

Биоорганическая химия, 2023, T. 49, № 2, стр. 153-164
Синтез и биологическая активность эфиров на основе циклоалкендикарбоновых кислот
А. А. Фирстова 1, *, Е. Р. Кофанов 1, М. И. Ковалева 2
1 Ярославский государственный технический университет
150023 Ярославль, Московский просп., 88, Россия
2 Ярославский государственный университет им. П.Г. Демидова
150003 Ярославль, ул. Советская, 14,
Россия
* E-mail: firstova.a.a@mail.ru
Поступила в редакцию 01.06.2022
После доработки 16.06.2022
Принята к публикации 12.08.2022
- EDN: GHIOTR
- DOI: 10.31857/S0132342323010086
Аннотация
Синтезированы неописанные ранее эфиры на основе N-замещенных имидов циклоалифатических карбоновых кислот с высокими выходами, представлены эффективные методы их получения. Проведены токсикологические исследования полученных соединений с использованием различных тест-объектов (Chlorella vulgaris, Allium cepa, Drosophila melanogaster). Показано, что исследованные соединения не активны по отношению к D. melanogaster, увеличивают частоту мутаций у C. vulgaris и обладают способностью индуцировать хромосомные перестройки у A. cepa. Синтезированные соединения могут использоваться при разработке препаратов с противоопухолевым эффектом.
ВВЕДЕНИЕ
Одно из перспективных направлений синтеза биологически активных веществ – образование эфирной или сложноэфирной группы, что позволяет конструировать соединения, содержащие в молекулах одновременно с фармакофорными фрагментами соответствующих карбоновых кислот структурные элементы спиртов. Достаточно высокая химическая устойчивость этих групп к гидролизу, алкоголизу и аммонолизу при нейтральных значениях pH, а также к воздействию биологических сред позволяет обоснованно считать их эффективным инструментом в области создания перспективных лекарственных препаратов.
В научной литературе приводятся многочисленные примеры применения эфиров карбоновых кислот. Описаны способы получения противоопухолевых препаратов, которые показали положительную динамику терапии в исследованиях [1, 2]. В настоящее время актуален поиск принципиально новых подходов к противоопухолевой терапии. Стабилизация опухолевого процесса или поддержание “дремлющего” состояния опухоли могут представлять многообещающие возможности для достижения цели лечения. Так, в статьях [3–5] приведены примеры применения имидов карбоновых кислот в качестве противоопухолевых препаратов.
Известны исследования влияния эфиров карбоновых кислот на глюкокортикоиды [6]. Глюкокортикоиды регулируют глюконеогенез в печени, их содержание в крови увеличивается при синдроме Кушинга, который вызывает метаболические нарушения, такие как висцеральные ожирение, нарушение толерантности к глюкозе, атеросклероз, дислипидемия и гипергликемия [7, 8]. Нормализация уровня глюкокортикоидов может изменить особенности метаболических синдромов [9]. Подавление действия глюкокортикоидов может быть осуществлено ингибиторами, описанными в статьях [10–14].
Одной из интересных областей применения имидов карбоновых кислот в последние годы стало проектирование миметиков вторичной структуры, имеющих конформационно жесткие непептидные структурные каркасы, которые служат идеальными каркасами для поддержки боковой цепи функциональных элементов с желаемой геометрией [15, 16]. В статье [17] продемонстрировано использование эндо-цис-(2S,3R)-бицикло[2.2.1]гепт-5-ена, имеющего непептидный молекулярный каркас, для создания аналогов пептидов.
Еще одно интересное применение имидов карбоновых кислот и их производных – использование в качестве регуляторов роста растений. Так, описаны препараты [18], имитирующие действие гормона растений гиббереллина, который представляет собой тетрациклический дитерпеноид, регулирующий прорастание семян, удлинение стеблей, расширение листьев и развитие цветков растений. Миметики гиббереллина, в частности некоторые имиды карбоновых кислот, способны формировать связь с рецептором так же, как и сам гормон [19–24].
Исходя из литературных данных, нами была поставлена цель работы – синтез N-замещенных имидов циклоалкендикарбоновых кислот, их дальнейшая модификация в сложные эфиры различными способами и исследование биологической активности этих соединений.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез эфиров карбоновых кислот на основе циклоалкендикарбоновых кислот. Нами был проведен поиск наиболее удобного метода получения эфиров на основе N-замещенных имидов карбоновых кислот, позволяющего получать продукты с высоким выходом и имеющего минимальное количество стадий. Это необходимо для увеличения эффективности по сравнению методами, описанными в литературе.
Ранее в научных статьях упоминались некоторые структуры ((XIV), (XVI), (XVIII)). Однако способы, предлагаемые авторами, экономически и энергозатратны. Так, например, в работе [25] был проведен синтез имидов кипячением в уксусной кислоте, и затем синтезированы эфиры с использованием дициклогексилкарбодиимида (DCC), 4-диметиламинопиридина (DMAP) в хлористом метилене в течение 12 ч.
В статье [26] приведен способ получения N‑малеилпроизводных взаимодействием малеинового ангидрида и аминокислоты в ледяной уксусной кислоте и последующей циклизации N-малеиламинокислот в толуоле с добавлением триэтиламина, полученные продукты очищали хроматографическими методами.
В статьях [27–29] в качестве субстратов применяли эфиры аминокислот – более дорогостоящие по сравнению с аминокислотами.
В результате анализа литературы нами были разработаны методы, позволяющие синтезировать предложенные соединения из доступного сырья и без применения особых условий (схема 1 ).

Схема 1 . Синтез N-замещенных имидов циклоалкендикарбоновых кислот (III–X) и эфиров (XI–XVIII) на их основе: i – аминокислота, AcOH, кипячение; ii – H2SO4, EtOH; iii – 1) аминокислота, CHCl3, кипячение; 2) TolSO2Cl, K2CO3, тетрабутиламмоний иодид, EtOH.
На первом этапе работы применяли двухстадийный синтез эфиров, предполагающий получение N-замещенного имида циклоалкендикарбоновой кислоты и далее этерификацию карбоксильной группы. По нашему мнению, наиболее перспективный метод получения N-замещенных имидов карбоновых кислот – взаимодействие ангидридов дикарбоновых кислот (I) и (II) с различными хиральными α-аминокислотами (в работе использовали Gly, L-Val, L-Phe, L-Ala, L-Ile) в уксусной кислоте при кипячении [30]. Данная методика позволяет получать продукты с высоким выходом и не требует дополнительных манипуляций по очистке.
На втором этапе было предложено несколько способов образования сложных эфиров N-замещенных циклоалкендикарбоновых кислот. Один из традиционных методов – взаимодействие карбоновых кислот и спиртов, протекающее в условиях кислотного катализа (например, с использованием серной кислоты) [31]. Применяемая методика известная и легко воспроизводимая, но имеет ряд недостатков: 1) необходимо использовать спирт в 10-кратном избытке (как реагент и растворитель); 2) требуется отводить выделяющуюся воду из-за обратимости реакции и возможного гидролиза; 3) длительность реакции и невысокий выход.
Поэтому применяли альтернативный способ, в котором использовали в качестве растворителя хлороформ при активном перемешивании. Таким образом, реакция шла на межфазном пространстве, однако высокого выхода целевых соединений достичь не удалось. Это объясняется низкой скоростью химической реакции, которая в данном методе определялась скоростью диффузии реагентов через границу раздела фаз (в нашем случае вода–хлороформ).
Для упрощения метода синтеза и решения проблемы низкой скорости реакции был предложен метод однореакторного синтеза с использованием катализатора межфазного переноса [32]. Известно, что реакция взаимодействия ангидридов карбоновых кислот и аминокислот идет при кипячении в растворителе. В методе однореакторного синтеза в качестве растворителя использовали хлороформ. Таким образом, на первой стадии получали имид карбоновой кислоты и далее без выделения вводили четвертичную аммонийную соль в качестве катализатора межфазного переноса [33, 34]. В данном случае выбор каталитической системы основывался на следующих утверждениях: эффективность больших четвертичных катионов доказана и в сравнении с ионами меньшего размера существенно выше; наличие в четвертичной аммониевой соли алкильного фрагмента также увеличивает каталитическую активность [33]. Данный метод позволял получать эфиры в мягких условиях и с высоким выходом [34]. Таким образом, метод межфазного катализа позволяет простым и удобным способом решить проблему растворимости и реакционной способности в синтезе эфиров карбоновых кислот.
Биологическая активность соединений. Нами были отобраны синтезированные соединения с предположительной высокой биологической активностью, селективностью и структурным разнообразием. Для этих структур проведена первичная оценка биологических эффектов.
В настоящее время разработано более 200 методов оценки генотоксикантов, но не существует универсального. Несмотря на высокую чувствительность, экономичность и эффективность, один метод позволяет регистрировать только один тип генетических нарушений. Поэтому для исследования генетических эффектов необходимо использовать систему токсикогенетических методов [35].
Для исследования были выбраны следующие токсикогенетические методы: тест учета мутагенности, митотоксичности и токсичности у лука Allium cepa (L.), тест учета выживаемости и видимых мутаций у Chlorella vulgaris (Bejer) и тест учета доминантных летальных мутаций (ДЛМ) у Drosophila melanogaster (Meigen). Каждый из методов позволяет регистрировать различные типы нарушений: нарушение пролиферации (A. cepa, C. vulgaris), любые летальные мутации в половых клетках (дрозофила), хромосомные аберрации в соматических клетках (A. cepa). Этот набор методов позволяет регистрировать широкий спектр нарушений.
Для биологического тестирования использовали соединения (XI), (XIV), (XV), (XVII) и (XVIII) в виде растворов в 0.25%-ном водном этаноле. Данный растворитель был выбран ввиду лучшей растворимости соединений. В качестве контролей использовали воду и 0.25%-ный водный этанол. Предварительная оценка токсичности растворителя показала, что его использование не приводит к увеличению митотического индекса, а снижение пролиферативной активности выражено в незначительной степени и, следовательно, не оказывает отрицательного воздействия на тест-системы.
Для оценки общей токсичности соединений проводили исследование с использованием тест-объекта A. cepa. В данном методе определяли отклонения средней длины корней A. cepa и сравнивали контрольные и опытный варианты (рис. 1). Также определяли способность препарата модифицировать пролиферативную активность. Для этого оценивали митотический индекс (рис. 2) и соотношение средних фазных индексов (рис. 3).
Рис. 1.
Влияние концентрации исследуемых соединений на длину корней Allium cepa в опыте по оценке общей токсичности. Обозначения на рис. 1–6: К – контроль (дистиллированная вода), Раствор – 0.25%-ный водный этанол.

Рис. 2.
Влияние концентрации исследуемых соединений на митотический индекс (%) в меристеме Allium cepa в опыте по оценке общей токсичности.

Рис. 3.
Влияние концентрации препаратов на фазные индексы в меристеме Allium cepa в опыте по оценке токсичности.

При сравнении контрольного варианта с растворителем (0.25%-ный водный раствор этанола) было выявлено снижение пролиферативной активности.
При оценке влияния различных концентраций исследуемых препаратов показано, что для соединений (XI) и (XVII) длина корней и митотический индекс уменьшаются по сравнению с контролем. Высокие концентрации соединений (XIV), (XV) и (XVIII) приводят к гибели корней, при этом низкие концентрации этих препаратов не влияют на митотический индекс, стимулируется рост длины корней.
Анализ фазных индексов позволяет выявить механизм нарушения пролиферации, данные представлены на рис. 3. Фазные индексы отражают соотношение клеток, находящихся на данной стадии, к сумме делящихся клеток. Можно отметить, что профазный индекс составляет >40%, следовательно, профаза – самая длительная фаза митоза в меристеме корней лука, что согласуется с литературными данными [35]. В профазе происходит подготовка к делению, компактизация хроматина и пр. При воздействии соединений (XIV), (XV) и (XVIII) с концентрацией 0.1% наблюдается полная задержка роста, что говорит о их токсичности. В остальных случаях заметных изменений фазных индексов по сравнению с контрольными вариантами не обнаружено.
На следующем этапе анализировали мутагенную активность препарата. Хромосомные аберрации выявляли с помощью анателофазного метода. Регистрировали такие нарушения, как отставший от полюсов хромосомный материал (фрагменты, мосты, отставшие хромосомы). На рис. 4 видно, что для каждого препарата прослеживается способность индуцировать хромосомные аберрации. При этом линейной зависимости частоты хромосомных аберраций (ХА) от концентрации исследуемых соединений не отмечено, необходимы дополнительные исследования.
Рис. 4.
Влияние концентрации препаратов на частоту хромосомных аберраций в меристеме Allium cepa в опыте по оценке мутагенности.

Далее была проведена оценка токсичности соединений с использованием в качестве тест-объекта одноклеточной водоросли C. vulgaris. С одной стороны, C. vulgaris – растительный тест-объект, поэтому она имеет преимущества растительных тест-систем. С другой стороны, C. vulgaris относится к микроорганизмам и обладает высокой чувствительностью, экономичностью и высокой скоростью размножения, что позволяет в короткие сроки получить результаты токсикогенетических экспериментов. Данным методом можно оценивать не только мутагенность препаратов (учет частоты мутантных макроколоний), но и митотоксичность (определение выживаемости колоний). При этом метод не дает возможность дифференцировать цитотоксический (гибель клеток) и цитостатический (нарушение деления клеток) эффекты.
Результаты исследования представлены на рис. 5 и 6. По полученным данным можно сделать вывод, что исследованные препараты повышают частоту мутаций в среднем в 1.5 раза по сравнению с контролем. При этом исследуемые препараты достоверно не снижают выживаемость колоний C. vulgaris.
Рис. 5.
Влияние концентраций препаратов на частоту мутаций Chlorella vulgaris в опыте по оценке мутагенности.
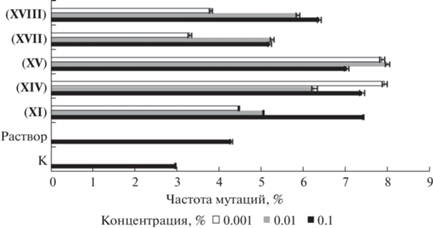
Рис. 6.
Влияние концентраций препаратов на выживаемость Chlorella vulgaris в опыте по оценке токсичности.

Плодовая мушка дрозофила – один из самых распространенных объектов, используемых для выявления генотоксического действия различных факторов. Эффективность ее использования для исследований обусловлена такими особенностями, как непродолжительный цикл развития и высокая плодовитость, известно большое число генов с легкоразличимыми признаками, описаны практически все виды мутаций и др. Известно, что дрозофила может использоваться для выявления мутагенов и промутагенов [36].
В исследовании с использованием тест-объекта D. melanogaster не наблюдалось увеличения частоты индуцированных доминантных летальных мутаций (ДЛМ), что показывает отсутствие генотоксичности соединений (XI), (XIV), (XVII) и (XVIII). Таким образом, исследованные концентрации препаратов (XI), (XIV), (XVII) и (XVIII) не обладают мутагенной активностью (табл. 1). Cоединение (XV) незначительно увеличивало частоту индуцированных доминантных летальных мутаций (ДЛМ).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованы коммерчески доступные реактивы марки “ч” или “х.ч.” отечественного производства и фирмы Sigma-Aldrich (США). Растворители перед использованием осушали и перегоняли по известным методикам [37].
Спектроскопия. Спектры 1Н- и 13С-ЯМР (δ, м.д.; J, Гц) регистрировали на приборе MSL300 (Bruker, Германия) с рабочей частотой 300 и 75.5 МГц соответственно. Спектры записывали для растворов анализируемых соединений в DMSO-d6 относительно остаточных протонов растворителя.
ИК-спектры (ν, см–1) регистрировали на приборе ИК-Фурье Spectrum RX1 (Perkin Elmer, Великобритания) на пластинах KBr в вазелиновом масле.
Масс-спектры высокого разрешения регистрировали на приборе MicrOTOF-II (Bruker Daltonics, Германия), метод ионизации – электрораспыление (ESI), температура источника ионизации – 180°С. Элюент ацетонитрил.
ТСХ осуществляли на пластинках Sulifol 201S в системе петролейный эфир–толуол–ацетон– уксусная кислота, 100 : 60 : 100 : 2 (об.).
Температуру плавления соединений определяли на приборе Electrothermal IA 9300 Series (Electrothermal Engineering Ltd., Великобритания).
N-Замещенные имиды алициклических дикарбоновых кислот (III–X) (общая методика) [30]. В круглодонную колбу, снабженную обратным холодильником, помещали 50 ммоль алициклического ангидрида (I) или (II), 55 ммоль аминокислоты (Gly, L-Val, L-Phe, L-Ala, L-Ile) и 15 мл уксусной кислоты, кипятили в течение 4 ч. Затем реакционную смесь выливали в воду (100 мл). Полученный осадок отфильтровывали, промывали водой и сушили при 40°С.
(1,3-Диоксо-1,3,3а,4,7,7a-гексагидро-2H-изоиндол-2-ил)уксусная кислота (III). Выход 93%. Т. пл. 69–72°С. ИК: 2724, 2671, 923 (OH); 1791, 1757, 1684 (C=O, имид); 1633 (С=С); 1178 (C–O). 1Н-ЯМР: 2.37 (4H, м, CH2), 3.22 (2H, м, HC–C=O), 4.04 (2H, с, NCH2), 5.87 (2H, м, HC=CH), 13.07 (1Н, с, COOH). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 210.076. Вычислено для С10H11NO4: 210.068.
(3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)уксусная кислота (IV). Выход 83%. Т. пл. 139–143°С. ИК: 2725, 2669, 920 (OH); 1791, 1758, 1680 (C=O, имид); 1643 (С=С); 1128 (C–O). 1Н‑ЯМР: 1.58 (2Н, м, CH2-мостик), 2.51 (1Н, м, HC–C=O), 3.26 (1Н, м, HC–C=O), 3.44 (2Н, м, CH), 3.89 (2Н, м, NCH2), 6.02 (2H, м, HC=CH), 12.95 (1Н, с, COOH). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 222.088. Вычислено для С11H11NO4: 222.068.
2-(1,3-Диоксо-3a,4-дигидро-1H-изоиндол-2(3H,-7H,7aH)-ил)-3-метилбутановая кислота (V). Выход 60%. Т. пл. 123–127°С. ИК: 2728, 2661, 2594, 901 (OH); 1769, 1744, 1673 (C=O, имид); 1192 (C–O). 1Н-ЯМР: 1.20 (3Н, м, CH3), 1.50 (3Н, м, CH3), 1.80 (1Н, м, HC–C=O), 2.51 (1Н, м, HC–C=O), 3.42 (2Н, м, CH2), 3.72 (1Н, м, CHN), 4.10 (2Н, м, CH2), 4.45 (1Н, м, CH), 6.03 (2Н, м, HC=CH), 12.61 (1Н, с, СООН). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 252.125. Вычислено для С13H17NO4: 252.115.
2-(3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)-3-метилбутановая кислота (VI). Выход 73%. Т. пл. 105–108°С. ИК: 2728, 2603, 901 (OH); 1769, 1706, 1683 (C=O, имид); 1682 (С=С); 1192 (С–О). 1Н-ЯМР: 0.84 (6Н, м, CH3). 1.31 (2Н, м, CH2-мостик), 1.63 (1Н, м, HC–C=O), 1.81 (1Н, м, HC–C=O), 3.37 (1Н, м, CHN), 3.42 (2Н, м, CH), 4.41 (1Н, м, CH), 6.03 (2Н, м, CH=CH), 12.86 (1Н, с, COOH). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 264.128. Вычислено для С14H17NO4: 264.115.
2-(1,3-Диоксо-3a,4-дигидро-1H-изоиндол-2(3H,-7H,7aH)-ил)-3-фенилпропановая кислота (VII). Выход 60%. Т. пл. 123–127°С. ИК: 2728, 2661, 2594, 901 (OH); 1769, 1744, 1673 (C=O, имид); 1192 (C–O). 1Н-ЯМР: 2.08 (2H, м, CH2-Ph). 2.33 (1H, м, HC–C=O), 3.18 (1H, м, HC–C=O), 3.43 (4H, м, CH2), 4.85 (1H, м, CHN), 5.57 (2H, м, HC=CH), 7.12 (2Н, д, J 7.0, Ar), 7.27 (1Н, д, J 7.5 Ar), 7.29 (2Н, т, J 14.2, Ar), 13.01 (1Н, с, СООН). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 300.124. Вычислено для С17H17NO4: 300.115.
2-(3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)-3-фенилпропановая кислота (VIII). Выход 74%. Т. пл. 136–139°С. ИК: 2721, 2661, 929 (OH); 1769, 1751 (C=O, имид); 1684 (С=С); 1603, 1398 (Ar); 1169 (С–О). 1Н-ЯМР: 1.42 (2H, м, CH2-Ph). 2.49 (2H, м, CH2-мостик), 3.11 (1H, м, HC–C=O), 3.19 (1H, м, HC–C=O), 3.33 (2H, м, CH), 4.86 (1H, м, CHN), 5.24 (1H, м, HC=CH), 5.67 (1H, м, HC=CH), 7.12 (2Н, д, J 7.0, Ar), 7.20 (1Н, д, J 7.5, Ar), 7.26 (2Н, т, J 14.2, Ar), 13.02 (1Н, с, СООН). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 312.123. Вычислено для С18H17NO4: 312.115.
2-(1,3-Диоксо-3a,4-дигидро-1H-изоиндол-2(3H,7H,7aH)-ил)-3-метилпентановая кислота (IX). Выход 80%. Т. пл. 132–134°С. ИК: 2591, 950 (OH); 1701 (C=O, имид); 1616 (С=С); 1263 (С–О–). 1Н‑ЯМР: 0.79 (6Н, м, CH3), 1.28 (1Н, м, CH), 1.67 (2Н, м, CH2), 1.97 (1Н, м, HC–C=O), 2.22 (1Н, м, HC–C=O), 3.17 (4Н, м, CH2), 4.49 (1Н, м, CHN), 5.84 (2Н, м, HC=CH), 12.77 (1Н, с, COOH). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 266.139. Вычислено для С14H19NO4: 266.131.
2-(3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)-3-метилпентановая кислота (X). Выход 93%. Т. пл. 103–105°С. ИК: 2723, 2656, 2597, 904 (OH); 1746, 1706 (C=O, имид); 1677 (С=С); 1190 (С–О). 1Н-ЯМР: 0.71 (3Н, м, CH3), 0.93 (3Н, м, CH3), 1.56 (2Н, м, CH2-мостик), 2.23 (1Н, м, CH), 2.34 (2Н, м, CH2), 3.37 (2Н, м, HC–C=O), 3.45 (2Н, м, CH), 4.07 (1Н, м, CHN), 6.01 (2Н, м, HC=CH), 11.95 (1Н, с, COOH). Масс-спектр (ИЭР), m/z: найдено: [M + H]+ 278.135. Вычислено для С15H19NO4: 278.131.
Сложные эфиры N-замещенных циклоалкендикарбоновых кислот (XI–XVIII). Метод 1. Синтез сложных эфиров N-замещенных циклоалкендикарбоновых кислот с использованием кислотного катализатора. В колбу, снабженную мешалкой и обратным холодильником, помещали 100 ммоль этилового спирта и 10 ммоль N-замещенной циклоалкендикарбоновой кислоты (III–X). Затем к полученному раствору добавляли каталитические количества серной кислоты. Реакционную смесь при перемешивании кипятили в течение 4 ч. Полученный раствор выливали в холодную воду, эфир экстрагировали хлороформом.
Метод 2. Одноректорный синтез сложных эфиров N-замещенных циклоалкендикарбоновых кислот. В колбе, снабженной мешалкой и обратным холодильником, растворяли в хлороформе 50 ммоль алициклического ангидрида (I) или (II). После полного растворения добавляли 55 ммоль аминокислоты (Gly, L-Val, L-Phe, L-Ala, L-Ile) и кипятили в течение 2 ч. Далее в реакционную смесь вносили 5 ммоль четвертичной аммониевой соли, а затем 5 ммоль 4-метилбензол-1-сульфонилхлорида и 20 ммоль карбоната калия, кипятили в течение 40 мин. По истечении времени реакции добавляли этиловый спирт (50 ммоль) и выдерживали массу еще 45 мин. Осадок отфильтровывали, фильтрат отгоняли.
Этиловый эфир (1,3-диоксо-1,3,3а,4,7,7а-гексагидро-изоиндол-2-ил)уксусной кислоты (XI). С12H15NO4. Выход 38%. Масло. ИК: 1748 (С=O, сложный эфир); 1709 (C=O, имид); 1210 (C–O–). 1Н-ЯМР: 1.20 (2H, м, СH2–O), 1.51 (3H, м, CH3), 2.26 (2H, м, CH2), 3.10 (2H, м, HC–C=O), 4.16 (2H, м, NCH2), 4.76 (2H, м, CH2), 5.93 (2H, м, НC=CH). Масс-спектр (ИЭР), m/z: найдено для С12H15NO4: 237.100. Вычислено для С12H15NO4: 237.100.
Этиловый эфир (3,5-диоксо-4-азатрицикло-[5.2.1.02,6]дец-8-ен-4-ил)уксусной кислоты (XII). С13H15NO4. Выход 55%. Масло. ИК: 1748 (С=О, сложный эфир); 1707 (C=O, имид); 1240 (C–O). 1Н-ЯМР: 1.25 (2H, м, СH2–O), 1.45 (3H, м, CH3), 1.62 (2Н, м, CH2-мостик), 3.11 (1Н, м, HC–C=O), 3.30 (1Н, м, HC–C=O), 3.67 (2Н, м, CH), 4.02 (2Н, м, NCH2), 6.10 (2H, м, HC=CH). Масс-спектр (ИЭР), m/z: найдено для С13H15NO4: 249.100. Вычислено для С13H15NO4: 249.100.
Этиловый эфир 2-(1,3-диоксо-3a,4-дигидро-1H-изоиндол-2(3H,7H,7aH)-ил)-3-метилбутановой кислоты (XIII). С15H21NO4. Выход 89%. Масло. ИК: 1768 (С=О, сложный эфир); 1719 (C=O, имид); 1210 (C–O–). 1H-ЯМР: 1.03 (3H, м, CH3), 1.08 (2H, м, O–CH2), 1.34 (3H, м, СН3), 2.29 (1Н, м, HC–C=O), 2.37 (1Н, м, HC–C=O), 3.03 (3Н, м, CH3-эфир), 3.14 (4Н, м, CH2), 4.20 (1H, м, CHN), 4.39 (1Н, м, CH), 5.89 (1H, м, НC=CH), 5.67 (1H, м, НC=CH). Масс-спектр (ИЭР), m/z: найдено для С15H21NO4: 279.147. Вычислено для С15H21NO4: 279.147.
Этиловый эфир 2-(3,5-диоксо-4-азатрицикло-[5.2.1.02,6]дец-8-ен-4-ил)-3-метилбутановой кислоты (XIV). С16H21NO4. Выход 36%. Масло. ИК: 1746 (С=О, сложный эфир); 1710 (C=O, имид); 1214 (C–O–). 1Н-ЯМР: 0.84 (6Н, м, CH3), 1.21 (2H, м, O–CH2), 1.39 (2Н, м, CH2-мостик), 1.53 (1Н, м, HC–C=O), 1.81 (1Н, м, HC–C=O), 3.24 (3H, м, CH3-эфир), 3.38 (1Н, м, CHN), 3.55 (2Н, м, CH), 4.55 (1Н, м, CH), 6.02 (2Н, м, НC=CH). Масс-спектр (ИЭР), m/z: найдено для С16H21NO4: 291.147. Вычислено для С16H21NO4: 291.148.
Этиловый эфир 2-(1,3-диоксо-3a,4-дигидро-1H-изоиндол-2(3H,7H,7aH)-ил)-3-фенилпропановой кислоты (XV). С19H21NO4. Выход 56%. Масло. ИК: 1758 (С=О, сложный эфир); 1715 (C=O, имид); 1212 (C–O–). 1Н-ЯМР: 1.30 (3H, м, CH3), 1.55 (2H, м, CH2-Ph). 2.43 (1H, м, HC–C=O), 3.28 (1H, м, HC–C=O), 3.55 (4H, м, CH2), 4.30 (2Н, м, O–СH2), 4.85 (1H, м, CHN), 6.05 (2H, м, HC=CH), 7.12 (2Н, д, J 7.2, Ar), 7.29 (3Н, м, Ar). Масс-спектр (ИЭР), m/z: найдено для С19H21NO4: 327.146. Вычислено для С19H21NO4: 327.147.
Этиловый эфир 2-(3,5-диоксо-4-азатрицикло-[5.2.1.02,6]дец-8-ен-4-ил)-3-фенилпропановой кислоты (XVI). С20H21NO4. Выход 75%. Масло. ИК: 1748 (С=О, сложный эфир); 1709 (C=O, имид); 1603 (Ar); 1210 (C–O–). 1H-ЯМР: 1.24 (3H, м, CH3), 1.43 (2H, м, CH2-Ph), 3.16 (1H, м, HC–C=O), 3.24 (1H, м, HC–C=O), 3.44 (2H, м, CH2-мостик), 3.46 (1H, м, CHN), 3.48 (1H, м, CH), 4.22 (2Н, м, O–СH2), 4.94 (1H, м, CH), 5.73 (1H, д, CH=CH), 5.76 (1H, д, CH=CH), 7.23 (2Н, д, J 7.0, Ar), 7.26 (3Н, м, Ar). Масс-спектр (ИЭР), m/z: найдено для С20H21NO4: 339.147. Вычислено для С20H21NO4: 339.147.
Этиловый эфир 2-(1,3-диоксо-3a,4-дигидро-1H-изоиндол-2(3H,7H,7aH)-ил)-3-метилпентановой кислоты (XVII). С16H23NO4. Выход 45%. Масло. ИК: 1758 (С=О, сложный эфир); 1715 (C=O, имид); 1211 (C–O–). 1Н-ЯМР: 0.79 (6Н, м, CH3), 1.34 (1Н, м, CH), 1.52 (3Н, м, СН3-эфир), 1.67 (2Н, м, CH2), 2.26 (2Н, м, O–CH2), 2.61 (2Н, м, HC–C=O), 3.10 (2Н, м, CH2), 4.16 (2Н, м, CH2), 4.75 (1Н, м, CHN), 5.90 (2Н, м, HC=CH). Масс-спектр (ИЭР), m/z: найдено для С16H23NO4: 293.1625. Вычислено для С16H23NO4: 293.1627.
Этиловый эфир 2-(3,5-диоксо-4-азатрицикло-[5.2.1.02,6]дец-8-ен-4-ил)-3-метилпентановой кислоты (XVIII). С17H23NO4. Выход 82%. Масло. ИК: 1758 (С=О, сложный эфир); 1705 (C=O, имид); 1209 (C–O–). 1Н-ЯМР: 0.71 (6Н, м, CH3), 1.41 (2H, м, O–CH2), 1.62 (2Н, м, CH2-мостик), 2.33 (1Н, м, CH), 2.40 (2Н, м, CH2), 3.35 (3H, м, CH3-эфир) 3.40 (2Н, м, HC–C=O), 3.45 (2Н, м, CH), 4.10 (1Н, м, CHN), 6.01 (2Н, м, HC=CH). Масс-спектр (ИЭР), m/z: найдено для С17H23NO4: 305.163. Вычислено для С17H23NO4: 305.162.
Исследование генотоксичности с использованием Allium-теста [35]. В качестве объекта исследования использовали лук репчатый Allium cepa (L.) сорта Штуттгартер Ризен (ООО “Лукаморе”, Россия). Для проведения опытов отбирали выровненный материал – типичные для используемого сорта луковицы одинакового размера. Луковицы помещали в емкости с растворами исследуемых соединений (в концентрациях 0.1, 0.01 и 0.001%) и контрольными растворами – дистиллированной водой и 0.25%-ным водным этанолом. Материал проращивали на свету в течение 3 сут. Далее с каждой луковицы срезали корни и промывали их водой. Затем определяли прорастание корней и их длину. Для каждой концентрации растворов исследуемых соединений проводили пять опытов. Ошибка при определении длины корней растений составляла 1.1–2.2%. Полученный материал (корни) фиксировали при помощи фиксатора Кларка (96%-ный этиловый спирт с ледяной уксусной кислотой, 3 : 1) в течение 3 сут. Перед окрашиванием корни отмывали от спирта в воде для лучшего прокрашивания препарата, а затем помещали в краситель – 2%-ный ацетоорсеин. Тигель с красителем и корни нагревали в пламени спиртовки до появления паров на покровном стекле. Окрашивание производили минимум 40 мин. Далее готовили давленые препараты корневой меристемы. Корни отмывали от красителя в 45%-ной уксусной кислоте. От корней отрезали кончик длиной 2–3 мм, помещали на предметное стекло в каплю 45%-ной уксусной кислоты, накрывали покровным стеклом и производили давление препарата до образования монослоя клеток. Препараты анализировали с использованием микроскопа МИКРОМЕД-1 (Микромед, Россия; увеличение 400×). На препаратах наблюдали мелкие меристематические клетки с хорошо прокрашенными ядрами. Учитывали делящиеся клетки на всех фазах митоза, отдельно регистрировали нормальные клетки на стадиях анафазы и телофазы, а также клетки на этих фазах с хромосомными аберрациями. Далее рассчитывали митотический индекс, частоту ХА и проводили статистическую обработку результатов. Для определения достоверности различий между средними значениями контрольных (дистиллированная вода и 0.25%-ный водный этанол) и опытного вариантов использовали t-критерий Стьюдента. Отклонение считали достоверным при p < 0.05. На рис. 1–6 все отклонения между контрольными и опытными вариантами достоверные.
Исследование частоты видимых мутаций с использованием Chlorella vulgaris. Для изучения генотоксического влияния использовали одноклеточную зеленую водоросль Chlorella vulgaris (Bejer.) штамма ЛАРГ-1, полученного из Института общей генетики им. Н.И. Вавилова РАН. Штамм ЛАРГ-1 – термофильный мутант, полученный из штамма Б-45, выделенного из озера Байкал. Готовили суспензию клеток хлореллы на дистиллированной воде. Осуществляли серию разведений для достижения плотности клеток 105 кл./мл. Клетки подсчитывали в камере Горяева. Затем в сухую пробирку вносили 1 мл рабочей суспензии хлореллы и 1 мл исследуемого раствора соединения с концентрациями 0.1, 0.01 и 0.001%. Пробирку оставляли на 2 ч для контакта с соединением, периодически перемешивая содержимое пробирки путем встряхивания. После этого в чашки Петри высевали по 0.1 мл суспензии, равномерно распределяли клетки на поверхности среды шпателем. В контрольном варианте суспензия хлореллы взаимодействовала с 1 мл дистиллированной воды. Во всех вариантах опыт проводили в трехкратной повторности. Инкубацию культуры осуществляли в люминостате (Флора-1, Россия) в течение 10 сут при 25°С. Учет видимых мутаций проводили с использованием стереомикроскопа Альтами СМ07 (Альтами, Россия) с увеличением от 15× до 40×. Регистрировали все выросшие колонии. Колонии, измененные по форме, цвету или размерам, учитывали как мутантные [35].
Исследование генотоксического эффекта методом доминантных летальных мутаций у Drosophila melanogaster. Для исследования использовали линию дикого типа D-32 Drosophila melanogaster (Meigen) из коллекции Института биология развития им. Н.К. Кольцова РАН (Москва, Россия). Постановку эксперимента проводили по стандартной методике. Самцы получали изучаемые препараты перорально. Воздействию изучаемых соединений (в концентрации 0.1, 0.01 и 0.001%) подвергали 3-дневных самцов, которых помещали в стаканчики на стеклянные фильтры, опущенные в растворы изучаемых концентраций препаратов в 5%-ном растворе сахарозы. Время экспозиции – 3 сут. Изучаемые препараты обновляли ежесуточно. Параллельно с опытом ставили контрольный эксперимент – самцов поили 5%‑ным раствором сахарозы. Далее подопытных самцов скрещивали с виргинными самками. Затем самцов отбирали. Оплодотворенных самок помещали в колбы с голодным агаром, который меняли каждые 12 ч. Проводили учет отложенных яиц, через 48 ч инкубации подсчитывали число яиц с невылупившимися личинками. Определяли частоту ДЛМ (D, %) [35].
ЗАКЛЮЧЕНИЕ
Разработаны методы синтеза эфиров карбоновых кислот на основе циклоалкендикарбоновых кислот, позволяющие получать продукты в мягких условиях и с высоким выходом. Данными методами получены восемь новых эфиров карбоновых кислот с циклоалкенильным фрагментом.
Анализ генотоксической активности полученных соединений, проведенный с использованием системы токсикогенетических методов, показал, что исследованные соединения по-разному влияют на различные тест-объекты (тест учета мутагенности, митотоксичности и токсичности у лука Allium cepa (L.), тест учета доминантных летальных мутаций (ДЛМ) у Drosophila melanogaster (Meigen) и тест учета выживаемости и видимых мутаций у Chlorella vulgaris (Bejer)). Изученные эфиры не увеличивали частоту ДЛМ у дрозофилы, но при этом достоверно увеличивали частоту мутаций у одноклеточной зеленой водоросли хлореллы. A. cepa чувствителен к исследуемым соединениям, при 0.1%-ной концентрации соединений зарегистрирована острая токсичность; при 0.01%-ной и 0.001%-ной концентрациях наблюдался нормальный рост корневой системы, несмотря на некоторое снижение пролиферативной активности. При этом частота ХА достоверно увеличивалась, т.е. исследуемые соединения не активны по отношению к многоклеточному животному – дрозофиле и обладают способностью индуцировать хромосомные перестройки у растений. Таким образом, исследованные соединения обладают специфическим действием в различных тест-системах. Поэтому при оценке генотоксической активности вновь синтезированных соединений необходимо всегда использовать систему методов, регистрирующих различные типы генетических нарушений.
С начала развития лекарственной противоопухолевой терапии (40-е гг. XX века) и по настоящее время главным образом используются агенты, механизм действия которых основан на ингибировании пролиферации опухолевых клеток путем нарушения целостности молекул ДНК или блокирования процессов транскрипции и митоза [38]. Несмотря на существование в клинической практике более 100 противоопухолевых препаратов, эффективность большинства из них недостаточна, и спектр онкологических заболеваний, чувствительных к химиотерапии, ограничен. Поэтому остается актуальным вопрос о разработке новых более активных препаратов, а также поиск соединений, эффективных при опухолях с первичной и приобретенной резистентностью к лекарственной терапии. Представленные в статье соединения могут найти применение в качестве препаратов с противоопухолевым действием, но необходимы доклинические исследования.
Список литературы
Yabuno T., Konishi N., Nakamura M., Tsuzuki T., Tsunoda S., Sakaki T., Hiasa Y. // J. Neurooncol. 1998. V. 36. P. 105–112. https://doi.org/10.1023/a:1005878402133
Perez-Tomas R. // Curr. Med. Chem. 2006. V. 13. P. 1859–1876. https://doi.org/10.2174/092986706777585077
Fu X., Palomar A.J., Hong E.P. // J. Nat. Prod. 2004. V. 67. P. 1415–1418. https://doi.org/10.1021/np0499620
Bailly C., Carrasco C., Joubert A. // Biochemistry. 2003. V. 42. P. 4136–4150. https://doi.org/10.1021/bi027415c
Richardson M.B., Gabriel K.N., Garcia J.A. // Bioconjugate Chem. 2020. V. 31. P. 1449−1462. https://doi.org/10.1021/acs.bioconjchem.0c00143
Yang H., Dou W., Lou J., Leng Y., Shen J. // Bioorg. Med. Chem. Lett. 2008. V. 18. P. 1340–1345. https://doi.org/10.1016/j.bmcl.2008.01.020
Arnaldi G., Angeli A., Atkinson A.B., Bertagna X., Cavagnini F., Chrousos G.P., Fava G.A., Findling J.W., Gaillard R.C., Grossman A.B., Kola B., Lacroix A., Mancini T., Mantero F., Newell-Price J., Nieman L. K., Sonino N., Vance M.L., Giustina A., Boscaro M.J. // J. Clin. Endocrinol. Metab. 2003. V. 88. P. 5593–5602. https://doi.org/10.1210/jc.2003-030871
Grundy S.M., Brewer H.B., Cleeman J.I., Smith S.C., Lenfant C. // Circulation. 2004. V. 109. P. 433–438. https://doi.org/10.1161/01.CIR.0000111245.75752.C6
Inagaki K., Otsuka F., Miyoshi T., Watanabe N., Suzuki J., Ogura T., Makino H. // Endocr. J. 2004. V. 51. P. 201–206. https://doi.org/10.1507/endocrj.51.201
Diederich S., Grossmann C., Hanke B., Quinkler M., Herrmann M., Bahr V., Oelkers W. // Eur. J. Endocrinol. 2000. V. 142. P. 200–207. https://doi.org/10.1530/eje.0.1420200
Alberts P., Nilsson C., Selen G., Engblom L.O.M., Edling N.H.M., Norling S., Klingstrom G., Larsson C., Forsgren M., Ashkzari M., Nilsson C.E., Fiedler M., Bergqvist E., Ohman B., Bjorkstrand E., Abrahmsen L.B. // Endocrinology. 2003. V. 144. P. 4755–4762. https://doi.org/10.1210/en.2003-0344
Gu X., Dragovic J., Koo G.C., Koprak S.L., LeGrand C., Mundt S.S., Shah K., Springer M.S., Tan E.Y., Thieringer R., Hermanowski-Vosatka A., Zokian H.J., Balkovec J.M., Waddell S.T. // Bioorg. Med. Chem. Lett. 2007. V. 17. P. 2838–2843. https://doi.org/10.1016/j.bmcl.2005.08.052
Miguet L., Zhang Z., Barbier M., Grigorov M.G. // J. Comput. Aided Mol. Des. 2006. V. 20. P. 67–81. https://doi.org/10.1007/s10822-006-9037-3
Schuster D., Maurer E.M., Laggner C., Nashev L.G., Wilckens T., Langer T., Odermatt A. // J. Med. Chem. 2006. V. 49. 3454–3466. https://doi.org/10.1021/jm0600794
Gierasch L.M. // Annu. Rev. Biochem. 1992. V. 61. P. 387–418. https://doi.org/10.1146/annurev.bi.61.070192.002131
Liskamp R.M. // Recl. Trav. Chim. Pays-Bas. 1994. V. 113. P. 1–19. https://doi.org/10.1002/recl.19941130102
Gante J. // Angew. Chem. Int. Ed. Engl. 1994. V. 33. P. 1699–1720. https://doi.org/10.1002/anie.199416991
Jiang K., Shimotakahara H., Luo M., Otani M., Nakamura H., Moselhy S.S., Abualnaja K.O., Labeed Al-Malki A., Kumosani T. A., Kitahata N., Takeshi N., Nakajima M., Asami T. // Bioorg. Med. Chem. Lett. 2017. V. 27. P. 3678–3682. https://doi.org/10.1016/j.bmcl.2017.07.012
Jiang K., Otani M., Shimotakahara H., Yoon J.-M., Park S.-H., Miyaji T., Nakano T., Nakamura H., Nakajima M., Asami T. // Plant Physiology. 2017. V. 173. P. 825–835. https://doi.org/10.1104/pp.16.00937
Gupta R., Chakrabarty S.K. // Plant Signal Behav. 2013. V. 8. P. 255–264. https://doi.org/10.4161/psb.25504
Rodaway S.J., Gates D.W., Brindle C. // Plant Growth Regul. 1991. V. 10. P. 243–259. https://doi.org/10.1007/BF00024415
Gallego-Giraldo C., Hu J., Urbez C., Gomez M.D., Sun T.-P., Perez-Amador M.A. // Plant J. 2014. V. 79. P. 1020–1032. https://doi.org/10.1111/tpj.12603
Chandler P.M., Harding C.A., Ashton A.R., Mulcair M.D., Dixon N.E., Mander L.N. // Mol. Plant. 2008. V. 1. P. 285–294. https://doi.org/10.1093/mp/ssn002
Sukiran N.A., Pollastri S., Steel P.G., Knight M.R. // Plant Direct. 2022. V. 26. P. 394–398. https://doi.org/10.1002/pld3.398
Wang X., Baloch S.K., Ma L., Wang X., Shi J., Zhu Y., Wu F., Pang Y., Lu G., Qi J., Gu H., Yang Y. // RSC Adv. 2015. V. 5. P. 31759–31767. https://doi.org/10.1039/C5RA01872B
Yang J., Sun W., He Z., Yu Ch., Bao G., Li Y., Hong L., Wang R. // Org. Lett. 2018. V. 20. P. 7080–7084. https://doi.org/10.1021/acs.orglett.8b03020
Verma S.M., Singh R.M. // J. Org. Chem. 1975. V. 40. P. 897–901. https://doi.org/10.1021/jo00895a019
Bodtke A., Otto H.-H. // Pharmazie. 2005. V. 60. P. 803–813.
Ranganathan D., Haridas V., Kurur S., Thomas A., Madhusudanan K.P., Nagaraj R., Kunwar A.C., Sarma A.V.S., Karle I.L. // J. Am. Chem. Soc. 1998. V. 120. P. 8448–8460. https://doi.org/10.1021/ja980143+
Фирстова А.А., Кофанов Е.Р., Закшевская В.М., Ковалева М.И. // Биоорг. химия. 2019. Т. 45. С. 204–213. [Firstova A.A., Kofanov E.R., Zakshevskaya V.M., Kovaleva M.I. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 204–213.] https://doi.org/10.1134/S0132342319030023
Травень В.Ф. // Органическая химия: учебник для вузов; в 2 т. Москва: ИКЦ “Академкнига”, 2004.
Островский В.А. // Соросовский образовательный журнал. 2000. Т. 6. № 11. С. 30–34.
Межфазный катализ: химия, катализаторы и применение / Под ред. Старкс Ч.М. Москва: Химия, 1991. 157 с.
Jaszay Z.M., Petnehazy I., Tokke L. // Synthesis. 1989. P. 745–747. https://doi.org/10.1055/s-1989-27380
Прохорова И.М., Ковалева М.И., Фомичева А.Н. // Генетическая токсикология: учебное пособие. Ярославль, 2005. 132 с.
Легостаева Т.Б., Ингель Ф.И., Антипанова Н.А., Ююрченко В.В., Юрцева Н.А., Котляр Н.Н. // Гигиена и санитария. 2010. № 4. С. 47–52.
Груммитт О. // Синтезы органических препаратов. Москва: Издательство иностранной литературы, 1952.
Безбородова О.А., Панкратов А.А., Немцова Е.Р., Венедиктова Ю.Б., Воронцова М.С., Енгалычева Г.Н., Сюбаев Р.Д. // Ведомости Научного центра экспертизы средств медицинского применения. 2020. Т. 10. № 2. С. 96–110.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия