была локализована в участках, кодирующих С-концевые фрагменты молекул линкерных гистонов, для связывания которых с ДНК важна в первую очередь не конкретная аминокислотная последовательность, а скорее характер боковых цепей составляющих их аминокислот [50, 51].
Гораздо более вероятной является гипотеза о нарушениях в результате мутаций взаимодействия Н1 с регуляторными белками или системами эпигенетической регуляции, которые могут происходить как по механизму утраты, так и по механизму приобретения функции (рис. 2г). Так, было установлено, что часть обнаруженных мутаций в областях генов, кодирующих С-концевые фрагменты Н1, приводила к ослаблению или полному исчезновению взаимодействия линкерного гистона с метилтрансферазой DNMT3B [45], что может нарушать осуществляемые этой метилтрансферазой эпигенетические регуляторные функции [52]. Тем не менее имеющихся на сегодня данных недостаточно для того, чтобы установить, в какой именно мере обнаруженные мутации являются причиной злокачественного перерождения, а частота самих этих мутаций зачастую крайне невелика [47, 49], поэтому вывод об этом следует делать только на основании результатов функциональных исследований.
ВЗАИМОДЕЙСТВИЕ ЛИНКЕРНЫХ ГИСТОНОВ С РЕГУЛЯТОРНЫМИ БЕЛКАМИ
На сегодняшний день достоверно известно, что варианты гистона Н1 взаимодействуют по меньшей мере с несколькими важными белками, регулирующими активность хроматина, среди которых – регуляторы транскрипции, системы посттрансляционной модификации и архитектурные белки. В то же время результаты связывания белков тотального клеточного экстракта с иммобилизованным Н1.0 указывают на то, что список этих белков намного шире и включает в себя также белки, участвующие в сплайсинге мРНК, синтезе рРНК и регуляции трансляции [53]. Необходимо отметить, что даже такая высокоспецифичная методика поиска возможных лигандов, как связывание с иммобилизованным белком, в случае с Н1 может оказаться неспособной выявить все взаимодействующие белки в силу того, что необходимые для взаимодействия эпитопы могут быть свернуты должным образом только у Н1, связавшегося с ДНК.
Следует также рассматривать возможность того, что как глобальные, так и локальные изменения количества связанного с хроматином Н1 возможны не только в результате изменений его экспрессии, но и в результате активности регуляторных белков. Так, было установлено, что при нокауте гена опухолевого репрессора PTEN, участвующего в регуляции активности протеинкиназы В [54], но имеющего также и ряд функций, не связанных с фосфатазной активностью [55], происходит диссоциация Н1 и декомпактизация хроматина клетки [56]. Диссоциацию Н1 можно вызвать также обработкой оплодотворенных яйцеклеток X. laevis PHD-доменом белка SSRP1, входящего в регуляторный комплекс FACT1 [7], что вызывает глобальное усиление интенсивности транскрипции хроматина и ускоряет прохождение клеточного цикла. Интересно, что похожий механизм действия был обнаружен для аналогичного линкерным гистонам белка D. melanogaster dBigH1, нокаут которого нарушал эмбриональное развитие за счет преждевременной дерепрессии транскрипции хроматина клеток эмбриона [8]. Снижение количества Н1, и особенно его варианта Н1.0, характерного как для высокодифференцированных клеток, так и для регенерирующих тканей [57]. Предпочтительное снижение содержания Н1.0 в клетках регенерирующих тканей вызвано, судя по всему, в первую очередь существованием обратной зависимости между интенсивностью транскрипции гена Н1.0 и скоростью прохождения клеточного цикла [58].
Важно отметить, что при общем повышении содержания Н1.0 в клетке в результате повышенной экспрессии увеличение количества связанного Н1.0 происходит в разных участках хроматина неравномерно, предпочтительно в области повторяющихся последовательностей [59], что, судя по всему, соответствует его локализации и в клетках дикого типа [60]. Дифференциальная локализация в хроматине была обнаружена также для гистона Н1.2, содержание которого в клетках рака молочной железы T47D значительно снижено вблизи точек начала транскрипции неактивных генов, но существенно повышено в их промоторных областях [61]. Эти данные позволяют предполагать, что одним из важных последствий злокачественной трансформации является не только общая декомпактизация хроматина, но и перераспределение вариантов линкерных гистонов (рис. 2в).
Выше уже было упомянуто, что в культуре клеток рака яичника OVCAR-3 гистон Н1.3 является селективным ингибитором онкогенной нкРНК Н19 [38]. В последнее время выявлено еще несколько механизмов регуляции активности генов, взаимодействующих с линкерными гистонами. Так, было показано, что регуляторный белок PARP-1 конкурирует с гистоном Н1 на ароматазном промоторе в клетках рака молочной железы 3T3-L1; гистон Н1 в результате такого взаимодействия оказывается поли-АДФ-рибозилирован [62]. Примечательно, что к усилению экспрессии с ароматазного промотора приводила не только повышенная экспрессия PARP-1, но и ингибирование его активности PJ34. Эффект от такой обработки терялся в присутствии ингибиторов гистондеацетилаз класса I/IIa, специфичных к гистону Н1. Таким образом, обнаруженное регуляторное действие PARP-1 на ароматазный промотор в клетках 3Т3-L1 обладает, судя по всему, сложным механизмом действия, зависящим от дозы компонентов и контекста хроматина.
Недавно было установлено, что активация сигнальных путей, регулируемых протоонкогеном Ras, приводит к подавлению фосфорилирования гистонов Н1.4 и Н1.5 и ускоряет рост и миграцию клеток глиомы и мелкоклеточного рака легких [63, 64]. Подавление фосфорилирования разных вариантов линкерного гистона происходило в результате работы разных внутриклеточных сигнальных путей – протеинкиназы В в случае Н1.5 и МАР-киназ в случае Н1.4. Было продемонстрировано, что усиление фосфорилирования этих линкерных гистонов приводит к подавлению пролиферации раковых клеток и снижению экспрессии генов, активируемых при усилении активности Ras, что свидетельствует в пользу того, что именно посттрансляционная модификация Н1.4 и Н1.5 является одной из мишеней соответствующих регуляторных путей. Важно отметить, что в обоих случаях фосфорилируемые аминокислоты находились в N-концевых участках линкерных гистонов – Thr10 в случае Н1.5 и Ser36 в случае Н1.4, что открывает новые подходы к установлению функций этого фрагмента, о регуляторном значении которого в настоящий момент известно немного (рис. 1).
Протоонкоген MTA1, связанный с развитием метастаз, ингибирует фосфорилирование С-концевого фрагмента другого варианта линкерного гистона, Н1.2 [65], а также вызывает перераспределение Н1 в хроматине клеток гепатоцитарной аденокарциномы [66]. Было установлено, что Н1 предпочтительно связывается с областями хроматина с пониженным содержанием МТА1, а повышенная экспрессия последнего приводит к диссоциации Н1 и деконденсации хроматина [66]. Помимо диссоциации Н1, повышенная экспрессия МТА1 вызывала также снижение содержания Н1.2T146ph (рис. 1) путем ускорения протеосомной деградации ДНК-протеинкиназы (DNA-PK), приводя к усилению пролиферационной активности клеток гепатоклеточной карциномы [65]. При помощи иммунопреципитации хроматина было продемонстрировано, что фосфорилированный Н1.2 связывается с промоторными областями мишеней МТА1, среди которых – металлопротеазы внеклеточного матрикса ММР-7 и ММР-9 и циклин D1.
Еще одна посттрансляционая модификация линкерного гистона, вовлеченная в канцерогенез, – метилирование Н1.4 в клетках чешуйчатоклеточной карциномы головы и шеи [67]. Было продемонстрировано, что метилтрансфераза WHSC1 в культуре таких клеток монометилирует гистон Н1.4 по 85-му остатку лизина, расположенному в его глобулярном домене (рис. 1). Такое метилирование ускоряло пролиферацию и повышало содержание Н1.4 в области гена белка ОСТ4, усиливая его экспрессию; этот белок является важным звеном канцерогенеза для многих типов раковых опухолей, а также важнейшим маркером стволовых свойств клеток [68].
Была выявлена связь между гистоном Н1 и цитохромом р53, который является противоопухолевым супрессором. ДНК-хеликаза CHD8 формирует с р53 и гистоном Н1 трехкомпонентный комплекс, привлекая последний к регуляторным элементам, чувствительным к р53, и препятствуя таким образом активируемому р53 апоптозу [69]. Примечательно, что CHD8 способна привлекать Н1 еще и для подавления активности генов, регулируемых β-катенином, что препятствует развитию злокачественных опухолей [70].
Злокачественная трансформация связана не только с дерегуляцией экспрессии генов и пролиферации клеток, но и с изменениями механических свойств клеток – клетки злокачественных опухолей менее жесткие, чем клетки других типов [71], что может способствовать их миграции через внеклеточный матрикс, усиливая их метастатическую активность. Среди компонентов клетки наибольшим препятствием для такого перемещения является ядро, от структуры хроматина в котором зависит эффективность миграции клеток [72]. Было продемонстрировано, что ингибирование экспрессии архитектурного белка хроматина HMGA1, конкурирующего с линкерными гистонами за сайты связывания [73], приводило к изменению фенотипа клеток культуры высокоинвазивного рака молочной железы MDA-MB-231, для которых характерна высокая интенсивность экспрессии гена HMGA1, с мезенхимного на эпителиальный и увеличивало их жесткость [74]. Повышенная экспрессия HMGA в клетках культуры низкоинвазивной опухоли MCF7, в норме HMGA1 почти не содержащих, приводила к обратному эффекту. Ингибирование экспрессии HMGA1 в клетках культуры MDA-MB-231 сопровождалось перераспределением Н1 внутри клеточного ядра: кластеры Н1 становились крупнее, их общее количество и плотность падали. Было также показано, что содержание в клетке HMGA1 регулирует степень фосфорилирования линкерных гистонов: для культур с изначально высоким содержанием HMGA1, экспрессирующих только Н1.2 и Н1.4, было показано общее снижение количества фосфорилированных линкерных гистонов и исчезновение трифосфорилированных форм с существенным сокращением количества моно- и дифосфорилированных; для культуры клеток с изначально низким содержанием HMGA1 его сверхэкспрессия давала обратный эффект. Примечательно, что эта культура экспрессировала еще два варианта Н1: Н1.3 и Н1.5, для которых был выявлен тот же эффект. Помимо изменений в фосфорилировании линкерных гистонов, содержание HMGA1 влияет также на общую интенсивность экспрессии генов всех гистонов клетки, включая линкерные [75].
С развитием методик изучения белок-белковых взаимодействий появляется все больше свидетельств в пользу того, что линкерные гистоны обладают, помимо основной архитектурной, еще и целым набором регуляторных функций, осуществляемых при участии других белков [76]. Их изучение позволит расширить понимание функциональных особенностей вариантов Н1 и механизмов канцерогенеза (рис. 3).

ВАРИАНТЫ ЛИНКЕРНЫХ ГИСТОНОВ КАК ПРОГНОСТИЧЕСКИЕ МАРКЕРЫ
Поскольку гистон Н1 является одним из наиболее распространенных архитектурных белков хроматина, за исключением гистонов нуклеосомного ядра, а в различных культурах раковых клеток были обнаружены разнообразные мутации и нарушения экспрессии этих белков, целесообразно рассмотреть линкерные гистоны в качестве прогностических маркеров для развития опухолей. Самым очевидным кандидатом на роль такого маркера выглядит гистон Н1.0, высокое содержание которого характерно для высокодиффференцированных клеток. Так, на основании анализа наборов данных от пациентов с мультиформной глиобластомой, раком груди, меланомой, раком печени, почек и глиомы низкой степени было установлено, что низкое содержание Н1.0 коррелировало с негативным прогнозом течения заболевания [5]. Примечательно, что негативный прогноз на основании стратификации по содержанию Н1.0 никак не коррелировал с негативными прогнозами на основании других факторов, что свидетельствует в пользу самостоятельной значимости содержания Н1.0 как прогностического критерия. Тем не менее более позднее подобное исследование на более широком наборе данных не выявило явной зависимости между содержанием Н1.0 и типом клеток [77] – содержание Н1.0 в ряде раковых клеток оказалось выше, чем в контрольных клетках нормальных тканей. Такая ситуация может быть следствием того, что некоторые регуляторные элементы промотора гена Н1.0 являются общими с регуляторными элементами гена гистона Н4 и активируются при прохождении клеточного цикла [78], а также того, что экспрессия Н1.0 в S-периоде при переводе клеток в культуру несколько повышается [79]. Необходимо учитывать также внутриопухолевую гетерогенность по степени экспрессии Н1.0 – в результате глобального нарушения регуляции экспрессии генов дифференцированные опухолевые клетки могут накапливать больше Н1.0, чем необходимо.
Несмотря на то, что другие варианты линкерных гистонов менее очевидны в качестве прогностических маркеров, была предпринята попытка оценки корреляции между их содержанием и выживаемостью пациентов с раком груди [74]. Было установлено, что негативный прогноз по развитию характерен для опухолей с высоким содержанием гистонов Н1.2 и Н1.4. Высокоинвазивные клетки рака груди экспрессировали только эти варианты линкерных гистонов. Примечательно, что высокое содержание Н1.0 оказалось характерно для пациентов с положительным прогнозом.
Таким образом, несмотря на привлекательность Н1 в качестве прогностического маркера, полученных на настоящий момент сведений недостаточно для точного установления значимости изменения содержания конкретных вариантов линкерных гистонов для развития рака.
ЗАКЛЮЧЕНИЕ
Несмотря на значительное количество исследований участия Н1 в эпигенетической регуляции канцерогенеза, понимание соответствующих механизмов все еще ограничено. Во многом это обусловлено трудностью изучения конкретного физиологического значения того или иного варианта Н1, поскольку их функции по меньшей мере частично перекрываются. Тем не менее наблюдаемые глобальные изменения в экспрессии Н1 в раковых клеток, так же как и повторяющиеся мутации в соответствующих генах, свидетельствуют о важной роли этого белка при развитии по меньшей мере некоторых типов раковых опухолей. На фоне недостатка информации относительно функций Н1 ключевым вопросом относительно его участия в канцерогенезе является вопрос о том, выступают ли наблюдаемые в опухолях изменения, связанные с Н1, причиной злокачественной трансформации или же ее следствием. Поскольку функции Н1 многочисленны, ответ в каждом конкретном случае может быть разным. Так, глобальные изменения в экспрессии и распределении Н1, скорее всего, являются следствием канцерогенеза, поскольку изменения содержания Н1 в клетке могут оказывать на разные противоопухолевые механизмы различное воздействие. Изменения посттрансляционных модификаций линкерных гистонов и их взаимодействий с регуляторными белками также, видимо, являются следствием канцерогенеза, т.к. в большинстве случаев гистон Н1 был мишенью онкогенных белков. Изучение влияния посттрансляционных модификаций линкерных гистонов на метаболизм раковых клеток поможет выявить физиологические функции отдельных его вариантов и фрагментов его структуры.
Мутации Н1 больше подходят на роль причин злокачественного перерождения, поскольку могут как препятствовать посттрансляционной модификации, так и нарушать взаимодействие с регуляторными белками, как по механизму утраты функции, так и по механизму приобретения. Изучение таких мутаций – еще один важный потенциальный источник знаний о функциях вариантов линкерных гистонов в организме. Расширение понимания роли Н1 в различных клеточных процессах позволит гораздо точнее интерпретировать значимость тех или иных изменений в содержании линкерного гистона в опухолевых клетках и точнее прогнозировать развитие опухолей на основании использования Н1 в сочетании с другими прогностическими маркерами.
Список литературы
Kornberg R.D. // Science. 1974. V. 184. P. 868–871. https://doi.org/10.1126/science.184.4139.868
Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J. // J. Mol. Biol. 2002. V. 319. P. 1097–1113. https://doi.org/10.1016/S0022-2836(02)00386-8
Botrugno O.A., Santoro F.,Minucci S. // Cancer Lett. 2009. V. 280. P. 134–144. https://doi.org/10.1016/j.canlet.2009.02.027
Fraga M.F., Ballestar E., Villar-Garea A., Boix-Chornet M., Espada J., Schotta G., Bonaldi T., Haydon C., Ropero S., Petrie K., Iyer N.G., Perez-Rosado A., Calvo E., Lopez J.A., Cano A., Calasanz M.J., Colomer D., Piris M.A., Ahn N., Imhof A., Caldas C., Jenuwein T., Esteller M. // Nat. Genet. 2005. V. 37. P. 391–400. https://doi.org/10.1038/ng1531
Torres C.M., Biran A., Burney M.J., Patel H., Henser-Brownhill T., Cohen A.S., Li Y., Ben-Hamo R., Nye E., Spencer-Dene B., Chakravarty P., Efroni S., Matthews N., Misteli T., Meshorer E., Scaffidi P. // Science. 2016. V. 353. P. aaf1644. https://doi.org/10.1126/science.aaf1644
Scaffidi P. // Biochim. Biophys. Acta. 2016. V. 1859. P. 533–539. https://doi.org/10.1016/j.bbagrm.2015.09.008
Falbo L., Raspelli E., Romeo F., Fiorani S., Pezzimenti F., Casagrande F., Costa I., Parazzoli D., Costanzo V. // Nat. Commun. 2020. V. 11. P. 1345. https://doi.org/10.1038/s41467-020-15180-5
Perez-Montero S., Carbonell A., Moran T., Vaquero A., Azorin F. // Dev. Cell. 2013. V. 26. P. 578–590. https://doi.org/10.1016/j.devcel.2013.08.011
Fan L., Roberts V.A. // Proc. Natl. Acad. Sci. USA. 2006. V. 103. P. 8384–8389. https://doi.org/10.1073/pnas.0508951103
Gajiwala K.S., Burley S.K. // Curr. Opin. Struct. Biol. 2000. V. 10. P. 110–116. https://doi.org/10.1016/s0959-440x(99)00057-3
Subirana J.A. // Biopolymers. 1990. V. 29. P. 1351–1357. https://doi.org/10.1002/bip.360291003
Roque A., Teruel N., Lopez R., Ponte I., Suau P. // J. Struct. Biol. 2012. V. 180. P. 101–109. https://doi.org/10.1016/j.jsb.2012.07.004
Roque A., Iloro I., Ponte I., Arrondo J.L., Suau P. // J. Biol. Chem. 2005. V. 280. P. 32 141–32 147. https://doi.org/10.1074/jbc.M505636200
Roque A., Ponte I., Suau P. // J. Phys. Chem. B. 2009. V. 113. P. 12 061–12 066. https://doi.org/10.1021/jp9022579
Fang H., Clark D.J., Hayes J.J. // Nucleic. Acids. Res. 2012. V. 40. P. 1475–1484. https://doi.org/10.1093/nar/gkr866
Fang H., Wei S., Lee T.H., Hayes J.J. // Nucleic. Acids. Res. 2016. V. 44. P. 9131–9141. https://doi.org/10.1093/nar/gkw586
Vyas P., Brown D.T. // J. Biol. Chem. 2012. V. 287. P. 11 778–11 787. https://doi.org/10.1074/jbc.M111.312819
Happel N., Doenecke D. // Gene. 2009. V. 431. P. 1–12. https://doi.org/10.1016/j.gene.2008.11.003
Pan C., Fan Y. // Biochim. Biophys. Acta. 2016. V. 1859. P. 496–509. https://doi.org/10.1016/j.bbagrm.2015.12.002
Zhou B.R., Jiang J., Feng H., Ghirlando R., Xiao T.S., Bai Y. // Mol. Cell. 2015. V. 59. P. 628–638. https://doi.org/10.1016/j.molcel.2015.06.025
Bednar J., Garcia-Saez I., Boopathi R., Cutter A.R., Papai G., Reymer A., Syed S.H., Lone I.N., Tonchev O., Crucifix C., Menoni H., Papin C., Skoufias D.A., Kurumizaka H., Lavery R., Hamiche A., Hayes J.J., Schultz P., Angelov D., Petosa C., Dimitrov S. // Mol. Cell. 2017. V. 66. P. 384–397. https://doi.org/10.1016/j.molcel.2017.04.012
Ozturk M.A., Pachov G.V., Wade R.C., Cojocaru V. // Nucleic. Acids. Res. 2016. V. 44. P. 6599–6613. https://doi.org/10.1093/nar/gkw514
Orrego M., Ponte I., Roque A., Buschati N., Mora X., Suau P. // BMC Biol. 2007. V. 5. P. 22. https://doi.org/10.1186/1741-7007-5-22
Talasz H., Sapojnikova N., Helliger W., Lindner H., Puschendorf B. // J. Biol. Chem. 1998. V. 273. P. 32 236–32 243. https://doi.org/10.1074/jbc.273.48.32236
Clausell J., Happel N., Hale T.K., Doenecke D., Beato M. // PLoS One. 2009. V. 4. P. e0007243. https://doi.org/10.1371/journal.pone.0007243
Th’ng J.P., Sung R., Ye M., Hendzel M.J. // J. Biol. Chem. 2005 V. 280. P. 27 809–27 814. https://doi.org/10.1074/jbc.M501627200
Conn K.L., Hendzel M.J., Schang L.M. // J. Virol. 2008. V. 82. P. 8629–8646. https://doi.org/10.1128/JVI.00616-08
Fan Y., Sirotkin A., Russell R.G., Ayala J., Skoultchi A.I. // Mol. Cell Biol. 2001. V. 21. P. 7933–7943. https://doi.org/10.1128/MCB.21.23.7933-7943.2001
Sirotkin A.M., Edelmann W., Cheng G., Klein-Szanto A., Kucherlapati R., Skoultchi A.I. // Proc. Natl. Acad. Sci. USA. 1995. V. 92. P. 6434–6438. https://doi.org/10.1073/pnas.92.14.6434
Lin Q., Sirotkin A., Skoultchi A.I. // Mol. Cell Biol. 2000. V. 20. P. 2122–2128. https://doi.org/10.1128/mcb.20.6.2122-2128.2000
Rabini S., Franke K., Saftig P., Bode C., Doenecke D., Drabent B. // Exp. Cell Res. 2000. V. 255. P. 114–124. https://doi.org/10.1006/excr.1999.4767
Fan Y., Nikitina T., Morin-Kensicki E.M., Zhao J., Magnuson T.R., Woodcock C.L., Skoultchi A.I. // Mol. Cell Biol. 2003. V. 23. P. 4559–4572. https://doi.org/10.1128/mcb.23.13.4559-4572.2003
Sancho M., Diani E., Beato M., Jordan A. // PLoS Genet. 2008. V. 4. P. e1000227. https://doi.org/10.1371/journal.pgen.1000227
Medrzycki M., Zhang Y., McDonald J.F., Fan Y. // Front. Biosci. (Landmark Ed). 2012. V. 17. P. 396–406. https://doi.org/10.2741/3934
Bond U.M., Yario T.A., Steitz J.A. // Genes Dev. 1991. V. 5. P. 1709–1722. https://doi.org/10.1101/gad.5.9.1709
Wang Z.F., Sirotkin A.M., Buchold G.M., Skoultchi A.I., Marzluff W.F. // J. Mol. Biol. 1997. V. 271. P. 124–138. https://doi.org/10.1006/jmbi.1997.1166
Pehrson J.R., Cole R.D. // Biochemistry. 1982. V. 21. P. 456–460. https://doi.org/10.1021/bi00532a006
Medrzycki M., Zhang Y., Zhang W., Cao K., Pan C., Lailler N., McDonald J.F., Bouhassira E.E., Fan Y. // Cancer Res. 2014. V. 74. P. 6463–6473. https://doi.org/10.1158/0008-5472.CAN-13-2922
Scaffidi P., Misteli T. // Nat. Cell Biol. 2011. V. 13. P. 1051–1061. https://doi.org/10.1038/ncb2308
Tillo D., Hughes T.R. // BMC Bioinformatics. 2009. V. 10. P. 442. https://doi.org/10.1186/1471-2105-10-442
Anderson J.D., Widom J. // Mol. Cell Biol. 2001. V. 21. P. 3830–3839. https://doi.org/10.1128/MCB.21.11.3830-3839.2001
Gambara G., Gaebler M., Keilholz U., Regenbrecht C.R.A., Silvestri A. // Front. Pharmacol. 2018. V. 9. P. 77. https://doi.org/10.3389/fphar.2018.00077
Qiu L., Hu X., Jing Q., Zeng X., Chan K.M., Han J. // J. Genet. Genomics. 2018. V. 45. P. 227–236. https://doi.org/10.1016/j.jgg.2018.04.004
Justin N., Zhang Y., Tarricone C., Martin S.R., Chen S., Underwood E., De Marco V., Haire L.F., Walker P.A., Reinberg D., Wilson J.R., Gamblin S.J. // Nat. Commun. 2016. V. 7. P. 11316. https://doi.org/10.1038/ncomms11316
Li H., Kaminski M.S., Li Y., Yildiz M., Ouillette P., Jones S., Fox H., Jacobi K., Saiya-Cork K., Bixby D., Lebovic D., Roulston D., Shedden K., Sabel M., Marentette L., Cimmino V., Chang A.E., Malek S.N. // Blood. 2014. V. 123. P. 1487–1498. https://doi.org/10.1182/blood-2013-05-500264
Okosun J., Bodor C., Wang J., Araf S., Yang C.Y., Pan C., Boller S., Cittaro D., Bozek M., Iqbal S., Matthews J., Wrench D., Marzec J., Tawana K., Popov N., O’Riain C., O’Shea D., Carlotti E., Davies A., Lawrie C.H., Matolcsy A., Calaminici M., Norton A., Byers R.J., Mein C., Stupka E., Lister T.A., Lenz G., Montoto S., Gribben J.G., Fan Y., Grosschedl R., Chelala C., Fitzgibbon J. // Nat. Genet. 2014. V. 46. P. 176–181. https://doi.org/10.1038/ng.2856
Landau D.A., Wu C.J. // Genome Med. 2013. V. 5. P. 47. https://doi.org/10.1186/gm451
Lohr J.G., Stojanov P., Lawrence M.S., Auclair D., Chapuy B., Sougnez C., Cruz-Gordillo P., Knoechel B., Asmann Y.W., Slager S.L., Novak A.J., Dogan A., Ansell S.M., Link B.K., Zou L., Gould J., Saksena G., Stransky N., Rangel-Escareno C., Fernandez-Lopez J.C., Hidalgo-Miranda A., Melendez-Zajgla J., Hernandez-Lemus E., Schwarz-Cruz y Celis A., Imaz-Rosshandler I., Ojesina A.I., Jung J., Pedamallu C.S., Lander E.S., Habermann T.M., Cerhan J.R., Shipp M.A., Getz G., Golub T.R. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. P. 3879–3884. https://doi.org/10.1073/pnas.1121343109
Sjoblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S.D., Willis J., Dawson D., Willson J.K., Gazdar A.F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B.H., Bachman K.E., Papadopoulos N., Vogelstein B., Kinzler K.W., Velculescu V.E. // Science. 2006. V. 314. P. 268–274. https://doi.org/10.1126/science.1133427
Hansen J.C., Lu X., Ross E.D.,Woody R.W. // J. Biol. Chem. 2006. V. 281. P. 1853–1856. https://doi.org/10.1074/jbc.R500022200
Lu X., Hamkalo B., Parseghian M.H., Hansen J.C. // Biochemistry. 2009. V. 48. P. 164–172. https://doi.org/10.1021/bi801636y
Yang S.M., Kim B.J., Norwood Toro L., Skoultchi A.I. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 1708–1713. https://doi.org/10.1073/pnas.1213266110
Kalashnikova A.A., Winkler D.D., McBryant S.J., Henderson R.K., Herman J.A., DeLuca J.G., Luger K., Prenni J.E., Hansen J.C. // Nucleic. Acids. Res. 2013. V. 41. P. 4026–4035. https://doi.org/10.1093/nar/gkt104
Lee Y.R., Chen M., Pandolfi P.P. // Nat. Rev. Mol. Cell Biol. 2018. V. 19. P. 547–562. https://doi.org/10.1038/s41580-018-0015-0
Shen W.H., Balajee A.S., Wang J., Wu H., Eng C., Pandolfi P.P., Yin Y. // Cell. 2007. V. 128. P. 157–170. https://doi.org/10.1016/j.cell.2006.11.042
Chen Z.H., Zhu M., Yang J., Liang H., He J., He S., Wang P., Kang X., McNutt M.A., Yin Y., Shen W.H. // Cell Rep. 2014. V. 8. P. 2003–2014. https://doi.org/10.1016/j.celrep.2014.08.008
Gjerset R., Gorka C., Hasthorpe S., Lawrence J.J., Eisen H. // Proc. Natl. Acad. Sci. USA. 1982. V. 79. P. 2333–2337. https://doi.org/10.1073/pnas.79.7.2333
Happel N., Warneboldt J., Hanecke K., Haller F., Doenecke D. // Cell Cycle. 2009. V. 8. P. 2226–2232. https://doi.org/10.4161/cc.8.14.8982
Cao K., Lailler N., Zhang Y., Kumar A., Uppal K., Liu Z., Lee E.K., Wu H., Medrzycki M., Pan C., Ho P.Y., Cooper G.P., Jr., Dong X., Bock C., Bouhassira E.E., Fan Y. // PLoS Genet. 2013. V. 9. P. e1003417. https://doi.org/10.1371/journal.pgen.1003417
Millan-Arino L., Izquierdo-Bouldstridge A., Jordan A. // Biochim. Biophys. Acta. 2016. V. 1859. P. 510–519. https://doi.org/10.1016/j.bbagrm.2015.10.013
Millan-Arino L., Islam A.B., Izquierdo-Bouldstridge A., Mayor R., Terme J.M., Luque N., Sancho M., Lopez-Bigas N., Jordan A. // Nucleic. Acids. Res. 2014. V. 42. P. 4474–4493. https://doi.org/10.1093/nar/gku079
Kaiser A., Kruger T., Eiselt G., Bechler J., Kniemeyer O., Huber O., Schmidt M. // Cells. 2020. V. 9. P. 427. https://doi.org/10.3390/cells9020427
Shi S., Zhang J., Liu M., Dong H., Li N. // Artif. Cells Nanomed. Biotechnol. 2019. V. 47. P. 2343–2351. https://doi.org/10.1080/21691401.2019.1624558
Sang B., Sun J., Yang D., Xu Z., Wei Y. // Artif. Cells. Nanomed. Biotechnol. 2019. V. 47. P. 2882–2890. https://doi.org/10.1080/21691401.2019.1638795
Li Y.H., Zhong M., Zang H.L., Tian X.F. // Front. Oncol. 2020. V. 10. P. 567. https://doi.org/10.3389/fonc.2020.00567
Liu J., Wang H., Ma F., Xu D., Chang Y., Zhang J., Wang J., Zhao M., Lin C., Huang C., Qian H., Zhan Q. // Mol. Oncol. 2015. V. 9. P. 218–235. https://doi.org/10.1016/j.molonc.2014.08.007
Saloura V., Vougiouklakis T., Bao R., Kim S., Baek S., Zewde M., Bernard B., Burkitt K., Nigam N., Izumchenko E., Dohmae N., Hamamoto R., Nakamura Y. // Neoplasia. 2020. V. 22. P. 283–293. https://doi.org/10.1016/j.neo.2020.05.002
Zeineddine D., Hammoud A.A., Mortada M., Boeuf H. // Am. J. Stem. Cells. 2014. V. 3. P. 74–82.
Nishiyama M., Oshikawa K., Tsukada Y., Nakagawa T., Iemura S., Natsume T., Fan Y., Kikuchi A., Skoultchi A.I., Nakayama K.I. // Nat. Cell Biol. 2009. V. 11. P. 172–182. https://doi.org/10.1038/ncb1831
Nishiyama M., Skoultchi A.I., Nakayama K.I. // Mol. Cell Biol. 2012. V. 32. P. 501–512. https://doi.org/10.1128/MCB.06409-11
Luo Q., Kuang D., Zhang B., Song G. // Biochim. Biophys. Acta. 2016. V. 1860. P. 1953–1960. https://doi.org/10.1016/j.bbagen.2016.06.010
Gerlitz G., Bustin M. // Trends. Cell Biol. 2011. V. 21. P. 6–11. https://doi.org/10.1016/j.tcb.2010.09.002
Catez F., Yang H., Tracey K.J., Reeves R., Misteli T., Bustin M. // Mol. Cell Biol. 2004. V. 24. P. 4321–4328.
Senigagliesi B., Penzo C., Severino L.U., Maraspini R., Petrosino S., Morales-Navarrete H., Pobega E., Ambrosetti E., Parisse P., Pegoraro S., Manfioletti G., Casalis L., Sgarra R. // Int. J. Mol. Sci. 2019. V. 20. P. 2733. https://doi.org/10.3390/ijms20112733
Celona B., Weiner A., Di Felice F., Mancuso F.M., Cesarini E., Rossi R.L., Gregory L., Baban D., Rossetti G., Grianti P., Pagani M., Bonaldi T., Ragoussis J., Friedman N., Camilloni G., Bianchi M.E., Agresti A. // PLoS Biol. 2011. V. 9. P. e1 001 086. https://doi.org/10.1371/journal.pbio.1001086
Kalashnikova A.A., Rogge R.A., Hansen J.C. // Biochim. Biophys. Acta. 2016. V. 1859. P. 455–461. https://doi.org/10.1016/j.bbagrm.2015.10.004
Wang T., Chuffart F., Bourova-Flin E., Wang J., Mi J., Rousseaux S., Khochbin S. // Front. Med. 2019. V. 13. P. 289–297. https://doi.org/10.1007/s11684-018-0667-3
Khochbin S., Wolffe A.P. // Eur. J. Biochem. 1994. V. 225. P. 501–510. https://doi.org/10.1111/j.1432-1033.1994.00501.x
Grunwald D., Khochbin S., Lawrence J.J. // Exp. Cell Res. 1991. V. 194. P. 174–179. https://doi.org/10.1016/0014-4827(91)90350-4
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия