

Биоорганическая химия, 2023, T. 49, № 1, стр. 48-64
Липосомы с набором Т-клеточных эпитопов вируса SARS-CoV-2 как прототип вакцинной конструкции
Д. С. Третьякова 1, А. С. Алексеева 1, Н. Р. Онищенко 1, И. А. Болдырев 1, Н. С. Егорова 1, Д. В. Васина 2, В. А. Гущин 2, А. С. Чернов 3, Г. Б. Телегин 3, В. А. Казаков 3, К. С. Плохих 4, М. В. Коновалова 1, Е. В. Свирщевская 1, Е. Л. Водовозова 1, *
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 ФГБУ “Национальный исследовательский центр эпидемиологии и микробиологии имени почетного академика Н.Ф. Гамалеи” Минздрава России
123098 Москва, ул. Гамалеи, 18, Россия
3 Филиал Института биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
142290 Пущино, Институтская ул., 8, Россия
4 ФГБУ “Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Академика Курчатова, 1, Россия
* E-mail: elvod@lipids.ibch.ru
Поступила в редакцию 04.07.2022
После доработки 10.07.2022
Принята к публикации 14.07.2022
- EDN: POMUEE
- DOI: 10.31857/S0132342322060252
Аннотация
На основании анализа публикаций по результатам полногеномного иммуноинформационного анализа Т-клеточных эпитопов вируса SARS-CoV-2 (уханьский штамм), а также ряда клинических исследований иммунодоминантных эпитопов среди выздоравливающих после перенесенного заболевания COVID-19 пациентов, отобраны и синтезированы потенциальные нонамерные эпитопы CD8+-Т-лимфоцитов из состава структурных, вспомогательных и неструктурных белков вируса (13 пептидов) и 15-мерный эпитоп CD4+-Т-лимфоцитов из S-белка. Пять композиций из 6–7 пептидов включали в липосомы из яичного фосфатидилхолина и холестерина (размер ~200 нм), полученные методом экструзии. После двукратной подкожной иммунизации конвенциональных мышей оценивали активацию клеточного звена иммунитета по уровню синтеза цитокинов спленоцитами in vitro в ответ на стимуляцию соответствующими пептидными композициями. Лучший результат, свидетельствующий о формировании специфического клеточного иммунитета в ответ на вакцинацию, отмечен у одной из липосомальных формуляций. Проверка протективной эффективности этой формуляции на инфекционной модели мышей показала положительную динамику частоты встречаемости гиалиноподобных мембран в просвете альвеол, а также коррекцию выраженности микроциркуляторных нарушений. Последнее обстоятельство потенциально может способствовать снижению тяжести заболевания и предупреждению неблагоприятных его исходов. Отрабатывается метод получения препаратов липосом с пептидными композициями для длительного хранения.
ВВЕДЕНИЕ
Традиционные вакцины для лечения и профилактики инфекционных заболеваний представляют собой живые ослабленные или инактивированные/убитые патогены. Они не нуждаются в адъювантах (костимуляторах), т.к. содержат не только антигены, но и другие компоненты бактерий или вирусов, которые эффективно активируют несколько компонентов врожденной иммунной системы. Однако такие вакцины могут вызывать аллергические и аутоиммунные реакции [1]. Так называемые субъединичные вакцины содержат обычно только поверхностные антигены (белки или пептиды) патогенов, что снижает не только аллергенность вакцин, но и их иммуногенность. Упаковка антигенов в частицы сравнимых с вирусами или бактериями размеров (от сотен нанометров до нескольких микрон) позволяет свести к минимуму риски описанных нежелательных реакций, а также обеспечивает защиту антигенов от преждевременной деградации и способствует созданию антигенного депо [2, 3]. Захваченные антиген-презентирующими клетками (APC, дендритные клетки и макрофаги) частицы подвергаются внутриклеточному процессингу, и, в зависимости от пути расщепления, антигены презентируются на поверхности плазмалеммы для распознавания “свой-чужой” Т-лимфоцитам в виде эпитопов – пептидных фрагментов длиной 9 аминокислотных остатков (а.о.) в комплексе с белками главного комплекса гистосовместимости I класса (MHCI) или фрагментов длиной 14–20 а.о. в комплексе с белками MHС II класса (MHCII). Комплексы “пептид–MHCI” стимулируют наивные CD8+-Т-лимфоциты и превращают их в цитотоксические Т-лимфоциты (CTLs), ответственные за клеточный иммунитет и уничтожение инфицированных клеток. Комплексы “пептид–МНСII” активируют наивные CD4+-Т-лимфоциты, которые пролиферируют и дифференцируются в субпопуляции так называемых хелперных клеток TH1 и/или TH2, в зависимости от типа инфекции [4]. Клетки TH1 секретируют различные цитокины, активирующие и регулирующие CTLs. Таким образом, для формирования клеточного иммунитета необходимо стимулировать как CD8+-, так и CD4+-Т-лимфоциты.
Липосомы, будучи адъювантами как таковые, представляют интерес в качестве носителей антигенов благодаря своей высокой биосовместимости и пластичности. Способность липосом индуцировать иммунный ответ на антигены, инкапсулированные во внутренний объем или ассоциированные с поверхностью, впервые была описана Аллисон и Грегориадисом [5]. В зависимости от состава и структуры они могут одновременно активировать различные пути трансдукции сигнала и вызывать специфический Т- и/или В-клеточный ответ: антигены, экспонированные на поверхности липосом, могут стимулировать В-лимфоциты, вызывая гуморальный иммунный ответ, и индуцировать Т-клеточные реакции; инкапсулированные антигены, для которых требуется внутриклеточное разрушение липосом, способны индуцировать СTLs. В настоящее время применяются липосомальные вакцины против вирусов гриппа и гепатита А, малярии и вируса Varicella zoster (Inflexal®, Epaxal®, Mosquirix® и Shingrix® соответственно); целый ряд липосомальных препаратов проходит клинические испытания в качестве профилактических и лечебных вакцин против малярии, гриппа, туберкулеза, ВИЧ, лихорадки Денге [6–10].
Адъювантные свойства липосом можно усилить или направить по пути того или иного типа иммунного ответа с использованием иммуностимуляторов – специфических лигандов, которые вызывают активацию рецепторов АРС, распознающих патоген-ассоциированные молекулярные паттерны (PAMPs) [11]. Одними из таких PAMPs являются неметилированные CpG-мотивы бактериальных ДНК, которые гораздо реже содержатся в хромосомах эукариот, поэтому синтетические СpG-ODN обладают иммуностимулирующей активностью. СpG-мотивы распознаются рецептором TLR9, который экспрессируется в мембране эндосомальных компартментов множества иммунокомпетентных клеток, включая В-клетки, моноциты, NK-клетки, дендритные клетки и макрофаги [12, 13]. В результате стимулируется выработка провоспалительных цитокинов и хемокинов, повышается экспрессия МНСII и костимуляторных молекул (CD40, CD80, CD83, CD86). Синтетические СpG-ODN с успехом применяются при конструировании липосомальных вакцин, причем иммуностимулятор коинкапсулируют во внутренний водный объем липосом либо адсорбируют на катионных липосомах [13].
Пептидные вакцины безопаснее вакцин на основе полноразмерных антигенов или других молекул патогенного происхождения, которые могут содержать онкогенные последовательности [14]. В ряде доклинических исследований показана иммуногенность вакцин на основе липосом с инкапсулированными или связанными с поверхностью специфическими пептидами [13, 15–18] (при совместном введении с СpG-ODN). Авторы [15, 16] конъюгировали с поверхностью липосом нонамерные Т-клеточные эпитопы нуклеокапсидного [15] и неструктурного белка полипротеин 1a [16] коронавируса SARS (SARS-CoV). В первом случае четыре CTL-эпитопа, рестриктированные HLA-A*0201 (HLA-A2 – один из наиболее распространенных аллельных вариантов молекул MHCI), были идентифицированы с использованием трансгенных мышей и рекомбинантного аденовируса, экспрессирующего 8 предсказанных in silico эпитопов; два липосомальных пептида стимулировали специфический Т-клеточный ответ, а одна из этих вакцин вызвала у мышей клиренс вируса осповакцины, экспрессирующего эпитопы SARS-CoV [15]. Во втором случае из 30 предсказанных пептидов восемь значительно стимулировали ответ CTLs и были конъюгированы с липосомами. Из них 7 были активны против вируса in vivo, причем один вариант вакцины индуцировал образование клеток памяти [16]. Другие авторы [17] исследовали различные методы инкапсулирования в катионные липосомы разнообразных по физико-химическим свойствам 24-членных пептидов, содержащих последовательность SIINFEKL (иммунодоминантный CTL-эпитоп овальбумина), на предмет достижения эффективной загрузки липосом; липосомы эффективно доставляли пептиды в дендритные клетки с последующей SIINFEKL-специфической активацией CD8+-T-клеток. При конструировании вакцины против вируса гриппа типа А [18] в липосомы инкапсулировали одновременно 10 консервативных пептидов структурных и неструктурных белков, из которых 5 нонамерных эпитопов были вычислены in silico как потенциальные для пандемического свиного гриппа H1N1. В сочетании с кристаллическим уратом натрия вакцинация поросят в интраназальной аэрозольной форме вызвала увеличение частоты вирус-специфических ТН-клеток, клеток памяти, стимулировала ответ CTLs, что в итоге привело к частичной защите животных от лихорадки и поражения легких
В связи с пандемией новой коронавирусной инфекции COVID-19 мировое научное сообщество приступило к ускоренной разработке вакцин, способных активировать как гуморальное, так и клеточное звенья иммунитета. Однако данных о вирулентности белков, кодируемых вирусным геномом, все еще недостаточно. Важным представляется поиск наиболее иммуногенных эпитопов не только в составе спайк-белка (S-белка) вируса SARS-CoV-2, но и других мембранных белков и белков вирусного капсида и нуклеопротеина. Судя по первым опубликованным данным иммуноинформатического анализа полноразмерного генома вируса, в ранжированном списке нонамерных эпитопов CTLs и 15-мерных эпитопов хелперных CD4+-Т-клеток (TН-клеток), общих по всем аллелям МНС и охватывающим все преобладающие супертипы HLA (лейкоцитарный антиген человека) в мировой популяции, эпитопы S-белка могут уступать по иммуногенности эпитопам других белков вируса, в том числе неструктурных [18–20]. Целью настоящего исследования стала разработка прототипа вакцинной конструкции на основе липосом, содержащих композицию Т‑клеточных эпитопов из первичных структур различных белков вируса SARS-CoV-2. В случае обнаружения иммуногенных эпитопов, не относящихся к S-белку, появляется перспектива разработки вакцин для лечения и профилактики COVID-19, эффективность которых будет мало зависеть от мутаций вирусного генома.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Выбор Т-клеточных эпитопов для получения вакцинных конструкций. Для выбора пептидов был проанализирован массив опубликованных результатов полногеномного иммуноинформатического анализа Т-клеточных эпитопов вируса SARS-CoV-2 [19–21] и результатов ряда клинических исследований иммунодоминантных эпитопов среди выздоравливающих после перенесенного заболевания COVID-19 пациентов [22–26]. На основании сопоставления всех данных были выбраны и синтезированы 14 потенциальных эпитопов: нонамерные CTL-эпитопы из S-белка, белка оболочки (Е-белок), мембранного белка (М-белок), нуклеокапсидного белка (N-белок), вспомогательных белков orf6, orf8 и неструктурных белков orf1a, orf1b, orf3a, а также 15-мерный Т-хелперный эпитоп S-белка. Перечень синтезированных пептидов приведен в табл. 1.
Таблица 1.
Синтезированные Т-клеточные эпитопы и их физико-химические свойства
| № | Эпитоп | Белок | Локация а.о. | Гидро-
фобность, % а.о.* |
pI** | Консерва-тивность | Число ответивших пациентов |
Частота ответов | HLA-аллели | Литература |
|---|---|---|---|---|---|---|---|---|---|---|
| CTL-эпитопы | ||||||||||
| Структурные белки | ||||||||||
| 1 | VGYLQPRTF | S | 267–275 | 44.44 | 9.82 | 1 | 5 | 0.714 | MHC-II покрытие 100%, MHC-I покрытие 65% |
[19–21, 24, 26]*** |
| 2 | YVYSRVKNL | E | 56–64 | 33.33 | 10.06 | 0.997 | 12 | 0.545 | MHC-I покрытие 74%; HLA-DRB1*04:01/HLA-DRB1*11:01 | [20, 25] |
| 3 | KTFPPTEPK | N | 361–369 | 44.44 | 9.84 | 0.998 | 10 | 1 | HLA-A*03:01/HLA-A*11:01 | [21, 22–24, 25] |
| 4 | KAYNVTQAF | N | 266–274 | 44.44 | 9.69 | 0.999 | 2 | 0.286 | MHC-I покрытие 74% | [20, 22, 24] |
| 5 | ATEGALNTPK | N | 134–143 | 40 | 6.92 | 0.997 | 13 | 0.812 | HLA-A*11:01 | [23, 25] |
| 6 | SPRWYFYYL | N | 105–113 | 44.44 | 9.44 | 1 | 4 | 0.8 | HLA-B*07:02 | [22–24, 26] |
| 7 | GMSRIGMEV | N | 316–324 | 44.44 | 6.94 | 0.999 | 12 | 0.522 | HLA-DRB1*01:01/HLA-DRB1*04:01/HLA-DRB1*07:01/HLA-DRB1*11:01 | [21, 24, 25] |
| 8 | ATSRTLSYYК | M | 171–179 | 20 | 9.58 | 0.981 | 3 | 0.6 | HLA-A*01:01 | [23] |
| Вспомогательные белки | ||||||||||
| 9 | KVSIWNLDY | orf6 | 23–31 | 44.44 | 6.66 | 0.995 | 6 | 0.857 | Из консенсусной последовательности CTL/ТH-лимфоцитов: HLVDFQVTIAEILLIIMR TFKVSIWNLDYIINLII | [19, 24, 26] |
| 10 | IQYIDIGNY | orf8 | 71–79 | 33.33 | 3.14 | 0.999 | 1 | 0.143 | – | [19, 24, 26] |
| Неструктурные белки | ||||||||||
| 11 | VLWAHGFEL | Orf1b | 1708–1716 | 66.67 | 5.17 | 1 | 2 | 0.286 | Покрытие 85% мировой популяции | [24, 26] |
| 12 | TTDPSFLGRY | Orf1a | 1637–1646 | 30 | 6.67 | 0.998 | 15 | 0.882 | HLA-A*01:01 | [23, 24, 25, 26] |
| 13 | FTSDYYQLY | Orf3a | 207–215 | 22.22 | 3.14 | 0.999 | 5 | 1 | HLA-A*01:01 | [23, 24, 26] |
| Эпитопы TН-лимфоцитов | ||||||||||
| 14 | SYGFQPTNGV- GYQPY | S | 494–508 | 26.67 | 5.83 | — | — | — | Хорошо презентируется комплексами MHCI/MHCII и теоретически предсказан как эпитоп В-клеток; локализация рядом с рецептор-связывающим доменом S-белка | [20] |
* https://www.peptide2.com/N_peptide_hydrophobicity_hydrophilicity.php. ** http://isoelectric.org/index.html. *** Полужирным шрифтом выделены ссылки на данные клинических исследований.
В соответствии с физико-химическими свойствами пептидов (табл. 1) были подобраны условия их растворения в высокой концентрации (5–7 мМ, т.е. в диапазоне ~7–11 мг/мл, в зависимости от числа пептидов в композиции и молекулярной массы пептида), чтобы при пассивном инкапсулировании смесей пептидов в липосомы загрузка каждого из них была достаточной для проявления потенциальной иммуногенности. В среде 20 мМ фосфатного буфера рН поддерживался в интервале 6.0–7.2. В буфере содержался изотонический раствор сахарозы вместо хлорида натрия как для повышения растворимости пептидов, так и для обеспечения возможности получения липосомальных вакцин длительного хранения, поскольку сахароза является криопротектантом. Пептиды 6 и 11 растворяли в органической фазе вместе с липидами при формировании липосом. К сожалению, не удалось подобрать условия растворения пептида 9 (KVSIWNLDY) вспомогательного белка orf6: по данным иммуноинформатического анализа [19], этот пептид относится к консенсусной последовательности эпитопов CTLs и TН-лимфоцитов, и он был выявлен как иммуногенный у шести из семи переболевших пациентов [24, 26].
Получение липосом с композициями пептидов. Исходя из принадлежности пептидов к различным вирусным белкам, а также необходимости сочетания в вакцине эпитопов цитотоксических и хелперных Т-клеток, были составлены 4 композиции по 6 пептидов для включения в липосомы L1–L4, где пептиды 6 и 11 встраивались в липидный бислой, а остальные инкапсулировались во внутренний водный объем, и композиция из семи водорастворимых пептидов для инкапсулирования в липосомы L5 (табл. 2; композицию для липосом L5 подбирали исходя из результатов первого цикла экспериментов, см. следующий раздел). Липосомы формировали на основе яичного фосфатидилхолина (ePC) и холестерина (Chol), ePC–Chol, 67 : 33 (мол. %), методом экструзии. При таком соотношении липидных компонентов создается конденсированная прочная мембрана липосом (так называемая жидкокристалическая упорядоченная фаза липидного бислоя Lo [27]), способная предотвратить преждевременную потерю пептидов из внутреннего водного объема.
Таблица 2.
Характеристики липосом с инкапсулированными пептидами
| Образец | Пептиды (обозначение композиции) | Диаметр (нм) ± ± SD1 | PDI ± SD1 | Пептиды вне липосом (%) ± ± SD2 | Включение в липосомы, %5 |
|---|---|---|---|---|---|
| Эксперимент I | |||||
| LК | – | 141.4 ± 2.1 | 0.047 ± 0.026 | – | – |
| L1 | 1, 2, 3, 5, 11, 14 (P1) | 107.5 ± 0.8 | 0.096 ± 0.024 | 40.4 ± 1.73 | 59.6 |
| L2 | 1, 2, 4, 6, 8, 14 (P2) | 123.0 ± 0.6 | 0.076 ± 0.009 | 51.9 ± 3.33 | 48.1 |
| L3 | 5, 7, 10, 11, 12, 14 (P3) | 113.4 ± 1.3 | 0.073 ± 0.018 | 57.0 ± 4.03 | 43 |
| L4 | 3, 7, 10, 11, 13, 14 (P4) | 101.0 ± 0.8 | 0.095 ± 0.004 | 48.0 ± 4.93 | 52 |
| Эксперимент II | |||||
| LК | – | 193.9 ± 1.4 | 0.092 ± 0.015 | – | – |
| L1 | 1, 2, 3, 5, 11, 14 (P1) | 201.9 ± 2.5 | 0.081 ± 0.017 | 66.5 ± 6.14 | 33.5 |
| L5 | 1, 2, 3, 5, 8, 12, 14 (P5) | 191.7 ± 1.7 | 0.080 ± 0.022 | 47.2 ± 7.64 | 52.8 |
1 По данным измерений на установке Brookhaven 90PLUS Particle Size Analyzer (Brookhaven Instruments Corp., США). 2 Рассчитано без учета пептидов 6 и 11, встроенных в мембрану липосом, по формуле: (масса невключившихся пептидов в смывах после ультрафильтрации)/(масса исходно взятых пептидов для инкапсулирования в липосомы) × 100. 3 По данным измерения модифицированным методом Лоури невключившихся пептидов в смывах после ультрафильтрации, n = 8. 4 По данным измерения оптической плотности при 273 нм в смывах после ультрафильтрации, n = 3–5. 5 Рассчитано по формуле: 100 − (пептиды вне липосом).
Для получения высокой загрузки пептидов в липосомы в ходе пассивного инкапсулирования концентрация липидов в водной фазе должна быть максимально высокой. Мы применили методику, описанную в работе [18], где смесь компонентов мембраны липосом лиофилизовали из трет-бутанола и затем гидратировали минимальным объемом растворов пептидов, достигая концентрации до 200 мг/мл по липидам. После прохождения нескольких циклов замораживания-оттаивания получается чрезвычайно густая суспензия, которая подвергается многократной экструзии через мембраны с порами 200, а затем 100 нм [18]. Размеры липосом по-разному влияют на эффективность вакцинации в зависимости от способа введения (подкожно, внутримышечно, внутрикожно), состава компонентов, этиологии инфекции и т.д. [7–10], и они могут варьировать от 100–150 нм до 1 мкм и более. Поэтому мы проводили экструзию через поры 200 нм с целью получения максимальной эффективности загрузки липосом. Тем не менее размеры полученных липосом оказались значительно меньше заданных (табл. 2, эксперимент I). При концентрации липидов 200 мг/мл (после экструзии за счет потерь на мембране концентрация липидов 140 мг/мл) получить более-менее однородные моноламеллярные везикулы размером порядка 200 нм вряд ли возможно, исходя из стереометрических расчетов. Кроме того, растворенные пептиды также повлияли на процесс формирования везикул: липосомы без пептидов, контрольные (LК), все же чуть крупнее остальных образцов (~141 нм против 101–123 нм). Когда в процедуру получения липосом было внесено изменение – перед экструзией суспензию с концентрацией 200 мг/мл разбавляли в 2 раза, – размеры липосом стали сравнимы с размерами пор 200 нм (табл. 2, эксперимент II).
При определении эффективности включения пептидов в липосомы была использована ультрафильтрация в варианте ступенчатой диафильтрации: дисперсии концентрировали примерно в 2 раза, разбавляли до исходного объема, вновь концентрировали и затем еще дважды повторяли цикл. По данным измерений концентрации невключившихся пептидов в фильтратах с помощью модифицированного метода Лоури [28] либо по оптической плотности в пике поглощения смесей пептидов при 273 нм, эффективность загрузки липосом составила 40–60% от исходно взятого количества пептидов (табл. 2). Важно также отметить, что расчеты проведены без учета пептидов 6 и 11, которые, вероятнее всего, остаются внедренными в мембрану липосом, и можно считать, что загрузка в липосомы L1–L4 на самом деле выше примерно на 16% (~1/6 часть пептидных композиций).
Невысокая загрузка при пассивном инкапсулировании во внутренний водный объем наноразмерных липосом характерна для растворов любых субстанций в случае отсутствия электростатических либо каких-либо иных специфических взаимодействий с липидной матрицей. Например, в работах [29–31] приведены значения эффективности включения пептидов в липосомы 25–40%. Ограничения включения связаны с малым внутренним объемом липосом и пределом концентрации коллоидного раствора, с одной стороны, и с невысокой растворимостью пептидов в водной фазе – с другой. Авторы [32, 33] определили высокое включение пептидов – более 80% – для липосом размера ~200 нм по флуоресценции BODIPY-меченого пептида после диализа липосомальной дисперсии. Однако по данным других авторов [17], различные пептиды включались в катионные липосомы размера несколько меньше 200 нм лишь с эффективностью 30–40% (несмотря на то, что электростатические взаимодействия должны способствовать загрузке соответствующих пептидов), как показал ВЭЖХ-анализ экстрагированных из липосом пептидов. Причем эффективность включения флуоресцентно-меченых аналогов этих же пептидов варьировала в интервале 9–50% (после ультрафильтрации в таких же концентраторах, какие использованы в нашей работе) [17]. В работе [18] эффективность включения различных по полярности и размерам – от нонамерных до 34-мерных – пептидов в липосомы из соевого лецитина и холестерина диаметра 130–140 нм составила 54–92% по данным флэш-гель-хроматографии (невключившиеся пептиды в элюате определяли по поглощению при 223 нм). Судя по приведенным данным публикаций, различные методы анализа эффективности загрузки липосом пептидами могут давать противоречивые результаты. При получении субъединичных вакцин невключившиеся антигены (белки, пептиды) обычно не отделяют, чтобы избежать потерь целевого материала за счет сорбции при очистке методами гель-фильтрации или ультрафильтрации/диафильтрации и последующей необходимости концентрирования [18, 29–33]. Технологичный в плане потерь метод тангенциальной фильтрации не применим при работе с аналитическими количествами растворов, как в нашем случае [34]. Поэтому мы не отделяли невключившиеся пептиды от липосом в расчете на получение достоверного биологического эффекта от инкапсулирования в липосомы какой-либо из композиций пептидов.
Оценка специфической эффективности композиций пептидов и липосомальных формуляций. Поскольку отбор пептидов осуществлен на основе известных Т-клеточных эпитопов человека, требовалось оценить возможность их распознавания Т-клетками мышей. С этой целью спленоциты интактных мышей инкубировали 7 дней с пептидными композициями Р1–Р4. В состав клеток селезенки входит ~20–25% СD4+-Т-клеток, 10–13% CD8+-Т-клеток, 35–40% В-клеток, 4–7% натуральных киллеров и 5–8% макрофагов. Макрофаги относятся к APC и могут представлять антигены Т-клеткам. Среди Т-клеток специфических клонов мало, в связи с чем анализ проводили для 1 млн спленоцитов в каждой лунке и в 24 повторностях, что позволяет оценить частоту специфических клонов среди 24 млн клеток. На рис. 1а–1г приведены данные по распознаванию композиций пептидов Р1–Р4 спленоцитами интактных мышей. В качестве контроля использовали аналогичное количество клеток без антигенов. За вычетом пролиферации спленоцитов без добавления пептидов в группах Р1–Р4 выявили 15, 5, 13 и 13 положительных клонов соответственно (рис. 1а–1г). Эти данные позволяют предположить, что часть отобранных пептидов может распознаваться Т-клетками мышей. Соответственно, полученные формуляции мы использовали для иммунизации мышей.
Рис. 1.
Характеристика иммунного ответа на композиции пептидов Р1–Р4 и их липосомальные формуляции L1–L4. (а–г) – Спонтанная пролиферация (МТТ-метод) спленоцитов (106/лунку) интактных мышей в ответ на композиции Р1 (а), Р2 (б), Р3 (в) и Р4 (г); среднее значение + стандартное отклонение пролиферации спленоцитов без добавления антигенов отмечено линией; n – число позитивных клонов; (д) – продукция in vitro IFN-γ спленоцитами мышей, иммунизированных различными препаратами, в ответ на специфические антигены Р1–Р4. Достоверные отличия (p < 0.05, критерий Манна–Уитни) отмечены скобками.
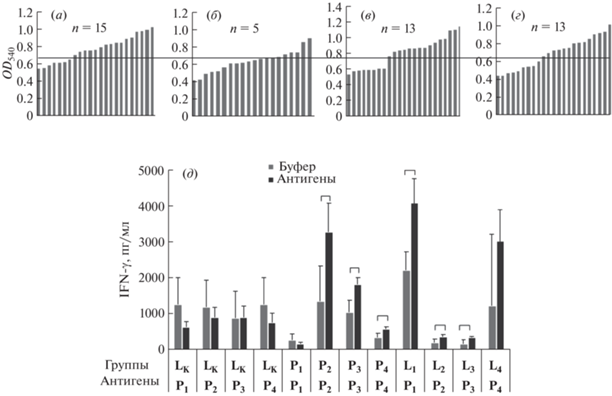
Для оценки иммуногенности полученных пептидных и липосомальных формуляций Р1–Р4, L1−L4 мышам линии C57BL/6 препараты вводили в подушечку задней лапы 2 раза с интервалом в 3 недели. Через неделю после последней иммунизации забирали селезенки и культивировали спленоциты с соответствующими пептидными композициями (Р1–Р4) в течение 24 ч. Активацию клеточного звена иммунитета оценивали по уровню синтеза IFN-γ в супернатантах спленоцитов иммунизированных мышей. На рис. 1д LK соответствует группе контроля, которой вводили липосомы, не несущие пептидов, но содержащие CpG-ODN. Поскольку такие липосомы могут быть иммуностимуляторами врожденного иммунитета, анализ продукции IFN-γ проводили при культивировании со всеми пулами пептидов Р1–Р4. Действительно, в ответ на иммунизацию LK спленоциты отвечали продукцией IFN-γ, однако специфического ответа на пептиды не было. Дополнительно в исследование включали группу, иммунизированную буфером на основе сахарозы, в котором вводили липосомальные формуляции с пептидами. В этой группе продукция IFN-γ была значительно ниже (данные не приведены). В группах мышей, иммунизированных пептидными композициями и липосомальными формуляциями с пептидами, наблюдали антиген-специфическую продукцию IFN-γ при иммунизации пептидными композициями Р2, Р3, Р4 и всеми липосомальными формуляциями. Максимальные ответы наблюдали в группах P2, Р3, L1 и L4. Однако в группе L4 наблюдалась значительная вариабельность в ответе на буфер, что привело к отсутствию достоверных отличий с ответом на пептиды. В целом, полученные результаты свидетельствуют о формировании специфического клеточного иммунитета в ответ на вакцинацию липосомальными формуляциями L1–L4, а также пептидными композициями Р2–Р4 (рис. 1д).
Как было показано ранее, подкожная вакцинация трансгенных мышей против коронавируса SARS-CoV липосомами с ковалентно связанными пептидами — нонамерными CTL-эпитопами вирусного нуклеокапсида (N-белка) [15] и полипротеина 1а (рр1а) [16], рестриктированными HLA-A*0201, индуцировала образование антиген-специфических CD8+-T-клеток. Также при вакцинации трансгенных мышей липосомами с инкапсулированными нонамерными Т-эпитопами вируса гепатита С и СpG-ODN авторы получили продукцию IFN-γ в тесте ELISPOT около 2000 пг/мл [33]. А в работе [18], при исследовании кандидатной пептидной вакцины против гриппа типа А на свиньях, уровень IFN-γ, продуцируемого фракцией мононуклеарных клеток периферической крови, достигал 1000–1500 пг/мл (в тесте ELISA). В связи с этим результаты, полученные для формуляции L1, можно считать сопоставимыми и перспективными для иммунизации с точки зрения получения Т-клеточного ответа.
Для дальнейшей работы была отобрана формуляция L1, для которой была выявлена достоверная разница между контролем и ответом in vitro на антиген. Из композиций Р2 и Р3 в следующем цикле экспериментов были отобраны пептиды для составления новой формуляции L5.
При составлении новой пептидной композиции P5 за основу был взят состав композиции P1, с изменениями: вместо пептида 11 были введены пептиды 8 и 12. Последние входили в состав композиций P2 и P3 (рис. 1д), которые проявили иммуногенность при вакцинации нелипосомальными растворами в первом цикле экспериментов. Таким образом, в липосомальной формуляции L5 пептиды в липидный бислой не включались, и эффективность загрузки липосом составила 52.8% (табл. 2).
Анализ распознавания пептидов интактными спленоцитами провели для формуляции P5, а также P1 в качестве контроля. Результаты подтвердили ранее полученные данные о совместимости части из отобранных пептидов с молекулами МНС мышей (рис. 2а, 2г).
Рис. 2.
Характеристика иммунного ответа на композиции пептидов P1 и P5 и липосомальные формуляции L1 (а–в) и L5 (г–е). Спонтанная пролиферация спленоцитов (106/лунку) интактных мышей в ответ на композиции Р1 (а) и Р5 (г); среднее + стандартное отклонение значения пролиферации спленоцитов без антигенов обозначено линией; позитивные клоны отмечены скобками. Концентрация IL-6 и TNF-α в сыворотках мышей, иммунизированных препаратами LK, L1 или P1 (б), LK, L5 или P5 (д); Инт. – интактный контроль. Пролиферация спленоцитов мышей, иммунизированных препаратами LK, L1 или P1 (в), LK, L5 или P5 (е), в ответ на индивидуальные пептиды. Достоверные отличия (p < 0.05, критерий Манна–Уитни) отмечены скобками.

Далее полученные формуляции использовали для иммунизации мышей. Сыворотки собирали через неделю после второй иммунизации и оценивали уровень цитокинов в крови. Для анализа использовали мультиплексный тест, включавший 10 цитокинов (IFN-γ, интерлейкины 2, 4, 6, 9, 10, 13, 17, 22 и TNF-α). Среди 10 цитокинов в сыворотке зарегистрировали только три: IFN-γ, IL-6 и ТNF-α. Уровень IFN-γ в крови был низким (данные не приведены). Иммунизация формуляциями Р1 и L1 привела к повышению уровня как IL-6, так и TNF-α (рис. 2б) по сравнению с их уровнем в крови интактных мышей и контрольной группы LK. Иммунизация формуляцией L5 также вызывала повышение уровней IL-6 и TNF-α (рис. 2д), но в меньшей степени, чем в случае L1.
Спленоциты иммунных мышей стимулировали in vitro индивидуальными пептидами. Общие для всех препаратов пептиды 1, 2, 3 и 5 распознавались спленоцитами после иммунизации липосомами L1 (рис. 2в), L5 и композицией пептидов Р5 (рис. 2е). При иммунизации липосомами L5 распознавались также пептиды 8, 12 и 14. В целом при иммунизации липосомальными формуляциями Т-клеточный ответ формировался эффективнее, чем при иммунизации композициями пептидов.
Уровень цитокинов в надосадочных средах спленоцитов иммунных мышей при стимуляции индивидуальными пептидами анализировали с помощью мультиплексного метода. Как и в сыворотке крови, из 10 факторов выявили только три: IFN-γ, IL-6 и TNF-α. Общий уровень IFN-γ был значительно ниже, чем в первом эксперименте, что можно объяснить различием методов определения. В первом эксперименте использовали иммуноферментную систему для определения концентрации IFN-γ, во втором случае использовали мультиплексный цитометрический анализ. Продукция IL-6 не коррелировала с антиген-специфическим ответом (данные не приведены). Продукция IFN-γ и TNF-α представлена на рис. 3.
Рис. 3.
Продукция in vitro IFN-γ (а, б) и TNF-α (в, г) спленоцитами мышей, иммунизированных контрольными липосомами LK, композициями пептидов P1 и P5 и липосомальными формуляциями L1 и L5, в ответ на специфические пептиды. Достоверные отличия (p < 0.05, критерий Манна–Уитни) отмечены скобками.
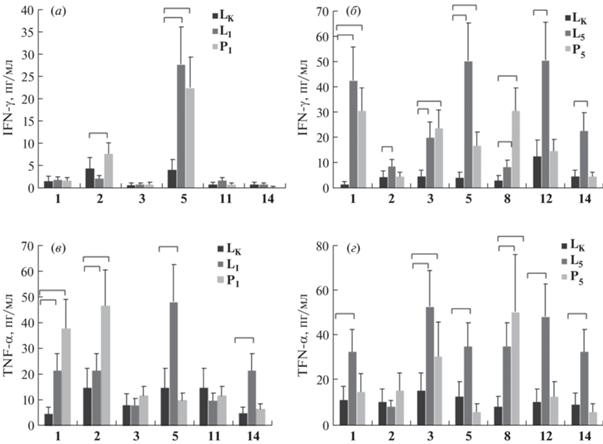
Т-клетки в ответ на пептиды 1, 2, 5 и 14 в группе мышей, иммунизированных липосомами L1, и на все пептиды из формуляции L5, за исключением пептида 2, продуцировали IFN-γ и TNF-α (рис. 3).
Высокогидрофобный пептид 11 из неструктурного белка Orf1b (табл. 1) был отобран по результатам клинических исследований, хотя частота иммунных ответов в мультиплексном тесте по идентификации антигенной специфичности Т-клеточных рецепторов была не столь высока (2 ответа из семи) [24, 26]. Судя по выработке IFN-γ спленоцитами иммунизированных конвенциональных мышей, не все композиции с пептидом 11 проявили иммуногенность (например, L3, рис. 1а). Кроме того, этот пептид не вызывал значительного усиления пролиферации спленоцитов по сравнению с остальными пептидами композиции P1, в отличие от пептида 2 белка вирусной оболочки Е (рис. 2в), и не распознавался Т-клетками в мультиплексном варианте анализа (рис. 3а, 3в). Тем не менее при замене в композиции пептида 11 на два гидрофильных пептида (пептиды 8 из М-белка и 12 из неструктурного Orf1b) в сыворотках иммунизированных мышей уровень цитокинов IL-6 и TNF-α уменьшился (ср. рис. 2б и 2д). Поэтому для исследования протективной эффективности на инфекционной модели мышей hACE2 была выбрана липосомальная формуляция L1.
Проверка протективной эффективности формуляции L1 на инфекционной модели мышей. Одной из характеристик эффекта вакцинации является протективный эффект. Для его оценки использовали генномодифицированных мышей линии C57BL/6JTgTn(CAG-human ACE2-IRES-Luciferase-WPRE-polyA) (сокращенно hАСЕ2), несущих гуманизированный рецептор ангиотензин-превращающего фермента-2 (АСЕ2), обеспечивающего проникновение в клетку штаммов коронавируса SARS-CoV-2, а также вирусов NL63 и SARS-CoV. Данная модель мышей может использоваться в исследованиях протективной эффективности препаратов, предназначенных для профилактики и лечения коронавирусной инфекции COVID-19 [35].
В настоящей работе проведена двукратная иммунизация самцов мышей hАСЕ2 с интервалом в 21 день липосомами L1 (n = 6) и контрольными липосомами LК (n = 4). По истечении двух недель после второй иммунизации мышей заражали летальной дозой (3 lg БОЕ) SARS-CoV-2 (уханьский штамм).
Среднее время жизни до гибели в группах не отличалось. После заражения вирусом все животные погибли в течение 4–5 дней (контрольную группу иммунизировали буфером, n = 3). Отсутствие различий в продолжительности жизни у самцов мышей различных групп объясняется, вероятно, изначально высокой дозой заражения SARS-CoV-2.
У всех мышей забирали легкие и проводили гистологический анализ. Результаты патоморфологического исследования легких зараженных мышей показали, что в контроле без лечения в легких мышей hACE2, зараженных уханьским штаммом SARS-CoV-2 в дозе 3 lg БОЕ, отмечалось диффузное выраженное полнокровие крупных сосудов, а также сосудов микроциркуляторного русла с явлениями стаза и сладжа эритроцитов (рис. 4а, 4б). В стенках и в просвете альвеол отмечали, как правило, умеренную диффузную мононуклеарную инфильтрацию с примесью немногочисленных нейтрофилов. В просвете отдельных альвеол в разных долях органа встречались гиалиноподобные мембраны и транссудат. В просвете большинства бронхов обращало на себя внимание выраженное слущивание респираторного эпителия с начальным разрушением целостности стенки бронхов.
Рис. 4.
Гистология легких трансгенных мышей hACE2, зараженных уханьским штаммом SARS-CoV-2 в дозе 3 lg БОЕ без лечения (а, б) и на фоне иммунизации с введением контрольных липосом LК (в, г) и формуляции L1 (д, е). Диффузное полнокровие крупных сосудов с явлениями стаза и сладжа эритроцитов отмечено стрелками, слущивание респираторного эпителия с начальным разрушением целостности стенки бронхов – звездочками. Шкала соответствует 200 (а, в, д) и 50 (б, г, е) мкм. Окрашивание гематоксилином и эозином.
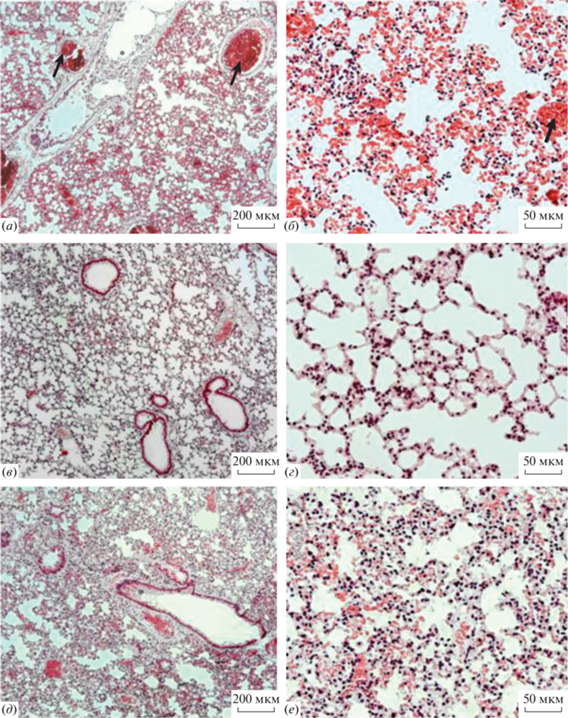
У животных, иммунизированных липосомами LК во всех долях легких не обнаруживали транссудат в легочных ацинусах (рис. 4в, 4г). Кроме того, у 3 из 4 животных отсутствовали выраженные микроциркуляторные нарушения и полнокровие крупных сосудов легких. У 2 из 4 самцов мышей данной группы отмечали снижение степени альтерации респираторного эпителия бронхов. Гиалиноподобные мембраны в данной группе животных встречались в 3 из 4 случаев со средним оценочным баллом 1.25 против 2.00 у самцов мышей без лечения. При этом выраженность мононуклеарной и нейтрофильной инфильтрации стенок и просвета альвеол была сопоставимой с таковой среди животных, не получавших лечения и предварительной иммунизации.
На фоне иммунизации животных препаратом L1 выраженных микроциркуляторных нарушений в легких не обнаруживали в 2/3 случаев, средний оценочный балл по данному признаку соответствовал 3.17 против 4.00 у животных без терапии (рис. 4д, 4e). Гиалиноподобные мембраны среди животных, получавших L1, также встречались реже (1.33 против 2.00 соответственно). Однако по остальным признакам каких-либо отличий от “фонового” течения заболевания не наблюдали.
Таким образом, введение липосом – как контрольных, так и с пептидами – улучшало состояние легочной функции, хотя и не предупреждало гибель животных, что связано с использованием летальной дозы вируса. Определенный протективный эффект липосом, как частиц, сравнимых по размеру с вирусами, связан, по-видимому, с активацией врожденной иммунной системы. Поскольку основной задачей была индукция адаптивного иммунитета с помощью специфических пептидов, отсутствие эффекта связано с быстро протекающим инфекционным процессом. Меньший эффект пептидной формуляции липосом по сравнению с контрольными липосомами может быть связан с активацией адаптивного иммунитета, на что расходуются энергетические ресурсы организма. Для выявления роли адаптивного иммунитета в дальнейшем следует использовать сублетальные дозы вируса.
Липосомальные формуляции длительного хранения. Формуляции липосом L1 и L5 исследовали на возможность получения препаратов длительного хранения. Дисперсии липосом подвергали лиофилизации, а затем восстанавливали регидратацией соответствующим объемом воды. Данные о размерах липосом до лиофилизации и после восстановления, а также о содержании в них пептидов представлены в табл. 3.
Таблица 3.
Характеристики образцов липосом до и после лиофилизации
| Образец | До лиофилизации | После лиофилизации и регидратации | ||||
|---|---|---|---|---|---|---|
| диаметр (нм) ± ± SD1 | PDI ± SD1 | включение в липосомы, %2 | диаметр (нм) ± ± SD1 | PDI ± SD1 | включение в липосомы, %2 | |
| LК | 211.3 ± 2.1 | 0.103 ± 0.022 | – | 187.9 ± 0.6 | 0.093 ± 0.028 | – |
| L1 | 220.6 ± 1.7 | 0.114 ± 0.029 | 33.53 | 188.9 ± 1.7 | 0.099 ± 0.028 | 49.23 |
| L5 | 212.0 ± 1.3 | 0.088 ± 0.026 | 52.8 | 185.3 ± 1.5 | 0.070 ± 0.022 | 51.9 |
1 По данным измерений на установке Brookhaven 90PLUS Particle Size Analyzer (Brookhaven Instruments Corp., США). 2 Рассчитано по формуле: 100 − (пептиды вне липосом). По данным измерения оптической плотности при 273 нм в смывах после ультрафильтрации, n = 3–5. 3 Рассчитано без учета пептида 11, встроенного в мембрану липосом.
Можно заключить, что после лиофилизации обе формуляции липосом не претерпели значительных изменений размеров. Содержание же инкапсулированных пептидов в образце L1 существенно увеличилось. Мы предположили, что такое изменение может быть связано с нарушением целостности мембраны липосом при лиофилизации/регидратации и перераспределением пептидов между липосомами и раствором неинкапсулированных пептидов. Структуру липосом анализировали с помощью криогенной просвечивающей электронной микроскопии (рис. 5).
Рис. 5.
Криоэлекронные микрофотографии контрольных липосом LK и липосомальных формуляций пептидов L1 и L5 до (а) и после лиофилизации и регидратации (б). Масштабный отрезок 50 нм.

Действительно, в образце L1 в ходе лиофилизации/регидратации от липосом отделяются мелкие частицы, вероятнее всего, сгустки-осадки гидрофобного пептида 11 (рис. 5б, центр). Возможно, данный пептид изначально присутствовал в липидном бислое в виде агрегатов, которые и высаживаются как достаточно крупные образования, наблюдаемые на изображениях, полученных в электронном микроскопе. Для липосом L5, которые содержат только водорастворимые пептиды, отдельных невезикулярных частиц не наблюдается. Таким образом, лиофилизация в данном случае является подходящим методом для длительного хранения вакцинных конструкций.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реагенты. Пептиды получены твердофазным синтезом с применением стратегии Fmoc/трет-бутил на тритилхлорид-полистирольном полимере как описано ранее [36]. Олигонуклеотид CpG-ODN 1826 (TCCATGACGTTCCTGACGTT), специфический для TLR9 мышей, любезно предоставлен д-ром В.А. Гущиным (“НИЦЭМ им. Н.Ф. Гамалеи”). Использовали фосфатидилхолин из яичного желтка (еРС, Lipoid E PC S) и холестерин (Chol) производства Lipoid GmbH (Heidelberg, ФРГ) квалификации USP (United States Pharmacopeia); сахарозу и этилендиаминтетрауксусную кислоту (EDTA) производства Panreac (USP, Испания); сефарозу CL-4B (Pharmacia, США); Na2HPO4, NaH2PO4 и KH2PO4 квалификации ACS (Хеликон, Россия); остальные реагенты производства фирм Sigma и Flow Laboratories (CША). Растворители очищали стандартными методами; упаривание проводили в вакууме при температурах не выше 40°С.
Получение липосомальных формуляций пептидов. Индивидуальные пептиды в виде солей с трифторуксусной кислотой растворяли (кроме пептидов 6 и 11) в фосфатном буфере с изотоническим раствором сахарозы PB-Suc, рН 7.2 (6.25 мМ Na2HPO4, 1.3 мМ NaH2PO4, 1.2 мМ KH2PO4, 1 мМ EDTA, 240 мМ сахароза, H2Odd). Затем готовили растворы смесей пептидов, где конечная концентрация каждого из пептидов составляла 1 мМ (состав композиций см. в табл. 2) и, при необходимости, титровали 1 н. NaOH до pH ~ 6.3–7.0. Растворы замораживали в жидком азоте (–196°С) и хранили при –20°С до применения.
Яичный фосфатидилхолин и холестерин растворяли в трет-бутаноле в мольном соотношении 67 : 33. В случае формуляций L1, L2, L3 и L4 в трет-бутанольный раствор липидов добавляли 0.5 мкмоль пептидов 11, 6, 11 и 11 соответственно. Растворы замораживали и лиофилизовали в течение 12 ч при давлении ~3 Па (лиофильная сушилка ИНЕЙ‑4; ИБП РАН, Россия). Лиофилизованные липидно-пептидные смеси гидратировали растворами пептидных композиций в течение 2 ч при периодическом встряхивании. После гидратации суспензию липосом, 200 мг/мл по липидам, подвергали 10-кратной процедуре замораживания (–196°С) – оттаивания (водяная баня, 40°С) – встряхивания (вортекс FV-2400, Biosan, Латвия), разбавляли в 2 раза буфером PB-Suc и продавливали последовательно по 10 раз через поликарбонатные мембранные фильтры Whatman Nuclepore (Cytiva, США) с размерами пор 400 и 200 нм на установке Mini-Extruder (Avanti Polar Lipids, США). Концентрацию ePC в липосомальных дисперсиях определяли с помощью ферментативного колориметрического метода (набор Phospholipids, Sentinel Diagnostics, Италия): 3 мкл образца и 150 мкл рабочего раствора (фосфолипаза D, >1500 ед./л; холиноксидаза, >7500 ед./л; 4‑аминоантипирин, 1.2 мM; пероксидаза, >7000 ед./л; TES-буфер, 50 мM, pH 7.6; гидроксибензойная кислота 12 мM; EDTA, 1.3 мM; азид натрия, <0.1%) добавляли в лунку 96-луночного планшета, инкубировали при 37°C 10 мин и измеряли оптическую плотность при 540 нм с помощью микропланшетного фотометрa Multiscan FC (ThermoFisher Scientific, США); количество ePC в образцах определяли по калибровочной кривой для дисперсий ePC в PB-Suc. Для получения контрольных липосом LК лиофилизованную смесь ePC–Chol (67 : 33, мольн.) гидратировали буфером PB-Suc и проводили экструзию, как описано выше.
Размер липосом определяли методом динамического лазерного светорассеяния на установке Brookhaven 90PLUS Particle Size Analyzer (Brookhaven Instruments Corp., США). Проводили по меньшей мере 3 измерения разбавленных дисперсий липосом (50 мкг липидов/мл PBS) с использованием гелий-неонового лазера, λ = = 633 нм, под углом 90°, 3 цикла по 1 мин. Липосомальные формуляции в концентрациях, использованных для вакцинаций (~40 мг/мл по суммарным липидам), сохраняли стабильность не менее трех недель при 4–8°С.
Определение количества невключившихся в липосомы пептидов. Образцы липосомальных формуляций L1 и L5 (500 мкл, ~8.5 мг/мл липидов) разбавляли в 2 раза буфером PB-Suc и помещали в предварительно промытые H2Odd концентраторы Vivaspin 2 (300 000 MWCO, Sartorius, ФРГ). Центрифугировали 35 мин при 2300 об/мин, ~1000 g (CM-6M, ELMI, Латвия). Затем измеряли оптическую плотность в прошедшем объеме буфера (~400–500 мкл) при 273 нм (СФ-2000, ОКБ-Спектр, Россия). Описанные этапы промывки и измерений проводили 3 раза. Молярные коэффициенты экстинкции смесей пептидов, рассчитанные по Tyr, составили 1536 и 1646 M–1 см–1 для композиций Р1 (без гидрофобного пептида 11) и Р5 соответственно. Суммировали количество свободных пептидов в трех фильтратах.
При определении количества пептидов в смывах по Лоури [28] в образцах L1–L4 для построения градуировочной функции готовили растворы смесей соответствующих пептидов (P1–P4). При использовании для этой цели стандартных растворов альбумина получали завышенные (до 3 раз) значения концентраций пептидов.
Криогенная просвечивающая электронная микроскопия. Для подготовки образцов использовали медные поддерживающие сетки с отверстиями в аморфной пленке углерода (Lacey C only, 01895-F/ 01896-F, Ted Pella, США), гидрофилизованные в тлеющем разряде на установке PELCO easiGlow (Ted Pella, США) при следующих условиях: время обработки образца – 25 с, сила тока – 0.20 мА, остаточное давление в камере – 0.26 мбар. На сетку наносили 3 мкл образца и с помощью автоматизированной системы Vitrobot Mark IV (Thermo Fisher Scientific, Waltham, MA, США) удаляли фильтровальной бумагой излишки раствора в течение 2.5 с при влажности в камере 95–100% и температуре 4°С, затем проводили витрификацию. Образцы исследовали с помощью криогенного просвечивающего электронного микроскопа Titan Krios 60-300 (Thermo Fisher Scientific, Waltham, MA, США), оборудованного устройством прямого детектирования электронов Falcon II и корректором сферических аберраций CEOS Image Corrector, работающего под управлением программного обеспечения EPU. Основные параметры получения данных: ускоряющее напряжение 300 кВ, номинальное увеличение 37 000×, время экспозиции 4 с, дефокусировка от –3 до –5 мкм.
Иммунизация мышей. Самок мышей линии C57BL/6 весом 18–20 г, полученных из питомника “Столбовая” (Россия), содержали в конвенционных условиях без ограничения в воде и корме. Все исследования и процедуры по рутинному уходу за животными проводили в соответствии с Международными руководящими принципами биомедицинских исследований на животных в ФГБУ “НИЦЭМ им Н.Ф. Гамалеи” Минздрава России, протокол Комитета по биомедицинской этике № 5 от 19.03.2021, и в ИБХ РАН, протокол Институтской комиссии по контролю за содержанием и использованием животных № 325 от 24.05.2021.
Формуляции L1–L5, пептидные композиции Р1–Р5 и контроль PB-Suc вводили в подушечки задних лап по 50 мкл в каждую 2 раза с интервалом в 21 день. Общее содержание пептидов в дозе – 120–130 мкг. Все препараты, кроме контроля PB-Suc, содержали иммуностимулятор CpG-ODN, 75 мкг. В каждой группе было по семь мышей. Через неделю после второй иммунизации у мышей под изофлурановым наркозом забирали кровь из орбитального синуса в гепаринизированные пробирки, получали плазму, которую хранили замороженной до анализа. Мышей умерщвляли методом цервикальной дислокации, стерильно забирали селезенки.
МТТ-анализ пролиферации спленоцитов. Селезенки интактных или иммунных мышей гомогенизировали в физиологическом растворе, центрифугировали при 1000 об/мин 7 мин, осадок обрабатывали 0.83%-ным хлористым аммонием для лизирования эритроцитов, отмывали в физиологическом растворе дважды центрифугированием и переводили в культуральную среду на основе RPMI-1640 с добавлением 7% фетальной телячьей сыворотки, пенициллина-стрептомицина-глутамина (ПанЭко, Россия). Спленоциты вносили в плоскодонные 96-луночные планшеты в количестве 106 кл./лунку в 200 мкл среды. В лунки вносили антигены в количестве 20–30 мкг. Планшеты инкубировали 72 ч. В течение последних 3 ч в каждую лунку добавляли по 10 мкл МТТ (5 мг/мл). После инкубации культуральную среду удаляли и в каждую лунку добавляли 100 мкл ДМСО. Планшеты инкубировали при встряхивании в течение 15 мин для растворения формазана. Оптическую плотность измеряли на спектрофотометре Titertek (Великобритания) при 540 нм. Результаты анализировали с помощью пакета Excel (Microsoft). Данные приведены в виде оптической плотности (OD).
Анализ спонтанного ответа на пептиды. Для оценки распознавания отобранных пептидов спленоцитами мышей ставили 24 или 48 реплик спленоцитов, полученных от одной мыши, с антигенами (смеси пептидов P1, Р2, Р3, Р4, Р5) в концентрации 160 мкг/мл и без антигенов (контроль), как описано выше.
Анализ антиген-специфического ответа после иммунизации мышей препаратами P1–Р4 и L1–L4 (эксперимент I). Определение концентрации IFN-γ, продуцируемого спленоцитами в ответ на стимуляцию пептидными композициями P1–Р4, проводили с использованием IGRA-теста (interferon-gamma release assay) в соответствии с описанной методикой [37]. Спленоциты засевали в плотности 107 кл./мл в 100 мкл ростовой культуральной среды RPMI-1640 с добавлением 2 мМ L-глутамина, 10% фетальной телячьей сыворотки, 1× антибиотика-антимикотика, 0.05 мМ 2-меркаптоэтанола (ПанЭко, Россия). Клетки инкубировали в CO2-инкубаторе при 37°С, 5% CO2, влажность 100% в течение 1.5–2 ч. В лунки вносили антигены P1, Р2, Р3, Р4 в концентрации 10 мкг/мл, положительный контроль конканавалин А (10 мкг/мл), отрицательный контроль (PB-Suc). Каждую опытную группу стимулировали соответствующей композицией пептидов, группы контроль PB-Suc и LK стимулировали отдельно каждой композицией пептидов. Каждую группу также стимулировали положительным и отрицательным контролями. Стимуляцию проводили в двух повторностях.
Культуральные планшеты инкубировали в СО2-инкубаторе 20 ч, отбирали культуральную среду и определяли в ней концентрацию IFN-γ (пг/мл) с использованием коммерческого набора IFN gamma Mouse ELISA Kit (Thermo Fisher, США) согласно инструкции производителя.
Анализ антиген-специфического ответа на пептиды (эксперимент II). Для оценки ответа на пептиды пролиферацией и продукцией цитокинов спленоциты иммунизированных мышей каждой группы (n = 7) стимулировали пептидами, входящими в состав соответствующей формуляции, в дозе 25–45 мкг. Эксперименты ставили в трех повторностях на каждый пептид. Через 24 ч из лунок забирали по 50 мкл, переносили их в новые планшеты, планшеты хранили при –60°С до анализа. В качестве контроля использовали спленоциты интактных мышей, мышей, иммунизированных буфером PBSuc или контрольными липосомами LK. Анализ пролиферации проводили через 72 ч, как описано выше.
Комплексный анализ цитокинов (эксперимент II). Стандартную панель магнитных бус для анализа цитокинов мыши: IFN-γ, IL-2, 4, 6, 9, 10, 13, 17, 22 и TNF-α (Biolegend, США) – использовали для анализа белков в плазме крови и надосадках спленоцитов по протоколу производителя с использованием проточного цитометра MACSQuant Tyro Sorter (Miltenei, Германия).
Статистический анализ. Статистический анализ проводили с использованием программного обеспечения Excel и t-критерия Стьюдента. Значения сравнения при р < 0.05 считали статистически значимыми.
Проверка протективной эффективности формуляции L1. В работе использовали самцов гуманизированных мышей C57BL/6-TgTn(CAG-humanACE2-IRES-Luciferase-WPRE-polyA) в возрасте 14–16 недель. Животных содержали в стандартных условиях Питомника лабораторных животных ФИБХ РАН (Уникальная научная установка “Био-модель” ИБХ РАН), имеющего международную аккредитацию AAALACi. Все эксперименты и манипуляции были одобрены Институтской комиссией по уходу и использованию животных (№ 757/22 от 17.02.2022). Всех животных обязательно проверяли на наличие целевого гена hACE2, экспрессию которого анализировали методом ОТ-ПЦР. Иммунизацию животных выполняли двукратно (в 1-й и 21-й дни), вводя в подушечки задних лап буфер PB-Suc (n = 3), контрольные липосомы LК (n = 4) и препарат L1 с добавлением 75 мкг CpG-ODN (n = 6), 100 мкл (2 × 50). По данным динамического светорассеяния, размер липосом LК составил 207.9 ± 5.7 нм (PDI 0.096 ± ± 0.042), липосом L1 – 207.5 ± 1.8 нм (PDI 0.093 ± ± 0.021).
Заражение мышей вирусом SARS-CoV-2. Через 36 дней после первой иммунизации всех мышей C57BL/6-TgTn(CAG-humanACE2-IRES-Luciferase-WPRE-polyA) передавали в специализированную лабораторию уровня ABSL-3 (Сергиев Посад), где заражали уханьским штаммом SARS-CoV-2. Инфицирование проводили с помощью интраназального введения вируса SARS-CoV-2 в дозе 3 lg БОЕ в объеме 20 мкл физиологического раствора [38]. У погибших в течение срока наблюдения мышей экстирпировали сердечно-легочный комплекс и после двухне-дельной инактивации в 10%-ном растворе фор-малина отправляли на гистологическое исследо-вание.
Гистология. Фиксированные в 10%-ном растворе нейтрального формалина легкие промывали в проточной воде, дегидратировали в этиловых спиртах восходящей концентрации и заливали в парафин. Парафиновые срезы толщиной 4–5 мкм, окрашенные гематоксилином и эозином, изучали с помощью микроскопa AxioScope.A1 (Carl Zeiss, Германия). Микрофотографии гистологических препаратов получали с помощью камеры высокого разрешения Axiocam 305 color (Carl Zeiss, Германия) и программного обеспечения ZEN 2.6 lite (Carl Zeiss, Германия). Оценку выраженности тех или иных патоморфологических признаков проводили по пятибалльной шкале, где 0 – отсутствие признака (в пределах нормы), 1 – минимальная степень выраженности, 2 – слабая, 3 – средняя (умеренная), 4 – выраженная, 5 – тяжелая [39].
ЗАКЛЮЧЕНИЕ
Показано, что подкожная вакцинация липосомами из природных липидов улучшает легочную функцию, по-видимому, благодаря активации врожденного иммунитета, но при летальной дозе вируса не защищает полностью. Липосомы, несущие набор Т-клеточных эпитопов вируса SARS-CoV-2, могут послужить основой для создания вакцины как минимум для профилактики хронического течения и средней тяжести инфекционного заболевания. При выборе эпитопов цитотоксических и хелперных Т-лимфоцитов для составления пептидных композиций следует брать за основу результаты клинических анализов выздоравливающих пациентов и уже во вторую очередь учитывать данные иммуноинформатического анализа. Иммунодоминантные эпитопы, вычисленные in silico, зачастую занимают первые позиции просто в силу своей высокой гидрофобности, и презентация их в комплексах МНСI или МНСII может быть затруднена. Кроме того, гидрофобные пептиды должны быть включены в липидный бислой, что создает определенные трудности при формировании липосом, а также может привести к разрушению последних в ходе лиофилизации из-за нарушения жидкокристаллической структуры мембраны. Оптимизация методики получения липосом с Т‑клеточными эпитопами, пригодных для длительного хранения, будет предметом наших дальнейших исследований.
Список литературы
Delany I., Rappuoli R., De Gregorio E. // EMBO Mol. Med. 2014. V. 6 (6). P. 708–720.
De Temmerman M.-L., Rejman J., Demeester J., Irvine D.J., Gander B., De Smedt S.C. // Drug Discov. Today. 2011. V. 16 (13-14). P. 569–582.
Reed S., Orr M., Fox C. // Nat. Med. 2013. V. 19. P. 1597–1608.
Kawai T., Akira S. // Nat. Immunol. 2010. V. 11. P. 373–384.
Allison A., Gregoriadis G. // Nature. 1974. V. 252 (5480). P. 252.
Schwendener R.A. // Ther. Adv. Vaccines. 2014. V. 2 (6). P. 159–182.
Perrie Y., Crofts F., Devitt A., Griffiths H.R., Kastner E., Nadella V. // Adv. Drug. Deliv. Rev. 2016. V. 99 (Pt A). P. 85–96.
Nisini R., Poerio N., Mariotti S., De Santis F., Fraziano M. // Front. Immunol. 2018. V. 9. P. 155.
Bernasconi V., Norling K., Bally M., Höök F., Lycke N.Y. // J. Immunol. Res. 2016. V. 2016. P. 5482087.
Третьякова Д.С., Водовозова Е.Л. // Биол. мембраны. 2022. Т. 39. С. 85–106. [Tretiakova D.S., Vodovozova E.L. // Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2022. V. 16 (1). P. 1–20.]
Gayed P.M. // Yale J. Biol. Med. 2011. V. 84 (2). P. 131–138.
Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. // Nature. 2000. V. 408. P. 740–745.
Lee Y., Lee Y.S., Cho S.Y., Kwon H.J. // Adv. Protein Chem. Struct. Biol. 2015. V. 99. P. 75–97.
Purcell A.W., McCluskey J., Rossjohn J. // Nat. Rev. Drug. Discov. 2007. V. 6 (5). P. 404–414.
Ohno S., Kohyama S., Taneichi M., Moriya O., Hayashi H., Oda H., Mori M., Kobayashi A., Akatsuka T., Uchida T., Matsui M. // Vaccine. 2009. V. 27. P. 3912–3920.
Kohyama S., Ohno S., Suda T., Taneichi M., Yokoyama S., Mori M., Kobayashi A., Hayashi H., Uchida T., Matsui M. // Antiviral Res. 2009. V. 84. P. 168–177.
Heuts J., Varypataki E.M., van der Maaden K., Romeijn S., Drijfhout J.W., van Scheltinga A.T., Ossendorp F., Jiskoot W. // Pharm. Res. 2018. V. 35. P. 207.
Dhakal S., Cheng X., Salcido J., Renu S., Bondra K., Lakshmanappa Y.S., Misch C., Ghimire S., Feliciano-Ruiz N., Hogshead B., Krakowka S., Carson K., McDonough J., Lee C.W., Renukaradhya G.J. // Int. J. Nanomedicine. 2018. V. 13. P. 6699–6715.
Mishra S. // R. Soc. Open Sci. 2020. V. 7. P. 201141.
Fast E., Altman R.B., Chen B. // Potential T-cell and B‑cell Epitopes of 2019-nCoV. bioRxiv preprint. This version posted March 18, 2020. https://doi.org/10.1101/2020.02.19.955484
Kalita P., Padhi A.K., Zhang K.Y.J., Tripathi T. // Microb. Pathogenesis. 2020. V. 145. P. 104236.
Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., Chia W.N., Chen M.I.-C., Wang L.-F., Ooi E.E., Kalimuddin S., Tambyah P.A., Low J.G.-H., Tan Y.-J., Bertoletti A. // Nature. 2020. V. 584. P. 457–462. https://doi.org/10.1038/s41586-020-2550-z
Ferretti A.P., Tomasz Kula T., Wang Y., Nguyen D.M.V., Weinheime A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., Whitton H.J., Virbasius A., Olivier K.J., Jr., Buckner L.R., Alistar A.T., Whitman E.D., Bertino S.A., Chattopadhyay S., MacBeath G. // Immunity. 2020. V. 53. P. 1095–1107.e3. https://doi.org/10.1016/j.immuni.2020.10.006
Snyder T.M., Gittelman R.M., Klinger M. // Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. medRxiv preprint. This version posted August 4, 2020. https://doi.org/10.1101/2020.07.31.20165647
Nelde A., Bilich T., Heitmann J.S., Maringer Y., Salih H.R., Roerden M., Lübke M., Bauer J., Rieth J., Wacker M., Peter A., Hörber S., Traenkle B., Kaiser P.D., Rothbauer U., Becker M., Junker D., Krause G., Strengert M., Schneiderhan-Marra N., Templin M.F., Joos T.O., Kowalewski D.J., Stos-Zweifel V., Fehr M., Rabsteyn A., Mirakaj V., Karbach J., Jäger E., Graf M., Gruber L.-C., Rachfalski D., Preuß B., Hagelstein I., Märklin M., Bakchoul T., Gouttefangeas C., Kohlbacher O., Klein R., Stevanović S., Rammensee H.-G., Walz J.S. // Nat. Immunol. 2021. V. 22. P. 74–85. https://doi.org/10.1038/s41590-020-00808-x
Quadeer A.A., Ahmed S.F., McKay M.R. // Cell Rep. Med. 2021. V. 2. P. 100312. https://doi.org/10.1016/j.xcrm.2021.100312
Mouritsen O.G., Jorgenson K. // Chem. Phys. Lipids. 1994. V. 73. P. 3–25.
Markwell M., Haas S., Bieber L., Tolbert N.E. // Anal. Biochem. 1978. V. 210. P. 206–210.
Chen W., Huang L. // Mol. Pharm. 2008. V. 5. P. 464–471.
Mansourian M., Badiee A., Jalali S.A., Shariat S., Yazdani M., Amin M., Jaafari M.R. // Immunol. Lett. 2014. V. 162. P. 87–93.
Schmidt S.T., Foged C., Korsholm K.S., Rades T., Christensen D. // Pharmaceutics. 2016. V. 8. P. 7.
Ludewig B., Barchiesi F., Pericin M., Zinkernagel R.M., Hengartner H., Schwendener R.A. // Vaccine. 2001. V. 19 (1). P. 23–32.
Engler O.B., Schwendener R.A., Dai W.J., Wolk B., Pichler W., Moradpour D., Brunner T., Cerny A. // Vaccine. 2004. V. 23 (1). P. 58–68.
Dalwadi G, Benson HA, Chen Y. // Pharm. Res. 2005. V. 22 (12). P. 2152–2162.
Dedoni S., Avdoshina V., Camoglio C., Siddi C., Fratta W., Scherma M., Fadda P. // Molecules. 2022. V. 27 (13). P. 4142.
Kryukova E.V., Egorova N.S., Kudryavtsev D.S., Lebedev D.S., Spirova E.N., Zhmak M.N., Garifulina A.I., Kasheverov I.E., Utkin Y.N., Tsetlin V.I. // Front. Pharmacol. 2019. V. 10. P. 748.
Tkachuk A.P., Gushchin V.A., Potapov V.D., Demidenko A.V., Lunin V.G., Gintsburg A.L. // PLoS One. 2017. V. 12 (4). P. e0176784.
Chernov A.S., Minakov A.A., Kazakov V.A., Rodionov M.V., Rybalkin I.N., Vlasik T.N., Yashin D.V., Saschenko L.P., Kudriaeva A.A., Belogurov A.A., Smirnov I.V., Loginova S.Y., Schukina V.N., Savenko S.V., Borisevich S.V., Zykov K.A., Gabibov A.G., Telegin G.B. // Inflamm. Res. 2022. V. 71 (5–6). P. 627–639.
Mann P.C., Vahle J., Keenan C.M., Baker J.F., Bradley A.E., Goodman D.G., Harada T., Herbert R., Kaufmann W., Kellner R., Nolte T., Rittinghausen S., Tanaka T. // Toxicol. Pathol. 2012. V. 40 (Suppl. 4). P. 7S–13S.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия