

Биоорганическая химия, 2022, T. 48, № 5, стр. 569-579
Синтез и исследование противовоспалительной активности новых производных пиримидина – ингибиторов изоформ циклооксигеназ
Ю. З. Хазимуллина 1, *, А. Р. Гимадиева 1, В. Р. Хайруллина 2, Л. Ф. Зайнуллина 3, Ю. В. Вахитова 3, А. Г. Мустафин 1, 2
1 Уфимский Институт химии – обособленное структурное подразделение ФГБНУ “Уфимский федеральный исследовательский центр” РАН
450054 Уфа, просп. Октября, 71, Россия
2 ФГБОУ ВО “Башкирский государственный университет”
450076 Уфа, ул. Заки Валиди, 32, Россия
3 Институт биохимии и генетики – обособленное структурное подразделение
ФГБНУ “Уфимский федеральный исследовательский центр” РАН
450054 Уфа, просп. Октября, 71, Россия
* E-mail: yulialion91@mail.ru
Поступила в редакцию 02.11.2021
После доработки 10.12.2021
Принята к публикации 22.12.2021
- EDN: ZJVBFP
- DOI: 10.31857/S0132342322050104
Аннотация
Методом молекулярного докинга проведено моделирование комплексообразования 17 производных урацила с циклическими и ациклическими сера- и кислородсодержащими заместителями в пиримидиновом цикле с активными центрами изоформ циклооксигеназ (COX). Из набора протестированных соединений выявлены два соединения-лидера, представляющие собой конъюгаты 5-гидрокси-1,3,6-триметилурацила с N-фталил-L-аминокислотами, которые могут быть эффективными ингибиторами изоформ COX с повышенной селективностью действия в отношении фермента COX-2, индуцируемого при воспалительных процессах в организме. Осуществлен синтез этих соединений и проведены их биологические испытания в условиях in vivo на четырех моделях воспаления, вызванного введением каррагинана, лидокаина, яичного белка и формалина. Установлено, что конъюгаты 5-гидрокси-1,3,6-триметилурацила с N-фталилаланином и N-фталилметионином обладают выраженной противовоспалительной активностью и по эффективности противовоспалительного действия сопоставимы с лекарственным препаратом Ортофеном. Приведена оценка изоэнзим-специфического ингибирования ферментов-изоформ COX, выявлена выраженная противовоспалительная активность полученных соединений.
ВВЕДЕНИЕ
Производные пиримидина, модифицированные по положениям N1, N3, C5 и C6, обладают широким спектром фармакологической активности, включая противоопухолевую, иммуномодулирующую, гепатопротекторную, а также антиокислительную и противовоспалительную активность в сочетании с умеренной или низкой токсичностью [1–6]. В настоящее время ведется активный поиск новых лекарственных средств с выраженным противовоспалительным действием среди производных данного класса органических соединений [7].
Известно, что противовоспалительное действие органических соединений может быть реализовано в живых системах по нескольким механизмам: ингибирование фосфолипазы А2, циклооксигеназы-2 (COX-2) или обеих изоформ циклооксигеназ (COX-1 и COX-2), 5-липоксигеназы, лейкотриен-А4-гидролазы и т.д. [8–15]. При этом в научной медицинской литературе имеется большое количество данных о том, что использование противовоспалительных лекарственных средств стероидной природы, снижающих каталитическую активность фосфолипазы А2, способствует развитию выраженных побочных эффектов, таких как увеличение индекса массы тела, тяжелые депрессивные состояния и др. [14–17]. В этой связи в настоящее время ведется активный поиск нестероидных противовоспалительных лекарственных средств. Однако при разработке нестероидных противовоспалительных лекарственных препаратов (НПВП) нового поколения актуальной проблемой оказывается исследование селективности их действия в отношении изоформ COX.
COX представляет собой гемсодержащий фермент, который катализирует реакцию биосинтеза простагландинов и тромбоксанов из арахидоновой кислоты [18–26]. Он существует в двух изоформах: циклооксигеназа-1 (COX-1) и циклооксигеназа-2 (COX-2) [20–25], которые гомологичны друг другу на 60%. Известно, что COX-1 – это конститутивный фермент, присутствующий в тканях млекопитающих практически повсеместно [18–24]. В тромбоцитах COX-1 обеспечивает превращение арахидоновой кислоты в тромбоксан [18–22]. Ингибирование каталитической активности COX-1 при приеме неселективных НПВП приводит к повреждению стенки желудка и развитию язв [22–24]. COX-2 в обычных условиях присутствует в мозге и корковом слое почек [18–20]. В других тканях COX-2 индуцируется при воспалении [20–24]. Кроме того, в литературе имеется множество фактов, подтверждающих участие COX-2 в канцерогенезе кишечника и молочных желез [18–24]. Все вышеперечисленные данные позволяют заключить, что поиск соединений, обладающих выраженной противовоспалительной активностью и высокой селективностью в отношении COX-2, оказывается также нерациональным. В этой связи разработка новых эффективных НПВП, характеризующихся умеренным фармакологическим профилем и умеренной активностью в отношении COX-2, можно считать актуальной научно-практической задачей.
К настоящему времени в научной литературе накоплен значительный объем количественной информации по эффективности ингибирования активности изоформ COX разными классами биологически активных веществ. В этих условиях эмпирический поиск органических соединений, обладающих повышенной избирательностью действия в отношении COX-2, без привлечения методов компьютерной химии представляет собой трудновыполнимую задачу, требующую значительных временных затрат [25–32]. По этим же причинам синтез новых биологически активных веществ, основанный исключительно на интуитивном опыте исследователя, также нерационален. Современным и более эффективным способом решения научных и практических задач поиска новых эффективных потенциальных лекарств среди разных классов гетероциклических соединений оказывается привлечение методов виртуального скрининга, базирующихся на изучении стерической комплементарности органических соединений с активными центрами ферментов и рецепторов. Эти методы позволяют уже на начальных стадиях создания потенциальных лекарственных соединений предсказывать их биологическую активность [27–32] и, следовательно, отбирать соединения-лидеры для дальнейших доклинических и клинических испытаний.
Цель настоящей работы – изучение возможности ингибирования каталитической активности изоформ COX производными пиримидина, модифицированными по положениям N1, N3, C5 и C6, с гетероциклическими и ациклическими заместителями, селективности действия этих соединений в отношении COX-1 и COX-2, а также их противовоспалительной активности in silico и in vivo.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Молекулярный докинг. На первом этапе скрининговых исследований нами была изучена стерическая комплементарность и аффинность соединений (I–XVII) (табл. 1) (соединения (I–XV) синтезированы ранее [33]) к активным центрам изоформ COX методом молекулярного докинга с использованием программы AutoDock 4.2 (Scripps Research, США). Результаты этих исследований приведены в табл. 2. Анализ данных показал, что из 17 протестированных соединений только лиганды (XVI) и (XVII), содержащие в качестве заместителей в положении R3 достаточно объемный полярный 2-замещенный изоиндолин-1,3-дионовый фрагмент, характеризуются сравнительно высокими численными значениями свободных энергий связывания с активными центрами изоформ COX, сопоставимыми с аналогичными характеристиками для референсных ингибиторов данных ферментов – действующих веществ НПВП Диклофенак, Флурбипрофен и Целекоксиб. Программа AutoDock 4.2 присваивает достаточно высокие численные значения оценочных функций этим соединениям благодаря их способности образовывать водородные связи, а также участию в образовании полярных и Т‑стэкинговых взаимодействий с активными центрами изоформ COX. В частности, положение соединений (XVI) и (XVII) в активном центре COX-1 и COX-2 стабилизируют водородные связи и полярные взаимодействия с Tyr355 и Met522, а также Т-стэкинговые взаимодействия с His90, Tyr355 и Trp387 (рис. 1). На основании этих данных можно предположить, что структуры моделируемых соединений (XVI) и (XVII) способны в пуле изоформ COX замещать арахидоновую кислоту. Установлено, что вследствие достаточно высокого структурного сходства они позиционируются в активных центрах данных ферментов в том же кластере, что и структуры действующего вещества НПВП Целекоксиб и соединения SC-558 (рис. 2). Кроме того, анализ данных табл. 3 позволяет заключить, что оба соединения-лидера будут преимущественно ингибировать каталитическую активность COX-2, и, следовательно, не исключено, что в доклинических испытаниях они могут обнаружить наряду с противовоспалительным выраженный кардиотоксический эффект.
Таблица 1.
Производные пиримидина, модифицированные по положениям N1, N3, C5 и C6
 |
||||
|---|---|---|---|---|
| Соединение | R1 | R2 | R3 | R4 |
| (I) | H | CH3 | OH | CH3 |
| (II) | CH3 | CH3 | OH | CH3 |
| (III) | CH3 | CH3 | OCH3 | CH3 |
| (IV) | –CH2CH2SC4H9-i | –CH2CH2SC4H9-i | H | CH3 |
| (V) | –CH2CH2SC4H9-i | –CH2CH2SC4H9-i | –OCH2CH2SC4H9-i | CH3 |
| (VI) | –CH2CH2SC4H9-i | –CH2CH2SC4H9-i | OH | CH3 |
| (VII) | H | –CH2CH2SC4H9-i | H | CH3 |
| (VIII) | CH3 | CH3 | –OCH2CH2SC4H9 | CH3 |
| (IX) | –CH2CH2SC4H9 | CH3 | –OCH2CH2SC4H9 | CH3 |
| (X) | –CH2CH2SOC4H9-i | –CH2CH2SOC4H9-i | H | CH3 |
| (XI) | H | –CH2CH2SOC4H9-i | H | CH3 |
| (XII) | –CH2CH2SOC4H9-i | –CH2CH2SOC4H9-i | OH | CH3 |
| (XIII) | –CH2CH2SOC4H9-i | –CH2CH2SOC4H9-i | –OCH2CH2SOC4H9-i | CH3 |
| (XIV) | –CH2CH2SOC4H9 | CH3 | –OCH2CH2SOC4H9 | H |
| (XV) | CH3 | CH3 | –OCH2CH2SOC4H9 | H |
| (XVI) | CH3 | CH3 | 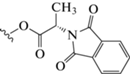 |
CH3 |
| (XVII) | CH3 | CH3 | 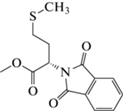 |
CH3 |
Таблица 2.
Результаты докинга активных центров COX-1 и COX-2
| Соединение или препарат | COX-1 | COX-2 | ||
|---|---|---|---|---|
| свободная энергия связывания, ккал/моль | количество докинг- решений в первом кластере | свободная энергия связывания, ккал/моль | количество докинг- решений в первом кластере | |
| (I) | –4.70 | 9 | –4.60 | 10 |
| (II) | –5.05 | 8 | –4.90 | 11 |
| (III) | –5.04 | 8 | –4.90 | 8 |
| (IV) | –5.87 | 11 | –6.71 | 10 |
| (V) | –5.00 | 10 | –6.03 | 6 |
| (VI) | –6.20 | 9 | –5.76 | 10 |
| (VII) | –6.01 | 9 | –6.28 | 12 |
| (VIII) | –5.89 | 11 | –5.78 | 12 |
| (IX) | –6.03 | 10 | –6.42 | 12 |
| (X) | –6.53 | 10 | –6.88 | 13 |
| (XI) | –6.51 | 9 | –6.56 | 13 |
| (XII) | –4.00 | 8 | –6.66 | 12 |
| (XIII) | –4.00 | 8 | –3.90 | 9 |
| (XIV) | –6.09 | 10 | –7.15 | 10 |
| (XV) | –6.31 | 10 | –6.22 | 5 |
| (XVI) | –8.45 | 12 | –9.67 | 10 |
| (XVII) | –7.98 | 10 | –9.58 | 8 |
| Целекоксиб | –10.01 | 12 | –10.27 | 20 |
| SC-558 | – | – | –10.09 | 20 |
| Диклофенак | –8.41 | 14 | –7.55 | 10 |
| Флурбипрофен | –8.01 | 19 | – | – |
| Арахидоновая кислота | –5.80 | 10 | –6.45 | 10 |

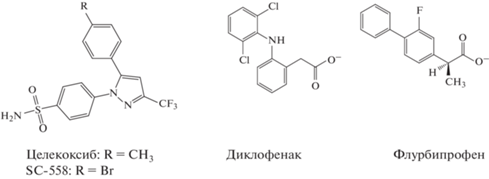
Таблица 3.
Влияние конъюгатов (XVI) и (XVII) на активность COX-1 и COX-2
| Соединение | IC50, мкM |
|---|---|
| COX-1 | |
| (XVI) | 95.5 ± 3.4 |
| (XVII) | 1.0 ± 0.1 |
| COX-2 | |
| (XVI) | ~60.4 ± 7.9 |
| (XVII) | 11.7 ± 4.6 |
Остальные лиганды, содержащие ациклические сера- и кислородсодержащие заместители в положениях R1, R2 и R3, образуют меньшее число водородных связей (преимущественно с Arg120, Met522 и Val523) по сравнению с соединениями (XVI) и (XVII) и характеризуются низкими значениями оценочной функции. Следует ожидать, что в условиях in vivo они могут не обнаружить противовоспалительного действия и по этой причине в дальнейшем не рассматривались.
Таким образом, результаты оценки стерической комплементарности и аффинности соединений (XVI) и (XVII) к активным центрам изоформ COX свидетельствуют о том, что оба соединения перспективны для дальнейших исследований in vivo. Мы осуществили синтез этих соединений и провели испытания их противовоспалительной активности in vivo.
Синтез хлорангидридов N-фталоилзащищенных аминокислот с 5-гидрокси-1,3,6-триметилурацилом. Конъюгаты 5-гидрокси-1,3,6-триметилурацила с аминокислотами аланином и метионином (XVI) и (XVII) были получены ацилированием 5‑гидрокси-1,3,6-триметилурацила хлорангидридами соответствующих N-фталимидзaщищенных аминокислот. Реакцию проводили в хлористом метилене СH2Cl2 в присутствии K2CO3 при комнатной температуре (схема 1 ).
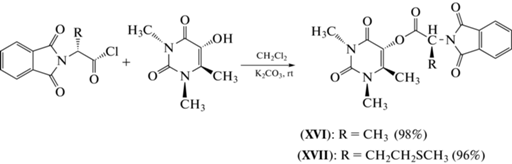
Схема 1 . Получение конъюгатов 5-гидрокси-1,3,6-триметилурацила с N-фталоил-защищенными аминокислотами.
Индивидуальность синтезированных соединений подтверждена методом тонкослойной хроматографии, структуры синтезированных соединений доказаны на основании спектральных данных. Так, в спектре 1Н-ЯМР всех полученных соединений, зарегистрированных в дейтерированном хлороформе, наблюдается появление сигналов протонов ароматического ядра фталильной группы в области 7.74 и 7.86 м.д., сигнала протона метиленовой группы в области 5.2 м.д. Исчезновение сигналов протона OH-группы доказывает образование продуктов реакции.
Противовоспалительная активность соединений (XVI) и (XVII). Противовоспалительная активность соединений (XVI) и (XVII) была изучена in vivo на белых мышах обоих полов в сравнении с Ортофеном (лекарственным препаратом противовоспалительного действия, входящим в число жизненно необходимых и важнейших лекарственных средств) на четырех моделях воспаления, вызванного введением формалина, каррагинана, яичного белка и лидокаина. Результаты этих исследований приведены в табл. 4. Полученные данные позволяют заключить, что конъюгаты (XVI) и (XVII) обладают выраженным противовоспалительным эффектом в дозе 50 мг/кг и по эффективности противовоспалительного действия сопоставимы с Ортофеном на всех моделях воспаления.
Таблица 4.
Противовоспалительная активность конъюгатов (XVI) и (XVII) in vivo на разных моделях воспаления
| Соединение или препарат | Увеличение отека лапок, % | Угнетение воспаления, % |
|---|---|---|
| 1) Каррагинановая модель | ||
| (XVI) | 37.9 ± 3.2 | 29.0 |
| (XVII) | 38.5 ± 5.0 | 27.9 |
| Ортофен | 36.8 ± 1.2 | 31.1 |
| Контроль | 53.4 ± 4.5 | – |
| 2) Лидокаиновая модель | ||
| (XVI) | 36.9 ± 3.4 | 32.3 |
| (XVII) | 38.2 ± 1.8 | 29.9 |
| Ортофен | 38.1 ± 1.5 | 30.1 |
| Контроль | 54.5 ± 5.0 | – |
| 3) Белковая модель | ||
| (XVI) | 43.4 ± 2.1 | 36.3 |
| (XVII) | 44.2 ± 4.1 | 35.1 |
| Ортофен | 40.2 ± 4.0 | 41.0 |
| Контроль | 68.2 ± 5.7 | – |
| 4) Формалиновая модель | ||
| (XVI) | 37.2 ± 2.0 | 35.0 |
| (XVII) | 38.6 ± 3.5 | 32.6 |
| Ортофен | 33.3 ± 2.7 | 42.0 |
| Контроль | 57.3 ± 3.5 | – |
Изоэнзим-специфического ингибирование COX-1 и COX-2. Как известно, COX-1 участвует в регуляции клеточного гомеостаза. COX-2 регулирует биосинтез простагландинов при остром воспалении и служит ключевым ферментом противовоспалительной активности для НПВП. Для оценки селективности действия соединений (XVI) и (XVII) в отношении изоформ COX-1 и COX-2 использовали коммерческий набор COX Fluorescent Inhibitor Screening Assay Kit (Cayman Chemical, США), позволяющий оценить ингибирование двух изоформ фермента во внеклеточной системе. Согласно полученным данным, для соединения (XVI) в концентрации 0.1, 1, 10 и 100 мкМ обнаружено концентрационно-зависимое снижение активности COX-1; при концентрации соединения (XVI) 95.5 ± 3.4 мкМ установлено ингибирование фермента на 50%. Зависимый от концентрации эффект в отношении COX-1 показан и для конъюгата (XVII), который уже в концентрации 1.0 ± 0.1 мкМ на 50% ингибирует работу фермента. Кроме того, соединение (XVI) в концентрации 60.4 ± 7.9 мкМ, а соединение (XVII) в концентрации 11.7 ± 4.6 мкМ в 2 раза ингибируют активность COX-2 (табл. 3). Следовательно, селективность в отношении изоформ COX-2/COX-1 для соединения (XVI) составляет 0.63, для соединения (XVII) – 11.7, для диклофенака – 0.68 [34].
Таким образом, соединения (XVI) и (XVII) перспективны для дальнейших доклинических испытаний в условиях in vivo с целью разработки на их основе потенциальных лекарственных средств с выраженным противовоспалительным действием и повышенной избирательностью ингибирующего действия в отношении COX-1 и COX-2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование. Анализ структур синтезированных соединений выполняли на оборудовании ЦКП “Химия” Института органической химии им. Н.Д. Зелинского РАН. Спектры 1H- и 13C-ЯМР (δ, м.д.) регистрировали на импульсных спектрометрах AMX-300 (Bruker, Германия) с рабочей частотой 300.13 МГц (1H) и 75.47 МГц (13С) при постоянной температуре образца 298 К в CDCl3. Внутренний стандарт – тетраметилсилан (ТМС). Оптическую активность измеряли на поляриметре 241 MC (Perkin-Elmer, США) в трубке длиной 1 дм. Температуру плавления определяли на микростолике Boetius (Nagema, Германия). Тонкослойную хроматографию проводили на пластинах Sorbfil ПТСХ-АФ-В (ЗАО “Сорбполимер”, Краснодар, Россия), используя систему растворителей СHCl3–EtOH 9 : 1. Пятна веществ обнаруживали парами иода, нингидриновым проявителем. Колоночную хроматографию проводили на силикагеле MN Kieselgel 60 (Macherey-Nagel, Германия).
Молекулярный докинг. Молекулярный докинг структур производных пиримидина (I–XVII) с активными центрами изоформ COX проводили с использованием программы AutoDock 4.2 (Scripps Research, США). В качестве моделей белков COX-1 и COX-2 выбрали, соответственно, цепи В и А макромолекул с кодами 3n8x и 1pxx в банке данных белков PDB (http://www.rcsb.org). Молекулы белков в ходе расчетов были жесткими, в то время как молекулы лигандов – подвижными. Размер трехмерного бокса, в котором проводили молекулярный докинг лигандов, во всех случаях составлял 50 шагов с разметкой шага 0.375 Å. За центр бокса принимали положение естественного субстрата данных ферментов – арахидоновой кислоты и структур референсных ингибиторов изоформ COX, представляющих собой активные компоненты нестероидных НПВП Целекоксиб, Флурбипрофен, Диклофенак и соединения SC-558 (рис. 1). Поиск биологически активных конформаций осуществляли на основе ламарковского генетического алгоритма с параметрами по умолчанию, за исключением угла вращения вокруг ординарных связей и вращательного движения молекул, которые были равны 30°. Решения докинга кластеризовали на основе величины RMSD = 2.0 Å. Оценку эффективности связывания лигандов (I–XVII) с белками проводили по полуэмпирической оценочной функции, заложенной в программе AutoDock 4.2 (http://autodock.scripps.edu/), при наложении силового поля AMBER [35].
Общая методика получения конъюгатов (XVI) и (XVII). К раствору 0.17 г (0.001 моль) 5-гидрокси-1,3,6-триметилурацила в 10 мл сухого хлористого метилена присыпали 0.14 г (0.0015 моль) К2СО3. Затем при перемешивании без доступа влаги в реакционную смесь добавляли 0.0015 моль хлорангидридов соответствующих N-фталиламинокислот в 10 мл сухого хлористого метилена. Реакционную смесь выдерживали с перемешиванием при 20–22°С в течение 24 ч. Образовавшиеся продукты в виде осадка отфильтровывали. Затем фильтрат упаривали в вакууме водоструйного насоса. Остаток хроматографировали на колонке с силикагелем, элюируя хлороформом.
1,3,6-Триметил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил-N-фталилаланинат (XVI). Бежевое кристаллическое вещество. Выход 360 мг (98%), т. пл. 312°С (EtOH). Спектр 1Н-ЯМР: 1.79 (д, 3Н, СН-СН3); 2.18 (с, 3H, С6-СН3); 3.13 (c, 3Н, N1-CH3); 3.42 (с, 3H, N3-CH3); 5.81 (д, 1H, СН); 7.58 (t, 1H, C2(Ar)-H); 7.58 (t, 1H, C3(Ar)-H); 7.81 (t, 1H, C1(Ar)-H); 7.81 (t, 1H, C4(Ar)-H). Спектр 13С-ЯМР: 12.60 (С6-СН3); 15.01 (CHСН3); 28.68 (N3-CH3); 31.91 (N1-CH3); 54.69 (СНCH3); 110.46 (С5); 123.57 (C1(Ar)); 123.57 (C4(Ar)); 132.53 (C5(Ar)); 132.53 (C6(Ar)); 133.90 (C2(Ar)); 133.90 (C3(Ar)); 146.86 (С2=О); 157.85 (С4=О); 166.77 (2С=О); 169.42 (С6); 180.11 (С=O).
1,3,6-Триметил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил-N-фталилметионинат (XVII). Темно-бежевое кристаллическое вещество. Выход 451 мг (96%), т. пл. >320°С (EtOH). Спектр 1Н-ЯМР: 2.08 (c, 3Н, S-СН3); 2.43 (м, 2Н, СH(СН2)SCH3); 2.51 (м, 2Н, СH(СН2)SCH3); 2.66 (c, 3Н, С6-СН3); 3.11 (c, 3Н, N1-CH3); 3.26 (c, 3H, N3-CH3); 6.86 (м, 1H, СH(СН2)SCH3); 7.53 (t, 1H, C2(Ar)-H); 7.53 (t, 1H, C3(Ar)-H); 7.84 (t, 1H, C1(Ar)-H); 7.84 (t, 1H, C4(Ar)-H). Спектр 13С‑ЯМР: 14.08 (СH(СН2)SCH3); 18.51 (C6); 27.80 (N3-CH3); 29.04 (СH(СН2)SCH3); 29.36 (N1-CH3); 30.63 (СH(СН2)SCH3); 56.23 (СH(СН2)SCH3); 110.27 (С5); 123.68 (C1(Ar)); 123.68 (C4(Ar)); 131.41 (C5(Ar)); 133.00 (C2(Ar)); 133.00 (C3(Ar)); 148.40 (С2=О); 157.65 (С4=О); 169.32 (2С=О); 168.96 (C6); 181.81 (С=O).
Противовоспалительная активность соединений (XVI) и (XVII). Противовоспалительную активность конъюгатов (XVI) и (XVII) в сравнении с Ортофеном изучали in vivo на четырех моделях воспаления, вызванного введением каррагинана, лидокаина, яичного белка и формалина [36].
Опыты проводили на белых мышах Mus albus officinarum обоего пола (n = 96) (возраст 30 дней, масса 18–20 г, получены из питомника факультета биотехнологий и ветеринарной медицины ФГБОУ ВО “Башкирский государственный аграрный университет”), разделенных на 16 групп по 6 животных в каждой. Каррагинановое, лидокаиновое, белковое и формалиновое воспаление у мышей вызывали субплантарным введением 50 мкл флогогена (патогенного раздражителя, вызывающего воспалительную реакцию) – 1%-ного раствора каррагинана, 2%-ного раствора лидокаина, 15%-ного раствора яичного белка, 2%-ного раствора формалина – в переднюю левую лапку. Антифлогистическую активность исследуемых соединений оценивали по угнетению отека воспаленной лапки мышей, вызванного введением флогогена. Соединения (XVI) и (XVII), а также Ортофен в указанных концентрациях (табл. 4) вводили внутрижелудочно в объеме 300 мкл за 1 ч до введения флогогена, сразу после введения флогогена и далее через 1 и 2 ч после введения флогогена. Животным контрольной группы по той же схеме вместо исследуемых веществ вводили физиологический раствор. Величину отека лапок измеряли онкометрически [37] через 3 ч после введения флогогена.
Противовоспалительную активность рассчитывали по формуле:
Изоэнзим-специфическое ингибирование COX-1 и COX-2. Метод определения изоэнзим-специфического ингибирования COX-1 и COX-2 с помощью набора COX Fluorescent Inhibitor Screening Assay Kit (Cayman Chemical, США) основан на реакции превращения арахидоновой кислоты в нестабильный простагландин G2 (PGG2). Образовавшийся PGG2 реагирует с флуоресцентным субстратом (ADHP), превращая последний в резоруфин. Резоруфин обладает высокой флуоресцентной активностью, которая детектируется при 535–590 нм.
Валидацию набора COX Fluorescent Inhibitor Screening Assay Kit проводили с помощью ингибиторов COX-1 (SC-560) и COX-2 (DuP-697), поставляемых в наборе. При концентрации, равной IC50, данные соединения показали ~50%-ное ингибирование ферментов: (54.38 ± 0.62)% для SC-560 (кат. № 760159, ингибитор COX-1, IC50 = 5 нM) и (57.38 ± 8.23)% для DuP-697 (кат. № 760158, ингибитор COX-2, IC50 = 25 нM). Процент ингибирования ферментов при инкубации с соединениями (XVI) и (XVII) рассчитывали по формуле:
Базовая активность фермента – 0 мкM соединения. Селективность в отношении COX-2 или COX-1 рассчитывали по соотношению IC50 соединения для данных изоформ.
Воздействие изучаемых соединений на активность COX-1 и COX-2 оценивали в трех независимых экспериментах. Среднее арифметическое по общей выборке данных и стандартную ошибку среднего (SEM) находили с помощью модуля описательной статистики в программе Statistica 6.1 (StatSoft. Inc., США). Расчет IC50 активности ферментов проводили с помощью модуля нелинейной регрессии с использованием логарифма концентраций и нормализованных значений процента ингибирования (log(inhibitor) vs. response) в программе GraphPad Prism (версия 10; GraphPad Software Inc., США).
ЗАКЛЮЧЕНИЕ
На основании результатов вычислительных экспериментов с использованием программы AutoDock 4.2, реализующей базовые принципы метода молекулярного докинга, из 17 протестированных производных урацила с циклическими и ациклическими сера- и кислородсодержащими заместителями в пиримидиновом цикле для синтеза отобраны два соединения-лидера (XVI) и (XVII), которые теоретически могут быть эффективными ингибиторами обеих форм COX, а следовательно, обладать выраженным противовоспалительным действием in vivo. Эти соединения представляют собой конъюгаты 5-гидрокси-1,3,6-триметилурацила с N-фталил-L-аминокислотами. Сравнительный анализ свободных энергий связывания, рассчитанных с использованием скоринг-функции AutoDock 4.2, позволил предположить, что оба этих соединения будут отличаться повышенной селективностью действия в отношении COX-2. Хлорангидридным методом осуществлен синтез соединений (XVI) и (XVII), методом хроматографии доказана индивидуальность полученных соединений, структура соединений подтверждена с помощью ЯМР. В результате биологических испытаний in vivo на мышах на четырех моделях воспаления, вызванного введением каррагинана, лидокаина, яичного белка и формалина, показано, что полученные конъюгаты (XVI) и (XVII) действительно обладают выраженной противовоспалительной активностью, сопоставимой с лекарственным препаратом Ортофеном. Однако результаты оценки изоэнзим-специфического ингибирования ферментов изоформ COX свидетельствуют о том, что только соединение (XVII) оказалось селективным ингибитором COX-2, в то время как соединение (XVI) характеризуется повышенной селективностью действия в отношении COX-1 и, следовательно, в клинических условиях может обнаружить выраженный гастро- и нефротоксический эффект. Оба соединения перспективны для дальнейших исследований в качестве потенциальных противовоспалительных лекарственных средств.
Список литературы
Grabovskiy S.A., Murinov Yu.I., Kabal’nova N.N. // Curr. Org. Chem. 2012. V. 16. P. 2389–2393. https://doi.org/10.2174/138527212803520056
Мышкин В.А., Ибатуллина Р.Б., Савлуков А.И., Бакиров А.Б., Сергеева С.А. // Антиоксидантные эффекты производных пиримидина и бензимидазола при острых отравлениях. Уфа: ООО ПКП “ДАР”, 2003.
Мышкин В.А., Бакиров А.Б. // Экспериментальная коррекция химических поражений печени производными пиримидина. Уфа: ООО ПКП “ДАР”, 2002.
Камилов Ф.Х., Лазарева Д.Н., Плечев В.В. // Пиримидины и их применение в медицине. Уфа: Изд. БГМИ, 1992.
Лазарева Д.Н., Алехин Е.К., Плечев В.В., Тимербулатов В.М., Плечева Д.В. // Иммурег. Уфа: Изд-во БГМУ, НПО Башбиомед, 2004.
Кривоногов В.П., Толстиков Г.А., Муринов Ю.И., Куковинец А.Г., Шакирова А.М., Сорокина Г.А., Селезнев Л.Г., Витвицкая А.С., Бриль А.С., Казаков В.П., Караваев А.Д., Зарудий Ф.С., Лазарева Д.Н., Комиссаров В.Д., Ахунов И.Р. // Патент RU2000298С1, опубл. 07.09.1993.
Isobe Y., Tobe M., Inoue Y., Isobe M., Tsuchiya M., Hayashi H. // Bioorg. Med. Chem. 2003. V. 11. P. 4933–4940. https://doi.org/10.1016/j.bmc.2003.09.012
Curtis-Prior P. // The Eicosanoids. John Wiley & Sons, Chichester, 2004.
Dennis E.A. // J. Biol. Chem. 1994. V. 269. P. 13057–13060.
Samy R.P., Gopalakrishnakone P., Chow V.T.K. // Bioinformation. 2012. V. 8. P. 4857. https://doi.org/10.6026/97320630008048
Flower R.J., Blackwell G.J. // Nature. 1979. V. 278. P. 456–459. https://doi.org/10.1038/278456a0
Henderson W.R., Oslund R.C., Bollinger J.G., Ye X., Tien Y.T., Xue J., Gelb M.H. // J. Biol. Chem. 2011. V. 286. P. 28049–28055. https://doi.org/10.1074/jbc.M111.235812
Berg O.G., Gelb M.H., Tsai M.D., Jain M.K. // Chem. Rev. 2001. V. 101. P. 2613–2654. https://doi.org/10.1021/cr990139w
Farooqui A.A., Ong W.-Y., Horrocks L.A. // Pharm. Rev. 2006. V. 58. P. 591–620. https://doi.org/10.1124/pr.58.3.7
Ong W.Y., Farooqui T., Kokotos G., Farooqui A.A. // ACS Chem. Neurosci. 2015. V. 6. P. 81431. https://doi.org/10.1007/s12035-020-02093-z
Sapolsky R.M. // J. Neurosci. 1986. V. 6. P. 2240–2244. https://doi.org/10.1523/jneurosci.06-08-02240.1986
Mruwat R., Cohen Y., Yedgar S. // Imunnotherapy. 2013. V. 5. P. 315–317. https://doi.org/10.2217/imt.13.18
Zarghi A., Arfaei S. // Iran. J. Pharm. Res. 2011. V. 10. P. 655–683.
Marnett L.J. // Cancer Prev. Res. (Phyla). 2009. V. 2. P. 288–290. https://doi.org/10.1158/1940-6207.capr-09-0033
Smith W.L., DeWitt D.L., Garavito R.M. // Annu. Rev. Biochem. 2000. V. 69. P. 145–182. https://doi.org/10.1146/annurev.biochem.69.1.145
Greenhough A., Smartt H.J.M., Moore A.E., Roberts R.R., Williams A.C., Kaidi C.P.A. // Carcinogenesis. 2009. V. 30. P. 377–386. https://doi.org/10.1016/j.ebiom.2015.01.016
Charlier C., Michaux C. // Eur. J. Med. Chem. 2003. V. 38. P. 645–659. https://doi.org/10.1016/s0223-5234(03)00115-6
Vane J.R., Bakhle Y.S., Botting R.M. // Annu. Rev. Pharmacol. Toxicol. 1998. V. 38. P. 97–120. https://doi.org/10.1146/annurev.pharmtox.38.1.97
Cha Y.I., DuBois R.N. // Annu. Rev. Med. 2007. V. 58. P. 239–252. https://doi.org/10.1146/annurev.med.57.121304.131253
Khairullina V.R., Gerchikov A.Ja., Taipov I.A., Boegel H., Zarudii F.S. // Pharm. Chem. J. 2011. V. 45. P. 539546. https://doi.org/10.1007/s11094-011-0675-y
Khairullina V.R., Taipov I.A., Gerchikov A.Ja., Zarudii F.S. // Pharm. Chem. J. 2012. V. 46. P. 553–564. https://doi.org/10.1007/s11094-012-0846-5
Хайруллина В.Р., Герчиков А.Я., Зарудий Ф.С. // Вестник Башкирского университета. 2014. № 19. С. 417422.
Sliwoski G., Kothiwale S., Meiler J., Lowe E.W. // Pharmacol. Rev. 2014. V. 66. P. 334–395. https://doi.org/10.1124/pr.112.007336
Jansen J.M., Amaro R.E., Cornell W., Tseng Y.Ja., Walters W.P. // Future Med. Chem. 2014. V. 4. P. 1893–1896. https://doi.org/10.4155/fmc.12.137
Khayrullina V.R., Gerchikov A.Ya., Lagunin A.A., Zarudii F.S. // Biochemistry (Mosc.). 2015. V. 80. P. 74–86. https://doi.org/10.1134/s0006297915010095
Gerchikov A.Ya., Vasil’ev M.N., Khayrullina V.R., Tsypysheva I.P., Zarudii F.S. // Pharm. Chem. J. 2015. V. 49. P. 582–586. https://doi.org/10.1007/s11094-015-1333-6
Khayrullina V.R., Taipov I.A., Gerchikov A.Ya., Vasil’ev M.N., Zarudii F.S., Boegel H. // Pharm. Chem. J. 2014. V. 48. P. 317–322. https://doi.org/10.1007/s11094-014-1102-y
Кривоногов В.П., Толстиков Г.А., Муринов Ю.И., Зарудий Ф.С., Лазарева Д.Н., Исмагилова А.Ф., Волкова С.С., Сахаутдинова Г.М., Афзалетдинова Н.Г., Хисамутдинов Р.А., Спирихин Л.В., Кривоногова И.М. // Хим.-фарм. журнал. 1993. Т. 27. С. 41–44.
Тринус Ф.П., Мохорт Н.А., Клебанов Б.М. // Нестероидные противовоспалительные средства. Киев: Здоров’я, 1975.
Wang J., Wolf R.M., Caldwell J.M., Kollman P.A., Case D.A. // J. Comp. Chem. 2004. V. 25. P. 1157–1174. https://doi.org/10.1002/jcc.20035
Dannhardt G., Kiefer W. // Eur. J. Med. Chem. 2001. V. 36. P. 109–126. https://doi.org/10.1016/s0223-5234(01)01197-7
Хабриев Р.У. Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. 2-изд., перераб. и доп. М.: ОАО Изд-во “Медицина”, 2005.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия