

Биоорганическая химия, 2023, T. 49, № 1, стр. 3-22
Мускариновые и никотиновые холинорецепторы в регуляции сердечно-сосудистой системы
А. В. Осипов 1, А. С. Аверин 2, Э. Р. Шайхутдинова 3, И. А. Дьяченко 3, В. И. Цетлин 2, Ю. Н. Уткин 2, *
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 ФГБУН “Институт теоретической и экспериментальной биофизики” РАН
142290 Пущино, ул. Институтская, 3, Россия
3 Филиал ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
142290 Пущино, просп. Науки, 6, Россия
* E-mail: utkin@ibch.ruyutkin@yandex.ru
Поступила в редакцию 28.07.2022
После доработки 16.08.2022
Принята к публикации 23.08.2022
- EDN: GGQIOX
- DOI: 10.31857/S0132342323010219
Аннотация
В регуляции сердечно-сосудистой системы (ССС) участвует множество различных рецепторов и ионных каналов, регулирующих ионные потоки. Функционирование ССС происходит с участием механизмов нервной и гуморальной регуляции, и в обоих случаях в процессах регуляции принимают участие холинорецепторы разных семейств и подтипов, имеющих различную локализацию. Показано, что холинорецепторы располагаются на мембранах клеток непосредственно сердца и кровеносных сосудов; и в данном обзоре рассматриваются механизмы регуляции функций ССС с участием только тех холинорецепторов, которые находятся в ткани сердца и сосудов. В целом как мускариновые, так и никотиновые холинорецепторы широко представлены в тканях ССС. При этом мускариновые холинорецепторы в общем вовлечены в регуляцию сосудистого тонуса и сократительной способности сердца, а никотиновые холинорецепторы участвуют в основном в регуляции ряда важных патофизиологических процессов, напрямую затрагивающих функционирование ССС. Регуляция функционирования холинорецепторов может рассматриваться в качестве дополнения к существующим способам лечения заболеваний ССС, включая такие заболевания, как атеросклероз и сердечная недостаточность. Обсуждается использование блокаторов и активаторов холинорецепторов для изучения и/или лечения патологических состояний ССС.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ ..........................................................3
ХОЛИНОРЕЦЕПТОРЫ......................................4
ХОЛИНОРЕЦЕПТОРЫ В СЕРДЦЕ...................6
Мускариновые холинорецепторы в сердце...............6
Никотиновые холинорецепторы в сердце................8
Холинергические рецепторы в регуляции функций сердца......................................................9
Аутокринная ненейрональная холинергическая система сердца......................................................12
ХОЛИНОРЕЦЕПТОРЫ В КРОВЕНОСНЫХ СОСУДАХ..........................12
Мускариновые холинорецепторы в кровеносных сосудах ...........................................12
Никотиновые холинорецепторы в кровеносных сосудах ...........................................14
ЗАКЛЮЧЕНИЕ..................................................18
СПИСОК ЛИТЕРАТУРЫ..................................18
ВВЕДЕНИЕ
Сердечно-сосудистая система (ССС) – одна из важнейших систем органов большинства животных, обеспечивающая организм необходимыми веществами и удаляющая продукты жизнедеятельности клеток. Органы ССС – сердце и кровеносные сосуды, их функционирование поддерживает гомеостаз организма в постоянно изменяющихся условиях. Физиологическая функция сердца и сосудов контролируется хорошо организованным взаимодействием различных ионных каналов, насосов (АТPаз) и обменников, регулирующих градиенты ионов Na+, Ca2+ и K+. Из ионов, участвующих в сложной работе сердца, кальций считается, пожалуй, самым важным. Этот ион имеет решающее значение для процесса, называемого электромеханическим сопряжением и позволяющего камерам сердца сокращаться и расслабляться. Сердечные K+-каналы – ключевые участники реполяризации сердца, противодействующие деполяризующим потокам Na+ и Ca2+. Ионы K+, в отличие от Na+ и Ca2+, проводятся множеством различных каналов, различающихся кинетикой активации/деактивации, а также своим вкладом в разные фазы потенциала действия.
Согласованное функционирование ССС происходит посредством механизмов нервной и гуморальной регуляции. Главную роль в нервной регуляции играет автономная нервная система, осуществляющая регуляцию через парасимпатические и симпатические нервные волокна в составе блуждающего нерва и симпатических стволов спинного мозга. Передача сигналов осуществляется в основном посредством холинергических и адренергических механизмов. В сердце парасимпатическая стимуляция, осуществляемая холинергическим путем, приводит к отрицательным эффектам: инотропному, хронотропному и дромотропному. Холинорецепторы расположены в основном во внешней мембране кардиомиоцитов, и именно их активация приводит к снижению насосной функции сердца. Симпатическая стимуляция, осуществляемая адренергическим путем, напротив, приводит к увеличению производительности сердца. За некоторым исключением все кровеносные сосуды, содержащие в своей стенке гладкомышечные клетки, иннервируются норадренергическими и холинергическими нервными волокнами из симпатического отдела автономной нервной системы. Медиаторы, поступающие в кровь, осуществляют гуморальную регуляцию ССС. Существует целый ряд таких веществ, однако и здесь большую роль играют как адреналин, усиливающий сердечную деятельность, так и ацетилхолин, ослабляющий деятельность сердца. При гуморальной регуляции тонуса сосудов наиболее мощным сосудосуживающим действием обладают адреналин и норадреналин. Сосудорасширяющее действие присуще ацетилхолину и некоторым другим соединениям (брадикинин, простагландины, гистамин и др.). Помимо того, существует так называемая ненейрональная холинергическая система, обладающая как аутокринными, так и паракринными свойствами. Так, некоторые ненейрональные клетки способны синтезировать и высвобождать ацетилхолин, а другие клетки, не имеющие ни симпатической, ни парасимпатической иннервации (как, например, эндотелиальные клетки кровеносных сосудов), несут на своей поверхности рецепторы ацетилхолина. Приведенное довольно схематичное описание регуляции деятельности ССС, в которой участвует множество более тонких механизмов, дает, однако, представление о существенной роли холинергической системы в этом процессе [1].
ХОЛИНОРЕЦЕПТОРЫ
Существует два больших семейства рецепторов, управляемых ацетилхолином. Это так называемые мускариновые холинорецепторы (мХР), которые активируются как ацетилхолином, так и мускарином и блокируются атропином, и никотиновые холинорецепторы (нХР), которые активируются ацетилхолином или никотином и блокируются некоторыми алкалоидами (например, d-тубокурарином) и нейротоксинами животных ядов (например, белковыми α-нейротоксинами змей или α-конотоксинами, нейротоксическими пептидами из ядовитых морских моллюсков рода Conus).
мХР представляют собой метаботропные рецепторы, относятся к семейству рецепторов, связанных с G-белками (GPCR), и передают сигналы в клетку посредством взаимодействия с этими белками. Имеется пять подтипов мХР: M1, M2, M3, M4 и M5, обладающих высоким сходством аминокислотных последовательностей (64–68% идентичных аминокислотных остатков), но разными молекулярными и сигнальными свойствами [2, 3]. Так, в результате взаимодействия с определенными G-белками они выполняют различные физиологические функции. мХР разделяют на две группы в зависимости от типа G-белков, с которыми они взаимодействуют. Рецепторы М1, М3 и М5 связаны с белками Gq/11, которые опосредуют активацию фосфолипазы C, а рецепторы М2 и М4 связаны с белками Gi/o, которые опосредуют ингибирование аденилатциклазы (AЦ) и, таким образом, вызывают снижение уровня циклического аденозинмонофосфата (cAMP) [4]. Данные рентгеноструктурного анализа и криоэлектронной микроскопии показывают, что структурно мХР представляют собой интегральные мембранные белки, которые содержат семь трансмембранных спиралей [5, 6] (рис. 1а). Внеклеточный лиганд-связывающий домен состоит из нескольких петель, содержащих консервативную дисульфидную связь, которая стабилизирует структуру рецептора. Внутриклеточный домен взаимодействует с G-белками и другими сигнальными молекулами и осуществляет внутриклеточную передачу сигналов.
Рис. 1.
(а) – Пространственная структура мХР М2-типа, установленная методом рентгеноструктурного анализа [5]. В связывающем центре рецептора находится антагонист хинуклидил-3-бензилат (выделен зеленым цветом и окружностью); (б) – пространственная структура нХР типа α7 человека, определенная методом криоэлектронной микроскопии в липидном окружении [9]. В связывающем центре рецептора находится агонист эпибатидин (показан зеленым цветом, в одном из связывающих центров выделен окружностью).
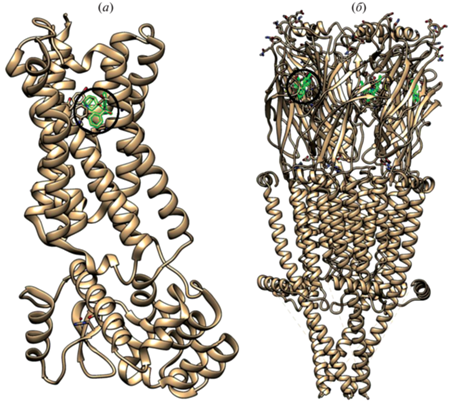
нХР – лиганд-управляемые ионные каналы, относящиеся к семейству Cys-петельных рецепторов, которое включает также ионотропные рецепторы гамма-аминомасляной кислоты (ГАМК), глициновые и серотониновые 5-HT3-рецепторы [7, 8]. нХР представляют собой пентамеры, включающие четыре различных типа субъединиц (α1–10, β1–4, γ либо ϵ, δ). Они существуют в виде гомомеров (со всеми субъединицами одного типа, например, (α7)5) или в виде гетеромеров, по крайней мере, с одной α- и одной β-субъединицей из пяти субъединиц, которые комбинируются в различных вариантах, например, (α1)2β1γδ, (α4)3(β2)2, (α4)2(β2)3, α4α6β3(β2)2. Сборки из пяти субъединиц образуют множество различных подтипов, которые имеют общую базовую структуру, но обладают специфическими фармакологическими и функциональными свойствами. Субъединицы нХР формируют пору, проницаемую для ионов Na+, K+ и Ca2+. Способность нХР изменять уровень внутриклеточного кальция приводит к активации различных внутриклеточных механизмов, которые могут играть ключевую роль в дальнейшей передаче сигналов. Все субъединицы имеют похожее строение и состоят из большого N-концевого внеклеточного домена, за которым следуют три гидрофобных трансмембранных домена (M1–M3), большая цитоплазматическая петля между доменами M3 и M4, четвертый гидрофобный трансмембранный домен (M4) и короткий внеклеточный С-концевой домен (рис. 1б). Трансмембранные домены M1–M4 расположены концентрическими слоями вокруг центральной поры: домен M2 выстилает пору, M1 и M3 защищают M2 от окружающего липидного двойного слоя, а M4 наиболее экспонирован в липидный бислой [9].
Холинорецепторы могут располагаться в различных отделах нервной и эндокринной систем, которые регулируют деятельность различных органов, в том числе ССС, при помощи медиаторов и гормонов, связывающихся с рецепторами иных типов. Поэтому холинорецепторы оказывают косвенное влияние на работу ССС, опосредованное, например, через ренин-ангиотензиновую систему [10]. Установлено, что никотин и другие агонисты нХР вызывают существенный подъем артериального давления (например, по данным Jutkiewicz et al. [11]). Однако эти эффекты опосредованы через нервную систему, и в процитированной работе они обусловлены активацией вегетативных ганглиев с участием некоторых подтипов нХР. В равной мере можно привести ряд примеров опосредованного влияния мХР на ССС. Так, исследование сонных артерий морских свинок показало, что М1 играет важную роль в модулировании высвобождения норадреналина, способствуя сужению сосудов [12]. М2, наоборот, задействован в опосредуемом карбахолом ингибировании высвобождения норадреналина, способствуя расслаблению мозговых артерий кошки [13]. М3-рецепторы, впрочем, также стимулируют выработку надпочечниками адреналина, который, в свою очередь, влияет на функцию сосудов при солечувствительной гипертензии [14].
Однако холинорецепторы располагаются также на мембранах клеток непосредственно сердца и кровеносных сосудов. В данном обзоре мы рассмотрим механизмы регуляции функций ССС с участием только тех холинорецепторов, которые сами находятся в ткани сердца и сосудов.
ХОЛИНОРЕЦЕПТОРЫ В СЕРДЦЕ
Хорошо известно, что парасимпатическая система, в которой основным нейромедиатором выступает ацетилхолин (АХ), играет существенную роль в регуляции сердечно-сосудистой функции. При нервной регуляции ССС сигнализация с участием холинорецепторов происходит в нейронных сетях парасимпатической нервной системы, а также между нейронными сетями и сердцем. Внутри нейронной сети преганглионарные волокна из продолговатого мозга, проходящие в блуждающем нерве, передают сигнал с участием нХР на плазматической мембране постганглионарных волокон. Затем эта стимуляция постганглионарных нервов приводит к секреции АХ, который связывается с рецепторами на плазматической мембране клеток сердца. Кроме того, установлено, что кардиомиоциты способны синтезировать и высвобождать АХ, и такая внутренняя холинергическая система известна как ненейрональная холинергическая система [15]. После высвобождения во внеклеточное пространство АХ может связываться с мХР или нХР и активировать специфические сигнальные пути в различных типах клеток ССС. Основные рецепторы АХ в сердце – мХР (M1–M5), которые гетерогенно распределены по всей ССС и выполняют в ней различные физиологические функции [16].
Мускариновые холинорецепторы в сердце
За последние годы в миокарде были обнаружены все пять известных типов мускариновых рецепторов, хотя их функциональная значимость сильно различается. В сердце преобладают изоформы подтипа М2.
Подтип М2. M2 мХР – основной мускариновый рецептор АХ в сердце млекопитающих. Он обильно экспрессируется в предсердиях и проводящей системе (синоатриальный и атриовентрикулярный узлы), тогда как его экспрессия в желудочках относительно низка по сравнению с предсердиями [16]. Исследования связывания радиолигандов в сердце человека подтверждают, что М2 имеет значительно более высокое содержание в предсердиях (до 2.5 раз) по сравнению с миокардом желудочков [16].
М2 мХР предпочтительно связывается с белками Gi, которые ингибируют аденилатциклазу (АЦ) и, таким образом, при активации ослабляют превращение АТР в cAMP. В целом стимуляция М2 оказывает прямое и косвенное влияние на функцию сердечной мышцы. М2, локализующиеся на постганглионарных симпатических нервных окончаниях, ингибируют высвобождение норадреналина, предотвращая активацию β-адренергических рецепторов миокарда. Это косвенное влияние, которое в данном обзоре не обсуждается. Прямое же влияние модулируется посредством взаимодействия с G-белком в мембранах кардиомиоцитов несколькими путями.
В первом пути М2 взаимодействует с α-субъединицей белка Gi, а βγ-субъединица белка Gi напрямую связывается с активируемым G-белком калиевым каналом внутреннего выпрямления (GIRK). Эти ионные каналы генерируют калиевый ток, активируемый ацетилхолином (IK,ACh). После активации канал становится проницаемым для K+, что приводит к ряду последствий, которые зависят от величины присущего конкретным клеткам мембранного потенциала во время диастолы: 1) к гиперполяризации в синоатриальном узле, что выражается в замедлении частоты сердечных сокращений; 2) к уменьшению продолжительности потенциала действия и эффективного рефрактерного периода у миоцитов предсердий, что делает эти клетки более восприимчивыми к преждевременным стимулам; 3) к снижению возбудимости клеток атриовентрикулярного узла, что замедляет распространение импульса от предсердий к желудочкам [17]. Каналы IK,ACh обнаружены и в желудочках, однако в гораздо меньшем количестве и с существенно меньшей восприимчивостью к стимуляции ацетилхолином, чем в предсердиях [18]. Считается, что М2, Gi и GIRK не имеют более никаких молекулярных посредников и для полноценного ответа должны физически взаимодействовать [17].
Во втором пути М2 модулирует cAMP-зависимый ответ на активацию β-адренергических рецепторов, которые активируют АЦ [19]. Здесь М2 связывается с βγ-субъединицей белка Gi, который посредством α-субъединицы ингибирует AЦ5/6, что приводит к снижению уровня внутриклеточного cAMP. Изменения в уровне cAMP в предсердиях напрямую влияют на каналы HCN (управляемые циклическими нуклеотидами гиперполяризационно-активируемые каналы водителя ритма, проницаемые для ионов Na+ и K+), а посредством активации протеинкиназы А – на кальциевые каналы L-типа. В желудочках эти изменения влияют на функционирование медленных выпрямляющих калиевых и хлорных каналов CFTR.
Однако есть еще и третий путь прямого влияния М2 на функцию сердца: М2 связывается с α‑субъединицей белка Gi, который своей βγ-субъединицей способствует активации АЦ4/7 β-адренорецептором, что приводит к усилению синтеза cAMP. Ингибиторное влияние М2 на синтез cAMP преобладает, оно выражено сильнее, но и заканчивается быстрее; активаторный эффект сохраняется дольше, поэтому сразу после прекращения раздражения блуждающего нерва или стимуляции ацетилхолином происходит резкое увеличение частоты сердечных сокращений и сократительной способности желудочков [19]. Кстати, ацетилхолин вызывает повышение уровня cAMP в миоцитах предсердий еще одним путем: М2 активирует фосфолипазу С для запуска фосфоинозитидного каскада, включающего, в свою очередь, каскад реакций с участием кальция/кальмодулина и протеинкиназы С. Это приводит к активации синтазы оксида азота (NOS); продукция оксида азота (NO) стимулирует выработку циклического гуанозинмонофосфата (cGMP) растворимой гуанилатциклазой. cGMP ингибирует фосфодиэстеразу-3, таким образом способствуя накоплению cAMP, и это приводит к отрицательному инотропному эффекту в отношении предсердий [20, 21].
Было отмечено, что М2 проявляют чувствительность к потенциалу, т.е. их сродство к лигандам изменяется в зависимости от потенциала мембраны [22]. Чувствительность М2 к потенциалу подразумевает, что сродство к АХ (и, следовательно, эффект АХ) изменяется в течение электрического цикла сердца. Моделирование этого процесса показало, что измененная чувствительность М2 к потенциалу может способствовать и возникновению, и развитию таких заболеваний, как мерцательная аритмия и неадекватная синусовая тахикардия [23].
В целом активация М2-рецептора приводит к снижению частоты сердечных сокращений (отрицательной хронотропии) и практически не влияет на сократительную способность сердца (отсутствие инотропного эффекта).
Подтип М3. М3 мХР преимущественно сопряжен с белком Gq. Этот белок передает сигнал от М3 к мембраносвязанному ферменту фосфоинозитидазе Cβ (фосфоинозитол-специфичной фосфолипазе С), при этом активация М3 усиливает активность фермента, вызывая образование фосфатидил-инозитол-4,5-дифосфата, который служит источником для образования двух посредников: инозитол-1,4,5-трифосфата и диацилглицерола. Эти две молекулы действуют как вторичные мессенджеры для мобилизации внутриклеточных запасов Ca2+ и активации протеинкиназы С.
Проведенные исследования показали, что подтип М3 также присутствует в клетках миокарда [24, 25]. Распределение М3 в сердце неравномерно; мРНК М3 была обнаружена в предсердиях и желудочках сердца человека, хотя и в меньшем количестве, чем мРНК М2. При этом экспрессия М3 в желудочках в 10 раз выше, чем в предсердиях. Кроме того, эксперименты по иммуноокрашиванию кардиомиоцитов желудочков человека показали, что концентрация М3 выше на вставочных дисках по сравнению с другими областями клеточной мембраны [26]. Предпочтительное распределение на вставочных дисках указывает на то, что М3 играет существенную роль в межклеточных коммуникациях кардиомиоцитов. Следует отметить, что имеется несколько функций М3-рецепторов сердца. Этот рецептор активирует не только путь протеинкиназы С, но и медленный компонент выпрямляющего калиевого тока, помогая таким образом регулировать частоту сердечных сокращений и реполяризацию кардиомиоцитов [27, 28]. Было также показано, что стимуляция М3-рецепторов вызывала выраженное укорочение длительности потенциала действия в предсердиях и желудочках функционирующего миокарда [29]. М3 присутствуют в водителе ритма и миокарде сердца мыши, где они опосредуют негативные холинергические эффекты, а именно замедление синусового ритма и укорочение потенциалов действия [29].
Подтип М1. М1 мХР обнаруживается в кардиомиоцитах предсердий и желудочков, но в разных количествах, а также, по-видимому, его содержание различается в зависимости от вида животного. Например, М1 был обнаружен как в желудочках [30], так и в предсердиях человека [18], а в предсердиях крыс линии Wistar – нет [16]. При этом локализация М1 в желудочках сердца обнаруживается у разных видов животных, хотя и в различных количествах. Было высказано предположение, что М1 в сердце человека играет определенную роль в гомеостатической регуляции. К тому же исследования на морских свинках показали, что активация М1 вызывает положительный инотропный эффект, который уравновешивает отрицательные эффекты активации М2 за счет усиления тока через Са2+-канал L-типа. Это в конечном итоге приводит к увеличению частоты и силы сокращений сердца [16]. Также для М1 мХР была показана возможность активации тока IK,Ach, который играет важную роль в патогенезе фибрилляции предсердий [31].
В согласии с вышесказанным, применение специфического блокатора М1 мХР пирензепина в высоких дозах приводит к тахикардии, но в малых дозах – к брадикардии. Поэтому блокированием М1 можно объяснить такой же парадоксальный двухфазный эффект низких и высоких доз неселективного антагониста мХР атропина [32]. Однако здесь следует быть осторожным в оценках, поскольку наиболее вероятный сценарий такой брадикардии – блокирование пресинаптического М1 блуждающего нерва, но не кардиомиоцитов; это облегчает высвобождение АХ и активацию им М2 сердца, сопровождающуюся вполне естественным замедлением ритма [32].
Подтип М4. Функция мХР М4 перекрывается с функцией М2, оба – рецепторы, связанные с белком Gi, и снижающие уровни cAMP. Имеющиеся к настоящему времени данные свидетельствуют о том, что М4, по-видимому, имеет большее значение в регуляции нервной системы, чем в сердечно-сосудистой, хотя мРНК, кодирующая М4, обнаружена в сердце. Данные, полученные с использованием тканей предсердий собаки, указывают на потенциальную роль М4 в контроле K+-каналов [33]. Недавнее исследование с использованием мышей, нокаутных по М4, показало, что рецепторы М4 могут также способствовать брадикардии, опосредованной рецептором М2 [16]. Было установлено, что тропикамид, ингибитор М4-подтипа, снижает частоту сердечных сокращений и минутный объем сердца у крыс, хотя величина ударного объема сердца увеличивается [34]. Однако необходимы дальнейшие исследования, чтобы уточнить, действительно ли (и каким именно образом) М4 участвует в функционировании ССС.
Подтип М5. мХР М5, как и другие члены этого семейства с нечетными номерами (М1 и М3), связан с белком Gq, однако данные о его функциональной роли в сердце весьма ограничены. Так, исследование экспрессии мРНК, кодирующей мХР у крыс, обнаружило мРНК М5 в предсердиях и желудочках сердца, где ее содержание составляет 5% от общей мРНК мХР [35]. Иммуноблоттинг с антителами, специфическими к определенному подтипу мХР, подтвердил присутствие белка М5 в препаратах мембран из тканей как предсердия, так и желудочка сердца человека. Плотность М5 была в ~5 раз ниже в ткани желудочка, чем в ткани предсердия. Одиночные миоциты желудочка окрашивались антителами к М5, и при конфокальной микроскопии характерная локализация М5 обнаружена на вставочных дисках [26].
Суммируя приведенные выше данные, можно заключить, что в сердце мХР подтипа М2 преобладают и играют наиболее важную роль в регуляции функционирования сердца.
Никотиновые холинорецепторы в сердце
Данные о наличии нХР в сердце немногочисленны. Тем не менее исследование онтогенетических изменений экспрессии α-субъединиц нХР в сердце крысы показало, что различные α-субъединицы нХР присутствуют на ненейрональных клетках сердца [36]. На всех постнатальных стадиях субъединица α3 была обнаружена только в предсердиях, в то время как субъединицы α2, α4, α5 и α7, по-видимому, экспрессируются и в предсердиях, и в желудочках. мРНК, кодирующая субъединицы α4, α5, α7 и α10, была обнаружена на всех стадиях развития. Ни на одной стадии развития не удалось получить однозначный сигнал для мРНК α9-субъединицы нХР. Иммуногистохимическое окрашивание показало наличие субъединицы α7 на нейронах сердца, фибробластах и кардиомиоцитах, а субъединиц α2 и α4 – на кардиомиоцитах с постнатальным перераспределением во вставочные диски. Эти данные свидетельствуют о ненейрональной экспрессии субъединиц нХР в сердце крысы [36]. В сердце крыс уровень экспрессии мРНК α7-субъединицы нХР измеряли с помощью ОТ-ПЦР и вестерн-блоттинга. Было показано наличие этого подтипа нХР в желудочках и предсердиях, при этом экспрессия в желудочках была существенно выше, чем в предсердиях [37]. Экспрессия мРНК практически всех субъединиц нХР, включая α2-, α3-, α4-, α5-, α6-, α7-, α9-, α10-, β1-, β2-, β4- и ε-субъединицы, была обнаружена методом количественной ПЦР в реальном времени в кардиомиоцитах новорожденных крыс и клетках H9c2 (клеточная линия, полученная из эмбриональной ткани сердца крыс BD1X) [38]. Однако по представленным в этой работе данным нельзя дать оценку уровням экспрессии отдельных субъединиц.
Использование 2-дезокси-2-[18F]фтор-D-глюкоз-A85380, избирательного радиолиганда α4β2-подтипа нХР, для визуализации этих рецепторов в сердце человека in vivo с применением позитронно-эмиссионной томографии выявило поглощение индикатора левым желудочком сердца [39]. Эти данные свидетельствуют о наличии α4β2-подтипа нХР в левом желудочке сердца человека. Однако полученное разрешение не позволило более точно локализовать эти рецепторы в конкретной ткани желудочка (ганглий, сосуд или мышца).
Активация нХР может приводить как к росту силы сокращения в левом желудочке [40, 41], так и к снижению силы сокращения предсердий [42]. Как обсуждалось выше, нХР участвуют в холинергической передаче в блуждающем нерве и вегетативных ганглиях, иннервирующих сердце. Поэтому иногда непросто дифференцировать воздействие холинергических лигандов на нХР самого сердца, исключив не только нХР синапсов, но и иные мишени. Так, никотин в физиологических условиях диффундирует через цитоплазматическую мембрану клетки и оказывает, причем в наномолярных концентрациях, токсическое воздействие на митохондрии, хотя сведения о возможном наличии нХР на мембранах митохондрий все еще недостаточно убедительны [43]. Например, в работе Nakatani et al. [42] отрицательные хронотропный и инотропный эффекты на изолированных мышцах предсердий крыс вызывались никотином в очень высоких концентрациях (0.3–1 мМ), но эти эффекты не предотвращались ни атропином (блокатор мХР), ни гексаметонием (блокатор ганглионарных нХР). Исходя из этого, авторы говорят о прямом влиянии никотина на мышцу сердца, но об участии нХР делают осторожное предположение, поскольку прямых доказательств (например, с использованием в опыте других блокаторов нХР) не приводят.
Холинергические рецепторы в регуляции функций сердца
Как обсуждалось выше, холинорецепторы играют существенную роль в различных сигнальных механизмах сердца, и наиболее ярко эта роль проявляется при некоторых патологических состояниях.
Когда говорят о регуляции ССС, обычно имеют в виду влияние на сократительную способность сердца и на сосудистый тонус. Как мы видим, такими регуляторами выступают мХР. Для нХР имеются лишь единичные сообщения о подобном прямом влиянии на ССС (в основном описывается косвенное). Однако нХР играют важную роль в осуществлении других физиологических и патологических процессов, которые специфичны именно для ССС (в частности, ангиогенез), или для которых эта система – основная мишень. Например, атеросклероз – это патология липидного обмена, но она связана с ССС и поражает в первую очередь ее, а все прочие проявления атеросклероза будут следствием этого поражения. Другой патологический процесс, который может затрагивать любые органы и системы организма, защитно-восстановительный – воспаление – так или иначе связан с целым рядом специфических состояний ССС (например, с инфарктом миокарда). Участвуя в различных патофизиологических процессах, нХР напрямую влияют на работу ССС, и мы считаем необходимым отразить этот аспект и здесь, и в разделе, посвященном регуляции функционирования кровеносных сосудов. Итак, холинергические лиганды могут предотвращать или ослаблять повреждения сердца, вызванные различными воздействиями. Эти лиганды выполняют свои функции, воздействуя на холинорецепторы, находящиеся в клетках парасимпатической нервной системы или непосредственно в клетках сердца.
Острый инфаркт миокарда (ОИМ) – наиболее частая причина острого повреждения миокарда и наиболее клинически значимая его форма. Считается, что основной метод лечения ОИМ – максимально быстрое восстановление адекватного коронарного кровотока (реперфузии) к ишемизированному миокарду. Между тем парадокс лечения заключается в том, что при восстановлении кровообращения в ишемизированной ткани доставка кислорода сопровождается образованием его активных форм, которые повреждают мембраны клеток. В результате этого возникает вторичное повреждение органа и развивается явление, названное ишемически-реперфузионным повреждением миокарда. Нарушение парасимпатической (вагусной) активности сердца – общий признак различных сердечно-сосудистых заболеваний, включая ОИМ.
Целый ряд исследований показывает, что увеличение секреции АХ при стимуляции блуждающего нерва защищает сердце, предотвращая ишемически-реперфузионное повреждение [44, 45] и повышая выживаемость в модели хронической сердечной недостаточности на животных [46, 47]. Стимуляция блуждающего нерва снижает частоту возникновения фибрилляции желудочков и уменьшает зону ОИМ в модели ишемии-реперфузии [48]. Кроме того, такая стимуляция защищает от дисфункции отдаленных сосудов миокарда, возникающей в результате ишемии-реперфузии, посредством холинергического противовоспалительного пути, в котором участвуют нХР α7-типа [49]. Недавно было также показано, что активация рецептора мХР M3 с помощью стимуляции блуждающего нерва модулирует митохондриальную динамику и улучшает митохондриальную функцию при ишемии миокарда, индуцированной изопротеренолом [50]. Более детально влияние активации блуждающего нерва на ишемически-реперфузионное повреждение миокарда рассмотрено в недавних обзорах [51, 52].
Что касается конкретных подтипов рецепторов, участвующих в кардиопротекции, то антагонисты М2 [53] или подавление экспрессии рецептора мХР М2 с помощью siRNA [54] устраняют эффекты АХ-индуцированной защиты в кардиомиоцитах, что свидетельствует о вовлеченности этого подтипа мХР в процесс кардиопротекции. М3-подтип мХР также играет защитную роль при сердечно-сосудистых заболеваниях [55]. Так, сообщалось, что активация рецептора М3 снижает гипертрофию сердца, индуцированную ангиотензином II, корректирует гемодинамическую дисфункцию сердца, ингибирует апоптоз клеток миокарда и уменьшает повреждение миокарда [56]. Фармакологическая активация М3-рецепторов также оказывает кардиопротекторное действие при ишемически-реперфузионном повреждении, что подтверждается использованием иодида 1,1-диметил-4-дифенилацетоксипиперидиния (4-DAMP), антагониста М3 [57]. Кроме того, гиперэкспрессия М3 в модели трансгенных мышей способствует снижению экспрессии аритмогенной микро-РНК miR-1, что, по некоторым данным, приводит к высвобождению субъединицы Kir2.1 соответствующего калиевого канала после ишемии-реперфузии. В итоге это приводит к увеличению калиевого тока внутреннего выпрямления и, как следствие, снижает частоту аритмий и смертельных исходов в модели ишемически-реперфузионного повреждения миокарда мыши [58].
Для выяснения роли нХР в ишемически-реперфузионном повреждении было исследовано влияние 3-(2,4-диметоксибензилиден)анабазеина (GTS21), селективного агониста нХР α7-типа, на функциональную активность сердца крысы in vivo и ex vivo. Введение GTS21 крысам, подвергшимся оперативной ишемии-реперфузии, или добавление этого реагента в перфузионный раствор изолированного сердца до ишемии приводило к значительному увеличению левожелудочкового давления и общего системного давления без влияния на частоту сердечных сокращений [59]. Совместное введение GTS21 с метилликаконитином, селективным антагонистом нХР α7-типа, блокировало эти положительные функциональные эффекты. In vivo введение GTS21 в начале реперфузии уменьшало размер инфаркта миокарда на 42% и количество активных форм кислорода в тканях на 62%. GTS21 нормализовал потенциал митохондриальной мембраны и концентрацию внутриклеточного АТР в культивируемых кардиомиоцитах, подвергшихся гипоксии/реоксигенации. Таким образом, активация нХР α7-типа обеспечивает терапевтический эффект при ишемически-реперфузионном повреждении сердца [59].
Уровень экспрессии мХР M2 и М4, а также нХР α7-типа при ишемически-реперфузионном повреждении измеряли с помощью ОТ-ПЦР и вестерн-блоттинга в изолированном сердце в течение 30 мин ишемии и 60 мин реперфузии [37]. Установлено, что до повреждения экспрессия M2 в левом желудочке была ниже, чем в предсердиях и правом желудочке, тогда как экспрессия нХР α7-типа была значительно выше в левом и правом желудочках, чем в предсердиях. После реперфузии экспрессия мРНК и белка M2 заметно увеличивалась в левом и правом желудочке, а экспрессия α7 нХР значительно усиливалась в левом желудочке. Экспрессия мРНК M4 заметно снижалась после ишемии и восстанавливалась до контрольного уровня после реперфузии в предсердиях, но уровень белка не менялся. Таким образом, увеличение уровня экспрессии M2 мХР и α7 нХР после реперфузии может быть компенсаторной реакцией на ишемически-реперфузионное повреждение миокарда [37].
Важный фактор ишемически-реперфузионного повреждения – клеточный окислительный стресс, один из ключевых медиаторов которого – пероксид водорода (Н2О2) [60]. Образование пероксида водорода при ишемически-реперфузионном повреждении приводит к перегрузке клеток Са2+ из-за усиления перекисного окисления липидов и окисления сульфгидрильных групп белков [61]. Следовательно, любой терапевтический подход, который может улучшить регуляцию и предотвратить перегрузку клеток Са2+, будет полезен для сердца, перенесшего реперфузию. Основываясь на предположении, что АХ может ослаблять изменение гомеостаза внутриклеточного Са2+, вызываемое H2O2 в кардиомиоцитах, было проведено исследование влияния АХ на внутриклеточную регуляцию кальция в изолированных кардиомиоцитах до или после их обработки H2O2 [62]. В миоцитах желудочков сердца крысы H2O2 значительно уменьшал как транзиторный вход внутриклеточного Са2+ в цитозоль, так и скорость его удаления из цитозоля в саркоплазматический ретикулум. Применение АХ до обработки H2O2 (но не после нее) ослабляло снижение транзиторного входа Са2+ и скорости его удаления. И атропин (блокатор мХР), и мекамиламин (блокатор нХР) значительно снижали защитное действие АХ от этого нарушения регуляции внутриклеточного Са2+. Более того, комбинация атропина и мекамиламина полностью устраняла защитное действие АХ. Таким образом, АХ ослабляет изменение гомеостаза внутриклеточного Са2+, индуцированное H2O2, активируя как мускариновые, так и никотиновые холинорецепторы [62].
Другой фактор, вызывающий гибель кардиомиоцитов и необратимые повреждения мышцы сердца, – это кардиотоксичность химиотерапевтических препаратов, в частности антрациклиновых антибиотиков, особенно доксорубицина. Важный патофизиологический признак кардиотоксичности доксорубицина – кардиальная вегетативная нейропатия и связанные с ней митохондриальная и клеточная дисфункции. Для исследования защитного действия лигандов холинорецепторов у крыс вызывали кардиотоксический эффект введением доксорубицина (3 мг/кг в день, 6 доз). Затем в течение 30 дней вводили либо агонист нХР α7 типа PNU-282 987 (3 мг/кг в день), либо агонист мХР бетанехол (12 мг/кг в день), либо оба агониста совместно. Активация α7 нХР способствовала слиянию митохондрий посредством усиления регуляции Mfn1-2 и ослабляла доксирубицин-индуцированную аутофагию, а активация мХР тормозила деление митохондрий и митофагию, что сопровождалось уменьшением митохондриального окислительного повреждения, а также апоптоза и воспаления [63].
Повреждение сердца может вызываться также вирусами. В частности, вирус Коксаки типа В3 инфицирует сердце и вызывает миокардит, сопровождающийся апоптозом кардиомиоцитов. Исследование защитной роли нХР при вирусном миокардите с использованием ПЦР в реальном времени показало, что уровень нХР, содержащего субъединицы α3β4, в кардиомиоцитах новорожденных крыс и клетках H9c2 (кардиомиобластах) возрастает при инфицировании вирусом Коксаки В3 [38]. Никотин при концентрации от 1 нМ до 1 мкМ ослаблял репликацию вируса в клетках H9c2 и кардиомиоцитах новорожденных крыс, однако при более высокой концентрации (10 мкМ) это влияние ослабевало. In vitro блокирование α3β4 нХР его специфическим ингибитором α-конотоксином AuIB отменяло опосредованную никотином защиту кардиомиоцитов от апоптоза, индуцированного вирусом, и это блокирование коррелирует с активацией пути PI3K/Akt и индукцией антиапоптотического белка сурвивина. In vivo у группы инфицированных мышей, получавших AuIB, были ниже выживаемость и хуже систолическая функция желудочков, а также более тяжелое воспаление, чем у группы мышей, получавших только никотин. Таким образом, α3β4 нХР участвуют в опосредованной никотином защите кардиомиоцитов от вирус-индуцированного апоптоза in vivo и in vitro [38].
Низкие дозы никотина способствуют аутофагии кардиомиоцитов новорожденных мышей, ингибируя апоптоз, но в высоких дозах никотин ингибирует аутофагию и способствует апоптозу. Более того, никотин в низких дозах повышал экспрессию гемоксигеназы-1, а нокдаун гена гемоксигеназы-1 отменял эффекты никотина на аутофагию и апоптоз. Селективный блокатор α7 нХР метилликаконитин ингибировал и экспрессию гемоксигеназы-1, и влияние никотина на аутофагию и апоптоз. Эти данные дополнительно подтверждают то, что активация α7 нХР никотином в низких дозах способствует аутофагии и ингибирует апоптоз кардиомиоцитов [64].
Суммарно представленные данные указывают на то, что активация холинорецепторов оказывает кардиопротекторное действие. Сам АХ как лекарственное средство не используется в силу целого ряда причин, включая быстрое расщепление в организме, множественность биологических эффектов и др., но в целом лиганды холинорецепторов могут рассматриваться в качестве защитных средств. Например, холин, взаимодействуя с холинорецепторами, проявляет множественные защитные эффекты против различных сердечно-сосудистых заболеваний, включая ОИМ [65], аритмии [66], сердечную гипертрофию [67, 68] и ишемически-реперфузионное повреждение [69]. Показано, что холин оказывает защитное действие против повреждения сосудов у крыс после ишемически-реперфузионного повреждения [70]. Эти защитные эффекты могут быть связаны с активацией рецептора М3 [57]. Исследование влияния холина на развитие сердечного фиброза показало, что он значительно ингибировал интерстициальный фиброз, и это благотворное действие было нейтрализовано 4‑DAMP, селективным антагонистом М3 мХР [71]. Роль M3 в пролиферации сердечных фибробластов подтверждена путем подавления экспрессии M3 с помощью специфической малой интерферирующей РНК. Уровень белка M3, определенный вестерн-блоттингом, был выше у мышей с сердечным фиброзом. Холин регулирует сердечный фиброз и связанные с ним заболевания сердца, возможно, воздействуя на M3 [71]. Нельзя исключать, что наблюдаемые положительные эффекты холина объясняются увеличением или облегчением синтеза ацетилхолина с последующим возрастанием способности нейронов к массированному (или более длительному) выбросу в синапсы уже ацетилхолина. В принципе, не важно, сам ли холин или его производное ацетилхолин в конечном итоге оказывает описываемые эффекты [70]. Важно, что участие мХР (предположительно М3) в рассматриваемых процессах действительно подтверждено Zhao et al. при помощи его блокатора 4-DAMP [71].
Впрочем, гиперактивация холинорецепторов, в том числе М2, может причинить вред ССС. Кардиопатия, как следствие болезни Шагаса, – наиболее частое осложнение хронической протозойной инфекции, вызываемой простейшим Trypanosoma cruzi. У 30% пациентов, инфицированных T. cruzi, заболевание развивается до состояния, которое может привести к сердечной недостаточности и внезапной смерти. Иммуноглобулины пациентов с хронической формой болезни распознают кислый аминокислотный кластер во второй внеклеточной петле (el2) мХР М2 сердца [72]. Эти остатки соответствуют общему сайту связывания различных аллостерических агентов. Характеристика взаимодействия рецептора с антителами выявила положительный кооперативный эффект между эндогенным лигандом и антителами, приводящий к брадикардии. Аллостерический антагонист М2 галантамин и пептид, соответствующий петле el2, блокировали этот эффект. Таким образом, при хронической кардиомиопатии, вызванной болезнью Шагаса, усиление ответа М2 на АХ за счет аллостерического взаимодействия с антителами может объяснить основные симптомы, о которых сообщается при этом заболевании, такие как синусовая брадикардия и блокада атриовентрикулярной проводимости [72].
Аутокринная ненейрональная холинергическая система сердца
Известно, что миокард желудочков имеет весьма немногочисленную автономную иннервацию, при этом аппликациям АХ присуща однозначно кардиопротекторная направленность и для желудочков сердца. Как стало очевидно в последнее десятилетие, кардиомиоциты, подобно холинергическим нейронам, способны к синтезу и высвобождению АХ, а также к обратному поглощению холина для синтеза АХ. Главный смысл ненейрональной холинергической системы сердца – в паракринном распространении сигнала, поступившего от водителя ритма, от первых кардиомиоцитов к следующим кардиомиоцитам уже без участия нервной ткани, причем с амплификацией и одновременным вовлечением все большего числа соседних кардиомиоцитов. Медиатор этой системы – АХ, а основным рецептором считается мускариновый рецептор М2. Функции системы – распространение потенциала действия, контроль частоты сердечных сокращений, включая циркадный ритм сердца, ограничительное влияние на энергетический метаболизм и гипертрофию миокарда [15]. Интересно, что эта система регулирует ангиогенез коронарных артерий при ОИМ и другие ответы на гипоксию, однако эти эффекты опосредуются опять же через М2, а не через нХР, как в случаях ангиогенеза в других органах.
Кроме АХ, кардиомиоциты способны продуцировать пептид катестатин – неконкурентный блокатор нХР, ингибирующий секрецию катехоламинов в нервной ткани. Защитный эффект катестатина при ишемически-реперфузионном повреждении подобен таковому карбамоилхолина и блокируется атропином и AF-DX116, селективным антагонистом М2 мХР, но не перензепином, антагонистом М1. Более детальные исследования молекулярного механизма показали, что катестатин связывается с рецептором M2 и активирует пути ERK1/2 и PI3 K/Akt, ингибируя таким образом апоптоз клеток, вызванный стрессом эндоплазматического ретикулума. Это приводит к ослаблению ишемически-реперфузионного повреждения. Из данных результатов следует, что за кардиопротекторный эффект катестатина отвечает не блокирование нХР, а активация им М2 мХР [73].
ХОЛИНОРЕЦЕПТОРЫ В КРОВЕНОСНЫХ СОСУДАХ
Следует отметить, что такие кровеносные сосуды, как артерии и вены, состоят из трех слоев, включая внутренний эндотелиальный слой, который играет важную роль в контроле артериального давления, регуляции компонентов свертывания крови и ангиогенезе, и средний слой, содержащий гладкие мышцы, которые контролируют диаметр сосуда. Клетки обоих слоев могут содержать холинорецепторы, регулирующие функциональную активность кровеносных сосудов [74, 75]. Проведенные исследования показали, что релаксация сосудов, вызванная АХ, полностью зависит от интактного эндотелия и опосредована релаксирующими факторами эндотелиального происхождения, включая оксид азота.
Мускариновые холинорецепторы в кровеносных сосудах
Агонисты мХР могут вызывать как сокращение, так и расслабление стенки кровеносного сосуда. Подтипы мХР и результирующий эффект различаются в зависимости от анатомического расположения конкретного кровеносного сосуда, от того, повреждена ли эндотелиальная оболочка кровеносного сосуда, и даже от вида животного [76]. В общем виде М3 и/или М5 клеток эндотелия сосудов посредством белка Gq активируют фосфолипазу С, что приводит к синтезу инозитолтрифосфата и диацилглицерола с активацией протеинкиназы C и последующим высвобождением внутриклеточного кальция, который активирует эпителиальную NO-синтазу eNOS, конститутивно экспрессируемую в эндотелиальных клетках. Сосудистые эндотелиальные клетки могут вызывать вазодилатацию, высвобождая множество факторов релаксации, но основной эффект стимуляции мускариновых рецепторов в коронарных сосудах человека – вазодилатация за счет высвобождения оксида азота (NO) из функционального эндотелия [77]. Затем это мощное сосудорасширяющее соединение диффундирует в гладкомышечное волокно, где стимулирует синтез cGMP растворимой гуанилатциклазой, который через активацию протеинкиназы G запускает процесс вазорелаксации.
Однако у человека и некоторых других видов активация мХР также может вызывать вазоконстрикцию за счет активации рецепторов, присутствующих на гладких мышцах кровеносных сосудов (в частности, легочной артерии) [78]. В общем виде стимуляция М1 и/или М3 гладкомышечных клеток приводит аналогичным путем к высвобождению уже их собственного внутриклеточного кальция и активации кальмодулин-зависимой киназы, которая фосфорилирует киназу легкой цепи миозина, что в итоге приводит к мышечному сокращению. В отсутствие функционального эндотелия эффект холинергической стимуляции гладкой мускулатуры становится незамаскированным, и в результате может наблюдаться вазоконстрикция [79].
Подтип М1. Рецептор М1 широко распространен по всей сосудистой системе, обнаруживается в эндотелии, артериях и легочных венах [80, 81]. Активация М1 в сосудистой сети способствует индукции вазодилатации. Вазодилатация зависит от интактного эндотелия из-за необходимой продукции NO. NO индуцирует вазодилатацию путем активации растворимой гуанилатциклазы, которая диффундирует в гладкомышечные клетки из эндотелия, что приводит к превращению GTP в cGMP. Затем cGMP активирует cGMP-зависимую протеинкиназу G, что приводит к удалению цитозольного Ca2+, ингибированию сократительного аппарата и стимулированию вазодилатации.
Характеристика подтипов мХР, вызывающих АХ-индуцированную эндотелий-независимую вазодилатацию в брыжеечных артериях крыс, показала, что в артериях с интактным эндотелием уровень экспрессии М1 и М3 был значительно выше, чем М2 [81]. Удаление эндотелия значительно снижало уровни экспрессии М2 и М3, но не М1.
В отличие от М3, М1 может вызывать дилатацию сосудов, не связанную с наличием эпителия. В перфузируемых мезентериальных сосудистых руслах с интактным эндотелием и активным тонусом экзогенный АХ (1, 10 и 100 нМ) вызывал концентрационно-зависимую и длительную вазодилатацию. В препаратах без эндотелия АХ в концентрации 1 нМ к релаксации не приводил, но АХ при 10 и 100 нМ вызывал длительную вазодилатацию, которая заметно блокировалась при обработке пирензепином (антагонист М1) или 4-DAMP (антагонист М1 и М3) совместно с гексаметонием (антагонист нХР), но не метоктрамином (антагонист М2 и М4). Эти результаты свидетельствуют о том, что в брыжеечных артериях крысы присутствует в основном мХР подтипа M1, и его (и/или M3-подтипа) активация вызывает эндотелий-независимую вазодилатацию [81].
Однако М1 также играет определенную роль в вазоконстрикции. В частности, активация рецепторов M1 в гладкомышечных клетках сосудов может вызывать вазоконстрикцию в отсутствие эндотелия [17]. Этот подтип связан с вазоконстрикцией мозговых артерий кошек [82], подкожных вен собак [83] и церебральных артериол мышей [84]. Фармакологическая характеристика подтипов мХР, опосредующих сокращение пупочной вены человека, выявила участие рецепторов М1 в данном процессе. И в этом случае вазомоторная активность, вызванная АХ, по-видимому, не модулируется эндотелиальными факторами [85].
Разработка специфических агонистов для рецепторов М1 может быть полезной для противодействия любому повышению артериального давления в малом круге кровообращения [74]. Интересно, что в этом круге имеется также и рецептор М3, а агонист М3 С1213 эффективен при легочной гипертензии [86]. Следует отметить еще один интересный факт о роли М1. На изолированных перфузируемых легких кроликов при моделировании тромбоэмболии легочной артерии в условиях инфузии ацетилхолина показано, что блокатор М1 пирензепин не дает в этих условиях сильно повышаться давлению в легочной артерии [87].
АХ вызывает заложенность носа за счет вазодилатации интраназальных задних собирательных вен в сочетании с вазоконтракцией экстраназальных вен оттока (дорсальная носовая вена и клиновидно-небная вена). Характеристика подтипов мХР, участвующих в индуцированном АХ расслаблении и сокращении носовых вен собак, показала, что подтипы М1 и М3 локализованы в гладкой мускулатуре вен обоих типов. Это означает, что АХ расслабляет интраназальные вены и сужает экстраназальные вены в первую очередь за счет мХР М1 и М3, что указывает на терапевтическую ценность М1/М3-специфических или высокоселективных антихолинергических средств при заложенности носа [88].
Подтип М2. Хотя мХР подтипа М2 в основном связан с сердцем, он также обнаруживается по всей сосудистой сети, особенно в эндотелии коронарных сосудов. Физиологический эффект, связанный с активацией рецепторов этого подтипа, заключается в расширении сосудов [16]. Сосудорасширяющий эффект проявляется за счет высвобождения NO. Использование карбахола или пирензепина позволяет предположить, что М1 и М2 могут быть обнаружены в правой коронарной артерии человека, что также было подтверждено авторадиографическими исследованиями сердца человека, хотя эти результаты предполагают, что М2 в основном экспрессируется в наружной оболочке коронарной артерии [16]. Методом ОТ-ПЦР гены рецепторов М2 и М3 были обнаружены в коронарных артериях человека [89]. Ни М1, ни М4 в исследованных образцах не экспрессировались.
Подтип М3. Рецепторы М3 также обнаружены в сосудистой системе, как в слое эндотелиальных клеток, так и в гладкомышечных клетках сосудов. Ген, кодирующий рецепторы М3, обнаружен в коронарных артериях человека [89]. Эти рецепторы, по-видимому, играют доминирующую роль в АХ-индуцированной вазодилатации большинства кровеносных сосудов [90]. М3 опосредует вазодилатацию в бедренной артерии и грудной аорте только за счет продукции оксида азота эпителием сосудов [90]. Однако ответ на АХ зависит от функциональной целостности эндотелия. В сосудистой системе активация рецепторов M3 и M5 в эпителиальных клетках вызывает вазорелаксацию, в то время как активация рецепторов M3 или M1 в гладкомышечных клетках сосудов в отсутствие эндотелия может вызывать вазоконстрикцию [17]. Так, у кошек активация М3 при интактном эндотелии ведет к вазодилатации, тогда как активация гладкой мускулатуры в отсутствие эндотелия вызывает вазоконстрикцию [91]. При патологиях, при которых эндотелиальный слой повреждается или удаляется, активация М3 на гладкомышечных клетках вызывает вазоконстрикцию [92]. АХ-индуцированная вазоконстрикция, опосредованная стимуляцией подтипа М3 рецепторов при удалении или разрушении эндотелиального слоя, описана для легочной артерии человека [80], перфорирующей ветви внутренней грудной артерии человека [79], коронарной артерии крупного рогатого скота [93] и коронарных артерий свиней [94]. Не исключается участие рецепторов М3 (вместе с М1) и в индуцированной АХ вазоконстрикции в кольцах пупочной вены человека [85].
Исследование мХР М3 в глазных артериях мыши с помощью ПЦР в реальном времени показало, что в артериях с интактным эндотелием имеется мРНК, кодирующая все пять подтипов мХР, но с наибольшим содержанием мРНК рецептора М3. В артериях с удаленным эндотелием рецепторы М1, М2 и М3 демонстрировали сходные уровни экспрессии мРНК, которые были выше, чем у рецепторов М4 и М5. АХ вызывал сужение сосудов в артериях с удаленным эндотелием, которое практически исчезало после инкубации с атропином, блокирующим мХР. Данные, полученные с использованием мышей, дефицитных по М3 (M3R–/–), свидетельствуют о том, что в глазных артериях мыши М3 опосредует холинергическую эндотелий-зависимую вазодилатацию и эндотелий-независимую вазоконстрикцию [92].
Рецепторы М3 локализованы в гладкой мускулатуре интраназальных задних собирательных вен и экстраназальных вен оттока (дорсальная носовая вена и клиновидно-небная вена), а также в эндотелии интраназальных вен носа собаки и, возможно, наряду с М1 и М2, участвуют в индуцированном АХ расслаблении интраназальных и сокращении экстраназальных вен [88].
Подтипы М4 и М5. Информация о наличии мХР подтипов М4 и М5 в кровеносных сосудах весьма ограничена. Так, использование селективного блокатора мХР М4-подтипа холинорецепторов тропикамида в опытах на крысах показало, что в зависимости от дозы у животных наблюдался целый ряд сосудистых эффектов. В частности, ингибирование М4 тропикамидом в дозах 1–100 мкг/кг (не превышающих порог селективности препарата) вызывает транзиторное и дозозависимое снижение артериального давления и общего периферического сосудистого сопротивления [34]. Интересно, что хотя сосудорасширяющий эффект тропикамида не органоспецифичный, он ярко проявляется только в отношении исследованных артерий; при этом скорость органного портального кровотока у большинства животных не изменяется, а у 25% крыс она временно незначительно возрастает [95]. На основании полученных данных авторы предполагают возможное участие М4 в ацетилхолин-индуцированной вазоконстрикции.
Что касается мХР подтипа М5, то сообщается, что этот рецептор опосредует холинергические реакции в церебральных кровеносных сосудах (данные получены с использованием нокаутных мышей M5R–/–) [96]. Воздействуя на М5 эндотелия сосудов, ацетилхолин дозозависимо вызывает повышение концентрации внутриклеточного Ca2+ в эндотелии через его резкий выброс из внутриклеточных депо с последующим поддержанием за счет входа внеклеточного Ca2+. Путь включает активацию фосфолипазы Сβ и синтез инозитолтрифосфата InsP3R3, что в конечном итоге приводит к выработке оксида азота и к эндотелий-зависимой вазодилатации [97]. Интересно, что результаты исследований с использованием мышей, нокаутных по М1, М3 и М5, показали, что за пределами церебральных сосудов М5, по-видимому, не играет значительной роли в вазодилатации [98].
Никотиновые холинорецепторы в кровеносных сосудах
Следует отметить, что в кровеносных сосудах обнаружены практически все субъединицы нХР. Так, экспрессия нХР α7-типа в кровеносных сосудах была первоначально описана в эндотелиальных клетках бычьей аорты [99]. Вскоре после этого α7 нХР были аналогичным образом идентифицированы в эндотелиальных клетках человека из микроциркуляторного русла и вен пуповины, где они вносят вклад в ангиогенный ответ на гипоксию и ишемию [100].
Распределение различных α-субъединиц нХР в артериальной системе крыс исследовали in situ с помощью ОТ-ПЦР и иммуногистохимии [101, 102]. Было обнаружено, что в эндотелиальных клетках экспрессируются субъединицы α3, α5, α7 и α10, что позволяет предположить формирование каналов, предпочтительно проницаемых как для кальция (гомопентамерные α7 нХР), так и для моновалентных катионов (гетеропентамеры, содержащие α3- и α5-субъединицы). Все подтипы α-субъединиц, кроме α9, экспрессировались гладкомышечными клетками сосудов с высокоспецифичным паттерном распределения по сосудистому дереву, в то время как каждая из субъединиц, кроме α9, была обнаружена в грудной аорте, внутрилегочные артериальные ветви содержали только иммунореактивную α7-субъединицу, а другие сосудистые русла занимали промежуточное положение по степени разнообразия экспрессируемых α-субъединиц [101].
Экспрессия α7 нХР также установлена в клетках гладкой мускулатуры аорты крыс [103], базилярной артерии морской свинки [104], церебральных артерий человека [105] и артерий пуповины [106]. Наличие субъединиц α3, α4, α5, α7 и α10 в гладкомышечных клетках показано в целом ряде работ (например, [99, 107]). Интересно, что α4-субъединица была обнаружена в брюшной аорте и не обнаруживается в восходящей аорте, легочном стволе, мышечных, почечных или и легочных артериях [101, 107]. α7-нХР экспрессируется на гладкомышечных клетках большинства сосудов, за исключением сосудов почек, и внутрилегочных артериях [107, 108]. В дополнение к указанным выше еще несколько работ показывают, что субъединицы α3-, α5-, α7-, α10- и β2–β4 также обнаруживаются в эндотелиальных клетках сосудов человека [99, 109, 110]. Кроме того, иммуногистохимическое окрашивание с использованием антител, специфичных к определенным субъединицам нХР, показало, что субъединицы α3, α4, α7, β2 и β4 экспрессируются эндотелиальными клетками пупочной вены человека [100]. Также было обнаружено, что эндотелиальные клетки коронарных микрососудов крысы экспрессировали мРНК субъединиц α2, α3, α4, α5 и α7, а также β2 и β4 нХР, в то время как мРНК β3‑субъединицы не была обнаружена [111].
Методом позитронно-эмиссионной томографии in vivo с использованием радиоактивно меченого 2-18F-фтор-3-[2(S)-2-азетидинилметокси]пиридина ([18F]-2-фтор-A85380), обладающего высоким сродством к нХР, содержащим β2-субъединицу, проведена визуализация артериальных нХР человека [112]. Наличие [18F]-2-фтор-A85380 количественно обнаруживалось в восходящей и нисходящей аорте, дуге аорты и сонных артериях. Было продемонстрировано специфическое поглощение индикатора стенками артерий. Значительно более высокие значения поглощения были обнаружены в нисходящей аорте [112].
Из приведенных данных следует, что в сосудистой системе нХР представлены существенно шире, чем мХР, но, в отличие от последних, они мало связаны с регуляцией сосудистого тонуса, хотя такие сведения также имеются.
В частности, при ингибировании мХР атропином нХР опосредуют вызванное АХ расслабление аорты крыс [113]. Как уже отмечалось, АХ вызывает эндотелий-зависимую релаксацию в аорте, и мХР играют существенную роль в этом процессе. Однако никотин индуцировал эндотелий-зависимую релаксацию как у гипертензивных, так и у нормотензивных крыс посредством активации α7 нХР. Таким образом, активация нХР может способствовать индуцированной АХ эндотелий-зависимой релаксации в аорте, т.е. эти рецепторы участвуют в эндотелий-зависимой регуляции сосудистого тонуса [113].
Следовательно, нХР играют значительную роль в осуществлении контроля нескольких важнейших для ССС физиологических и патофизиологических процессов.
Хорошо известна модулирующая роль эндотелиального α7 нХР как в физиологическом, так и в патологическом ангиогенезе [114, 115]. Проведенные исследования показали, что активация эндотелиального α7 нХР – существенна для процессов пролиферации, миграции, старения и выживания клеток эндотелия [100, 104, 115–117]. В эндотелии сосудов специфические ионотропные свойства α7 нХР (высокая проницаемость для Ca2+) и последующие Ca2+-опосредованные внутриклеточные каскады могут играть важную роль в физиологии (ангиогенез) и патологии (воспаление и атерогенез). Механизмы, лежащие в основе этих эффектов, включают повышение внутриклеточной концентрации Ca2+, активацию митоген-активируемой протеинкиназы, фосфатидилинозитол-3-киназы, эндотелиальной NOS и NF-κB, повышение активности сиртуина 1 и активацию циклина [100, 104, 115, 116, 118].
С использованием комбинации фармакологических и биохимических методов, а также флуоресцентной микроскопии показано, что α7 нХР в клетках эндотелия артерий крыс и эндотелия вен человека обнаруживаются при чрезвычайно низких уровнях экспрессии (∼50 фмоль/мг белка), однако никотин в концентрации 50 мкМ увеличивает экспрессию рецептора в ∼300 раз с одновременным увеличением его экспонированности на поверхности клетки, и этот эффект зависит от липидного состава мембраны клеток [119]. Воздействие никотина заметно стимулировало миграцию клеток и ускоряло заживление ран, этот процесс замедлялся в клетках, лишенных стерола. Ангиогенный эффект никотина усиливался при увеличении содержания холестерина в мембране.
Установлено, что нХР вовлечены в развитие ряда патологий и заболеваний ССС. Курение представляет собой один из наиболее важных факторов риска развития сердечно-сосудистых заболеваний, и никотин играет в этих процессах существенную роль. Никотин вызывает дисфункцию сосудов, изменяя вазореактивность посредством эндотелий-зависимых и/или эндотелий-независимых механизмов и приводя к клиническим проявлениям. Помимо этого, никотин индуцирует ремоделирование сосудов за счет своего влияния на пролиферацию, миграцию и продукцию матрикса как эндотелия сосудов, так и клеток гладкой мускулатуры сосудов [120]. Хотя начальные проявления сосудистой дисфункции, вызванной никотином, могут быть обнаружены не сразу, эти изменения вносят вклад в патогенез серьезных заболеваний, включая атеросклероз, аневризму брюшной аорты, ишемическую болезнь сердца и ОИМ [121–123].
Накопленные данные указывают на то, что вредное воздействие никотина в составе табачного дыма на артериальную стенку может быть опосредовано именно нХР [100, 108–111, 124]. Эти опосредующие эффекты нХР связаны с контролем пролиферации гладкомышечных клеток и ангиогенеза, пролиферации эндотелиальных клеток, среди прочего, путем индукции экспрессии эндотелиальных факторов роста и повышения уровня эндотелиальной NOS в эндотелиальных клетках. Ранее уже обсуждалось, что как эндотелиальные, так и гладкомышечные клетки сосудов экспрессируют множественные α- и β-субъединицы нХР [102, 111, 115, 125, 126], что делает сосудистую сеть прямой мишенью для никотина.
В частности, обнаружена существенная роль α7 нХР в развитии сосудистых патологий. Изменение активности и распределения в клетках этого типа нХР связаны с ангиогенезом (миграция ранозаживляющих клеток) и атерогенезом (изменение содержания холестерина) в эндотелиальных клетках. В гладкомышечных клетках сосудов активация α7 нХР связана с модуляцией миграции, подавлением окислительного стресса, ингибированием гиперплазии неоинтимы, аневризмы брюшной аорты и ремоделированием цитоскелета [127]. Интересно, что неоваскуляризация, миграция/пролиферация гладкомышечных клеток сосудов, ремоделирование сосудов и окислительный стресс способствуют возникновению и прогрессированию атеросклеротических бляшек [128].
Атеросклероз – медленно прогрессирующее хроническое воспалительное заболевание артерий большого и среднего калибра; он включает сложное рекрутирование иммунных клеток, накопление липидов и структурное ремоделирование сосудов. В нескольких типах клеток, участвующих в генезе и прогрессировании атеросклероза, включая макрофаги, дендритные клетки, Т- и В-клетки, эндотелиальные клетки сосудов и гладкомышечные клетки, обнаружена экспрессия нХР α7-типа. Кроме того, α7 нХР – важный регулятор воспаления, поскольку этот рецептор опосредует ингибирование синтеза провоспалительных цитокинов, что приводит к ослаблению атеросклеротического процесса. С другой стороны, активация α7 нХР способствует ангиогенезу и пролиферации гладкомышечных клеток сосудов, что может усиливать атеросклеротические изменения. Вследствие всех этих эффектов α7 нХР – один из ключевых элементов сложной патофизиологии атеросклероза и может представлять многообещающую мишень для лечения воспаления сосудов и атеросклероза [127].
Показано, что нарушение функции α7 нХР с помощью фармакологических антагонистов (например, α-бунгаротоксина или метиликаконитина) или с использованием методологии siРНК ослабляет ангиогенез в экспериментальных моделях ишемического повреждения [114, 124]. Таким образом, агенты, ингибирующие α7 нХР, могут быть полезны для подавления ангиогенеза бляшек при атеросклерозе [129].
Интересно, что нХР, содержащий α1-субъединицу, так называемый мышечный нХР, также может быть вовлечен в патогенез атеросклероза [6, 43]. Так, введение плазмидной ДНК, экспрессирующей siRNA для α1-субъединицы, мышам с дефицитом аполипопротеина Е (ApoE–/–), получавшим атеросклеротическую диету с высоким содержанием жиров, приводило к снижению уровней α1 нХР, значительному уменьшению площади атеросклеротического поражения и других проявлений атеросклероза [130, 131]. Следовательно, продукт гена α1-субъединицы нХР в стенке артерии обладает некоторым функционалом, и снижение его экспрессии замедляет развитие атеросклеротической бляшки.
Таким образом, нХР играют важную роль в процессах, способствующих развитию бляшек и включающих миграцию и пролиферацию гладкомышечных клеток, а также неоваскуляризацию бляшек [132]. Ранее уже обсуждалось использование меченного радиоактивным изотопом лиганда нХР для обнаружения нХР в ткани сонных артерий, дуги аорты и нисходящей аорты [112], а также сердца у человека in vivo [39]. Вполне возможно, что такие агенты могут быть использованы для визуализации атеросклеротических бляшек у пациентов.
Следовательно, никотиновый холинергический путь регулирует несколько компонентов атерогенеза, включая воспаление, фенотип гладкомышечных клеток, их пролиферацию и миграцию, а также неоваскуляризацию бляшек. Все это говорит о возможном использовании лигандов нХР в терапии атеросклероза.
ЗАКЛЮЧЕНИЕ
Холинорецепторы, как метаботропные мускариновые, так и ионотропные никотиновые, участвуют в важнейших физиологических и патофизиологических процессах, протекающих в сердечно-сосудистой системе (табл. 1). При этом разные подтипы холинорецепторов различным образом вовлечены в эти процессы. Для более точного описания таких процессов требуется намного больше информации о структуре и функционировании данных рецепторов. Такая информация может быть получена с применением лигандов, селективных к определенным подтипам холинорецепторов, в этом плане более удобны селективные блокаторы, “выключающие” тот или иной подтип рецептора. Поиск таких соединений – важная фундаментальная задача биоорганической химии.
Таблица 1.
Участие холинорецепторов в регуляции сердечно-сосудистой системы*
| Мускариновый холинорецептор. | Никотиновый холинорецептор. | |||||||
|---|---|---|---|---|---|---|---|---|
| Метаботропный рецептор, сопряженный с G-белком | Ионотропный рецептор – катионный канал | |||||||
| М1 | М2 | М3 | М4 | М5 | α1β1γδ | α3β4 | α7** | |
| Принципиальные локации в ССС | Миокард (желудочки), эндотелий и мышечная стенка сосудов |
Предсердия, проводящая система сердца, эндотелий коронарных сосудов | Миокард, водитель ритма, эндотелий и мышечная стенка сосудов | Предсердия, сосуды | Эндотелий церебральных сосудов | – | Кардиомиоциты | Проводящая система сердца, фибробласты, кардиомиоциты |
| Регулируемые процессы | Сократительная способность сердца | – | – | – | – | Холинергический противовоспалительный путь, ангиогенез, апоптоз | ||
| – | Реперфузия после ишемии миокарда | |||||||
| Паракринное распространение возбуждения, ангиогенез коронарных артерий | – | |||||||
| Тонус кровеносных сосудов | ||||||||
| Влияние на сердце | Увеличение частоты и силы сокращений сердца | Замедление ритма | Замедление синусового ритма и укорочение потенциалов действия | Брадикардия (возможно) | – | – | Защита кардиомиоцитов от вирус-индуцированного апоптоза | Кардиопротекция, ингибирование апоптоза кардиомиоцитов |
| Кардиопротекция | ||||||||
| Влияние на сосуды | Эндотелий-зависимая и независимая вазодилатация, вазоконстрикция некоторых сосудов | Эндотелий-зависимая вазодилатация | Эндотелий-зависимая вазодилатация, эндотелий-независимая вазоконстрикция | Вазоконстрикция | Вазодилатация | Развитие атеросклеротических бляшек | – | Пролиферация и выживание эндотелиальных и мышечных клеток сосудов, эндотелий-зависимая релаксация аорты, неоваскуляризация атеросклеротических бляшек |
| Общие агонисты*** | Ацетилхолин, карбахол, бетанехол | Ацетилхолин, никотин (кроме α9), холин | ||||||
| Селективные агонисты*** | – | Катестатин | С1213, холин | – | – | – | – | PNU-282987, GTS21 |
| Общие антагонисты*** | Мускарин | Катестатин, мекамиламин | ||||||
| Гексаметоний | ||||||||
| Селективные антагонисты*** | Пирензепин, 4-DAMP (менее предпочтительно) | Галантамин****, AF-DX116, метоктрамин |
4-DAMP (более предпочтительно) | Тропикамид, метоктрамин | – | α-Бунгаротоксин, тубокурарин | α-Конотоксин AuIB | Метиликаконитин, α-бунгаротоксин |
* Табл. 1 отражает не все сведения об ацетилхолиновых рецепторах; она суммирует данные, которые связаны с участием ацетилхолиновых рецепторов в регуляции ССС и обсуждаются в данном обзоре. ** В тканях сердца и сосудов описана экспрессия ряда других подтипов нХР (или их субъединиц), но их роль в регуляции ССС не установлена. *** В качестве примера приведены те соединения, которые обсуждаются в тексте, или наиболее широко известные препараты. **** Антихолинэстеразное вещество, одновременно выступающее аллостерическим ингибитором М2-подтипа мХР.
Для борьбы с некоторыми заболеваниями ССС, например, с аритмиями, медицина уже использует ряд лигандов холинорецепторов. Приведенные в обзоре результаты многочисленных экспериментов показывают, что положительное влияние на здоровье достигается чаще активацией рецепторов при соответствующих состояниях. Следовательно, поиск таких соединений, прежде всего активаторов (холиномиметиков), селективных к конкретным подтипам холинорецепторов, имеет уже и прикладное значение, поскольку будет способствовать решению ряда важных медицинских проблем. Среди них – лечение и профилактика аритмий, нарушений сосудистого тонуса, атеросклероза, миокардитов и других связанных с воспалением ССС нозологий.
Список литературы
Орлов Р.С., Ноздрачев А.Д. // Нормальная физиология. Глава 23. Сердечно-сосудистая система. Москва: ГЭОТАР-Медиа, 2009. С. 472–526.
Kostenis E., Zeng F.Y., Wess J. // Life Sci. 1999. V. 64. P. 355–362. https://doi.org/10.1016/s0024-3205(98)00574-8
Leach K., Simms J., Sexton P.M., Christopoulos A. // Handb. Exp. Pharmacol. 2012. V. 208. P. 29–48. https://doi.org/10.1007/978-3-642-23274-9_2
Hulme E.C., Birdsall N.J., Buckley N.J. // Annu. Rev. Pharmacol. Toxicol. 1990. V. 30. P. 633–673. https://doi.org/10.1146/annurev.pa.30.040190.003221
Haga K., Kruse A.C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W.I., Okada T., Kobilka B.K., Haga T., Kobayashi T. // Nature. 2012. V. 482. P. 547–551. https://doi.org/10.1038/nature10753
Maeda S., Qu Q., Robertson M.J., Skiniotis G., Kobilka B.K. // Science. 2019. V. 364. P. 552–557. https://doi.org/10.1126/science.aaw5188
Thompson A.J., Lester H.A., Lummis S.C. // Q. Rev. Biophys. 2010. V. 43. P. 449–499. https://doi.org/10.1017/S0033583510000168
Nys M., Kesters D., Ulens C. // Biochem. Pharmacol. 2013. V. 86. P. 1042–1053. https://doi.org/10.1016/j.bcp.2013.07.001
Noviello C.M., Gharpure A., Mukhtasimova N., Cabuco R., Baxter L., Borek D., Sine S.M., Hibbs R.E. // Cell. 2021. V. 184. P. 2121–2134. https://doi.org/10.1016/j.cell.2021.02.049
Oakes J.M., Fuchs R.M., Gardner J.D., Lazartigues E., Yue X. // Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018. V. 315. P. R895–R906. https://doi.org/10.1152/ajpregu.00099.2018
Jutkiewicz E.M., Rice K.C., Carroll F.I., Woods J.H. // Drug Alcohol Depend. 2013. V. 131. P. 284–297. https://doi.org/10.1016/j.drugalcdep.2012.12.021
Casado M.A., Marín J., Salaices M. // Naunyn Schmiedebergs Arch. Pharmacol. 1992. V. 346. P. 391–394. https://doi.org/10.1007/BF00171079
Alonso M.J., Arribas S., Marín J., Balfagón G., Salaices M. // Brain Res. 1991. V. 567. P. 76–82. https://doi.org/10.1016/0006-8993(91)91438-7
Deng A.Y., Huot-Marchard J.É., deBlois D., Thorin E., Chauvet C., Menard A. // Can. J. Cardiol. 2019. V. 35. P. 661–670. https://doi.org/10.1016/j.cjca.2018.12.029
Saw E.L., Kakinuma Y., Fronius M., Katare R. // J. Mol. Cell Cardiol. 2018. V. 125. P. 129–139. https://doi.org/10.1016/j.yjmcc.2018.10.013
Saternos H.C., Almarghalani D.A., Gibson H.M., Meqdad M.A., Antypas R.B., Lingireddy A., AbouAlaiwi W.A. // Physiol. Genomics. 2018. V. 50. P. 1–9. https://doi.org/10.1152/physiolgenomics.00062.2017
Harvey R.D. // In: Muscarinic Receptors. Handbook of Experimental Pharmacology. Berlin: Springer, 2012. V. 208. P. 299–316. https://doi.org/10.1007/978-3-642-23274-9_13
Brodde O.E., Michel M.C. // Pharmacol. Rev. 1999. V. 51. P. 651–690.
Harvey R.D., Belevych A.E. // Br. J. Pharmacol. 2003. V. 139. P. 1074–1084. https://doi.org/10.1038/sj.bjp.0705338
Sterin-Borda L., Echagüe A.V., Leiros C.P., Genaro A., Borda E. // Br. J. Pharmacol. 1995. V. 115. P. 1525–1531. https://doi.org/10.1111/j.1476-5381.1995.tb16646.x
Wang Y.G., Rechenmacher C.E., Lipsius S.L. // J. Gen. Physiol. 1998. V. 111. P. 113–125. https://doi.org/10.1085/jgp.111.1.113
Navarro-Polanco R.A., Moreno Galindo E.G., Ferrer-Villada T., Arias M., Rigby J.R., Sanchez-Chapula J.A., Tristani-Firouzi M. // J. Physiol. 2011. V. 589. P. 1741–1753. https://doi.org/10.1113/jphysiol.2010.204107
Moss R., Sachse F.B., Moreno-Galindo E.G., Navarro-Polanco R.A., Tristani-Firouzi M., Seemann G. // PLoS Comput. Biol. 2018. V. 14. P. e1006438. https://doi.org/10.1371/journal.pcbi.1006438
Wang H., Lu Y., Wang Z. // Auton. Autacoid Pharmacol. 2007. V. 27. P. 1–11. https://doi.org/10.1111/j.1474-8673.2006.00381.x
Patanè S. // Int. J. Cardiol. 2014. V. 177. P. 646–649. https://doi.org/10.1016/j.ijcard.2014.09.178
Wang H., Han H., Zhang L., Shi H., Schram G., Nattel S., Wang Z. // Mol. Pharmacol. 2001. V. 59. P. 1029–1036. https://doi.org/10.1124/mol.59.5.1029
Wang Z., Shi H., Wang H. // Br. J. Pharmacol. 2004. V. 142. P. 395–408. https://doi.org/10.1038/sj.bjp.0705787
Lymperopoulos A., Cora N., Maning J., Brill A.R., Sizova A. // FEBS J. 2021. V. 288. P. 2645–2659. https://doi.org/10.1111/febs.15771
Abramochkin D.V., Tapilina S.V., Sukhova G.S., Nikolsky E.E., Nurullin L.F. // Pflugers Arch. 2012. V. 463. P. 523–529. https://doi.org/10.1007/s00424-012-1075-1
Pérez C.C.N., Tobar I.D.B., Jiménez E., Castañeda D., Rivero M.B., Concepción J.L., Chiurillo M.A., Bonfante-Cabarcas R. // Pharmacol. Res. 2006. V. 54. P. 345–355. https://doi.org/10.1016/j.phrs.2006.07.001
Heijman J., Kirchner D., Kunze F., Chrétien E.M., Michel-Reher M.B., Voigt N., Knaut M., Michel M.C., Ravens U., Dobrev D. // Int. J. Cardiol. 2018. V. 255. P. 61–68. https://doi.org/10.1016/j.ijcard.2017.12.050
Poller U., Nedelka G., Radke J., Pönicke K., Brodde O.E. // J. Am. Coll. Cardiol. 1997. V. 29. P. 187–193. https://doi.org/10.1016/s0735-1097(96)00437-8
Shi H., Wang H., Wang Z. // Mol. Pharmacol. 1999. V. 55. P. 497–507.
Коваленко Н.Я., Мациевский Д.Д., Решетняк В.К. // Патологическая физиология и экспериментальная терапия. 2013. Т. 57. № 3. С. 23–26.
Krejcí A., Tucek S. // Mol. Pharmacol. 2002. V. 61. P. 1267–1272. https://doi.org/10.1124/mol.61.6.1267
Dvorakova M., Lips K.S., Brüggmann D., Slavikova J., Kuncova J., Kummer W. // Cell Tissue Res. 2005. V. 319. P. 201–209. https://doi.org/10.1007/s00441-004-1008-1
Li D.L., Liu B.H., Sun L., Zhao M., He X., Yu X.J., Zang W.J. // Clin. Exp. Pharmacol. Physiol. 2010. V. 37. P. 1114–1119. https://doi.org/10.1111/j.1440-1681.2010.05448.x
Li P., Yan Y., Shi Y., Cheng B., Zhan Y., Wang Q., Ye Q., Weng Y., Wu T., Wu R. // Oxid. Med. Cell Longev. 2019. V. 2019. P. e.9496419. https://doi.org/10.1155/2019/9496419
Bucerius J., Joe A.Y., Schmaljohann J., Gündisch D., Minnerop M., Biersack H.J., Wüllner U., Reinhardt M.J. // Clin. Res. Cardiol. 2006. V. 95. P. 105–109. https://doi.org/10.1007/s00392-006-0342-6
Brasch H., Iven H.B., Zetler G. // Naunyn-Schmiedeberg’s Arch. Pharmacol. 1977. V. 299. P. 259–265. https://doi.org/10.1007/BF00500318
Fenton R.A., Dobson J.G. // Am. J. Physiol. 1985. V. 49. P. H463–H469. https://doi.org/10.1152/ajpheart.1985.249.3.H463
Nakatani T., Nakashima T., Satoh H. // Gen. Pharmacol. 1994. V. 25. P. 865–873. https://doi.org/10.1016/0306-3623(94)90088-4
Malińska D., Więckowski M.R., Michalska B., Drabik K., Prill M., Patalas-Krawczyk P., Walczak J., Szymański J., Mathis C., Van der Toorn M., Luettich K., Hoeng J., Peitsch M.C., Duszyński J., Szczepanowska J. // J. Bioenerg. Biomembr. 2019. V. 51. P. 259–276. https://doi.org/10.1007/s10863-019-09800-z
Katare R.G., Ando M., Kakinuma Y., Arikawa M., Handa T., Yamasaki F., Sato T. // J. Thorac. Cardiovasc. Surg. 2009. V. 137. P. 223–231. https://doi.org/10.1016/j.jtcvs.2008.08.020
Calvillo L., Vanoli E., Andreoli E., Besana A., Omodeo E., Gnecchi M., Zerbi P., Vago G., Busca G., Schwartz P.J. // J. Cardiovasc. Pharmacol. 2011. V. 58. P. 500–507. https://doi.org/10.1097/FJC.0b013e31822b7204
Li M., Zheng C., Sato T., Kawada T., Sugimachi M., Sunagawa K. // Circulation. 2004. V. 109. P. 120–124. https://doi.org/10.1161/01.CIR.0000105721.71640.DA
Sun J., Lu Y., Huang Y., Wugeti N. // Int. J. Clin. Exp. Med. 2015. V. 8. P. 9334–9340.
Shinlapawittayatorn K., Chinda K., Palee S., Surinkaew S., Thunsiri K., Weerateerangkul P., Chattipakorn S., Ken-Knight B.H., Chattipakorn N.N. // Heart Rhythm. 2013. V. 10. P. 1700–1707. https://doi.org/10.1016/j.hrthm.2013.08.009
Zhao M., He X., Bi X.Y., Yu X.J., Gil Wier W., Zang W.J. // Basic Res. Cardiol. 2013. V. 108. P. 345. https://doi.org/10.1007/s00395-013-0345-1
Xue R.Q., Sun L., Yu X.J., Li D.L., Zang W.J. // J. Cell Mol. Med. 2017. V. 21. P. 58–71. https://doi.org/10.1111/jcmm.12938
Intachai K., Chattipakorn S.C., Chattipakorn N., Shinlapawittayatorn K. // Int. J. Mol. Sci. 2018. V. 19. P. 2466. https://doi.org/10.3390/ijms19092466
Liu L., Zhao M., Yu X., Zang W. // Neurosci. Bull. 2019. V. 35. P. 156–166. https://doi.org/10.1007/s12264-018-0286-7
Li D.L., Liu J.J., Liu B.H., Hu H., Sun L., Miao Y., Xu H.F., Yu X.J., Ma X., Ren J., Zang W.J. // J. Cell Physiol. 2011. V. 226. P. 1052–1059. https://doi.org/10.1002/jcp.22424
Miao Y., Zhou J., Zhao M., Liu J., Sun L., Yu X., He X., Pan X., Zang W. // Cell Physiol. Biochem. 2013. V. 31. P. 189–198. https://doi.org/10.1159/000343360
Hang P.Z., Zhao J., Qi J.C., Wang Y., Wu J.W., Du Z.M. // Curr. Drug Targets. 2013. V. 14. P. 372–377.
Liu Y., Wang S., Wang C., Song H., Han H., Hang P., Jiang Y., Wei L., Huo R., Sun L., Gao X., Lu Y., Du Z. // J. Transl. Med. 2013. V. 11. P. 209. https://doi.org/10.1186/1479-5876-11-209
Lu X.Z., Bi X.Y., He X., Zhao M., Xu M., Yu X.J., Zhao Z.H., Zang W.J. // Br. J. Pharmacol. 2015. V. 172. P. 5619–5633. https://doi.org/10.1111/bph.13183
Liu Y., Sun L., Pan Z., Bai Y., Wang N., Zhao J., Xu C., Li Z., Li B., Du Z., Lu Y., Gao X., Yang B. // Mol. Med. 2011. V. 17. P. 1179–1187. https://doi.org/10.2119/molmed.2011.00093
Mavropoulos S.A., Khan N.S., Levy A.C.J., Faliks B.T., Sison C.P., Pavlov V.A., Zhang Y., Ojamaa K. // Mol. Med. 2017. V. 23. P. 120–133. https://doi.org/10.2119/molmed.2017.00091
Monassier J.P. // Arch. Cardiovasc. Dis. 2008. V. 101. P. 491–500. https://doi.org/10.1016/j.acvd.2008.06.014
Dhalla N.S., Golfman L., Takeda S., Takeda N., Nagano M. // Can. J. Cardiol. 1999. V. 15. P. 587–593.
Palee S., Apaijai N., Shinlapawittayatorn K., Chattipakorn S.C., Chattipakorn N. // Cell Physiol. Biochem. 2016. V. 39. P. 341–349. https://doi.org/10.1159/000445628
Prathumsap N., Ongnok B., Khuanjing T., Arinno A., Maneechote C., Apaijai N., Chunchai T., Arunsak B., Shinlapawittayatorn K., Chattipakorn S.C., Chattipakorn N. // Transl. Res. 2022. V. 243. P. 33–51. https://doi.org/10.1016/j.trsl.2021.12.005
Xing R., Cheng X., Qi Y., Tian X., Yan C., Liu D., Han Y. // Biochem. Biophys. Res. Commun. 2020. V. 522. P. 1015–1021. https://doi.org/10.1016/j.bbrc.2019.11.086
Wang S., Han H.M., Jiang Y.N., Wang C., Song H.X., Pan Z.Y., Fan K., Du J., Fan Y.H., Du Z.M., Liu Y. // Clin. Exp. Pharmacol. Physiol. 2012. V. 39. P. 343–349. https://doi.org/10.1111/j.1440-1681.2012.05672.x
Liu Y., Sun H.L., Li D.L., Wang L.Y., Gao Y., Wang Y.P., Du Z.M., Lu Y.J., Yang B.F. // Can. J. Physiol. Pharmacol. 2008. V. 86. P. 860–865. https://doi.org/10.1139/Y08-094
Wang S., Han H.M., Pan Z.W., Hang P.Z., Sun L.H., Jiang Y.N., Song H.X., Du Z.M., Liu Y. // Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012. V. 385. P. 823–831. https://doi.org/10.1007/s00210-012-0740-4
Zhao Y., Wang C., Wu J., Wang Y., Zhu W., Zhang Y., Du Z. // Int. J. Biol. Sci. 2013. V. 9. P. 295–302. https://doi.org/10.7150/ijbs.5976
Zhao J., Su Y., Zhang Y., Pan Z., Yang L., Chen X., Liu Y., Lu Y., Du Z., Yang B. // Br. J. Pharmacol. 2010. V. 159. P. 1217–1225. https://doi.org/10.1111/j.1476-5381.2009.00606.x
Liu L., Lu Y., Bi X., Xu M., Yu X., Xue R., He X., Zang W. // Sci. Rep. 2017. V. 7. P. 42553. https://doi.org/10.1038/srep42553
Zhao L., Chen T., Hang P., Li W., Guo J., Pan Y., Du J., Zheng Y., Du Z. // Front. Pharmacol. 2019. V. 10. P. 1386. https://doi.org/10.3389/fphar.2019.01386
Hernandez C.C., Nascimento J.H., Chaves E.A., Costa P.C., Masuda M.O., Kurtenbach E., Campos D.E., Carvalho A.C., Gimenez L.E. // J. Recept. Signal Transduct. Res. 2008. V. 28. P. 375–401. https://doi.org/10.1080/10799890802262319
Liao F., Zheng Y., Cai J., Fan J., Wang J., Yang J., Cui Q., Xu G., Tang C., Geng B. // Sci. Rep. 2015. V. 16. P. 16590. https://doi.org/10.1038/srep16590
Walch L., Brink C., Norel X. // Therapie. 2001. V. 56. P. 223–226.
Radu B.M., Osculati A.M.M., Suku E., Banciu A., Tsenov G., Merigo F., Di Chio M., Banciu D.D., Tognoli C., Kacer P., Giorgetti A., Radu M., Bertini G., Fabene P.F. // Sci. Rep. 2017. V. 7. P. 5083. https://doi.org/10.1038/s41598-017-05384-z
Eglen R.M., Hegde S.S., Watson N. // Pharmacol. Rev. 1996. V. 48. P. 531–565.
Konidala S., Gutterman D.D. // Prog. Cardiovasc. Dis. 2004. V. 46. P. 349–373. https://doi.org/10.1016/j.pcad.2003.10.001
Walch L., Norel X., Leconte B., Gascard J.P., Brink C. // Therapie. 1999. V. 54. P. 99–102.
Pesić S., Grbović L., Jovanović A. // Pharmacology. 2002. V. 64. P. 182–188. https://doi.org/10.1159/000056169
Norel X., Walch L., Costantino M., Labat C., Gorenne I., Dulmet E., Rossi F., Brink C. // Br. J. Pharmacol. 1996. V. 119. P. 149–157. https://doi.org/10.1111/j.1476-5381.1996.tb15688.x
Tangsucharit P., Takatori S., Zamami Y., Goda M., Pakdeechote P., Kawasaki H., Takayama F. // J. Pharmacol. Sci. 2016. V. 130. P. 24–32. https://doi.org/10.1016/j.jphs.2015.12.005
Dauphin F., Ting V., Payette P., Dennis M., Hamel E. // Eur. J. Pharmacol. 1991. V. 207. P. 319–327. https://doi.org/10.1016/0922-4106(91)90006-4
O’Rourke S.T., Vanhoutte P.M. // J. Pharmacol. Exp. Ther. 1987. V. 241. P. 64–67.
Shimizu T., Rosenblum W.I., Nelson G.H. // Am. J. Physiol. 1993. V. 264. P. H665–H669. https://doi.org/10.1152/ajpheart.1993.264.3.H665
Pujol Lereis V.A., Hita F.J., Gobbi M.D., Verdi M.G., Rodriguez M.C., Rothlin R.P. // Br. J. Pharmacol. 2006. V. 147. P. 516–523. https://doi.org/10.1038/sj.bjp.0706654
Ahmed M., VanPatten S., Lakshminrusimha S., Patel H., Coleman T.R., Al-Abed Y. // Physiol. Rep. 2016. V. 4. P. e13069. https://doi.org/10.14814/phy2.13069
Евлахов В.И., Березина Т.П., Поясов И.З., Овсянников В.И. // Бюлл. эксперим. биол. мед. 2021. Т. 171. № 2. С. 159–163. https://doi.org/10.47056/0365-9615-2021-171-2-159-163
Lung M.A. // Am. J. Rhinol. Allergy. 2011. V. 25. P. e60–e65. https://doi.org/10.2500/ajra.2011.25.3604
Niihashi M., Esumi M., Kusumi Y., Sato Y., Sakurai I. // Angiology. 2000. V. 51. P. 295–300. https://doi.org/10.1177/000331970005100404
Bény J.L., Nguyen M.N., Marino M., Matsui M. // J. Cardiovasc. Pharmacol. 2008. V. 51. P. 505–512. https://doi.org/10.1097/FJC.0b013e31816d5f2f
Dauphin F., Hamel E. // Eur. J. Pharmacol. 1990. V. 178. P. 203–213. https://doi.org/10.1016/0014-2999(90)90476-M
Gericke A., Steege A., Manicam C., Böhmer T., Wess J., Pfeiffer N. // Invest. Ophthalmol. Vis. Sci. 2014. V. 55. P. 625–631. https://doi.org/10.1167/iovs.13-13549
Duckles S.P., Garcia-Villalon A.L. // J. Pharmacol. Exp. Ther. 1990. V. 253. P. 608–613.
Jaiswal N., Lambrecht G., Mutschler E., Tacke R., Malik K.U. // J. Pharmacol. Exp. Ther. 1991. V. 258. P. 842–850.
Коваленко Н.Я., Мациевский Д.Д., Решетняк В.К. // Бюлл. эксперим. биол. мед. 2013. Т. 156. № 12. С. 697–700.
Yamada M., Lamping K.G., Duttaroy A., Zhang W., Cui Y., Bymaster F.P., McKinzie D.L., Felder C.C., Deng C.X., Faraci F.M., Wess J. // Proc. Natl. Acad. Sci. USA. 2001. V. 98. P. 14096–14101. https://doi.org/10.1073/pnas.251542998
Zuccolo E., Laforenza U., Negri S., Botta L., Berra-Romani R., Faris P., Scarpellino G., Forcaia G., Pellavio G., Sancini G., Moccia F. // J. Cell. Physiol. 2019. V. 234. P. 4540–4562. https://doi.org/10.1002/jcp.27234
Gericke A., Sniatecki J.J., Mayer V.G., Goloborodko E., Patzak A., Wess J., Pfeiffer N. // Am. J. Physiol. Heart Circ. Physiol. 2011. V. 300. P. H1602–H1608. https://doi.org/10.1152/ajpheart.00982.2010
Conti-Fine B.M., Navaneetham D., Lei S., Maus A.D.J. // Eur. J. Pharmacol. 2000. V. 393. P. 279–294. https://doi.org/10.1016/S0014-2999(00)00036-34
Heeschen C., Weis M., Aicher A., Dimmeler S., Cooke J.P. // J. Clin. Invest. 2002. V. 110. P. 527–536. https://doi.org/10.1172/JCI14676
Brüggmann D., Lips K.S., Pfeil U., Haberberger R.V., Kummer W. // Histochem. Cell. Biol. 2002. V. 118. P. 441–447. https://doi.org/10.1007/s00418-002-0475-2
Brüggmann D., Lips K.S., Pfeil U., Haberberger R.V., Kummer W. // Life Sci. 2003. V. 72. P. 2095–2099. https://doi.org/10.1016/s0024-3205(03)00067-5
Wada T., Naito M., Kenmochi H., Tsuneki H., Sasaoka T. // Endocr. Rev. 2007. V. 148. P. 790–799. https://doi.org/10.1210/en.2006-0907
Li D.-J., Zhao T., Xin R.-J., Wang Y.-Y., Fei Y.-B., Shen F.-M. // Cell. Physiol. Biochem. 2014. V. 33. P. 468–478. https://doi.org/10.1159/000358627
Clifford P.M., Siu G., Kosciuk M., Levin E.C., Venkataraman V., D’Andrea M.R., Nagele R.G. // Brain Res. 2008. V. 1234. P. 158–171. https://doi.org/10.1016/j.brainres.2008.07.092
Lips K.S., Bruggmann D., Pfeil U., Vollerthun R., Grando S.A., Kummer W. // Placenta. 2005. V. 26. P. 735–746. https://doi.org/10.1016/j.placenta.2004.10.009
Gotti C., Clementi F. // Prog. Neurobiol. 2004. V. 74. P. 363–396. https://doi.org/10.1016/j.pneurobio.2004.09.006
Egleton R.D., Brown K.C., Dasgupta P. // Pharmacol. Ther. 2009. V. 121. P. 205–223. https://doi.org/10.1016/j.pharmthera.2008.10.007
Cooke J.P., Bitterman H. // Ann. Med. 2004. V. 36. P. 33–40. https://doi.org/10.1080/07853890310017576
Macklin K.D., Maus A.D., Pereira E.F., Albuquerque E.X., Conti-Fine B.M. // J. Pharmacol. Exp. Ther. 1998. V. 287. P. 435–439.
Moccia F., Frost C., Berra-Romani R., Tanzi F., Adams D.J. // Am. J. Physiol. Heart. Circ. Physiol. 2004. V. 286. P. H486–H491. https://doi.org/10.1152/ajpheart.00620.2003
Bucerius J., Manka C., Schmaljohann J., Mani V., Gündisch D., Rudd J.H., Bippus R., Mottaghy F.M., Wüllner U., Fayad Z.A., Biersack H.J. // JACC Cardiovasc. Imaging. 2012. V. 5. P. 528–536. https://doi.org/10.1016/j.jcmg.2011.11.024
Zou Q., Leung S.W., Vanhoutte P.M. // J. Pharmacol. Exp. Ther. 2012. V. 341. P. 756–763. https://doi.org/10.1124/jpet.112.192229
Cooke J.P., Ghebremariam Y.T. // Trends Cardiovasc. Med. 2008. V. 18. P. 247–253. https://doi.org/10.1016/j.tcm.2008.11.007
Wu J.C.F., Chruscinski A., Perez V.A.D.J., Singh H., Pitsiouni M., Rabinovitch M., Utz P.J., Cooke J.P. // J. Cell. Biochem. 2009. V. 446. P. 433–446. https://doi.org/10.1002/jcb.22270
Li D.-J., Huang F., Ni M., Fu H., Zhang L.-S., Shen F.-M. // Arterioscler. Thromb. Vasc. Biol. 2016. V. 36. P. 1566–1576. https://doi.org/10.1161/ATVBAHA.116.307157
Liu L., Wu H., Cao Q., Guo Z., Ren A., Dai Q. // Mediators Inflamm. 2017. V. 2017. P. 2401027. https://doi.org/10.1155/2017/2401027
Li X., Wang H. // Life Sci. 2006. V. 78. P. 1863–1870. https://doi.org/10.1016/j.lfs.2005.08.031
Peña V.B., Bonini I.C., Antollini S.S., Kobayashi T., Barrantes F.J. // J. Cell. Biochem. 2011. V. 112. P. 3276–3288. https://doi.org/10.1002/jcb.23254
Whitehead A.K., Erwin A.P., Yue X. // Acta Physiol. (Oxf). 2021. V. 231. P. e13631. https://doi.org/10.1111/apha.13631
Centner A.M., Bhide P.G., Salazar G. // Cells. 2020. V. 9. P. 1035. https://doi.org/10.3390/cells9041035
Li Z.Z., Dai Q.Y. // Mediators Inflamm. 2012. V. 2012. P. 103120. https://doi.org/10.1155/2012/103120
Gaemperli O., Liga R., Bhamra-Ariza P., Rimoldi O. // Curr. Pharm. Des. 2010. V. 16. P. 2586–2597. https://doi.org/10.2174/138161210792062894
Cooke J.P. // Life Sci. 2007. V. 80. P. 2347–2351. https://doi.org/10.1016/j.lfs.2007.01.061
Boswijk E., Bauwens M., Mottaghy F.M., Wildberger J.E., Bucerius J. // Methods. 2017. V. 130. P. 90–104. https://doi.org/10.1016/j.ymeth.2017.06.008
Vazquez-Padron R.I., Mateu D., Rodriguez-Menocal L., Wei Y., Webster K.A., Pham S.M. // Cardiovasc. Res. 2010. V. 88. P. 296–303. https://doi.org/10.1093/cvr/cvq213
Vieira-Alves I., Coimbra-Campos L.M.C., Sancho M., da Silva R.F., Cortes S.F., Lemos V.S. // Front Physiol. 2020. V. 11. P. 621769. https://doi.org/10.3389/fphys.2020.621769
Libby P., Buring J. E., Badimon L., Hansson G.K., Deanfield J., Bittencourt S., Tokgözoğlu L., Lewis E.F. // Nat. Rev. Dis. Primers. 2019. V. 5. P. 56. https://doi.org/10.1038/s41572-019-0106-z
Santanam N., Thornhill B.A., Lau J.K., Crabtree C.M., Cook C.R., Brown K.C., Dasgupta P. // Atherosclerosis. 2012. V. 225. P. 264–273. https://doi.org/10.1016/j.atherosclerosis.2012.07.041
Lee J., Cooke J.P. // Atherosclerosis. 2011. V. 215. P. 281–283. https://doi.org/10.1016/j.atherosclerosis.2011.01.003
Zhang G., Marshall A.L., Thomas A.L, Kernan K.A., Su Y., LeBoeuf R.C., Dong X.R., Tchao B.N. // Atherosclerosis. 2011. V. 215. P. 34–42. https://doi.org/10.1016/j.atherosclerosis.2010.07.057
Brown K.C., Lau J.K., Dom A.M., Witte T.R., Luo H., Crabtree C.M., Shah Y.H., Shiflett B.S., Marcelo A.J., Proper N.A., Hardman W.E., Egleton R.D., Chen Y.C., Mangiarua E.I., Dasgupta P. // Angiogenesis. 2012. V. 15. P. 99–114. https://doi.org/10.1007/s10456-011-9246-9
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия