

Биоорганическая химия, 2023, T. 49, № 2, стр. 165-177
Синтез и трансфицирующая активность дисульфидных поликатионных амфифилов
И. А. Петухов 1, П. А. Пучков 1, *, Н. Г. Морозова 1, М. А. Зенкова 2, М. А. Маслов 1, **
1 Институт тонких химических технологий им. М.В. Ломоносова, МИРЭА – Российский технологический университет
119571 Москва, просп. Вернадского, 86, Россия
2 Институт химической биологии и фундаментальной медицины СО РАН
630090 Новосибирск, просп. Академика Лаврентьева, 8, Россия
* E-mail: puchkov_pa@mail.ru
** E-mail: mamaslov@mail.ru
Поступила в редакцию 23.06.2022
После доработки 18.07.2022
Принята к публикации 12.08.2022
- EDN: GIHSNO
- DOI: 10.31857/S0132342323010232
Аннотация
Осуществлен синтез новых поликатионных амфифилов, содержащих в своей структуре дисульфидную группу. На основе полученных соединений и липида-хелпера 1,2-диолеоил-sn-глицеро-3-фосфатидилэтаноламина сформированы катионные липосомы, которые продемонстрировали отсутствие токсичности для клеток НЕК293 и HeLa и высокую эффективность доставки флуоресцентно-меченого олигодезоксирибонуклеотида. Эффективность доставки плазмидной ДНК pEGFP-C2 зависела от клеточной линии и структуры амфифила, при этом липосомы на основе тетракатионных амфифилов оказались наиболее эффективными трансфектантами, которые могут быть использованы для трансфекции эукариотических клеток in vitro, а также для проведения дальнейших биологических испытаний in vivo.
ВВЕДЕНИЕ
Быстрое развитие технологии рекомбинантных ДНК, методов переноса плазмидных ДНК в клетку и выяснение молекулярных основ многих заболеваний привело к возникновению новой области медицины – генной терапии. Этот метод лечения наследственных и приобретенных заболеваний основан на введении в клетки терапевтических нуклеиновых кислот (НК) с целью направленного устранения генетических дефектов или придания клеткам новых функций [1]. Главное условие успешного применения генной терапии – эффективная доставка НК в клетки-мишени и создание условий для ее длительного функционирования внутри клеток.
В настоящее время наиболее эффективными системами доставки НК считаются вирусные векторы [2], однако они имеют ряд серьезных недостатков. Это стимулирует разработку альтернативных подходов, одним из которых выступает липофекция – метод доставки НК с помощью катионных липосом (КЛ). К преимуществам катионных липосом относятся неинфекционность, способность переносить НК разного размера и защищать их от действия клеточных ферментов, а также стабильность при хранении и экономическая доступность [3]. В настоящее время КЛ, составляющие основу фармацевтических композиций, включающих НК, проходят клинические испытания для лечения ряда заболеваний [4], более того, КЛ используются в качестве платформ при создании мРНК-вакцин против COVID-19 [5]. Существенный недостаток известных на сегодняшний день систем доставки НК на основе липосом – их низкая эффективность, обусловленная наличием внеклеточных и внутриклеточных биологических барьеров [6], которые должен преодолеть НК-липидный комплекс (липоплекс), прежде чем НК проявит свою биологическую активность. К внеклеточным барьерам можно отнести неспецифическое взаимодействие липоплексов с компонентами крови и белками иммунной системы, в результате которых возможны дестабилизация липоплекса и преждевременное высвобождение НК, а основными внутриклеточными барьерами, приводящими к снижению эффективности доставки, выступают эндосомальная и ядерная мембраны.
Трансфицирующую активность катионного амфифила, а также его стабильность в биологических системах и токсичность определяет тип связывания гидрофобного и гидрофильного доменов. Устойчивые липиды с простой эфирной связью более токсичны по сравнению с ацильными липидами, которые легко гидролизуются в клетке эндогенными эстеразами. Наиболее удачное сочетание стабильности и токсичности амфифила обеспечивает карбамоильный линкер [7]. Кроме того, для увеличения эффективности высвобождения НК из эндосом и липоплекса в структуру катионных амфифилов вводят лабильные структурные модули, которые разрушаются под действием внутриклеточных факторов и агентов, приводя к дестабилизации липоплекса [8, 9]. Один из типов таких катионных амфифилов – липиды с дисульфидными связями, разрушаемые под действием внутриклеточных восстановителей (NADPH, глутатион, редуктазы) [10, 11].
Ранее в нашей лаборатории был синтезирован поликатионный амфифил Х2, в структуре которого остаток холестерина был присоединен к первичной аминогруппе спермина через карбамоильный линкер [12]. Целью данной работы стал синтез новых поликатионных амфифилов S1–S3 – аналогов амфифила Х2, содержащих в своей структуре дисульфидную группу. Амфифилы S1–S3 различаются линкером, связывающим спермин и гидрофобный домен (сложноэфирный, карбамоильный), длиной спейсера, а также количеством катионных аммонийных групп (трикатионные и тетракатионные амфифилы). В работе проведена предварительная оценка способности катионных липосом, сформированных из амфифилов S1–S3 и липида-хелпера 1,2-диолеоил-sn-глицеро-3-фосфатидилэтаноламина (DOPE), доставлять в эукариотические клетки короткие и протяженные нуклеиновые кислоты – синтетический олигодезоксирибонуклеотид и плазмидную ДНК (пДНК).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Для создания поликатионных амфифилов в качестве основных структурных компонентов были выбраны природные соединения – холестерин и спермин (рис. 1). Известно, что катионные амфифилы, содержащие в качестве гидрофобного остатка холестерин, обладают высокой трансфицирующей активностью, низкой токсичностью и применяются для исследования механизмов слияния искусственных мембран и структурно-функционального изучения сформированных на их основе липоплексов [13]. Природные полиамины, в том числе спермин, способны упаковывать ДНК в тороидальные и стержневые структуры, при этом метиленовые фрагменты, разделяющие атомы азота, играют важную роль во взаимодействии полиамина с двойной спиралью ДНК. Среди липофильных полиаминов производные спермина наиболее эффективно связывают и упаковывают ДНК и поэтому лучше переносят ее в клетки. На основе спермина были созданы некоторые коммерческие препараты для трансфекции [14–16].

Один из наиболее простых способов получения амфифилов с дисульфидной связью заключается в присоединении к молекуле холестерина спейсера, содержащего дисульфидную группу. Такие спейсеры (IIa, b) были получены с количественными выходами окислением перекисью водорода 2‑меркаптоуксусной и 3‑меркаптопропионовой кислот соответственно [17].
Ацилирование холестерина (I) дитиодиуксусной (IIa) и дитиодипропионовой кислотами (IIb) с использованием в качестве конденсирующего агента N,N'‑дициклогексилкарбодиимида (DCC) в присутствии N,N‑диметиламинопиридина (DMAP) приводило к получению производных холестерина (IIIa, b) со сложноэфирной связью между стероидным остатком и спейсером (рис. 2). Варьирование соотношениями реагентов показало, что использование двухкратного избытка дикарбоновых кислот (IIa, b) и 0.5 экв. DMAP приводит к образованию соединений (IIIa, b) с выходом 67 и 45% соответственно, тогда как при использовании других конденсирующих агентов (BOP, CDI, Boc2O) выход соединений (IIIa, b) не превышал 10%.

Для создания амфифила с карбамоильным линкером холестерин (I) обрабатывали 1.1-кратным избытком 1,1'‑карбонилдиимидазола (CDI), получая имидазолид (IV) [12], дальнейшее взаимодействие которого с избытком цистамина приводило к получению соединения (V) с выходом 59%.
Синтез дисульфидных трикатионных амфифилов (S1 и S2) осуществляли конденсацией три-Boc-защищенного спермина (VI) [18] и карбоксипроизводных холестерина (IIIa, b) с использованием различных активирующих агентов (рис. 3, табл. 1). Наибольший выход соединений (VIIa, b) был достигнут при использовании TBTU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат). После удаления Вос-защитных групп действием 4 н. хлористого водорода в диоксане получали трикатионные амфифилы (S1 и S2) с количественным выходом.
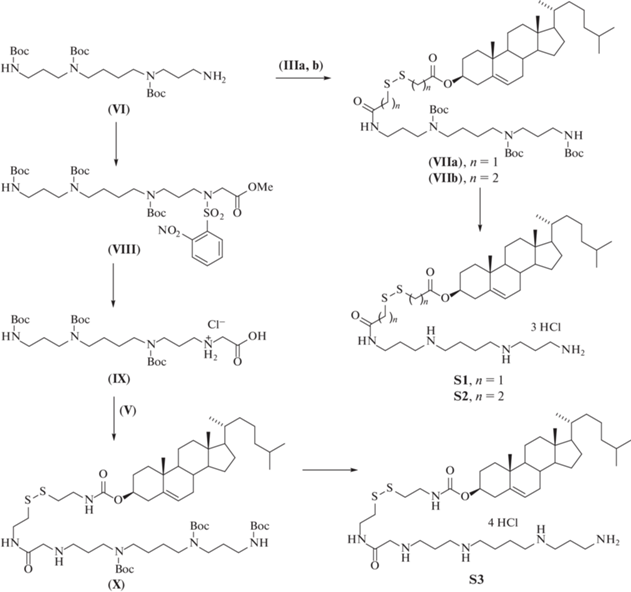
Таблица 1.
Оптимизация условий синтеза соединений (VIIa, b)
| Карбоксильная компонента | Конденсирую-щий агент | Выход соединений (VIIa, b), % |
|---|---|---|
| (IIIa) | BOP | 28 |
| DCC/DMAP | 27 | |
| TBTU | 60 | |
| (IIIb) | TBTU | 82 |
Для синтеза тетракатионного амфифила (S3) на первом этапе было получено карбоксипроизводное спермина (IX). Для этого проводили N‑сульфонилирование соединения (VI) под действием 1.2-кратного избытка 2-нитробензолсульфонилхлорида в присутствии триэтиламина, получая соответствующий амид, в результате последующего алкилирования которого метиловым эфиром бромуксусной кислоты получали соединение (VIII) с выходом 77% на две стадии. Последовательное удаление 2‑нитробензолсульфонильной группы под действием 10-кратного избытка тиофенола в присутствии карбоната цезия и омыление метилового эфира раствором гидроксида натрия приводило к образованию соединения (IX) с выходом 85% (рис. 3).
Целевой тетракатионный амфифил (S3) получали конденсацией полиаминного синтона (IX) и аминопроизводного холестерина (V) в присутствии TBTU. После хроматографической очистки на силикагеле соединение (X) было выделено с выходом 92%. В результате последующего удаления Boc-групп получали целевой амфифил S3 с выходом 81%.
Изучение свойств полученного ряда соединений позволяет оценить влияние структуры дисульфидных катионных амфифилов на их цитотоксичность и трансфицирующую активность. Для этого методом гидратации липидной пленки [19] были получены КЛ, состоящие из поликатионных амфифилов S1, S2 или S3 и липида-хелпера DOPE в соотношении 1 : 1 (мольн.). Описанные ранее катионные липосомы Х2-DOPE [20], не содержащие дисульфидную связь, использовали во всех экспериментах для сравнения.
Цитотоксичность КЛ оценивали с помощью МТТ-теста на трех линиях эукариотических клеток (HEK293, HeLa и ВНК21). Клетки инкубировали с КЛ в диапазоне концентраций 2.5–40.0 мкМ в течение 24 ч в присутствии 10% эмбриональной бычьей сыворотки (FBS). Для клеток HEK293 и HeLa значение IC50 (концентрация КЛ, при которой наблюдается гибель 50% клеток в популяции) было достигнуто только для липосом Х2-DOPE (табл. 2). Клетки ВНК21 оказались более чувствительны к цитотоксическому действию КЛ, особенно S1-DOPE и S3-DOPE, тогда как наименьшей цитотоксичностью в отношении всех использованных линий клеток обладали липосомы S2-DOPE, содержащие трикатионный амфифил с более длинным спейсером.
Способность КЛ образовывать липоплексы при различных соотношениях N/P (отношение количества аминогрупп катионного амфифила к количеству фосфатных групп НК) – это важная характеристика, которая может оказывать значительное влияние на эффективность трансфекции. Для изучения эффективности связывания КЛ с НК был использован метод задержки в геле [21]. Для всех КЛ при соотношении N/P 2/1 наблюдалось неполное включение плазмидной ДНК pEGFP-С2, кодирующей усиленный зеленый флуоресцентный белок, в состав липоплекса, о чем свидетельствует наличие полосы свободной ДНК, мигрирующей в геле (рис. 4). При увеличении соотношения N/P до 4/1 для липосом Х2-DOPE и S3-DOPE наблюдалось полное связывание пДНК, тогда как для липосом S1-DOPE и S2-DOPE полное связывание пДНК происходило при соотношениях N/P 6/1 и 8/1 соответственно. Таким образом, наименьшей эффективностью связывания обладают липосомы S2-DOPE: для этих КЛ полное связывание пДНК происходило только при N/P 8/1.
Рис. 4.
Связывание катионных липосом с пДНК при разных соотношениях N/P, определенное методом задержки в геле. Электрофореграмма 1.5%-ного агарозного геля после разделения комплексов липосом с пДНК. Окрашивание бромистым этидием. К – свободная пДНК.

В качестве объектов для доставки в эукариотические клетки были выбраны неспецифичный короткий 25-звенный олигодезоксирибонуклеотид, меченный флуоресцеином по 5'-концу (FITC-ODN), и пДНК pEGFP-С2. Липоплексы формировали при разных соотношениях N/P, а эффективность накопления/доставки НК в клетках анализировали с помощью проточной цитофлуориметрии, определяя и количество трансфицированных клеток, и уровень интенсивности флуоресценции клеток в популяции. Эксперименты проводили в присутствии 10% FBS в ростовой среде, поскольку известно, что наличие FBS в среде может влиять и на стабильность липоплексов, образованных липосомами, и на их взаимодействие с клетками [22]. Эффективность доставки НК в клетки сравнивали с коммерческим препаратом Липофектамином 2000 (Lf2000).
В случае доставки FITC-ODN увеличение соотношения N/P приводило к увеличению числа трансфицированных клеток: наибольшую эффективность трансфекции проявляли липосомы S2-DOPE и S3-DOPE при соотношении N/P 3/1 (рис. 5). Следует отметить, что процент клеток, трансфицированных FITC-ODN, и в случае дисульфидных липосом (S2-DOPE и S3-DOPE), и в случае липосом Х2-DOPE, нечувствительных к восстановлению, был примерно одинаковым, а средняя интенсивность флуоресценции в случае липосом S3-DOPE была существенно выше по сравнению со всеми остальными композициями, что говорит о более эффективном накоплении FITC-ODN в клетках.
Рис. 5.
Накопление флуоресцентно-меченого олигонуклеотида в клетках НЕК293 в составе комплексов с катионными липосомами при различных соотношениях N/P в присутствии сыворотки крови в ростовой среде. Процент трансфицированных клеток (а) и средняя интенсивность флуоресценции клеток в популяции (б) определяли методом проточной цитометрии. Стандартное отклонение не превышало 7–9%.

Результаты трансфекции клеток HEK293 пДНК показали, что КЛ на основе дисульфидных трикатионных амфифилов S1 и S2 неэффективно доставляют пДНК в клетки (рис. 6а, 6б). Наибольшая трансфицирующая активность наблюдалась для липосом S3‑DOPE и X2-DOPE при N/P 6/1, а дальнейшее увеличение количества амфифила в липоплексе приводило к снижению эффективности доставки.
Рис. 6.
Трансфекция клеток НЕК293 (а, б), HeLa (в, г) и ВНК21 (д, е) комплексами катионных липосом с пДНК (pEGFP-C2) при различных соотношениях N/P. Процент трансфицированных клеток (а, в, д) и среднюю интенсивность флуоресценции клеток в популяции (б, г, е) определяли методом проточной цитометрии через 44 ч после трансфекции клеток по уровню экспрессии EGFP. Стандартное отклонение не превышало 7–9%.

Аналогичный эксперимент по трансфекции клеток HeLa показал схожие результаты – наиболее эффективными были липосомы S3‑DOPE и X2-DOPE на основе тетракатионных амфифилов, причем эффективность доставки пДНК возрастала с увеличением количества катионных липосом в липоплексе. Тем не менее трансфицирующая активность КЛ S3‑DOPE и X2-DOPE была ниже таковой для Липофектамина 2000. Доставка пДНК в клетки ВНК21 была менее эффективной. Возможно, это связано с меньшей скоростью деления клеток ВНК21 по сравнению с клетками НЕК293 и HeLa [23]. В случае клеток BHK21 только липосомы S3-DOPE обеспечивали сопоставимый с Липофектамином 2000 уровень трансфекции по числу EGFP-положительных клеток, но меньшую среднюю интенсивность флуоресценции клеточной популяции.
Влияние длины спейсера (амфифилы S1 и S2) на эффективность доставки пДНК не было однозначно установлено. Различия в эффективности доставки разных типов НК обусловлены тем, что для FITC-ODN фиксируется сигнал флуоресценции после его проникновения в составе комплексов через плазматическую мембрану, тогда как для пДНК регистрируется интенсивность флуоресценции экспрессированного клетками белка EGFP после проникновения комплекса в цитозоль, высвобождения из эндосом и транспорта пДНК в ядро клетки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали растворители отечественного производства, а также DMF, карбонат калия, метиловый эфир бромуксусной кислоты, 2‑нитробензолсульфонилхлорид, триэтиламин, 4 н. раствор HCl в диоксане (Aldrich, США); TBTU, карбонат цезия (Fluka, США); тиофенол (Merck, Германия). ТСХ проводили на пластинках Kieselgel 60 F254 (Merck, Германия) в следующих системах: CHCl3–MeOH, 10 : 0.6 (А), 40 : 1 (Б), 10 : 1 (В); CHCl3–MeOH–PriNH2, 4 : 1 : 2 (Г); CHCl3–MeOH–CH3COOH, 200 : 1 : 0.3 (Д); CHCl3–MeOH–CH3COOH, 1 : 0.16 : 0.05 (Е); 1 : 0.12 : 0.05 (Ж). Пятна на хроматограммах обнаруживали с использованием раствора фосформолибденовая кислота–сульфат церия(IV) с последующим прогреванием при 120°С, паров хлора с последующим опрыскиванием раствором о‑толуидина или при УФ-облучении (254 нм). Колоночную хроматографию проводили на силикагеле Kieselgel 60 (0.040–0.063 мм; Merck, Германия). Спектры 1Н- и 13C-ЯМР регистрировали на импульсном Фурье-спектрометре DPX‑300 (Bruker, Германия) в CDCl3, если не указано иное (внутренний стандарт – тетраметилсилан). Значения химических сдвигов (δ) приведены в миллионных долях (м.д.), КССВ (J) – в герцах (Гц). Масс-спектры получали на времяпролетном масс-спектрометре Ultraflex (Bruker, Германия) методом лазерной десорбции/ионизации с использованием в качестве матрицы 2,5‑дигидроксибензойной кислоты.
Синтез имидазолида (IV) [12], N1,N4,N9-три-трет-бутоксикарбонил-1,12-диамино-4,9-диазадодекана (VI) [18], соединения (IIIa) [24] и амфифила Х2 [12] осуществляли согласно описанным ранее методам.
3-[3-(Холест-5-ен-3β-илоксикарбонил)пропил]дитиопропионовая кислота (IIIb). К раствору холестерина (I) (1.0 г, 2.59 ммоль) в безводном этилацетате (40 мл), охлажденному до 0°С, добавляли в атмосфере аргона дитиодипропионовую кислоту (1.09 г, 5.17 ммоль), DCC (1.067 мг, 5.17 ммоль), а затем DMAP (0.158 мл, 1.29 ммоль) и Et3N (0.54 мл, 3.88 ммоль). Через 12 ч реакционную массу фильтровали, промывали 3%-ным водным раствором соляной кислоты (3 × 30 мл), сушили над Na2SO4, фильтровали, растворители удаляли в вакууме. После хроматографии на силикагеле (CHCl3–MeOH–CH3COOH, 25 : 0.1 : 0.03) получили соединение (IIIb) в виде кристаллов белого цвета (0.681 г, 45%). Rf 0.40 (Д). Масс-спектр, m/z: 601.217 [М + Na]+, вычислено для C33H54O4S2 578.346 [М]+. Спектр 1Н-ЯМР: 0.67 (с, 3Н, C(13)Me), 0.85 (д, 3Н, J 6.6, C(25)Me), 0.86 (д, 3Н, J 6.6, C(25)Me), 0.91 (д, 3Н, J 6.5, C(20)Me), 1.01 (с, 3Н, C(10)Me), 0.95–1.70 (м, 21Н, протоны Chol), 1.75–2.10 (м, 5H, протоны Chol), 2.26–2.40 (м, 2Н, H2С(4)), 2.70 (т, 2Н, J 7.1, CH2COOChol), 2.79 (т, 2Н, J 7.5, CH2COOH), 2.92 (т, 2Н, J 7.1, CH2S), 2.92 (т, 3Н, J 7.5, CH2S), 4.52–4.78 (м, 1 Н, H(3)), 5.30–5.45 (м, 1Н, H(6)). Спектр 13С-ЯМР: 11.83, 18.69, 19.28, 21.00, 22.54, 22.80, 23.81, 24.26, 27.73, 27.98, 28.20, 31.82, 31.87, 32.70, 33.31, 33.86, 34.42, 35.76, 36.16, 36.54, 36.92, 38.04, 39.49, 39.69, 49.98, 56.11, 56.65, 74.54, 122.74, 139.45, 171.08, 177.59.
(Холест-5-ен-3β-ил)-N-{2-[(2-аминоэтил)дитио]этил}карбамата гидрохлорид (V). К суспензии цистамина (1.98 г, 13.00 ммоль) в безводной смеси растворителей CH2Cl2–Et3N–диоксан (30 : 10 : 10 мл) добавляли имидазолид (IV) (0.782 г, 1.63 ммоль) и перемешивали при кипении в течение 35 ч. Реакционную смесь фильтровали, промывали 3%‑ным водным раствором HCl (3 × 30 мл), сушили над Na2SO4, фильтровали, растворители удаляли в вакууме. После хроматографии на силикагеле (CHCl3–MeOH, 10 : 1) получили соединение (V) в виде кристаллов светло коричневого цвета (0.580 г, 59%). Rf 0.30 (В). Масс-спектр, m/z: 565.456 [М – Cl]+, вычислено для C32H57N2O2S2: 565.386 [М – Cl]+. Спектр 1Н-ЯМР: 0.61 (с, 3Н, C(13)Me), 0.79 (д, 3Н, J 6.6, C(25)Me), 0.80 (д, 3Н, J 6.6, C(25)Me), 0.85 (д, 3Н, J 6.2, C(20)Me), 0.94 (с, 3Н, C(10)Me), 0.87–1.62 (м, 21Н, протоны Chol), 1.65–2.07 (м, 5H, протоны Chol), 2.17–2.40 (м, 2Н, H2С(4)), 2.70–2.90 (м, 2Н, CH2NH2), 2.79 (т, 2Н, J 7.5, CH2COOH), 2.92 (т, 2Н, J 7.1, CH2S), 2.92 (т, 3Н, J 7.5, CH2S), 2.90–3.17 (м, 2Н, CH2NHCOOChol), 3.17–3.60 (м, 4Н, 2 CH2S), 4.20–4.58 (м, 1Н, H(3)), 5.22–5.40 (м, 1Н, H(6)), 5.43–5.70 (м, 1Н, NHCO), 7.00–7.50 (м, 3Н, NH2). Спектр 13С-ЯМР: 11.98, 18.84, 19.49, 21.19, 22.65, 22.90, 24.06, 24.41, 28.09, 28.35, 32.00, 35.81, 35.95, 36.34, 36.68, 37.14, 38.21, 38.75, 39.29, 39.63, 39.89, 50.11, 56.39, 56.81, 74.69, 122.65, 139.89, 156.44.
(Холест-5-ен-3β-ил)-N11,N16,N19-три-(трет-бутилоксикарбонил)-19-амино-6-оксо-3,4-дитиа-7,11,16-триазадодеканоат (VIIа). К раствору соединения (VI) (99 мг, 0.198 ммоль) в ДМФА (2.5 мл) последовательно добавляли соединение (IIIа) (0.131 г, 0.238 ммоль), TBTU (0.076 мг, 0.238 ммоль) и Et3N (0.033 мл, 0.238 ммоль). Через 1 ч растворители удаляли в вакууме, остаток хроматографировали на колонке с силикагелем, элюируя смесью CHCl3–MeOH, 100 : 1. Получили соединение (VIIa) в виде кристаллизующегося бесцветного масла (0.122 г, 60%). Rf 0.35 (Б). Спектр 1Н-ЯМР: 0.68 (с, 3Н, C(13)Me), 0.85 (д, 3Н, J 6.5, C(25)Me), 0.86 (д, 3Н, J 6.5, C(25)Me), 0.91 (д, 3Н, J 6.5, C(20)Me), 1.02 (с, 3Н, C(10)Me), 0.90–2.10 (м, 34Н, протоны Chol, 2 NCH2CH2CH2N, NCH2(CH2)2CH2N), 1.43 (с, 9H), 1.45 (c, 9H) и 1.50 (с, 9H, 3 C(CH3)3), 2.29–2.40 (м, 2Н, H2С(4)), 3.02–3.35 (м, 12H, 6 CH2N), 3.47 (c, 2H, SCH2COOChol), 3.54 (c, 2H, SCH2CONH), 4.35–4.75 (м, 1Н, H(3)), 5.27–5.40 (м, 1Н, H(6)). Спектр 13С-ЯМР: 11.85, 18.72, 19.27, 21.05, 22.53, 22.77, 23.83, 24.27, 25.91, 27.70, 27.99, 28.19, 28.44, 28.47, 29.67, 31.88, 31.91, 33.94, 35.77, 36.20, 36.60, 36.95, 37.68, 38.00, 39.52, 39.75, 41.04, 41.71, 42.67, 44.11, 46.85, 49.14, 50.08, 56.20, 56.72, 75.76, 79.64, 122.99, 139.31, 156.01, 167.95, 169.08.
(Холест-5-ен-3β-ил)-N13,N18,N 21-три-(трет-бутилоксикарбонил)-21-амино-8-оксо-4,5-дитиа-9,13,18-триазагенэйкозаноат (VIIb). Получали как соединение (VIIa), исходя из соединения (VI) (0.158 г, 0.314 ммоль), соединения (IIIb) (0.218 г, 0.377 ммоль), TBTU (0.121 г, 0.377 ммоль) и Et3N (0.053 мл, 0.377 ммоль). Получили соединение (VIIb) в виде кристаллизующегося бесцветного масла (0.178 г, 82%). Rf 0.4 (Б). Спектр 1Н-ЯМР: 0.68 (с, 3Н, C(13)Me), 0.85 (д, 3Н, J 6.5, C(25)Me), 0.86 (д, 3Н, J 6.5, C(25)Me), 0.91 (д, 3Н, J 6.5, C(20)Me), 1.02 (с, 3Н, C(10)Me), 0.91–2.05 (м, 34Н, протоны Chol, 2 NCH2CH2CH2N, NCH2(CH2)2CH2N), 1.46 (уш. с, 27H, 3 C(CH3)3), 2.27–2.37 (м, 2Н, H2С(4)), 2.58 (т, 2H, J 7.1, SCH2CH2COOChol), 2.70 (т, 2H, J 7.1, SCH2CH2CONH), 2.87–3.00 (м, 4H, 2 SCH2), 3.07–3.40 (м, 12H, 6 CH2N), 4.55–4.72 (м, 1Н, H(3)), 5.33–5.40 (м, 1Н, H(6)). Спектр 13С-ЯМР: 11.82, 18.67, 19.28, 20.98, 22.54, 22.80, 23.78, 24.24, 25.94, 27.17, 27.64, 27.72, 27.98, 28.19, 28.33, 28.41, 31.79, 31.86, 33.11, 33.82, 34.40, 35.75, 35.97, 36.13, 36.54, 36.91, 38.05, 39.47, 39.66, 42.94, 43.31, 46.66, 46.81, 49.95, 56.06, 56.63, 74.46, 79.92, 80.33, 122.74, 139.46, 157.02, 157.51, 170.66, 171.13.
(Холест-5-ен-3β-ил)-19-амино-6-оксо-3,4-дитиа-7,11,16-триазадодеканоат тригидрохлорид (S1). К раствору соединения (VIIa) (0.055 г, 0.053 ммоль) в 3 мл CH2Cl2 добавляли 4 н. HCl в диоксане (1 мл) и перемешивали 1 ч при 24°С. Растворители удаляли в вакууме, остаток промывали CHCl3. Получили соединение S1 в виде аморфных бежевых кристаллов (0.030 г, 67%). Rf 0.85 (Г). Масс-спектр, m/z: 735.623 [М – 3HCl + H]+, вычислено для C41H77Cl3N4O3S2: 735.528 [М – 3HCl + H]+. Спектр 1Н-ЯМР (CDCl3–CD3OD, 1 : 5): 0.67 (с, 3Н, C(13)Me), 0.85 (д, 3Н, J 6.5, C(25)Me), 0.86 (д, 3Н, J 6.5, C(25)Me), 0.92 (д, 3Н, J 6.5, C(20)Me), 1.03 (с, 3Н, C(10)Me), 0.91–2.05 (м, 34Н, протоны Chol, 2 NCH2CH2CH2N, NCH2(CH2)2CH2N), 2.30–2.41 (м, 2Н, H2С(4)), 2.91–3.21 (м, 12H, 6 CH2N), 3.46 (c, 2H, SCH2COOChol), 3.55 (c, 2H, SCH2CONH), 4.36–4.76 (м, 1Н, H(3)), 5.28–5.41 (м, 1Н, H(6)).
(Холест-5-ен-3β-ил)-21-амино-8-оксо-4,5-дитиа-9,13,18-триазагенэйкозаноат тригидрохлорид (S2). Получали как соединение S1, исходя из соединения (VIIb) (0.05 г, 0.073 ммоль). Получили соединение S2 в виде аморфных белых кристаллов (0.048 г, 76%). Rf 0.86 (Г). Масс-спектр, m/z: 763.725 [М – 3HCl + H]+, вычислено для C43H81Cl3N4O3S2: 763.560 [М – 3HCl + H]+. Спектр 1Н-ЯМР (CDCl3–CD3OD, 1 : 5): 0.68 (с, 3Н, C(13)Me), 0.84 (д, 3Н, J 6.5, C(25)Me), 0.85 (д, 3Н, J 6.5, C(25)Me), 0.90 (д, 3Н, J 6.5, C(20)Me), 1.01 (с, 3Н, C(10)Me), 0.90–2.06 (м, 34Н, протоны Chol, 2 NCH2CH2CH2N, NCH2(CH2)2CH2N), 2.27–2.37 (м, 2 Н, H2С(4)), 2.59 (т, 2H, J 7.1, SCH2CH2COOChol), 2.71 (т, 2H, J 7.1, SCH2CH2CONH), 2.88–3.21 (м, 16H, 2 SCH2, 6 CH2N), 4.55–4.72 (м, 1Н, H(3)), 5.33–5.40 (м, 1Н, H(6)).
Метил-N '-[N 4,N 9,N 12-три-(трет-бутоксикарбонил)-12-амино-4,9-диазадодец-1-ил]-N '-(2-нитрофенилсульфонил)аминоацетат (VIII). К охлажденному до 0°С раствору соединения (VI) (0.431 г, 0.858 ммоль) в безводном CH2Cl2 (5 мл) добавляли молекулярные сита 4 Å (1.0 г) Et3N (0.239 мл, 1.716 ммоль) и 2‑нитробензолсульфонилхлорид (0.228 г, 1.02 ммоль). Реакционную смесь перемешивали 1 ч при 24°С. Молекулярные сита отфильтровывали, промывали CH2Cl2, растворители удаляли в вакууме. Остаток хроматографировали на колонке с силикагелем, элюируя смесью CHCl3–MeOH (50 : 1). Получили N4,N 9,N12-три-(трет-бутоксикарбонил)-N1-(2-нитрофенилсульфонил)-1,12-диамино-4,9-диазадодекан в виде кристаллизующегося масла (0.507 г, 88%). Масс-спектр, m/z: 710.295 [М + Na]+, вычислено для C31H53N5NaO10S: 710.341 [М + Na]+. Спектр 1Н-ЯМР: 1.27 (уш. с, 31Н, 3 C(CH3)3 и NCH2(CH2)2CH2N), 1.52–1.71 (м, 4Н, 2 NCH2CH2CH2N), 2.92–3.11 (м, 8Н, 4 NCH2), 3.11–3.30 (м, 4Н, 2 NHCH2), 7.59–7.71 (м, 2Н), 7.71–8.89 (м, 1Н) и 8.00–8.11 (м, 1Н, C6H4). Спектр 13С-ЯМР: 25.96, 28.54, 28.59, 28.93, 37.91, 41.03, 43.78, 44.23, 46.82, 47.05, 79.16, 79.74, 80.02, 125.25, 130.93, 132.68, 133.51, 148.29, 156.17.
К раствору N 4,N 9,N 12-три-(трет-бутоксикарбонил)-N1-(2-нитрофенилсульфонил)-1,12-диамино-4,9-диазадодекана (0.271 г, 0.394 ммоль) в безводном ДМФА (3 мл) последовательно добавляли Cs2CO3 (0.128 г, 0.394 ммоль) и метиловый эфир бромуксусной кислоты (0.149 мл, 1.58 ммоль). Реакционную смесь перемешивали 3 ч при 75°С. К реакционной смеси добавляли 15 мл CH2Cl2, промывали 5%-ной лимонной кислотой (3 × 20 мл), сушили над Na2SO4, упаривали. Остаток хроматографировали на колонке с силикагелем, элюируя смесью CHCl3–MeOH (175 : 1). Получили соединение (VIII) в виде кристаллизующегося масла желтоватого цвета (0.270 г, 90%). Rf 0.51 (Б). Спектр 1Н-ЯМР: 1.26–1.46 (м, 4H, NCH2(CH2)2CH2N), 1.32 (c, 18H), 1.35 (c, 9H, 3 C(CH3)3) 1.46–1.60 (м, 2H), 1.60–1.75 (м, 2H, 2 NCH2CH2CH2N), 2.90–3.23 (м, 10H, 2 CH2NCH2, NHCH2), 3.23–3.37 (м, 2Н, NCH2CH2), 3.54 (c, 3H, OCH3), 4.10 (c, 2H, OC(O)CH2), 7.46–7.68 (м, 3H), 7.87–8.02 (м, 1H, C6H4). Спектр 13С-ЯМР: 25.87, 26.92, 28.46, 28.48, 37.63, 43.96, 44.49, 46.42, 46.75, 47.81, 52.27, 78.96, 79.59, 124.20, 130.86, 131.74, 133.13, 133.70, 148.04, 155.46, 156.07, 169.14.
2-[N4,N9,N12-Три-(трет-бутоксикарбонил)-12-амино-4,9-диазадодец-1-ил]аминоуксусной кислоты гидрохлорид (IX). К раствору соединения (VIII) (0.178 г, 0.235 ммоль) в ДМФА (2 мл) при перемешивании добавляли K2CO3 (0.036 г, 0.258 ммоль), а затем PhSH (0.241 мл, 2.35 ммоль). Через 1 ч к реакционной массе добавляли 10 мл CH2Cl2, промывали водой (15 мл), сушили над Na2SO4, упаривали. После хроматографии на силикагеле CHCl3–MeOH–25%-ный водный NH3 (5 : 0.1 : 0.01) получили метил-N'-[N4,N9,N12-три-(трет-бутоксикарбонил)-12-амино-4,9-диазадодец-1-ил]аминоацетат в виде кристаллизующегося масла (0.116 г, 86%). Rf 0.33 (А). Масс-спектр, m/z: 575.456 [М + H]+, вычислено для C28H54N4O8: 575.404 [М + H]+. Спектр 1Н-ЯМР: 1.25–1.48 (м, 4H, NCH2(CH2)2CH2N), 1.34 (c, 9H), 1.35 (c, 9H), 1.36 (c, 9 H, 3 C(CH3)3), 1.48–1.70 (м, 4H, 2NCH2CH2CH2N), 2.51 (т, 2H, J 7.0, NHCH2CH2), 2.91–3.25 (м, 10H, 2 CH2NCH2, NHCH2), 3.31 (c, 2H, OC(O)CH2), 3.63 (c, 3H, OCH3). Спектр 13С-ЯМР: 25.52, 25.93, 28.39, 28.93, 37.36, 43.75, 44.10, 44.82, 46.80, 50.73, 51.66, 78.77, 79.20, 79.43, 155.51, 155.99, 172.79.
К раствору метил-N'-[N4,N 9,N12-три-(трет-бутоксикарбонил)-12-амино-4,9-диазадодец-1-ил]аминоацетата (0.100 г, 0.174 ммоль) в MeOH (3 мл) при перемешивании добавляли NaOH (0.031 г, 0.783 ммоль) в 0.5 мл воды. Через 2 ч к реакционной массе добавляли 10 мл CH2Cl2, промывали 3%-ным водным раствором HCl (15 мл), сушили над Na2SO4, упаривали. Получили соединение (IX) в виде кристаллов белого цвета (105 мг, 100%). Rf 0.33 (Е). Масс-спектр, m/z: 561.476 [М – Cl]+, вычислено для C27H53N4O8: 561.386 [М – Cl]+. Спектр 1Н-ЯМР (CD3OD–CDCl3, 1 : 5): 1.17–1.40 (уш. с, 31H, NCH2(CH2)2CH2N, 3 C(CH3)3), 1.40–1.60 (м, 2H), 1.70–1.93 (м, 2Н, 2 NCH2CH2CH2N), 2.63–2.82 (м, 2H, CH2NH2CH2CO), 2.83–3.18 (м, 10H, 2 CH2NCH2, NHCH2), 3.30 (c, 2H, OC(O)CH2). Спектр 13С-ЯМР: 24.21, 25.10, 27.59, 27.64, 28.16, 36.74, 42.78, 43.29, 43.67, 44.19, 45.79, 46.29, 49.07, 78.47, 79.18, 79.60, 155.73, 155.80, 155.85, 169.73.
N 14,N 19,N 22-Три-(трет-бутоксикарбонил)-N1-(холест-5-ен-3β-илоксикарбонил)-1,22-диамино-8-оксо-3,4-дитиа-7,10,14,19-тетраазадокозан (X). К охлажденному до 0°С раствору соединения (IX) (100 мг, 0.167 ммоль) в ДМФА (5 мл) последовательно добавляли TBTU (0.134 мг, 419 ммоль), раствор соединения (V) (0.302 мг, 0.502 ммоль), DIEA (0.146 мл, 0.837 ммоль) в ДМФА (2.5 мл). Через 2 ч к реакционной массе добавляли 20 мл CH2Cl2, промывали 3%-ным водным раствором HCl (2 × 15 мл), сушили над Na2SO4, упаривали. После хроматографии на силикагеле CHCl3–MeOH (80 : 1) получили соединение (X) в виде кристаллизующегося масла (0.176 г, 92%). Rf 0.57 (Ж). Масс-спектр, m/z: 1107.867 [М – Cl]+, вычислено для C59H107N6O9S2: 1107.754 [М – Cl]+. Спектр 1Н-ЯМР: 0.64 (с, 3Н, C(13)Me), 0.83 (д, 3Н, J 6.6, C(25)Me), 0.84 (д, 3Н, J 6.6, C(25)Me), 0.88 (д, 3Н, J 6.5, C(20)Me), 0.97 (с, 3Н, C(10)Me), 0.85–1.70 (м, 36Н, протоны Chol, NCH2(CH2)2CH2N, NCH2CH2CH2NH2CH2CO), 1.40 (с, 9H), 1.41 (с, 9H) и 1.42 (с, 9H, 3 C(CH3)3), 1.70–2.07 (м, 7H, протоны Chol, NHCH2CH2CH2N), 2.20–2.39 (м, 2Н, H2С(4)), 2.55 (т, 2Н, J 6.5, CH2NH2CH2CO), 2.76–2.84 (м, 4Н, 2 CH2S), 3.01–3.32 (м, 10H, 2 CH2NCH2, NHCH2), 3.21 (c, 2H, CH2NH2CH2CO), 3.44 (кв, J 6.2, 2Н, CH2NHCOOChol), 3.57 (кв, J 6.3, 2Н, CH2OCONHCH2), 4.30–4.58 (м, 1Н, H(3)), 5.25–5.50 (м, 1Н, H(6)). Спектр 13С-ЯМР: 11.93, 18.80, 19.40, 21.12, 22.63, 22.88, 23.91, 24.36, 25.98, 28.06, 28.25, 28.29, 28.52, 28.54, 28.56, 31.97, 31.98, 35.85, 36.27, 36.64, 37.08, 37.82, 37.94, 38.51, 38.64, 39.59, 39.83, 44.11, 46.73, 47.07, 50.12, 56.24, 56.78, 74.57, 79.07, 79.53, 79.64, 122.56, 139.87, 156.17, 156.18, 156.19.
1,22-Диамино-N1-(холест-5-ен-3β-илоксикарбонил)-8-оксо-3,4-дитиа-7,10,14,19-тетраазадокозан (S3). К раствору соединения (X) (0.200 г, 0.146 ммоль) в 4 мл THF добавляли 4 н. HCl в диоксане (3 мл) и перемешивали 3 ч при 24°С. Растворители удаляли в вакууме, остаток промывали MeOH. Получили соединение S3 в виде белых кристаллов (0.155 г, 81%). Rf 0.35 (Г). Масс-спектр, m/z: 807.654 [М – 4HCl + H]+, вычислено для C44H86N6O3S2: 807.597 [М – 4HCl + H]+. Спектр 1Н-ЯМР (D2O): 0.65 (с, 3Н, C(13)Me), 0.84 (д, 3Н, J 6.6, C(25)Me), 0.85 (д, 3Н, J 6.6, C(25)Me), 0.87 (д, 3Н, J 6.5, C(20)Me), 0.96 (с, 3Н, C(10)Me), 0.85–1.70 (м, 38Н, протоны Chol, NCH2(CH2)2CH2N, NCH2CH2CH2NH2CH2CO), 1.70–2.07 (м, 5H, протоны Chol), 2.21–2.40 (м, 2Н, H2С(4)), 2.54 (т, 2Н, J 6.5, CH2NH2CH2CO), 2.77–2.84 (м, 4Н, 2 CH2S), 2.94–3.25 (м, 14H, 7 CH2N), 3.45 (кв, J 6.2, 2Н, CH2NHCOOChol), 3.56 (кв, J 6.3, 2Н, CH2OCONHCH2), 4.31–4.59 (м, 1Н, H(3)), 5.24–5.49 (м, 1Н, H(6)).
Катионные липосомы. К раствору поликатионного амфифила в смеси растворителей CНCl3–МеОН (4 : 1 об.) добавляли раствор 1,2-диолеоил-sn-глицеро-3-фосфатидилэтаноламина (DOPE, Avanti Polar Lipids, США) в хлороформе в соотношении поликатионный амфифил–DOPE 1 : 1 (мольн.). Растворители удаляли в вакууме водоструйного насоса, образовавшуюся липидную пленку сушили 3 ч в вакууме масляного насоса (0.01 Торр), после чего добавляли деионизированную воду и гидратировали 4 ч при 24°С (концентрация по поликатионному амфифилу составляла 1 мМ). Образовавшуюся дисперсию обрабатывали на ультразвуковой бане (Bandelin Sonorex Digitec DT 52H, Германия) 15 мин при 60–65°С. Полученные катионные липосомы хранили при 4°С в атмосфере аргона.
Нуклеиновые кислоты. 25-Звенный олигодезоксирибонуклеотид с аминогексильным линкером на 5′-конце (5′-TACAGTGGAATTGTATGCCTATTAT-3′) синтезировали фосфорамидатным методом и очищали с помощью ВЭЖХ (Институт химической биологии и фундаментальной медицины СО РАН, Новосибирск). Чистоту олигонуклеотида проверяли с помощью электрофореза в 20%-ном ПААГ с 8 M мочевиной, она составляла 95–98%. FITC-мечение олигонуклеотида проводили, как описано ранее [25]. FITC-ODN выделяли с помощью ВЭЖХ (Alliance, США), используя колонку Waters XTerra и линейный градиент концентрации ацетонитрила (0–30%) в 50 мМ NaClO4. Чистоту FITC-ODN проверяли с помощью электрофореза в 20%-ном ПААГ с 8 M мочевиной, она составляла 95–98%. Концентрацию олигонуклеотида измеряли с помощью спектрофотометра BioMate 3 (Termo Electron Corporation, США). Раствор FITC-ODN хранили при −20°C.
В экспериментах по доставке плазмидной ДНК использовали плазмиду pEGFP-C2 (Clontech, Германия), кодирующую усиленный зеленый флуоресцентный белок.
Комплексы катионных липосом с нуклеиновыми кислотами. Для проведения трансфекции эукариотических клеток комплексы получали следующим образом: растворы катионных липосом (25 мкл) и нуклеиновых кислот (25 мкл) с концентрацией, соответствующей определенному соотношению N/P, смешивали в среде Opti-MEM® (Thermo Fisher Scientific, США) и инкубировали 20 мин при 24°С.
Образование комплексов с ДНК исследовали с помощью электрофореза в 1.5%-ном агарозном геле в нативных условиях при напряжении электрического поля 20 В/см в течение 30−45 мин. Гель окрашивали бромистым этидием и визуализировали в УФ-свете (254 нм).
Клеточные культуры. Клетки почки эмбриона человека HEK293 и цервикальной аденокарциномы HeLa были получены из Банка клеточных культур Института цитологии РАН (Санкт-Петербург, Россия). Клетки почки эмбриона хомячка BHK21 были любезно предоставлены профессором В.С. Прасоловым (Институт молекулярной биологии РАН, Москва). Клеточные линии HEK293, HeLa и BHK21 культивировали в среде DMEM, содержавшей 10% FBS, 100 ед./мл пенициллина, 0.1 мг/мл стрептомицина и 0.25 мкг/мл амфотерицина, в атмосфере 5%-ного СО2 при 37°С. Клетки высаживали в 96-луночные (HeLa и BHK21 – по 3 × 103 клеток на лунку, HEK293 – по 1 × 104 клеток на лунку) или 24-луночные планшеты (HeLa и BHK21 – по 105 клеток на лунку, HEK293 – по 1.2−1.7 × 105 клеток на лунку) и давали им прикрепиться в течение 24 ч.
Цитотоксичность катионных липосом (МТТ-тест). Клетки HEK293, HeLa и BHK21 культивировали в 96-луночных планшетах, как описано выше. Через 24 ч клеточную среду заменяли растворами липосом в DMEM (150 мкл) в отсутствие сыворотки и инкубировали в атмосфере 5%-ного СО2 в течение 4 ч при 37°С, после чего в лунки добавляли по 16 мкл FBS и дополнительно инкубировали 20 ч при 37°С. По окончании инкубации к клеткам без смены среды добавляли раствор MTT (Sigma, США) до концентрации 0.5 мг/мл. Через 4 ч культуральную среду удаляли, кристаллы формазана растворяли в DMSO (100 мкл на лунку), определяли разницу между оптической плотностью при 620 и 570 нм (Multiscan RC, Labsystems, Финляндия). Поглощение в контроле (инкубация клеток в отсутствие КЛ) принимали за 100% живых клеток, количество живых клеток определяли относительно контроля. Значения IC50 (концентрация липосом, при которой наблюдается гибель 50% клеток) определяли путем экстраполяции концентрационной зависимости. Результаты представлены как средние значения для трех независимых экспериментов.
Трансфекция клеток липоплексами, сформированными катионными липосомами и нуклеиновыми кислотами. Клетки высаживали в 24-луночные планшеты, как описано выше, и давали им прикрепиться в течение 24 ч. В день трансфекции культуральную среду заменяли на среду DMEM (200 мкл) с 10%-ной сывороткой крови (без антибиотиков). К клеткам добавляли комплексы пДНК (конечная концентрация 2 мкг/мл) или FITC-ODN (конечная концентрация 1 мкМ) с катионными липосомами, сформированными при различных соотношениях N/P, в 50 мкл OPTI-MEM и инкубировали в атмосфере 5%-ного СО2 в течение 4 ч при 37°С. В случае FITC-ODN клетки анализировали сразу, в случае пДНК через 4 ч среду заменяли на DMEM с 10%-ной FBS (500 мкл) и дополнительно инкубировали при 37°С в течение 44 ч. Доставку нуклеиновых кислот с помощью Липофектамина 2000 (Thermo Fisher Scientific Inc., США) осуществляли согласно протоколу производителя (www.thermofisher.com).
Проточная цитометрия. Эффективность трансфекции клеток оценивали с помощью проточной цитометрии. После инкубации клетки промывали фосфатно-солевым буфером, обрабатывали раствором трипсина в этом же буфере (0.5 мг/мл) в течение 2 мин при 37°C для открепления с поверхности лунки и суспендировали в среде с сывороткой для ингибирования действия трипсина. Затем клетки переносили в пробирки, осаждали центрифугированием (Contron T42K, Centricon Instruments, Италия) при 1000 об/мин (200 g) в течение 10 мин, удаляли среду и дважды промывали фосфатно-солевым буфером. Затем клетки фиксировали в 600 мкл 2%-ного раствора формальдегида. Количество трансфицированных клеток и среднее значение интенсивности флуоресценции в клеточной популяции измеряли на цитофлуориметре Cytomics FC500 (Beckman Coulter, США) с использованием программы CXP Analysis (Beckman Coulter, США). В каждом образце анализировали не менее 20 тыс. клеток. Все экспериментальные точки были получены в результате трех независимых экспериментов. Стандартное отклонение не превышало 7−9%.
ЗАКЛЮЧЕНИЕ
Синтезированы новые дисульфидные амфифилы на основе холестерина и спермина, отличающиеся способом присоединения спермина к гидрофобному домену (сложноэфирный или карбамоильный линкер), длиной спейсера, а также количеством катионных аммонийных групп. Катионные липосомы на основе полученных соединений оказались нетоксичными для клеток НЕК293 и HeLa и полностью связывали плазмидную ДНК в НК-липидном комплексе. Липосомы S3-DOPE, содержащие тетракатионный дисульфидный амфифил, продемонстрировали высокую трансфицирующую активность, которая в зависимости от клеточной линии была сопоставима с эффективностью Липофектамина 2000 и липосом X2-DOPE, нечувствительных к восстановлению, или превышала таковую. Липосомы S3-DOPE могут быть использованы для трансфекции эукариотических клеток in vitro, а также проведения дальнейших биологических испытаний in vivo.
Список литературы
Sung Y.K., Kim S.W. // Biomater. Res. 2019. V. 23. P. 1–7. https://doi.org/10.1186/S40824-019-0156-z
Ni R., Zhou J., Hossain N., Chau Y. // Adv. Drug Deliv. Rev. 2016. V. 106. P. 3–26. https://doi.org/10.1016/J.ADDR.2016.07.005
Lan T., Que H., Luo M., Zhao X., Wei X. // Mol. Cancer. 2021. V. 21. P. 71. https://doi.org/10.1186/s12943-022-01550-8
Beltrán-Gracia E., López-Camacho A., Higuera-Ciapara I., Velázquez-Fernández J.B., Vallejo-Cardona A.A. // Cancer Nanotechnol. 2019. V. 10. P. 1–40. https://doi.org/10.1186/S12645-019-0055-Y
Patel R., Kaki M., Potluri V.S., Kahar P., Khanna D. // Hum. Vaccin. Immunother. 2022. V. 18. P. 2002083. https://doi.org/10.1080/21645515.2021.2002083
Gottfried L.F., Dean D.A. // Extracellular and Intracellular Barriers to Non-Viral Gene Transfer. In Novel Gene Therapy Approaches / Ed. Wei M. London: InTech, 2013. P. 75–88.
Puchkov P.A., Maslov M.A. // Pharmaceutics. 2021. V. 13. P. 920. https://doi.org/10.3390/pharmaceutics13060920
Yu C., Li L., Hu P., Yang Y., Wei W., Deng X., Wang L., Tay F.R., Ma J. // Adv. Sci. 2021. V. 8. P. 2100540.
Zhang X.X., McIntosh T.J., Grinstaff M.W. // Biochimie. 2012. V. 94. P. 42–58. https://doi.org/10.1016/j.biochi.2011.05.005
Chen X., Yang J., Liang H., Jiang Q., Ke B., Nie Y. // J. Mater. Chem. B. 2017. V. 5. P. 1482–1497. https://doi.org/10.1039/C6TB02945K
Liu J., Chang J., Jiang Y., Meng X., Sun T., Mao L., Xu Q., Wang M. // Adv. Mater. 2019. V. 31. P. 1–7. https://doi.org/10.1002/adma.201902575
Петухов И.А., Маслов М.А., Морозова Н.Г., Серебренникова Г.А. // Известия Академии наук. Серия химическая. 2010. № 1. С. 254–261.
Sebyakin Y.L., Budanova U.A., Guryeva L.Y. // Biochem. Suppl. Ser. A Membr. Cell Biol. 2007. V. 1. P. 212–218. https://doi.org/10.1134/S1990747807030038
Goyal P., Goyal K., Kumar S.G.V., Singh A., Katare O.P., Mishra D.N. // Acta Pharm. 2005. V. 55. P. 1–25.
Masotti A., Mossa G., Cametti C., Ortaggi G., Bianco A., Grosso N.D., Malizia D., Esposito C. // Colloids Surf. B Biointerfaces. 2009. V. 68. P. 136–144. https://doi.org/10.1016/J.COLSURFB.2008.09.017
Byk T., Haddada H., Vainchenker W., Louache F. // Hum. Gene Ther. 1998. V. 9. P. 2493–2502. https://doi.org/10.1089/hum.1998.9.17-2493
Shen G., Rajan R., Zhu J., Bell C.E., Pei D. // J. Med. Chem. 2006. V. 49. P. 3003–3011. https://doi.org/10.1021/jm060047g
Miller K.A., Kumar E.V.K.S., Wood S.J., Cromer J.R., Datta A., David S.A. // J. Med. Chem. 2005. V. 48. P. 2589–2599. https://doi.org/10.1021/jm049449j
Elsana H., Olusanya T.O.B., Carr-Wilkinson J., Darby S., Faheem A., Elkordy A.A. // Sci. Rep. 2019. V. 9. P. 1–17. https://doi.org/10.1038/s41598-019-51065-4
Maslov M.A., Kabilova T.O., Petukhov I.A., Morozova N.G., Serebrennikova G.A., Vlassov V.V., Zenkova M.A. // J. Control. Release. 2012. V. 160. P. 182–193. https://doi.org/10.1016/j.jconrel.2011.11.023
Aljaberi A., Spelios M., Kearns M., Selvi B., Savva M. // Colloids Surf. B Biointerfaces. 2007. V. 57. P. 108–117. https://doi.org/10.1016/j.colsurfb.2007.01.012
Sebastiani F., Yanez Arteta M., Lindfors L., Cárdenas M. // J. Colloid Interface Sci. 2022. V. 610. P. 766–774. https://doi.org/10.1016/J.JCIS.2021.11.117
Ivanković M., Ćukušić A., Gotić I., Škrobot N., Matijašić M., Polančec D., Rubelj I. // Biogerontology. 2007. V. 8. P. 163–172. https://doi.org/10.1007/s10522-006-9043-9
Tang F., Hughes J.A. // Bioconjug. Chem. 1999. V. 10. P. 791–796. https://doi.org/10.1021/BC990016I
Yong-Hee K., You Han Bae, Sung Wan Kim // J. Control. Release 1994. V. 28. P. 143–152. https://doi.org/10.1016/0168-3659(94)90161-9
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия