

Биоорганическая химия, 2023, T. 49, № 1, стр. 93-104
Получение комплексов фицина с карбоксиметилхитозаном и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном и изучение их структурных особенностей
Н. В. Малыхина 1, С. С. Ольшанникова 1, М. Г. Холявка 1, 2, *, А. В. Сорокин 1, 2, М. С. Лавлинская 1, 2, В. Г. Артюхов 1, Д. А. Файзуллин 3, Ю. Ф. Зуев 3
1 Воронежский государственный университет
394018 Воронеж, Университетская пл., 1, Россия
2 Севастопольский государственный университет
299053 Севастополь, ул. Университетская, 33, Россия
3 Казанский институт биохимии и биофизики, Федеральный исследовательский центр “Казанский научный центр” РАН
420111 Казань, ул. Лобачевского, 2/31, Россия
* E-mail: holyavka@rambler.ru
Поступила в редакцию 27.04.2022
После доработки 30.05.2022
Принята к публикации 01.06.2022
- EDN: FSCCFU
- DOI: 10.31857/S0132342322060173
Аннотация
Синтезированы производные хитозана – карбоксиметилхитозан и N-(2-гидрокси)пропил-3-триметиламмонийхитозан с молекулярными массами 200, 350 и 600 кДа. Получены комплексы фицина с хитозаном и его названными производными, зарегистрированы ИК-спектры хитозана, карбоксиметилхитозана и N-(2-гидрокси)пропил-3-триметиламмонийхитозана и их комплексов с фицином. Анализ спектров подтвердил образование конъюгатов между макромолекулами полисахаридов и фицина. Оптимальное соотношение содержания белка (0.7 мг/г носителя) и удельной активности (1590 ед./мг белка) выявлено при образовании комплекса фицина с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном с молекулярной массой 350 кДа. Эффективность комплексообразования фицина (по удельной каталитической активности) с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном (350 кДа) превышает таковую для хитозана (350 кДа) и карбоксиметилхитозана (350 кДа) в 2.4 и 9.8 раз соответственно. Методом молекулярного докинга изучены типы взаимодействий, энергии первого связывания, аминокислотный состав поверхностей фицина, которые в процессе комплексообразования взаимодействуют с носителем. Установлено, что связи и взаимодействия с хитозаном и его производными образуются в том числе с участием аминокислотных остатков, расположенных вблизи активного центра фицина (Cys25 и His162), что объясняет изменение протеолитической активности полученных комплексов. Комплексы фицина с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном растворимы в широком диапазоне рН среды и поэтому могут оказаться более перспективными, чем его комплексы с хитозаном, при разработке медицинских препаратов и биокатализаторов для пищевой, пивоваренной и кожевенной промышленности.
ВВЕДЕНИЕ
Фицин (КФ 3.4.22.3) – протеолитический фермент класса гидролаз, обладающий противоожоговыми, ранозаживляющими и противовоспалительными свойствами, которые обусловливают его применение в биотехнологии, медицине и косметологии. Фицин выделяют из латекса растений рода Ficus. Молекула фицина содержит две сульфгидрильные группы, из которых только одна находится в активном центре фермента. Молекулярная масса энзима составляет 23 кДа, рН-оптимум действия 7.0, температурный оптимум 60–65°С [1, 2].
Использование свободных форм ферментов в различных отраслях производства затруднено вследствие того, что они неустойчивы при хранении, а также подвержены действию внешних факторов. Эти проблемы можно преодолеть с помощью комплексообразования белка с полимерным носителем. Кроме того, изучение иммобилизованных ферментов и их систем способствует расширению теоретических знаний о путях регулирования структурно-функциональных свойств энзимов [3–6].
Попытки получить иммобилизованные формы фицина неоднократно описаны в литературе. В качестве носителей были использованы глиоксилагароза [7, 8], поливиниловый спирт [9], композитные материалы [10]. Эффективность иммобилизации фицина на этих носителях, рассчитанная как процент сохранения удельной каталитической активности фермента, составила 60, 74 и 90% соответственно.
Известно, что перспективными носителями для иммобилизации ферментов являются хитозан и его производные. Хитозан – это статистический сополимер, состоящий из остатков D-глюкозамина и N-ацетил-D-глюкозамина, связанных между собой 1,4-β-гликозидными связями. Важнейшие свойства данного полимера ‒ биодеградируемость, гипоаллергенность, антимикробная активность, противоязвенные и противоопухолевые свойства. Хитозан характеризуется способностью образовывать растворы с высокой вязкостью и обладает высокой хелато- и комплексообразующей способностью, возможностью формировать межмолекулярные водородные связи, а также реакционноспособными группами, обеспечивающими возможность его химических модификаций. В условиях рН-оптимума цистеиновых протеаз хитозан нерастворим в воде, что ограничивает доступность центров его макромолекул для взаимодействий с белком, поэтому представляет интерес рассмотреть в качестве полимеров для комплексообразования с фицином растворимые при рН 7.5 производные хитозана [11–13]. Карбоксиметилхитозан (КМХ) применяется в производстве высокотехнологичной косметики, хелатирующих агентов тяжелых металлов, агентов замедленного высвобождения лекарств, регуляторов роста растений, а также для очистки сточных вод [14]. N-(2-Гидрокси)пропил-3-триметиламмонийхитозан (ГПХ) – одно из производных хитозана, полученных путем алкилирования. Этот носитель менее подвержен действию рН среды и хорошо растворим в воде. Он нашел широкое применение во многих отраслях производства, например, в химической, фармацевтической промышленности, а также при получении наночастиц [15].
Предсказание особенностей взаимодействия фицина с молекулами полисахаридов помогает минимизировать эмпирические эксперименты по подбору оптимальной матрицы для комплексообразования. Молекулярный докинг позволяет предварительно оценить положение лиганда относительно поверхности молекулы фермента [16], а с помощью ИК-спектроскопии можно исследовать в молекулах белков и полисахаридов функциональные группы и другие фрагменты [17, 18]. Сочетание методов ИК-спектроскопии и гибкого молекулярного докинга позволяет выявлять функциональные группы модифицированных полисахаридов, а также аминокислотные остатки на поверхности молекулы фицина с указанием типов взаимодействий, образующих конъюгат энзим–носитель.
Целью данной работы явилось получение комплексов фицина с карбоксиметилхитозаном и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном и изучение их структурных особенностей.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез производных хитозана. На первом этапе работы по известным методикам с небольшими модификациями (см. “Эксперим. часть”) из коммерческого хитозана (ЗАО “Биопрогресс”, Россия) были синтезированы производные – карбоксиметилхитозан и N-(2-гидрокси)пропил-3-триметиламмонийхитозан с молекулярными массами 200, 350 и 600 кДа.
Получение комплексов фицина с полисахаридами. ИК-спектроскопия. Были получены комплексы протеолитического фермента фицина (Sigma, США) с полисахаридами: хитозаном и его синтезированными производными – карбоксиметилхитозаном и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном. Для подтверждения факта комплексообразования между молекулами фицина и полисахаридов были зарегистрированы и проанализированы ИК-спектры полученных образцов.
На рис. 1 представлены ИК-спектры хитозана и его комплекса с фицином. ИК-спектр полисахарида содержит следующие характеристические полосы. Полосы 2921 и 2875 см–1 относятся к валентным антисимметричным и симметричным колебаниям связей С–Н. Положение их типично для полисахаридов. Остаточные N-ацетильные группы проявляются полосами ~1645 (валентные С=О, амид I) и 1325 (валентные C–N, амид III) см–1. Полоса амид II (деформационные N–H), обычно присутствующая при 1550 см–1, в данном случае скрыта сильной близко расположенной полосой деформационных N–H первичных аминов на 1589 см–1. Деформационные колебания групп СН2 и СН3 проявляются при 1423 и 1375 см–1 соответственно. Узкая полоса на 1150 см–1 соответствует поглощению гликозидной связи С–О–С, а компоненты 1060 и 1025 см–1 относятся к валентным колебаниям связи С–О в структуре пиранозных колец [19, 20]. В ИК-спектре комплекса хитозана с фицином присутствуют описанные выше полосы, однако их относительная интенсивность и положение меняются из-за наложения спектра поглощения белка. Так, видимый максимум полосы амид I смещается к 1638 см–1, проявляются белковые полосы амид II и амид III. Из-за наложения амидных полос хитозана и белка трудно судить о характере взаимодействия по группам N–H и С=О. Однако в области поглощения связей С–О боковых групп пиранозных колец относительное падение интенсивности компоненты 1060 см–1 в спектре комплекса по сравнению с чистым хитозаном свидетельствует по меньшей мере о том, что взаимодействие с белком затрагивает боковые ОН-группы полисахарида.
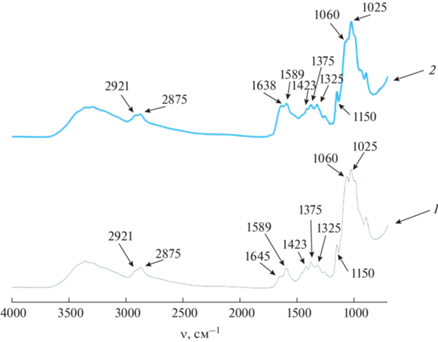
На рис. 2 представлены ИК-спектры карбоксиметилхитозана и его комплекса с фицином. По сравнению с немодифицированным хитозаном, в них проявились интенсивные полосы 1590, 1410 и 1320 см–1, обусловленные колебаниями карбоксильных групп νasCOO–, νsCOO– и ρwCH2 соответственно [21]. Дополнительная связь С–О–С в боковой цепи карбоксиметилхитозана проявляется компонентой 1120 см–1. Присутствие белка в спектре комплекса обнаруживается по повышенному поглощению при 1650 см–1 на склоне полосы 1590 см–1. Как и в случае немодифицированного хитозана, комплексообразование полисахарида с белком приводит к падению интенсивности компоненты 1060 см–1 относительно компоненты 1025 см–1. Указанные изменения подтверждают образование конъюгата между макромолекулами карбоксиметилхитозана и молекулами фицина.

На рис. 3 представлены ИК-спектры N-(2-гидрокси)пропил-3-триметиламмонийхитозана и его комплекса с фицином. ИК-спектр N-(2-гидрокси)пропил-3-триметиламмонийхитозана содержит следующие характеристические признаки, отличающие его от спектра немодифицированного хитозана: 2926 и 1478 см–1 – валентное и деформационное колебания С–Н триметиламмонийных групп, отсутствие полосы при 1590–1600 см–1 деформационного колебания первичных аминогрупп вследствие замены на вторичные амины, увеличенная интенсивность полосы валентных ОН-колебаний за счет дополнительной гидроксильной группы [22, 23]. Форма полосы колебаний групп в составе пиранозных остатков в сравнении с хитозаном изменений не претерпела. При этом, как и в предыдущих двух случаях, комплексообразование с белком приводит к падению интенсивности компоненты 1060 относительно 1025 см–1.
Рис. 3.
ИК-спектры N-(2-гидрокси)пропил-3-триметиламмонийхитозана (1) и его комплекса с фицином (2).

Содержание белка в комплексах фицина с полисахаридами и их удельная каталитическая активность. Анализ содержания белка в полученных препаратах показал, что наибольшее количество фицина образует комплекс с карбоксиметилхитозаном с молекулярной массой 200 кДа (рис. 4), при этом эффективность комплексообразования (по содержанию белка) составляет 49%. Однако фермент, вероятно, находится в каталитически “невыгодной” конформации или происходят процессы автолиза, что отрицательно сказывается на его протеолитической активности. Наибольшую удельную активность показали комплексы фицина с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном с молекулярной массой 350 кДа (рис. 5). Именно они, на наш взгляд, могут оказаться более перспективными, чем комплексы протеазы с хитозаном, для применения в промышленности и медицине, благодаря своей высокой каталитической активности и растворимости в широком диапазоне рН среды. Вероятно, соотношение 0.6–0.7 мг фермента на 1 г носителя оптимально для предотвращения процессов автолиза и приобретения фицином наиболее каталитически активной конформации. При увеличении этого соотношения до 9.7 мг на 1 г полимера удельная протеолитическая активность фицина снижается в 9.8 раз по сравнению с максимально измеренной (при соотношении фермент/полисахарид, равном 0.7 мг/г).
Рис. 4.
Содержание белка (мг/г носителя) в комплексах фицина с хитозаном и его производными: карбоксиметилхитозаном (КМХ) и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном (ГПХ). Над столбцами указана эффективность комплексообразования фицина (по содержанию белка), выраженная в процентах фермента в составе комплекса от его количества в растворе в процессе комплексообразования, принятого за 100%.

Рис. 5.
Удельная каталитическая активность (ед./мг белка) комплексов фицина с хитозаном и его производными: карбоксиметилхитозаном (КМХ) и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном (ГПХ). Над столбцами указана эффективность комплексообразования фицина (по удельной каталитической активности), выраженная в процентах сохранения удельной протеолитической активности фермента после иммобилизации по отношению к удельной каталитической активности фицина в растворе, принятой за 100%.

Механизмы комплексообразования между фицином и полисахаридами. Для проектирования активно и стабильно функционирующих биокатализаторов на основе комплексов ферментов и полисахаридов необходимо понимать механизм взаимодействия носителя с молекулами белка. Известно, что специфические взаимодействия возникают не одновременно. Сначала образуются различные “одноточечно” и “двухточечно” связанные состояния, а за ними следует появление связей по некоторой контактной линии и, наконец, по определенной контактной плоскости, при этом иммобилизация становится кинетически необратимой [24, 25].
Методом молекулярного докинга мы смоделировали первую “точку” связывания фицина с хитозаном, карбоксиметилхитозаном и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном. Были изучены типы взаимодействий, энергии первого связывания, аминокислотный состав поверхностей фицина, которые в процессе комплексообразования взаимодействуют с носителем (рис. 6–8, табл. 1). Из рисунков видно, что связи и взаимодействия с хитозаном и его производными образуются в том числе с участием аминокислотных остатков, расположенных вблизи активного центра фермента (Cys25 и His162), что, естественно, отражается на активности полученных комплексов. Энергии первого взаимодействия фицина с изучаемыми нами полисахаридами практически идентичны: с хитозаном и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном – 6.6, с карбоксиметилхитозаном – 6.5 ккал/моль.
Рис. 6.
Связи и взаимодействия между фицином и хитозаном (пунктирными линиями обозначены водородные связи, длина связей приведена в Å).

Рис. 7.
Связи и взаимодействия между фицином и карбоксиметилхитозаном (пунктирными линиями обозначены водородные связи, длина связей приведена в Å).
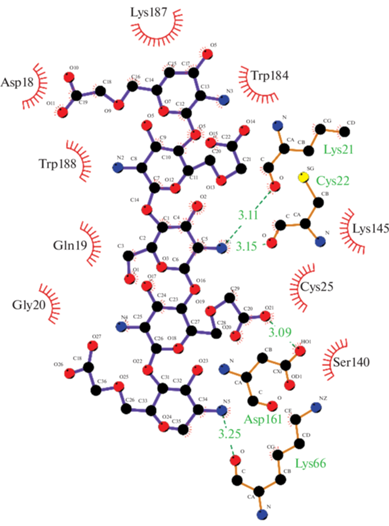
Рис. 8.
Связи и взаимодействия между фицином и N-(2-гидрокси)пропил-3-триметиламмонийхитозаном (пунктирными линиями обозначены водородные связи, длина связей приведена в Å).

Таблица 1.
Аминокислотные остатки фицина, формирующие связи и взаимодействия с хитозаном и его производными
| Энергия первого взаимодействия, ккал/моль | Аминокислотные остатки, формирующие | |
|---|---|---|
| водородные связи, и длина связи, Å | иные типы взаимодействий | |
| 1) Аминокислоты фицина, которые образуют связи и взаимодействия с хитозаном | ||
| −6.6 | Asn18, 2.90 Å Gly20, 3.13 Å Glu145, 3.01 Å Asp161, 2.85 Å Trp184, 3.06 Å |
Gln19, Ala139, Gly140, His162 |
| 2) Аминокислоты фицина, которые образуют связи и взаимодействия с карбоксиметилхитозаном | ||
| −6.5 | Lys21, 3.11 Å Cys22, 3.15 Å Lys66, 3.25 Å Asp161, 3.09 Å |
Asp18, Gln19, Gly20, Cys25, Ser140, Lys145, Trp184, Lys187, Trp188 |
| 3) Аминокислоты фицина, которые образуют связи и взаимодействия с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном | ||
| −6.6 | Gln19, 3.15 Å Gly20, 2.85 Å Cys22, 3.02 Å Lys145, 2.78 и 3.16 Å |
Asp18, Lys21, Gly23, Cys25, Lys66, Gly67, Gly68, Asn160, Asp161, Trp184, Trp188 |
Исходя из полученных нами данных, можно предположить, что путем введения в структуру хитозана новых функциональных групп можно модулировать механизмы комплексообразования и активность фицина, подбирая оптимальное микроокружение для аминокислот из его активного центра – Cys25 и His162.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объект исследования и носители для его иммобилизации. В качестве объекта исследования был выбран фицин из латекса растений рода Ficus (Sigma, США), субстратом для гидролиза служил азоказеин (Sigma, США), носителями для комплексообразования – хитозаны (ХТЗ) (ЗАО “Биопрогресс”, Россия) и их производные: карбоксиметилхитозан (КМХ), N-(2-гидрокси)пропил-3-триметиламмонийхитозан (ГПХ) с молекулярными массами 200, 350 и 600 кДа соответственно.
Синтез производных хитозана. Производные хитозана получали по известным методикам с небольшими модификациями. Синтез карбоксиметилхитозана (КМХ) осуществляли по следующей методике: навеску хитозана массой 3.00 г диспергировали в 65 мл изопропилового спирта, затем при помощи капельной воронки в течение 15 мин вводили водный раствор NaOH из расчета 13 моль NaOH на 1 моль хитозана. После этого по каплям в реакционную смесь добавляли спиртовой раствор монохлоруксусной кислоты из расчета 7 моль кислоты на 1 моль хитозана и перемешивали в течение 12 ч при комнатной температуре. Полученный твердый продукт отфильтровывали, суспендировали в метиловом спирте, нейтрализовали раствором уксусной кислоты, промывали этиловым спиртом и сушили в вакуумном сушильном шкафу до постоянной массы [26].
Выход продуктов, вычисленный как отношение массы полученного продукта к теоретически возможной, составлял 79–92%; степень замещения, рассчитанная из данных ИК-спектроскопии, составила 0.46, 0.54 и 0.78 для хитозана с молекулярной массой 600, 350 и 200 кДа соответственно. Существенное увеличение значения степени замещения для хитозана с молекулярной массой 200 кДа может быть объяснено не только влиянием стерического фактора, но и тем, что изначально этот коммерческий хитозан находился в форме порошка, в то время как два других – в форме “чешуек”. Карбоксиметилирование проводили в гетерогенных условиях, поэтому форма и размер частиц модифицируемого полимера оказывают значительное влияние на протекание химического процесса.
N-(2-Гидрокси)пропил-3-триметиламмонийхитозан (ГПХ) получали по следующей методике: навеску хитозана массой 3.00 г суспендировали в 30 мл дистиллированной воды в течение 30 мин при 85°С. Затем по каплям вводили глицидилтриметиламмоний хлорид (ГТМАХ) из расчета на 1 моль хитозана 3 моль ГТМАХ и выдерживали при 85°С в течение 10 ч. Продукт из реакционной смеси выделяли осаждением в ацетон, после чего трижды промывали этиловым спиртом и сушили в вакуумном сушильном шкафу до постоянной массы [27]. Выход продуктов составлял 62–74%; степень замещения, рассчитанная из данных ИК-спектроскопии, составила 0.24, 0.19 и 0.57 для хитозана с молекулярной массой 600, 350 и 200 кДа соответственно.
ИК-спектроскопия. Регистрацию ИК-спектров анализируемых образцов в виде сухих порошков без наполнителей осуществляли на спектрометре IR-Affinity 1 (Shimadzu Instruments, Япония) в режиме нарушенного полного внутреннего отражения (НПВО) с призмой из ZnSe, в диапазоне частот 700–4000 см–1, разрешение 4 см–1.
Получение комплексов фицина с полисахаридами. Комплексообразование фицина с хитозаном и его производными осуществляли следующим образом: к 1 г хитозана добавляли 20 мл раствора фермента (в концентрации 10 мг/мл 0.05 М глицинового буфера, рН 9.0), инкубировали в течение 2 ч. После окончания инкубации образовавшийся комплекс промывали с помощью диализа против 50 мМ Tris-HCl-буфера (pH 7.5) через целлофановую мембрану с размером пор 25 кДа до отсутствия в промывных водах белка, контроль осуществляли на спектрофотометре СФ-2000 (ОКБ Спектр, Россия) при λ = 280 нм. Полученные препараты лиофильно высушивали до постоянной массы и использовали для дальнейших исследований.
Определение содержания белка в комплексах фицина с полисахаридами и их удельной каталитической активности. Содержание белка в комплексах фицина с хитозаном и его производными определяли методом Лоури [29] со следующей модификацией: на первом этапе анализа осуществляли разрушение связей между полисахаридом и молекулами фермента. Для этого иммобилизованный фицин обрабатывали раствором K,Na-тартрата (в концентрации 20 мг/мл или 0.7 М), приготовленным на 1 М NaOH, при 50°С в течение 10 мин [30]. Отсутствие процессов разрушения фермента контролировали путем регистрации и анализа его спектра поглощения на спектрофотометре UV-2550PC (Shimadzu, Япония).
Определение протеолитической активности фермента проводили на субстрате азоказеине (Sigma, США) [31, 32]. К 50 мг образца добавляли 200 мкл Tris-HCl-буфера, pH 7.5, 800 мкл азоказеина (0.5% в 50 мМ Tris-HCl-буфере, pH 7.5) и инкубировали 2 ч при 37°C. Далее добавляли 800 мкл ТХУ (5%), инкубировали 10 мин при 4°C, затем центрифугировали в течение 3 мин при 11 700 g для удаления негидролизованного азоказеина. К 1200 мкл супернатанта добавляли 240 мкл 3%‑ного NaOH для нейтрализации кислоты, после чего измеряли оптическую плотность опытной пробы при 410 нм в 1-см кювете. Контрольная проба содержала 800 мкл азоказеина, 800 мкл ТХУ, 50 мг образца и 200 мкл Tris-HCl-буфера. За единицу каталитической активности принимали количество фицина (в мг белка), которое в условиях эксперимента гидролизует 1 мкмоль субстрата за 1 мин. Удельную протеолитическую активность фицина рассчитывали по формуле [33]:
где А – протеолитическая активность, мкмоль субстрата в мин на 1 мг белка; ΔD – изменение оптической плотности опытного образца по отношению к контрольному при 410 нм; V – общий объем пробы, мл; С – концентрация белка в пробе, мг/мл; 120 – время инкубации, мин; ε – коэффициент молярной экстинкции 0.5%-ного раствора азоказеина при 410 нм, равный 12 мМ–1 см–1; l – длина оптического пути, равная 1 см.Эффективность комплексообразования фицина по содержанию белка выражали в процентах фермента в составе комплекса от его количества в растворе в процессе комплексообразования, принятого за 100%; по удельной каталитической активности – в процентах сохранения удельной протеолитической активности фермента после иммобилизации по отношению к удельной каталитической активности фицина в растворе, принятой за 100%.
Статистическую значимость различий величин контрольных и опытных показателей определяли по t-критерию Стьюдента (при p < 0.05, n = = 8), поскольку все показатели характеризовались нормальным распределением. Полученные данные (рис. 4 и 5) представлены в виде средних значений ± доверительный интервал.
Молекулярный докинг. Подготовку структуры фицина (PDB ID: 4YYW, https://www.rcsb.org/ structure/4YYW) для докинга выполняли по схеме, стандартной для Autodock Vina (https://sourceforge.net/projects/autodock-vina-1-1-2-64-bit/), описанной авторами пакета на сайте: из входного файла PDB были удалены координаты атомов (и сами атомы) молекул растворителя, буфера и лигандов. Центр молекулы и параметры бокса (“ячейки”) задавали вручную, добиваясь того, чтобы молекула протеазы полностью была внутри расчетной области пространства [28].
Модели структур хитозана и его производных были нарисованы в молекулярном конструкторе HyperChem (https://hyperchem.software.informer.com), последовательно оптимизированы сначала в силовом поле AMBER, а потом квантово-химически в PM3 (Parametric Method 3). Лиганд в расчетах докинга имел максимальную конформационную свободу: допускалось вращение функциональных групп вокруг всех одинарных связей. Расстановку зарядов на молекулах полисахаридов и их протонирование/депротонирование проводили автоматически в пакете MGLTools 1.5.6 (https://ccsb.scripps.edu/mgltools/1-5-6/).
ЗАКЛЮЧЕНИЕ
Синтезированы производные хитозана – карбоксиметилхитозан и N-(2-гидрокси)пропил-3-триметиламмонийхитозан с молекулярными массами 200, 350 и 600 кДа. Проведено комплексообразование фицина с хитозаном и его производными. Анализ ИК-спектров подтвердил образование конъюгатов между макромолекулами названных полисахаридов и фицина. Оптимальное соотношение содержания белка (0.7 мг/г носителя) и удельной активности (1590 ед./мг белка) выявлено при образовании комплекса фицина с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном с молекулярной массой 350 кДа. По отношению к удельной каталитической активности фицина в растворе удельная протеолитическая активность полученных комплексов фермента с хитозаном (200 и 350 кДа) составила ~70%, с карбоксиметилхитозаном (350 и 600 кДа) находилась в пределах 20%, а с N-(2-гидрокси)пропил-3-триметиламмонийхитозаном (350 кДа) составила 166%, что делает данный комплекс перспективным для практического применения в пищевой, пивоваренной и кожевенной промышленности, а также в медицине. Методом молекулярного докинга установлено, что связи и взаимодействия с хитозаном и его производными образуются в том числе с участием аминокислотных остатков, расположенных вблизи активного центра фицина (Cys25 и His162), что объясняет изменение протеолитической активности полученных комплексов.
Список литературы
Holyavka M., Faizullin D., Koroleva V., Olshannikova S., Zakhartchenko N., Zuev Yu., Kondratyev M., Zakharova E., Artyukhov V. // Int. J. Biol. Macromol. 2021. V. 180. P. 161–176. https://doi.org/10.1016/j.ijbiomac.2021.03.016
Holyavka M.G., Kayumov A.R., Baydamshina D.R., Koroleva V.A., Trizna E.Y., Trushin M.V., Artyukhov V.G. // Int. J. Biol. Macromol. 2018. V. 115. P. 829–834. https://doi.org/10.1016/j.ijbiomac.2018.04.107
Wingard L.B., Berezin I.V., Klyosov A.A. // Enzyme Engineering. Future Directions. New York: Plenum Press, 1980. XIV, 522 p. https://doi.org/10.1007/978-1-4684-3719-5
Efremenko E.N., Lozinsky V.I., Sergeeva V.S., Plieva F.M., Makhlis T.A., Kazankov G.M., Gladilin A.K., Varfolomeyev S.D. // J. Biochem. Biophys. Methods. 2002. V. 51. P. 195–201. https://doi.org/10.1016/S0165-022X(01)00135-X
Efremenko E., Peregudov A., Kildeeva N., Perminov P., Varfolomeyev S. // Biocatalysis and Biotransformation. 2005. V. 23. P. 103–108. https://doi.org/10.1080/10242420500132474
Muronetz V.I., Zhang N.X., Bulatnikov I.G., Wang C.-C. // FEBS Lett. 1998. V. 426. P. 107–110. https://doi.org/10.1016/S0014-5793(98)00319-6
Siar E.-H., Zaak H., Kornecki J.F., Zidoune M.N., Barbosa O., Fernandez-Lafuente R. // Process Biochem. 2017. V. 58. P. 98–104. https://doi.org/10.1016/j.procbio.2017.04.009
Siar E.-H., Morellon-Sterling R., Zidoune M.N., Fernandez-Lafuente R. // Int. J. Biol. Macromol. 2020. V. 144. P. 419–426. https://doi.org/10.1016/j.ijbiomac.2019.12.140
Hayashi T., Hyon S.-H., Cha W.-I., Ikada Y. // Polym. J. 1993. V. 25. P. 489–497. https://doi.org/10.1295/polymj.25.489
Pan Y., Pang Y., Shi Y., Zheng W., Long Y., Huang Y., Zheng H. // Microchim. Acta. 2019. V. 186. P. 213. https://doi.org/10.1007/s00604-019-3331-y
Kulikov S.N., Tikhonov V.E., Bezrodnykh E.A., Lopatin S.A., Varlamov V.P. // Russ. J. Bioorg. Chem. 2015. V. 41. P. 57–62. https://doi.org/10.1134/S1068162015010100
Akpan E.I., Gbenebor O.P., Adeosun S.O., Cletus O. // In Handbook of Chitin and Chitosan. Chapter 5 / Eds. Gopi S., Thomas S., Pius Anitha. Elsevier, 2020. P. 131–164. https://doi.org/10.1016/b978-0-12-817970-3.00005-5
Gregorio C. // Environ. Chem. Lett. 2019. V. 17. P. 1623–1643. https://doi.org/10.1007/s10311-019-00901-0
Prashanth K.V.H., Tharanathan R.N. // Trends in Food Science & Technology. 2007. V. 18. P. 117–131. https://doi.org/10.1016/j.tifs.2006.10.022
Xu I., Du I., Huang R., Gao L. // Biomaterials. 2003. V. 24. P. 5015–5022. https://doi.org/10.1016/s0142-9612(03)00408-3
Trott O., Olson A.J. // J. Comput. Chem. 2010. V. 31. P. 455–461. https://doi.org/10.1002/jcc.21334
Barth A. // Biochim. Biophys. Acta. 2007. V. 1767. P. 1073–1101. https://doi.org/10.1016/j.bbabio.2007.06.004
Byler D.M., Susi H. // Biopolymers. 1986. V. 25. P. 469–487. https://doi.org/10.1002/bip.360250307
Wang Q.Z., Chen X.G., Liu N., Wang S.X., Liu C.S., Meng X.H., Liu C.G. // Carbohydrate Polymers. 2006. V. 65. P. 194–201. https://doi.org/10.1016/j.carbpol.2006.01.001
Mazancova P., Némethova V., Treľova D., Klescíkova L., Lacik I., Razga F. // Carbohydr. Polym. 2018. V. 192. P. 104–110. https://doi.org/10.1016/j.carbpol.2018.03.030
Wolpert M., Hellwig P. // Spectrochim. Acta Part A: Mol. Biomol. Spectroscopy. 2006. V. 64. P. 987–1001. https://doi.org/10.1016/j.saa.2005.08.025
Yang Y., Xing R., Liu S., Qin Y., Li K., Yu H., Li P. // Carbohydr. Polym. 2019. V. 205. P. 194–201. https://doi.org/10.1016/j.carbpol.2018.10.101
Wang C., Fan J., Xu R., Zhang L., Zhong S., Wang W., Yu D. // J. Mater. Sci. 2019. V. 54. P. 12522–12532. https://doi.org/10.1007/s10853-019-03824-x
Шкутина И.В., Стоянова О.Ф., Селеменев В.Ф., Меркулова Ю.Д. // Сорбционные и хроматографические процессы. 2004. Т. 4. № 4. С. 422–427.
Шкутина И.В., Стоянова О.Ф., Лунина В.В. // Сорбционные и хроматографические процессы. 2009. Т. 9. № 2. С. 247–253.
Chen S.C., Wu Y.C., Mi F.L., Lin Y.H., Yu L.C., Sung H.W. // J. Control. Release. 2004. V. 96. P. 285–300.
Gorshkova M., Volkova I., Alekseeva S., Molotkova N., Skorikova E., Izumrudov V. // Polymer Science Series A. 2011. V. 53. P. 57–66. https://doi.org/10.1134/S0965545X11010019
Abdullatypov A.V., Kondratyev M.S., Holyavka M.G., Artyukhov V.G. // Biophysics. 2016. V. 61. P. 565–571. https://doi.org/10.1134/S0006350916040023
Lowry O.H., Rosebrough N.J., Faar A.L., Randall R.J. // J. Biol. Chem. 1951. V. 193. P. 265–275.
Artyukhov V.G., Kovaleva T.A., Kholyavka M.G., Bityutskaya L.A., Grechkina M.V. // Appl. Biochem. Microbial. 2010. V. 46. P. 422–427. https://doi.org/10.1134/S0003683810040034
Sabirova A.R., Rudakova N.L., Balaban N.P., Ilyinskaya O.N., Demidyuk I.V., Kostrov S.V., Rudenskaya G.N., Sharipova M.R. // FEBS Lett. 2010. V. 584. P. 4419–4425. https://doi.org/10.1016/j.febslet.2010.09.049
Charney J., Tomarelly R.M. // J. Biol. Chem. 1947. V. 171. P. 501–505.
Coelho D.F., Saturnino T.P., Fernandes F.F., Mazzola P.G., Silveira E., Tambourgi E.B. // Biomed. Res. Int. 2016. P. 8409183. https://doi.org/10.1155/2016/8409183
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия