

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 1, стр. 15-31
Тромбоцитарные интегрины αIIbβ3: механизмы активации и кластеризации, роль в гетерогенности структуры тромба
В. Н. Канева a, b, c, А. А. Мартьянов a, b, c, Д. С. Морозова d, М. А. Пантелеев a, b, c, e, *, А. Н. Свешникова a, b, c, f
a Московский государственный университет им. М.В. Ломоносова, физический факультет,
119991 Москва, Ленинские горы, 1, стр. 2, Россия
b Национальный медицинский исследовательский центр им. Д. Рогачева,
117997 Москва, ул. Саморы Машела, 1, Россия
c Центр теоретических проблем физико-химической фармакологии РАН,
119991 Москва, ул. Косыгина, 4, Россия
d Московский государственный университет им. М.В. Ломоносова, факультет фундаментальной медицины,
119991 Москва, Ломоносовский просп., 27, к. 1, Россия
e Московский физико-технический институт, факультет биологической и медицинской физики,
141700 Долгопрудный, Институтский пер., 9, Россия
f Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский университет),
119991 Москва, ул. Трубецкая, 8, стр. 2, Россия
* E-mail: mapanteleev@yandex.ru
Поступила в редакцию 09.05.2018
После доработки 20.08.2018
Принята к публикации 05.08.2018
Аннотация
Гликопротеин IIb-IIIa, также известный как интегрин αIIbβ3, представляет собой ключевой адгезионный рецептор тромбоцитов. Эти молекулы являются самыми многочисленными (около 100 000 копий на клетку) трансмембранными рецепторами тромбоцитов и играют особую роль в тромбообразовании, так как механически обеспечивают образование их агрегатов. Для образования мостиков между тромбоцитами за счет связывания αIIbβ3 со своими основными лигандами – фибриногеном, фибрином и фактором Виллебранда – интегрин должен перейти в активированное состояние. Активация интегринов обеспечивается сложной сетью внутриклеточной сигнализации, в которой задействовано несколько вторичных мессенджеров (“inside-out” сигнализация). Как и другие интегрины, αIIbβ3 способны и к обратной передаче сигнала в клетку (“outside-in” сигнализация). Исследования последних лет показывают, что тромбы неоднородны по своей структуре: их внутренняя часть более плотная и стабильная, чем рыхлая и текучая внешняя оболочка. Поскольку агрегация тромбоцитов в первую очередь обеспечивается взаимодействием через интегрины, можно предположить, что именно особенности активации и кластеризации данных рецепторов влияют на формирование особенной архитектуры тромба. В настоящем обзоре систематизированы последние данные об активации и функционировании тромбоцитарных интегринов αIIbβ3, проведены параллели между гетерогенностью тромба и состоянием интегринов на поверхности тромбоцитов.
1. ВВЕДЕНИЕ
Интегрины – класс трансмембранных рецепторов, экспонируемых на поверхности клеток и отвечающих за образование связей между клетками [1]. Эти молекулы обеспечивают адгезию клеток крови к стенкам сосуда и образование прочных агрегатов клеток друг с другом [2]. Интегрины представляют собой нековалентно связанные гетеродимеры, состоящие из α- и β-цепей. В процессе активации α- и β-цепи могут менять положение друг относительно друга, что приводит к изменению способности интегринов связываться с лигандами. Таким образом, интегрины могут существовать в двух состояниях по отношению к лиганду – низкоаффинном и высокоаффинном. Иногда выделяют также промежуточную стадию активации интегринов [3, 4]. Как и прочие адгезионные рецепторы, интегрины тесно взаимодействуют с цитоскелетом для обеспечения прикрепления клеток как единого целого. Однако способность менять аффинность и передавать таким образом сигналы в обе стороны является уникальной чертой молекул этого класса.
Интегрины играют особую роль в гемостазе (остановке кровотечения) и тромбозе, так как обеспечивают образование агрегатов тромбоцитов – клеток крови, ответственных за поддержание целостности кровеносной системы [2, 5, 6]. При повреждении сосудистой стенки и нарушениях сосудистого эндотелия тромбоциты активируются в результате взаимодействия с белками внеклеточного матрикса (коллаген, фибронектин, ламинин) и/или растворимыми агонистами (ADP, тромбин, адреналин, тромбоксан) [7]. Известно, что из присутствующих на поверхности тромбоцитов трех семейств интегринов (с β1-, β2- и β3-цепями) – α2β1, α5β1, α6β1, αLβ2, αIIbβ3, ανβ3 – в высокоаффинное состояние при активации переходят интегрины αIIbβ3 (GPIIb-IIIa), присутствующие в количестве 100 000 на клетку [8–10].
В активированном состоянии αIIbβ3 связываются с фибриногеном, который в достаточных количествах присутствует в плазме крови, а также содержится в альфа-гранулах [11] и высвобождается при активации тромбоцитов. Также активированные интегрины αIIbβ3 могут связываться с молекулами фактора Виллебранда (vWF), который содержится в плазме крови и альфа-гранулах тромбоцитов и, кроме того, экспонируется активированными клетками эндотелия [12–14]. Другими лигандами αIIbβ3 являются витронектин, фибронектин и тромбоспондин, которые могут регулировать адгезию тромбоцитов в месте обнажения субэндотелиального матрикса в поврежденном сосуде [14]. Через мостики αIIbβ3-фибриноген/vWF-αIIbβ3 тромбоциты могут связываться друг с другом, образуя тромбы – агрегаты, перекрывающие повреждение или весь сосуд [6, 15].
Формирующийся в микрососуде тромб неоднороден по своей структуре. Так, наиболее активированные тромбоциты находятся ближе к месту повреждения. Они сами секретируют содержимое гранул, содержащих активаторы, способствуя дальнейшему росту тромба. Эти тромбоциты прочно связаны друг с другом и составляют плотное и стабильное ядро [16, 17]. Внешние тромбоциты “подактивированы”, они слабо связаны – эта часть называется оболочкой тромба. Оболочка нестабильна, она “обтекает” плотное внутреннее ядро. Можно предположить, что градиент плотности упаковки обеспечивается различиями в адгезионных силах между тромбоцитами, которые могут определяться увеличением прочности отдельных связей или локальным ростом их плотности. Интегрины αIIbβ3 – наиболее вероятные кандидаты на роль регуляторов этого процесса.
При активации тромбоцита повышение аффинности интегрина αIIbβ3 к его лигандам может происходить по классическому пути сигнализации “изнутри-наружу” (“inside-out”), а также, как считают некоторые авторы, активацию интегрина может индуцировать появление лиганда [18]. Для активации интегринов “изнутри” необходимо изменение субклеточной локализации нескольких белков, основные из которых талин-1, киндлин-3, а также малая GTP-аза Rap1b [19–22]. В активации (замещении GDP на GTP) последней задействованы два вторичных мессенджера внутриклеточной сигнализации тромбоцита – ионы кальция и фосфатидилинозитол-3,4,5-трисфосфат, которые появляются при активации тромбоцита. Цитоплазматические домены интегринов αIIbβ3 могут быть ассоциированы с талином-1 и киндлином, которые, в свою очередь, связаны с актиновым цитоскелетом. При возникновении продольных напряжений на мембране тромбоцита, обусловленных воздействием на актиновый цитоскелет, также может происходить активация интегринов [23]. Связывание αIIbβ3 с лигандами приводит к усилению активации тромбоцита – так называемая сигнализация “снаружи-внутрь” (“ouside-in”), которая в экспериментах выражается в виде “распластывания” тромбоцитов по поверхности, покрытой фибриногеном [18, 24]. Известно, что сигнализация от интегринов зависит от тирозинкиназы Syk, обычно активируемой рецепторами, содержащими ITAM-домен [25], однако так как у αIIbβ3 такого домена нет, то какой из ITAM-содержащих белков служит посредником сигнализации от интегринов и есть ли необходимость в таком посреднике, является предметом интенсивной дискуссии [26–29].
Кроме тромбоцитов, αIIbβ3 экспонируется на поверхности мегакариоцитов, тучных клеток, базофилов, а также клеток некоторых опухолей (меланом, плоскоклеточных карцином и т.д.) [30–36]. Несмотря на это, цепи β3 и αIIb часто служат маркерами тромбоцитов при использовании соответствующих антител (anti-CD61 и anti-CD41). Нарушения сигнализации “outside-in”/“nside-out” наравне с дефицитом интегрина αIIbβ3 на тромбоцитах (тромбастения Гланцмана) приводят к серьезным нарушениям свертывания крови: меноррагиям, носовым кровотечениям, желудочно-кишечным кровотечениям, послеродовым кровотечениям, а также к обильным послеоперационным кровотечениям [37, 38]. При тромбастении Гланцмана рост тромбов на коллагеновой подложке нарушен при любых скоростях сдвига, в отличие от синдрома Бернара–Сулье (дефицит тромбоцитарного рецептора к VWF GPIb) – при низких скоростях сдвига рост тромбов не нарушен [39].
Цель данного обзора – систематизация данных о функционировании тромбоцитарного интегрина αIIbβ3, а также оценка влияния сильной/слабой активации тромбоцитов на кластеризацию αIIbβ3 и роли активации тромбоцитарных интегринов αIIbβ3 в формировании гетерогенной структуры тромбов.
2. МОЛЕКУЛЯРНЫЕ СОБЫТИЯ ПРИ АКТИВАЦИИ αIIbβ3
2.1. Структура интегринов αIIbβ3
Интегрины – интегральные гликопротеины плазматической мембраны клеток, состоящие из α- и β-цепей. Каждая субъединица содержит длинный внеклеточный домен, трансмембранный регион, а также короткую цитоплазматическую аминокислотную последовательность. Внеклеточный домен αIIb цепи состоит из β-пропеллера – структуры из серии повторов, которые совместно образуют семиконечную (семь лезвий) кольцевую последовательность с отверстием в середине [40, 41]. Связанные с остатками аспартата катионы увеличивают жесткость связки между β-пропеллером и нижележащими доменами (бедро, домены calf1, calf2). Между бедром и доменом calf1 расположена “гибкая” гидрофобная последовательность, позволяющая менять положение αIIb цепи относительно β3-цепи [42–44] (рис. 1а).
Рис. 1.
Структура интегрина αIIbβ3. а – Внеклеточный домен закрытой формы αIIbβ3 (PDB: 3FCS). Показаны три металлсвязывающих сайта в домене I β-субъединицы; б – aминокислотные остатки вокруг металлсвязывающих доменов; в – открытая форма αIIbβ3, связанная с лигандом (PDB: 3FCS) [127].
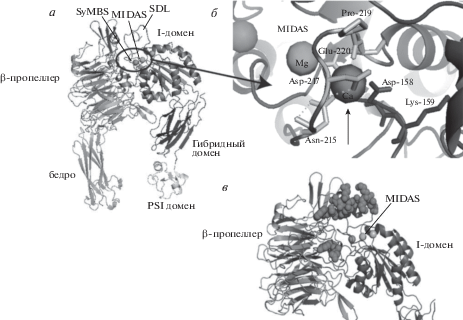
Цепь β3 состоит из А-домена, который благодаря аргинину (Arg261) “входит” в отверстие β-пропеллера αIIb-цепи [42, 45–47]. А-домен обладает несколькими металлсвязывающими доменами (MIDAS, ADMIDAS, LIMBS, см. рис. 1б) [48]. Через гибридный домен А-домен связан с несколькими EGF-доменами. EGF-домены обогащены остатками цистеина и благодаря дисульфидным мостикам образуют стержнеподобную структуру [49, 50]. Между EGF-доменами и гибридным доменом, как и в αIIb-цепи, расположена гибкая гидрофобная последовательность [46, 51].
Трансмембранный регион αIIbβ3 состоит из двух скрещенных спиралей. В трансмембранном домене αIIb также содержит последовательность GFFKR. В дезактивированном интегрине αIIbβ3 эта последовательность скрыта в плазматической мембране (рис. 1а), но при изменении конформации интегрина GFFKR оказывается в цитозоле и становится сайтом докинга для ряда белков эффекторов [53, 54] (рис. 1в).
Цитоплазматические домены обеих цепей, составляющих αIIbβ3, представляют собой короткие участки, содержащие несколько функциональных аминокислотных последовательностей, в частности NxxY [55, 56].
Интегрины αIIbβ3 могут связывать несколько различных лигандов: фибриноген, фибрин и VWF. Интегрины αIIbβ3 связываются с разными последовательностями в молекулах фибриногена и VWF: RGD – в VWF и KQAGDV в фибриногене. Важную роль в координации положения лигандов при связывании с αIIbβ3 также играют катионсвязывающие домены β3-цепи MIDAS и ADMIDAS [57].
Благодаря наличию “гибких” регионов в αIIb- и β3-цепях интегрины могут менять свою конформацию и переходить при активации из закрытого (низкоаффинного) состояния в открытое (высокоаффинное).
2.2. Примембранные события активации интегринов
Цитоплазматический домен цепи β3 содержит два NxxY-мотива, которые являются сайтами докинга для талина-Н и киндлина-3, приводящими к конформационным перестройкам интегрина αIIbβ3. В неактивном состоянии талин-Н связан с талином-R и не способен связаться с β3-цепью. Разделение двух субъединиц талина может происходить несколькими путями. При увеличении концентрации свободных ионов кальция в цитозоле активируется белок кальпаин, который разрезает связь между субъединицами, что делает талин-Н активным. С другой стороны, субъединицы талина могут быть разделены в результате связывания комплекса талина и RIAM [58, 59].
Свободные субъединицы талина-Н связываются с одним из мотивов NxxY своими FERM-доменами, что считается одним из важных этапов перехода интегрина αIIbβ3 в активное состояние. Талин-Н может прямо или посредством образования комплексов с винкулином связывать интегрины αIIbβ3 с актиновым цитоскелетом, что также способствует изменению формы тромбоцита. Талин-Н способен связываться с мембранными фосфоинозитидами (например, PIP2), что, наоборот, переведет его в неактивное состояние [60]. Роль талина показана в котрансфицированных талином и αIIbβ3 клетках CHO: в отсутствие талина интегрины αIIbβ3 не переходили в активное состояние [55, 61].
Другие важные участники активации тромбоцитарных интегринов αIIbβ3 – белки семейства киндлинов, без которых не работает ни “inside-out”, ни “outside-in” сигнализация [62, 63]. Подобно талину киндлины связываются FERM-доменом с NxxY-последовательностями в цитоплазматическом домене цепи β3, чаще с удаленным от мембраны мотивом. Также киндлины содержат PH-домен, который позволяет им одновременно связываться с мембранными фосфоинозитидами [62, 63]. Тем не менее, механизм, по которому киндлин-3 приводит к активации интегрина αIIbβ3 и его переходу в высокоаффинное к лиганду состояние, не уточнен [64].
С NxxY-мотивами β3-цепи могут также связываться филамин и белки ICAP, конкурирующие с талином и киндлином и блокирующие активацию интегринов αIIbβ3 [65]. Мигфиллин способен отводить филамин от цитоплазматического домена β3 и таким образом выступать в качестве кофактора активации интегринов, в частности, кофактора киндлина-2, что также показано на CHO-клетках.
С αIIb-цепью могут связываться CIB [66], ICln [67], Aup1 [68], фосфатаза PP1 [69], шарпин [70], однако их роли в активации интегринов не установлены.
2.3.1. GPCR-индуцированная активация
Большинство рецепторов на поверхности тромбоцитов передают внутриклеточный сигнал посредством семейства G-белков (GPCR-рецепторы) [71]. Такими рецепторами являются, например, PAR1, PAR4 (активируемые тромбином), P2Y1, P2Y12 (активируемые ADP) и т.д. Связывание одного из рецепторов G-белков с лигандом приводит к его ассоциации с гетеротримерным G-белком и замене GDP на GTP в α-субъединице G-белка, что вызывает диссоциацию всего комплекса и появление гетеродимера Gβɣ и Gα-GTP – активных переносчиков сигнала. Как Gβɣ, так и Gα-GTP класса “q” могут независимо друг от друга или совместно активировать фосфолипазу Cβ (PLCβ) [72]. Кроме того, Gα-GTP класса “i” и “z”, образующиеся при активации тромбоцита, ингибируют аденилатциклазу, снижая количество cAMP в тромбоците и тем самым усиливая активацию PLCβ [73]. В свою очередь, Gβγ, образующиеся в большом количестве при активации GPCR (до 50 000 на тромбоцит [74]), активируют PI3Kγ.
2.3.2. Индуцированная тирозинкиназами активация
При адгезии в области повреждения происходит активация тромбоцитарных рецепторов GP-VI, обусловленная взаимодействием с коллагеном – компонентом субэндотелиального матрикса. В результате запускается каскад внутриклеточных тирозинкиназ, основными элементами которого являются киназы SFK и Syk [76–78]. При связывании GP-VI с коллагеном SFK фосфорилирует два тирозинсодержащих мотива YxxL в Fcɣ-цепи, ассоциированной с GP-VI. Затем к ним присоединяются SH-2-домены Syk-киназы, переходящие при этом в полностью активное состояние. Активные Syk-киназы фосфорилируют крупный адаптерный белок LAT, к которому присоединяются PI3Kβ, а также PLCγ2 [79]. PI3Kβ и PI3Kγ определяют образование PIP3 из PIP2, который служит сайтом докинга для ряда белков, таких как Btk, а также упомянутый выше киндлин. Активированные PLCγ2 и PLCβ гидролизуют PIP2 до IP3 и DAG [25, 80, 81]. Таким же образом происходит передача сигналов и через другие рецепторы тромбоцитов, зависимые от каскада тирозинкиназ CLEC-2 и FcγRIIa, принимающих участие в более специфичных физиологических ситуациях [82, 83].
2.3.3. Rap1, RASA3, CalDAG-GEFI
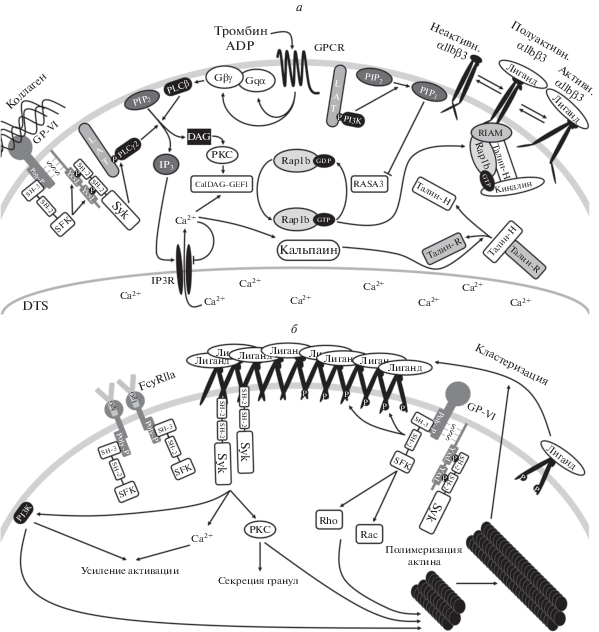
Образовавшиеся в результате активности PLC IP3 и DAG являются важнейшими вторичными мессенджерами при активации тромбоцитов. IP3, связываясь со своими рецепторами на поверхности плотной тубулярной системы (остатков эндоплазматического ретикулума мегакариоцитов, DTS), приводит к выходу свободных ионов кальция в цитозоль клетки [71, 84]. DAG активирует PKC – сериновую киназу, фосфорилирующую большое количество мишеней в клетке. Повышение концентрации свободных ионов кальция приводит к активации не только кальпаина, но и CalDAG-GEFI [85]. Этот белок катализирует реакцию диссоциации GDP в нуклеотидсвязывающем сайте Rap1b, что приводит к связыванию Rap1b с GTP. Активные Rap-1b вместе с адаптерами RIAM и талином-Н приводят к активации интегринов αIIbβ3 [22, 58, 86]. Rap1b-GTP спонтанно инактивируется в результате гидролиза GTP. Этот процесс может многократно ускоряться при связывании Rap1b c белками типа GAP (GTPase activating protein, белки, увеличивающие GTP-азную активность), среди которых в тромбоците в наибольшем количестве представлен белок RASA3 [87, 88]. RASA3, содержащий PH-домен, может связываться как с PIP2, так и с PIP3, что изменяет его субклеточную локализацию. Таким образом, при наработке PIP3, RASA3 покидает зону влияния интегринов, что увеличивает время жизни Rap1b-GTP и ведет к активации αIIbβ3 [87, 88]. Кроме того, существует гипотеза, что фосфорилирование CalDAG-GEFI протеинкиназой А (дезактивируемой при активации тромбоцита) делает его неспособным к замене GDP на GTP в сайте Rap1b. Таким образом блокируется внутренний путь активации интегринов αIIbβ3 в покоящихся тромбоцитах (см. рис. 2а) [59, 85].
2.3.4. Роль взаимодействия тромбоцитов с vWF в активации интегринов
При повреждении стенок сосуда происходит не только “обнажение” субэндотелиального матрикса, но и активация клеток сосудистого эндотелия. Активированные эндотелиальные клетки секретируют из телец Вайбеля-Паладе vWF – мультимерный белок, который может состоять из нескольких десятков мономеров размером 250 кДа каждый [89, 90]. Также известно, что vWF содержится в плазме и альфа-гранулах тромбоцитов [91]. Высокие сдвиговые скорости в потоке (наравне с возможными турбулентными потоками) приводят к тому, что связанные с коллагеном или эндотелием мультимеры vWF “разворачиваются”, что приводит к экспонированию их активных доменов А1, которые могут связываться с тромбоцитарным рецептором GPIb. Этот процесс приводит к первичной адгезии тромбоцитов и, возможно, к их дальнейшей активации по тирозинкиназному пути через Syk и SFK [92]. Также vWF связывается своими C1-доменами с активированными интегринами αIIbβ3, что приводит к развитию сигнализации “outside-in” (см. 2.4), усилению активации тромбоцита и дальнейшему образованию агрегата тромбоцитов в месте повреждения [93].
2.4. Усиление активации: сигнализация “outside-in”
После активации в высокоаффинном к лигандам состоянии интегрины αIIbβ3 связываются с фибрином, фибриногеном или vWF, что приводит к дополнительному усилениию активации тромбоцитов, так называемой “outside-in”-сигнализации [94]. Как уже сказано, цитоплазматические домены активированных интегринов связаны с актиновым цитоскелетом через комплексы талина и киндлина. При активации клеток происходит перераспределение большинства сигнальных молекул посредством диффузии или перестройки актинового цитоскелета, в результате чего интегрины могут образовывать гетерокластеры с другими рецепторами, в частности GPIb, GPVI, FcɣRIIА [24, 27, 28, 95]. Через свои SH3-домены киназы SFK связаны с полипролином в цитоплазматических участках данных рецепторов [96, 97]. Таким образом, активные SFK оказываются в непосредственной близости к активированным интегринам αIIbβ3. SFK фосфорилируют остатки тирозина в участке NxxY, что приводит к дополнительной активации Syk и SFK и усиливает активацию тромбоцитов. Многократно показано, что отсутствие GP-VI или FcɣRIIa приводит к нарушению активации интегринов αIIbβ3 и формирования тромбов у мышей. С другой стороны, кластеризованные интегрины αIIbβ3 могут активировать Syk-киназы и в отсутствие рецепторов GP-VI или FcɣRIIa [27, 28]. Более того, фосфорилирование остатков тирозина в цитоплазматическом домене β3-цепи не является необходимым для этого условием. Предполагается, что Syk может образовать связь с цитоплазматическим доменом β3-цепи, а затем активироваться по пути аутофосфорилирования и транс-аутофосфорилирования [29]. Получающаяся в результате активации “outside-in” положительная обратная связь многократно усиливает активацию тромбоцитов (см. рис. 2б).
2.5. Кластеризация интегринов и необратимое связывание фибриногена
В предыдущем разделе сказано, что для успешного усиления активации тромбоцита (реализация “outside-in”) необходимо образование кластеров интегринов. Однако роль кластеризации интегринов в тромбообразовании остается неясной. Кластеризацию интегринов можно наблюдать по связыванию экзогенного фибриногена человека, меченного флуоресцентной или радиоактивной меткой [98]. В экспериментах тромбоциты иммобилизуются на покрытом адгезионным белком покровном стекле из суспензии либо из цельной крови [89, 99].
На рис. 3 приведены фотографии, полученные в ходе изучения связывания отмытыми тромбоцитами FITC-меченного экзогенного фибриногена человека [100]. В первом эксперименте (рис. 3а, 3б) субстратом для адгезии тромбоцитов также был фибриноген. Можно наблюдать необратимое связывание фибриногена с кластерами интегринов (кластеры интегринов остаются связанными с тромбоцитами после отмывки буфером, см. рис. 3б), в то время как связь фибриногена с одиночными интегринами распадается при разбавлении суспензии (на рис. 3б кластеры интегринов более контрастны, чем на рис. 3а). Во втором эксперименте (рис 3в, 3г) субстратом для адгезии были антитела к рецептору PECAM1 [101]. Далее тромбоциты активировались ADP в концентрации 10 мкМ. Можно наблюдать, что в таких условиях образование кластеров коррелирует с распластыванием тромбоцита (на рис. 3в распластанный тромбоцит связывает фибриноген, а нераспластанный на рис. 3г – нет).
Рис. 3.
Связывание флуоресцентно меченного фибриногена с тромбоцитами человека, микроскопия. Приведены микрофотографии тромбоцитов, полученные в режиме дифференциально-интерференционного контраста (DIC, верхний ряд) и в режиме флуоресценции FITC (нижний ряд). а, б – Отмытые тромбоциты пропускали через проточную камеру с фибриногеном (100 мкг/мл) при помощи шприцевого насоса. После иммобилизации в течение 20 мин не прикрепившиеся к подложке тромбоциты смывали раствором Тироде. Затем через проточную камеру пропускали раствор с ADP (10 мкМ) и FITC-меченным фибриногеном (100 мкг/мл) и регистрировали связывание тромбоцитами меченого фибриногена (а). После этого пропускали буфер Тироде, фотографировали тромбоцит после отмывки (б). Необратимое связывание фибриногена наблюдали в виде “кластеров” с высокой интенсивностью флуоресценции. в, г – Отмытые тромбоциты пропускали через проточную камеру с антителами anti-PECAM1 (клон vm64) (20 мкг/мл) при помощи шприцевого насоса. После иммобилизации в течение 20 мин не прикрепившиеся к подложке тромбоциты смывали буфером Тироде. Через проточную камеру пропускали раствор с ADP (10 мкМ) и FITC-меченным фибриногеном (100 мкг/мл). В случае с активированными (распластанными) тромбоцитами наблюдали связывание фибриногена в флуоресцентном режиме (в). В случае с неактивированными тромбоцитами, имеющими только ламеллиподии, связывание фибриногена не наблюдалось (г).

На рис. 4 приведены данные проточной цитофлуориметрии. В первом эксперименте (рис. 4а) изучали связывание фибриногена после активации тромбином и TRAP-6 с последующим разбавлением буфером в 2 раза. Более слабый активатор (TRAP-6) вызывает снижение связывания при разбавлении. Предполагается, что при определенных условиях связывание фибриногена с интегринами, по крайней мере с некоторой их частью, обратимо. Во втором эксперименте тромбоциты (2 × 107/мл) активировали ADP с последующим разбавлением буфером (без фибриногена) в 20 раз и наблюдали четкое разделение тромбоцитов на две субпопуляции по связыванию меченого фибриногена (рис. 4б). Можно предположить, что одна из популяций (с низким уровнем активации интегринов) связывает фибриноген обратимо, а вторая – необратимо [100].
Рис. 4.
Связывание FITC-меченного фибриногена с тромбоцитами в разбавленной суспензии, проточная цитометрия. Выделение тромбоцитов происходило по протоколу, описанному ранее [71]. а – Непрерывная регистрация событий на проточном цитометре осуществлялась по протоколу, описанному в работах [71, 128]. По оси абсцисс – время наблюдения события (тромбоцита), по оси ординат – интенсивность флуоресценции FITC в условных единицах. Концентрация тромбоцитов 106 клеток/мл. Фибриноген был добавлен в начале эксперимента (не показано). Далее в суспензию были добавлены активаторы: тромбин (25 нМ, черная кривая), TRAP-6 (10 мкМ, серая кривая). После двукратного разбавления буфером без фибриногена наблюдалось падение уровня связывания в случае более слабого активатора – TRAP-6. б – Связывание фибриногена с тромбоцитами в разбавленной суспензии (2 × 107 клеток/мл) до и после активации ADP (10 мкM).
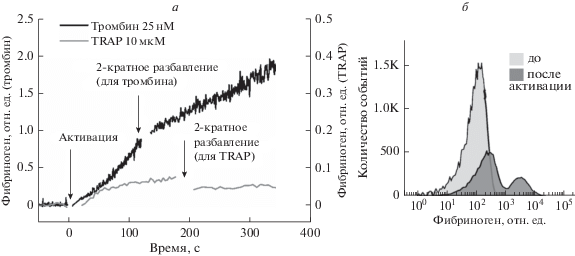
Эти результаты могут свидетельствовать о том, что существуют две степени активации тромбоцитов, соответствующие двум состояниям связывания лигандов интегринов: одиночные интегрины, обратимо связывающие фибриноген, и кластеры интегринов, необратимо связывающие фибриноген. Интересно, что в недавнем сообщении группы исследователей из Технологического университета Джорджии на конгрессе ISTH SSC независимо предложено существование нового “состояния неполной активации” тромбоцитов, индуцированного механорецепцией через гликопротеин Ib и соответствующего обратимому низкоаффинному связыванию фибриногена и фибронектина [102]. Однако этот вопрос требует дополнительных подтверждений, поскольку такие предположения противоречат ранее опубликованным данным [103–106] о связывании активированных тромбоцитов с фибриногеном или антителами к активированным рецепторам PAC-1 в суспензии. В этих работах сообщалось о существовании одного типа сайтов связывания фибриногена. Также необходимо учитывать, что ранее было показано, что в связанные с фибриногеном рецепторы находятся преимущественно в кластеризованном состоянии [107].
3. МЕХАНИКА ВЗАИМОДЕЙСТВИЙ αIIbβ3-ЛИГАНД
Связи между тромбоцитами должны быть достаточно прочными, чтобы растущий тромб сопротивлялся потоку. С использованием атомно-силовой микроскопии показано, что максимальные силы связи между клетками могут достигать несколько наноньютонов.
Механика связывания αIIbβ3 с различными лигандами была изучена в экспериментах Литвинова и соавт. [94, 109, 110]. Они использовали оптическую ловушку – сфокусированный лазерный луч, при помощи которого можно манипулировать небольшими диэлектрическими объектами. Оптическая ловушка позволяет измерить также силу, приложенную к удерживаемому объекту (она будет пропорциональна величине его отклонения от фокуса лазерного луча). В экспериментах использовали кварцевый шарик с нанесенными на него молекулами одного из лигандов αIIbβ3. Кварцевый шарик приводили в контакт с полимерным шариком, покрытым рецепторами αIIbβ3, выделенными из тромбоцитов человека [94, 110].
Изучение взаимодействия интегринов с фибриногеном показало, что распределение критической силы разрыва имеет две моды: оно является суммой убывающей экспоненты (до 20–50 пН) и распределения Гаусса с пиком в 70–80 пН. (рис. 5а) Бимодальная природа распределения сил говорит, возможно, о том, что комплекс αIIbβ3-фибриноген может существовать в двух состояниях с различной стабильностью [94].
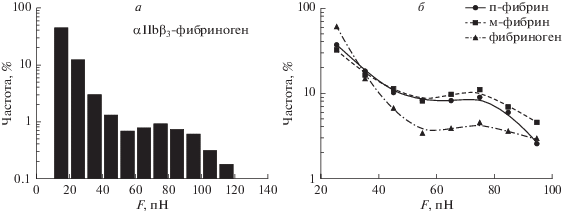
В экспериментах по взаимодействию интегринов с мономерным фибрином (м-фибрин) было выяснено, что вероятность образования связи при контакте в несколько раз выше, чем в случае фибриногена (рис. 5б). При этом гистограмма распределения сил разрыва связи м-фибрин–интегрин не имеет столь же ярко выраженных максимумов, а средняя сила значительно выше. Нужно иметь в виду, что это может свидетельствовать о том, что в некоторых случаях взаимодействие было мультимолекулярным, и критические силы для фибриногена и м-фибрина можно сравнивать лишь качественно. Следует также обратить внимание на то, что при ингибировании 1 мМ γC-додекапептидом (H12) вторая мода для критических связей αIIbβ3–фибриноген пропадала. При ингибировании 1 мМ cRGD и для фибриногена, и для м-фибрина оставалась лишь экспоненциально убывающая часть распределения. Это говорит о том, что при взаимодействии интегринов с различными лигандами меняется не только критическая сила, но и количество возможных стабильных состояний комплекса в данных условиях [94]. При взаимодействии αIIbβ3 с полимеризованным фибрином на графике распределения сил различимы два пика, при этом силы разрыва были в среднем выше, чем в случае других лигандов [110].
Таким образом, по критическим силам взаимодействия лиганды αIIbβ3 можно расположить в следующей последовательности: полимер фибрина > мономер фибрина > фибриноген (рис. 5б). Предполагается, что, комплекс интегрин-лиганд может существовать в двух состояниях с разной стабильностью. Тем не менее, нельзя сделать вывод о том, как меняются критические силы между тромбоцитами при различной степени активации, поскольку использовали выделенные из клеток рецепторы, оценить состояние которых не представляется возможным.
В более ранней работе Литвинова и соавт. [111] исследовали взаимодействие фибриногена с интегринами в мембране тромбоцитов. Распределение сил разрывов связи интегрин-лиганд для активированных ADP тромбоцитов также было бимодальным, но первый пик трактовали как неспецифическое взаимодействие, поскольку он выявлен и на неактивированных ADP тромбоцитах. Однако можно предположить, что на этих тромбоцитах присутствовали активированные интегрины, поскольку анализируемые тромбоциты были частично активированы – это видно на изображении, полученном методом сканирующей электронной микроскопии (тромбоциты шарообразные и имеют филоподии) [111]. Таким образом, этот результат нельзя интерпретировать однозначно. С другой стороны, об одном типе сайтов связывания сообщают более ранние работы [103–106], в которых исследовали связывание тромбоцитов, активированных различными индукторами, с фибриногеном или антителами к активированным рецепторам PAC-1 в суспензии.
4. ПРЕДПОЛАГАЕМАЯ РОЛЬ αIIbβ3 В СТРУКТУРЕ ТРОМБА
Развитие экспериментальных моделей индуцированного тромбообразования у мышей вывело исследования структуры тромба и динамики его роста на новый уровень. Используя флуоресцентное мечение и конфокальную микроскопию, можно с достаточным разрешением наблюдать детали процесса тромбообразования в поврежденном тонкостенном микрососуде.
Именно так показали, что микрососудистый тромб по своей структуре неоднороден (рис. 6): у самого основания формируется стабильная часть, в то время как внешние слои подвижны [7]. Кроме того, анализ диффузии флуоресцентно меченного альбумина показал, что внутри тромб более плотный (меньше альбумина проходит в промежутки между клетками), чем снаружи. Логично предположить, что гетерогенность тромба по плотности тромбоцитов происходит из гетерогенности по силам связи между тромбоцитами. Действительно, тромбоциты, а следовательно, и адгезионные рецепторы различаются по степени активации и доле активированных рецепторов (в расчете на тромбоцит) в разных частях тромба: положительные по P-селектину (дегранулированные) тромбоциты локализованы в основании тромба [7, 16]. Рядом с повреждением обнаруживается и фибрин. Плотную, стабильную, сильно активированную часть тромба называют ядром, а внешнюю рыхлую, текучую, состоящую из подактивированных тромбоцитов – оболочкой [7].
Рис. 6.
Модель микрососудистого тромба. Микрососудистый тромб по своей архитектуре гетерогенный: он состоит из плотного ядра, состоящего из сильно активированных тромбоцитов, и рыхлой шубы из слабо активированных клеток. Тромбоциты ядра секретируют ADP из плотных гранул. Плотность ядра приводит к тому, что диффузия тромбина и ADP из него затруднена (градиент тромбина показан темным цветом). Фибрин локализован у основания тромба. Предполагается, что интегрины αIIbβ3 на тромбоцитах ядра кластеризованы, что обеспечивает усиление активации (выход гранул, переход в прокоагулянтное состояние) и переход в необратимое состояние связей между тромбоцитами. Тромбоциты ядра имеют измененную форму, не показанную на рисунке с целью упрощения.
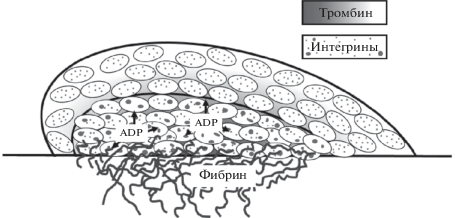
Считается, что для формирования ядра необходимо наличие тромбина – в присутствии ингибитора которого ядро не образуется, тромб нестабильный и постоянно срывается [7, 16]. Кроме того, предполагается, что неоднородность плотности упаковки тромбоцитов приводит к существованию градиента тромбина в тромбе – он в высокой концентрации локализован в ядре, что, с одной стороны, ограничивает рост тромба, а с другой, способствует полимеризации фибрина в уже сформированном ядре [7, 17].
Критичной для наличия оболочки тромба считается активация тромбоцитов через один из рецепторов к ADP, P2Y12. В экспериментах с добавлением с систему кангрелора (ингибитор P2Y12), размер оболочки был значительно уменьшен, в то время как ядро оставалось таким же. С другой стороны, у мышей с мутацией белка Gi (сделавшей их нечувствительными к инактивации RGS-белками, обеспечивающими отрицательную обратную связь при P2Y12-индуцированной сигнализации) оболочка была значительно увеличена, но изменения не затронули ядро. Таким образом, показано, что сигнализация от P2Y12 критична для формирования оболочки тромба [7]. Как упоминалось выше, P2Y12 ассоциирован с белком Gi и его активация приводит к инактивации протеинкиназы А и активации PI3Kγ, т.е. к дополнительному усилению активации тромбоцитарных интегринов “inside-out”. Вероятно, что в формировании рыхлой оболочки тромба значительную роль играет и другой медиатор вторичной активации тромбоцитов – тромбоксан А2.
Можно предположить, что концентрация ADP во внешних слоях тромба достаточна лишь для обратимой или частично необратимой активации ограниченного числа интегринов на поверхности отдельных тромбоцитов (слабая активация, аналогично рис. 4а, TRAP-6). Это может быть связано с тем, что из-за рыхлости оболочки из нее активно вытекают растворимые агонисты, или с тем, что требуемую концентрацию ADP не могут обеспечить ее источники (например, дегранулированные тромбоциты). Поэтому оболочка не так стабильна. Тромбин в основании тромба приводит к сильной активации тромбоцитов и наработке фибрина, также связывающегося с тромбоцитарными интегринами, а значит, уплотнению тромба. Вытекание тромбина из ядра затруднено, что и ограничивает его дальнейший рост.
5. ИНАКТИВАЦИЯ ИНТЕГРИНОВ В ПРОКОАГУЛЯНТНЫХ ТРОМБОЦИТАХ
При сильной активации [112] тромбоциты переходят в прокоагулянтное состояние, связанное с запрограммированной клеточной смертью [84] по типу некроза [113, 114]. При этом на мембранах экспрессируется отрицательно заряженный фосфатидилсерин, выступающий в качестве основы для сборки комплексов [115, 116], а также происходят другие специфические реакции [117, 118]. Интересно, что интегрины в этом прокоагулянтном состоянии не активны [119]. По-видимому, в ходе такой сверхактивации сначала происходит активация, а потом инактивация интегринов по не вполне ясному механизму [120]. Предполагается, что в эту инактивацию вносит вклад расщепление ассоциированных с интегрином белков цитоскелета кальпаином и сам по себе фосфолипидный скрамблинг [121], хотя в других работах ингибирование кальпаина никак не влияло на этот процесс [122]. Интересно, что прокоагулянтные тромбоциты при этом остаются ассоциированными с тромбом [123, 124]. Это взаимодействие обусловлено тем [125], что на их поверхности под действием трансглутаминаз и полимеризации фибрина [126] формируется оболочка – “шуба” из альфа-гранулярных белков, которые служат лигандами для интегринов соседних “нормальных” тромбоцитов.
6. ЗАКЛЮЧЕНИЕ
Интегрины αIIbβ3 – рецепторы тромбоцитов, обеспечивающие их стабильную агрегацию. Интегрины могут менять конформацию, при этом предполагается, что существует спектр состояний интегринов, обладающих различной аффинностью к лигандам – фибриногену, фибрину и фактору Виллебранда. Для перехода интегрина в высокоаффинное состояние необходим сигнал “изнутри”, который заключается в кальций- и фосфоинозитид-зависимой активации малой GTP-азы Rap1b. Как альтернативный путь активации в некоторых работах [43, 53] рассматривается перестройка актинового цитоскелета тромбоцита в области интегринов в результате механических напряжений. Слабая активация тромбоцита приводит к обратимому связыванию интегрина с лигандами (аналогично рис. 4а, TRAP-6), в то время как сильная активация приводит к необратимому связыванию с лигандами. Процесс кластеризации интегринов представляется необходимым и достаточным для развития сигнализации outside-in – усилению активации тромбоцита.
Способность интегринов находиться в различных состояниях (активированное и неактивированное) имеет важное значение при формировании гетерогенной структуры тромба. Для формирования ядра критично наличие тромбина, в то время как для формирования шубы – ADP. Можно предположить, что интегрины тромбоцитов оболочки, активированные ADP, слабо связываются через фибриноген, поэтому шуба нестабильна, так как ADP не может вызвать активацию и кластеризацию достаточного числа интегринов на тромбоцит. С другой стороны, на активированных тромбоцитах ядра большая доля интегринов кластеризована и необратимо связывается с фибриногеном или фибрином. Таким образом, плотность и стабильность внутренней части тромба обеспечивается тем, что комплекс кластеров интегринов-лигандов стабилен и может выдерживать большие силы, возможно, за счет увеличения локальной плотности рецепторов. Плотность ядра по тромбоцитам препятствует вытеканию из него тромбина, что, способствует его высокой локальной концентрации и полимеризации фибрина, а с другой, ограничивает рост ядра. Сильно активированные тромбоциты секретируют ADP из плотных гранул, что обеспечивает слабую активацию тромбоцитов оболочки (концепция изложена на рис. 6) [7, 16, 17].
Дальнейшие исследования требуют развития методов анализа критических сил, которые способны выдержать связанные с лигандом одиночные интегрины. На сегодняшний день это сделано только для одиночных тромбоцитарных интегринов in vitro, что позволяет оценить только порядок сил. Также нельзя достоверно сравнить значения сил для интегринов разной степени активации и оценить роль кластеризации.
Поскольку показано, что фибрин связывается с интегринами прочнее, чем фибриноген [110], необходимо проанализировать вклад этого взаимодействия в реальном тромбе. Это позволит сделать вывод о возможности использования фибрина в качестве мишени при необходимости дестабилизации тромбов.
Работа выполнена в рамках гранта Российского научного фонда (№ 17-74-20 045).
Список литературы
Bouvard D., Brakebusch C., Gustafsson E., Aszódi A., Bengtsson T., Berna A., Fässler R. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89 (3), 211–223.
Якименко А.О., Свешникова А.Н., Артеменко Е.О., Пантелеев М.А. 2014. Этот загадочный тромбоцит. Природа. (2002), 3–8.
Bennett J.S. 1990. The molecular biology of platelet membrane proteins. Semin. Hematol. 27 (2), 186–204.
Plow E.F., Byzova T. 1999. The biology of glycoprotein IIb-IIIa. Coron. Artery Dis. 10 (8), 547–551.
Stalker T.J., Newman D.K., Ma P., Wannemacher K.M., Brass L.F. 2012. Platelet Signaling. Handb. Exp. Pharmacol. (210), 59–85.
Panteleev M.A., Ananyeva N.M., Greco N.J., Ataullakhanov F.I., Saenko E.L. 2005. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J. Thromb. Haemost. 3 (11), 2545–2553.
Stalker T.J., Traxler E.A., Wu J., Wannemacher K.M., Cermignano S.L., Voronov R., Diamond S.L., Brass L.F. 2013. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 121 (10), 1875 –1885.
Niiya K., Hodson E., Bader R., Byers-Ward V., Koziol J.A., Plow E.F., Ruggeri Z.M. 1987. Increased surface expression of the membrane glycoprotein IIb/IIIa complex induced by platelet activation. Relationship to the binding of fibrinogen and platelet aggregation. Blood. 70 (2), 475–483.
Wagner C.L., Mascelli M.A., Neblock D.S., Weisman H.F., Coller B.S., Jordan R.E. 1996. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 88 (3), 907–914.
Stouffer G.A., Smyth S.S. 2003. Effects of thrombin on interactions between beta3-integrins and extracellular matrix in platelets and vascular cells. Arterioscler. Thromb. Vasc. Biol. 23 (11), 1971–1978.
Harrison P., Cramer E.M. 1993. Platelet alpha-granules. Blood Rev. 7 (1), 52–62.
Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. 2000. Ligand binding to integrins. J. Biol. Chem. 275 (29), 21785–21788.
Plow E.F., D’Souza S.E., Ginsberg M.H. 1992. Ligand binding to GPIIb-IIIa: A status report. Semin. Thromb. Hemost. 18 (3), 324–332.
Savage B., Cattaneo M., Ruggeri Z.M. 2001. Mechanisms of platelet aggregation. Curr. Opin. Hematol., 8 (5), 270–276.
Panteleev M.A., Ananyeva N.M., Ataullakhanov F.I., Saenko E.L. 2007. Mathematical models of blood coagulation and platelet adhesion: Clinical applications. Curr. Pharm. Des. 13 (14), 1457–1467.
Welsh J.D., Stalker T.J., Voronov R., Muthard R.W., Tomaiuolo M., Diamond S.L., Brass L.F. 2014. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 124 (11), 1808 LP–1815.
Tomaiuolo M., Stalker T.J., Welsh J.D., Diamond S.L., Sinno T., Brass L.F. 2014. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood. 124 (11), 1816 LP–1823.
Shattil S.J., Kim C., Ginsberg M.H. 2010. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 11, 288.
Buensuceso C., de Virgilio M., Shattil S.J. 2003. Detection of integrin alpha IIbbeta 3 clustering in living cells. J. Biol. Chem. 278 (17), 15217–15224.
Bunch T.A. 2010. Integrin alphaIIbbeta3 activation in Chinese hamster ovary cells and platelets increases clustering rather than affinity. J. Biol. Chem. 285 (3), 1841–1849.
Hato T., Pampori N., Shattil S.J. 1998. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin alphaIIb beta3. J. Cell Biol. 141 (7), 1685–1695.
Banno A., Ginsberg M.H. 2008. Integrin activation. Biochem. Soc. Trans. 36 (Pt 2), 229–234.
Böttcher R.T., Fässler R. 2014. Membrane tension drives ligand- independent integrin signaling. EMBO J. 33 (21), 2439–2442.
Shattil S.J., Newman P.J. 2004. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood. 104 (6), 1606–1615.
Moroi A.J., Watson S.P. 2015. Akt and mitogen-activated protein kinase enhance C-type lectin-like receptor 2-mediated platelet activation by inhibition of glycogen synthase kinase 3α/β. J. Thromb. Haemost. 13 (6), 1139–1150.
Lova P., Paganini S., Sinigaglia F., Balduini C., Torti M. 2002. A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J. Biol. Chem. 277 (14), 12009–12015.
Mangin P.H., Onselaer M.-B., Receveur N., Le Lay N., Hardy A.T., Wilson C., Sanchez X., Loyau S., Dupuis A., Babar A.K., Miller J.L., Philippou H., Hughes C.E., Herr A.B., Ariëns R.A., Mezzano D., Jandrot-Perrus M., Gachet C., Watson S.P. 2018. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 103 (5), 898–907.
Boylan B., Gao C., Rathore V., Gill J.C., Newman D.K., Newman P.J. 2008. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 112 (7), 2780–2786.
Antenucci L., Hytönen V.P., Ylänne J. 2018. Phosphorylated immunoreceptor tyrosine-based activation motifs and integrin cytoplasmic domains activate spleen tyrosine kinase via distinct mechanisms. J. Biol. Chem. 293 (13), 4591–4602.
Lawler J., Hynes R.O. 1989. An integrin receptor on normal and thrombasthenic platelets that binds thrombospondin. Blood. 74 (6), 2022–2027.
Rivas G.A., González-Rodríguez J. 1991. Calcium binding to human platelet integrin GPIIb/IIIa and to its constituent glycoproteins. Effects of lipids and temperature. Biochem. J. 276 (Pt 1), 35–40.
Sheldrake H.M., Patterson L.H. 2009. Function and antagonism of beta3 integrins in the development of cancer therapy. Curr. Cancer Drug Targets. 9 (4), 519–540.
Uzan G., Prenant M., Prandini M.H., Martin F., Marguerie G. 1991. Tissue-specific expression of the platelet GPIIb gene. J. Biol. Chem. 266 (14), 8932–8939.
Oki T., Kitaura J., Eto K., Lu Y., Maeda-Yamamoto M., Inagaki N., Nagai H., Yamanishi Y., Nakajima H., Kumagai H., Kitamura T. 2006. Integrin alphaIIbbeta3 induces the adhesion and activation of mast cells through interaction with fibrinogen. J. Immunol. 176 (1), 52–60.
Oki T., Eto K., Izawa K., Yamanishi Y., Inagaki N., Frampton J., Kitamura T., Kitaura J. 2009. Evidence that integrin alpha IIb beta 3-dependent interaction of mast cells with fibrinogen exacerbates chronic inflammation. J. Biol. Chem. 284 (45), 31463–31472.
Emambokus N.R., Frampton J. 2003. The glycoprotein IIb molecule is expressed on early murine hematopoietic progenitors and regulates their numbers in sites of hematopoiesis. Immunity. 19 (1), 33–45.
Nurden A.T., Pillois X., Wilcox D.A. 2013. Glanzmann thrombasthenia: State of the art and future directions. Semin. Thromb. Hemost. 39 (6), 642–655.
Di Minno G., Zotz R.B., d’Oiron R., Bindslev N., Di Minno M.N.D., Poon M.-C. 2015. The international, prospective Glanzmann Thrombasthenia Registry: Treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia. Haematologica. 100 (8), 1031–1037.
Tsuji S., Sugimoto M., Miyata S., Kuwahara M., Kinoshita S., Yoshioka A. 1999. Real-time analysis of mural thrombus formation in various platelet aggregation disorders: Distinct shear-dependent roles of platelet receptors and adhesive proteins under flow. Blood. 94 (3), 968–975.
Springer T.A. 1997. Folding of the N-terminal, ligand-binding region of integrin alpha-subunits into a beta-propeller domain. Proc. Natl. Acad. Sci. USA. 94 (1), 65–72.
Springer T.A. 2002. Predicted and experimental structures of integrins and beta-propellers. Curr. Opin. Struct. Biol. 12 (6), 802–813.
Xiong J.P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D.L., Joachimiak A., Goodman S.L., Arnaout M.A. 2001. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 294 (5541), 339–345.
Ma Y.-Q., Qin J., Plow E.F. 2007. Platelet integrin alpha(IIb)beta(3): Activation mechanisms. J. Thromb. Haemost. 5 (7), 1345–1352.
Campbell I.D., Humphries M.J. 2011. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 3 (3), a004994.
Zhu J., Luo B.-H., Xiao T., Zhang C., Nishida N., Springer T.A. 2008. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell. 32 (6), 849–861.
Luo B.-H., Carman C. V., Springer T.A. 2007. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Calvete J.J., Henschen A., Gonzalez-Rodriguez J. 1989. Complete localization of the intrachain disulphide bonds and the N-glycosylation points in the alpha-subunit of human platelet glycoprotein IIb. Biochem. J. 261 (2), 561–568.
Xiong J.-P., Stehle T., Goodman S.L., Arnaout M.A. 2003. Integrins, cations and ligands: Making the connection. J. Thromb. Haemost. 1 (7), 1642–1654.
Xiong J.-P., Mahalingham B., Alonso J.L., Borrelli L.A., Rui X., Anand S., Hyman B.T., Rysiok T., Muller-Pompalla D., Goodman S.L., Arnaout M.A. 2009. Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. J. Cell Biol. 186 (4), 589–600.
Beglova N., Blacklow S.C., Takagi J., Springer T.A. 2002. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9 (4), 282–287.
Takagi J., Petre B.M., Walz T., Springer T.A. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110 (5), 511–599.
Yang J., Ma Y.-Q., Page R.C., Misra S., Plow E.F., Qin J. 2009. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc. Natl. Acad. Sci. USA. 106 (42), 17729–17734.
Liu S., Calderwood D.A., Ginsberg M.H. 2000. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113 (Pt 2), 3563–3571.
Aylward K., Meade G., Ahrens I., Devocelle M., Moran N. 2006. A novel functional role for the highly conserved alpha-subunit KVGFFKR motif distinct from integrin alphaIIbbeta3 activation processes. J. Thromb. Haemost. 4 (8), 1804–1812.
Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T.A., Plow E.F., Qin J. 2002. A Structural Mechanism of Integrin alpha(IIb)beta(3) “Inside-Out” activation as Regulated by Its Cytoplasmic Face. Cell. 110 (5), 587–597.
Weljie A.M., Hwang P.M., Vogel H.J. 2002. Solution structures of the cytoplasmic tail complex from platelet integrin alpha IIb- and beta 3-subunits. Proc. Natl. Acad. Sci. USA. 99 (9), 5878–5883.
Calderwood D.A., Fujioka Y., de Pereda J.M., Garcia-Alvarez B., Nakamoto T., Margolis B., McGlade C.J., Liddington R.C., Ginsberg M.H. 2003. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA. 100 (5), 2272–2277.
Calderwood D.A., Zent R., Grant R., Rees D.J., Hynes R.O., Ginsberg M.H. 1999. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274 (40), 28071–28074.
Tadokoro S., Shattil S.J., Eto K., Tai V., Liddington R.C., de Pereda J.M., Ginsberg M.H., Calderwood D.A. 2003. Talin binding to integrin beta tails: A final common step in integrin activation. Science. 302 (5642), 103–106.
Martel V., Racaud-Sultan C., Dupe S., Marie C., Paulhe F., Galmiche A., Block M.R., Albiges-Rizo C. 2001. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276 (24), 21217–21227.
Wegener K.L., Partridge A.W., Han J., Pickford A.R., Liddington R.C., Ginsberg M.H., Campbell I.D. 2007. Structural basis of integrin activation by talin. Cell. 128 (1), 171–182.
D’Souza M.-A.M.A., Kimble R.M., McMillan J.R. 2010. Kindler syndrome pathogenesis and fermitin family homologue 1 (kindlin-1) function. Dermatol. Clin. 28 (1), 115–118.
Moser M., Legate K.R., Zent R., Fassler R. 2009. The tail of integrins, talin, and kindlins. Science. 324 (5929), 895–899.
Kammerer P., Aretz J., Fässler R. 2017. Lucky kindlin: A cloverleaf at the integrin tail. Proc. Natl. Acad. Sci. USA. 114 (35), 9234–9236.
Brunner M., Millon-Fremillon A., Chevalier G., Nakchbandi I.A., Mosher D., Block M.R., Albiges-Rizo C., Bouvard D. 2011. Osteoblast mineralization requires beta1 integrin/ICAP-1-dependent fibronectin deposition. J. Cell Biol. 194 (2), 307–322.
Shock D.D., Naik U.P., Brittain J.E., Alahari S.K., Sondek J., Parise L.V. 1999. Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem. J. 342 Pt 3, 729–735.
Larkin D., Murphy D., Reilly D.F., Cahill M., Sattler E., Harriott P., Cahill D.J., Moran N. 2004. ICln, a novel integrin alphaIIbbeta3-associated protein, functionally regulates platelet activation. J. Biol. Chem. 279 (26), 27286–27293.
Kato A., Kawamata N., Tamayose K., Egashira M., Miura R., Fujimura T., Murayama K., Oshimi K. 2002. Ancient ubiquitous protein 1 binds to the conserved membrane-proximal sequence of the cytoplasmic tail of the integrin alpha subunits that plays a crucial role in the inside-out signaling of alpha IIbbeta 3. J. Biol. Chem. 277 (32), 28934–28941.
Vijayan K.V., Liu Y., Li T.-T., Bray P.F. 2004. Protein phosphatase 1 associates with the integrin alphaIIb subunit and regulates signaling. J. Biol. Chem. 279 (32), 33039–33042.
Rantala J.K., Pouwels J., Pellinen T., Veltel S., Laasola P., Mattila E., Potter C.S., Duffy T., Sundberg J.P., Kallioniemi O., Askari J.A., Humphries M.J., Parsons M., Salmi M., Ivaska J. 2011. SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat. Cell Biol. 13 (11), 1315–1324.
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A. V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Horowitz L.F., Hirdes W., Suh B.-C., Hilgemann D.W., Mackie K., Hille B. 2005. Phospholipase C in living cells: Activation, inhibition, Ca2+ requirement, and regulation of M current. J. Gen. Physiol. 126 (3), 243–262.
Шатурный В.И., Шахиджанов С.С., Свешникова А.Н., Пантелеев М.А. 2014. Активаторы, рецепторы и пути внутриклеточной сигнализации в тромбоцитах крови. Биомед. химия. 60 (2), 182–200.
Burkhart J.M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., Zahedi R.P. 2012. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 120 (15), e73–e82.
Lian L., Wang Y., Draznin J., Eslin D., Bennett J.S., Poncz M., Wu D., Abrams C.S. 2005. The relative role of PLC and PI3K in platelet activation. Blood. 106 (1), 110–117.
Tsang E., Giannetti A.M., Shaw D., Dinh M., Tse J.K.Y., Gandhi S., Ho A., Wang S., Papp E., Bradshaw J.M. 2008. Molecular mechanism of the Syk activation switch. J. Biol. Chem. 283 (47), 32 650–32 659.
Watson S.P., Herbert J.M.J., Pollitt A.Y. 2010. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8 (7), 1456–1467.
Dunster J.L., Mazet F., Fry M.J., Gibbins J.M., Tindall M.J. 2015. Regulation of early steps of GPVI signal transduction by phosphatases: A systems biology approach. PLoS Comput. Biol. 11 (11), 1–26.
Pasquet J.M., Gross B., Quek L., Asazuma N., Zhang W., Sommers C.L., Schweighoffer E., Tybulewicz V., Judd B., Lee J.R., Koretzky G., Love P.E., Samelson L.E., Watson S.P. 1999. LAT is required for tyrosine phosphorylation of phospholipase cgamma2 and platelet activation by the collagen receptor GPVI. Mol. Cell. Biol. 19 (12), 8326–8334.
Moroi A.J., Watson S.P. 2015. Impact of the PI3-kinase/Akt pathway on ITAM and hemITAM receptors: Haemostasis, platelet activation and antithrombotic therapy. Biochem. Pharmacol. 94 (3), 186–194.
Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S.M., Moroi M., Andrews R.K., Berndt M.C., Watson S.P. 2002. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277 (24), 21561–21566.
Nieswandt B., Bergmeier W., Schulte V., Rackebrandt K., Gessner J.E., Zirngibl H. 2000. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcR gamma chain. J. Biol. Chem. 275 (31), 23 998–24 002.
May F., Hagedorn I., Pleines I., Bender M., Vogtle T., Eble J., Elvers M., Nieswandt B. 2009. CLEC-2 is an essential platelet activating receptor in hemostasis and thrombosis. Blood. 114 (16), 3464–3473.
Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2015. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol. BioSyst. 11 (4), 1052–1060.
Stefanini L., Bergmeier W. 2014. CalDAG-GEFI and platelet activation. Platelets. 21 (4), 239–243.
Stefanini L., Bergmeier W. 2016. RAP1-GTPase signaling and platelet function. J. Mol. Med. 94 (1), 13–19.
Stefanini L., Paul D.S., Robledo R.F., Chan E.R., Getz T.M., Campbell R.A., Kechele D.O., Casari C., Piatt R., Caron K.M., Mackman N., Weyrich A.S., Parrott M.C., Boulaftali Y., Adams M.D., Peters L.L., Bergmeier W. 2015. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Invest. 125 (4), 1419–1432.
Battram A.M., Durrant T.N., Agbani E.O., Heesom K.J., Paul D.S., Piatt R., Poole A.W., Cullen P.J., Bergmeier W., Moore S.F., Hers I. 2017. The phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) binder Rasa3 regulates phosphoinositide 3-kinase (PI3K)-dependent integrin αIIbβ3outside-in signaling. J. Biol. Chem. 292 (5), 1691–1704.
Lawrence M.B., McIntire L.V., Eskin S.G. 1987. Effect of flow on polymorphonuclear leukocyte/endothelial cell adhesion. Blood. 70 (5), 1284 LP-1290.
Turner N.A., Sartain S.E., Hui S.-K., Moake J.L. 2015. Regulatory components of the alternative complement pathway in endothelial cell cytoplasm, factor H and factor I, are not packaged in Weibel-Palade bodies. PLoS One. 10 (3), e0121994.
Gralnick H.R., Williams S.B., McKeown L.P., Magruder L., Hansmann K., VAIL M., Parker R.I. 1991. Platelet von Willebrand Factor. Mayo Clin. Proc. 66 (6), 634–640.
Yin H., Liu J., Li Z., Berndt M.C., Lowell C.A., Du X. 2008. Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood. 112 (4), 1139–1146.
Canobbio I., Balduini C., Torti M. 2004. Signalling through the platelet glycoprotein Ib-V–IX complex. Cell. Signal. 16 (12), 1329–1344.
Litvinov R.I., Farrell D.H., Weisel J.W., Bennett J.S. 2016. The platelet integrin αIIbβ3 differentially interacts with fibrin versus fibrinogen. J. Biol. Chem. 291 (15), 7858–7867.
Schaff M., Tang C., Maurer E., Bourdon C., Receveur N., Eckly A., Hechler B., Arnold C., de Arcangelis A., Nieswandt B., Denis C. V., Lefebvre O., Georges-Labouesse E., Gachet C., Lanza F., Mangin P.H. 2013. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. 128 (5), 541–552.
Wu Y., Span L.M., Nygren P., Zhu H., Moore D.T., Cheng H., Roder H., DeGrado W.F., Bennett J.S. 2015. The tyrosine kinase c-Src specifically binds to the active integrin αIIbβ3 to initiate outside-in signaling in platelets. J. Biol. Chem. 290 (25), 15825–15834.
Schmaier A.A., Zou Z., Kazlauskas A., Emert-Sedlak L., Fong K.P., Neeves K.B., Maloney S.F., Diamond S.L., Kunapuli S.P., Ware J., Brass L.F., Smithgall T.E., Saksela K., Kahn M.L. 2009. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc. Natl. Acad. Sci. USA. 106 (50), 21167–21172.
Peerschke E.I.B.I. 1995. Bound fibrinogen distribution on stimulated platelets: Examination by confocal scanning laser microscopy. Am. J. Pathol. 147, 678–687.
Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2016. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 14 (9), 1867–1881.
Morozova D., Demianova A., Canault M., Panteleev M., Alessi M.-C., Sveshnikova. A. 2018. Identification of platelet intracellular signalling pathways controlling the reversible and irreversible platelet aggregation. Res. Pract. Thromb. Haemost. 2 (S1), 46.
Mazurov A.V., Vinogradov D.V., Kabaeva N.V., Antonova G.N., Romanov Y.A., Vlasik T.N., Antonov A.S., Smirnov V.N. 1991. A monoclonal antibody, VM64, reacts with a 130 kDa glycoprotein common to platelets and endothelial cells: Heterogeneity in antibody binding to human aortic endothelial cells. Thromb. Haemost. 66 (4), 494—499.
Chen Y., Ju L., Jackson S., Zhu C. 2018. An intermediate state of integrin αIIbβ3 mediates platelet aggregation under disturbed flow. Res. Pract. Thromb. Haemost. 2 (S1), 43.
Plow E.F., Marguerie G.A. 1980. Participation of ADP in the binding of fibrinogen to thrombin-stimulated platelets. Blood. 56 (3), 553–555.
Plow E.F., Marguerie G.A. 1980. Induction of the fibrinogen receptor on human platelets by epinephrine and the combination of epinephrine and ADP. J. Biol. Chem. 255 (22), 10971–10977.
Shattil S.J., Hoxie J.A., Cunningham M., Brass L.F. 1985. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J. Biol. Chem. 260 (20), 11107–11114.
Bennett J.S., Vilaire G. 1979. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Invest. 64 (5), 1393–1401.
Isenberg W.M., Mcever R.P., Phillips D.R., Shuman M.A., Bainton D.F. 1989. Immunogold-surface replica study of ADP-induced ligand binding and fibrinogen receptor clustering in human platelets. Am. J. Anat. 85, 142–148.
Nguyen T.-H., Palankar R., Bui V.-C., Medvedev N., Greinacher A., Delcea M. 2016. Rupture forces among human blood platelets at different degrees of activation. Sci. Rep. 6, 25402.
Weisel J.W., Litvinov R.I. 2013. Mechanisms of fibrin polymerization and clinical implications. Blood. 121 (10), 1712–1719.
Höök P., Litvinov R.I., Kim O. V., Xu S., Xu Z., Bennett J.S., Alber M.S., Weisel J.W. 2017. Strong binding of platelet integrin αIIbβ3 to fibrin clots: Potential target to destabilize thrombi. Sci. Rep. 7 (1), 13001.
Litvinov R.I., Shuman H., Bennett J.S., Weisel J.W. 2002. Binding strength and activation state of single fibrinogen-integrin pairs on living cells. Proc. Natl. Acad. Sci. USA. 99 (11), 7426–7431.
Topalov N.N., Kotova Y.N., Vasil’ev S.A., Panteleev M.A. 2012. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 157 (1), 105–115.
Shakhidzhanov S.S., Shaturny V.I., Panteleev M.A., Sveshnikova A.N. 2015. Modulation and pre-amplification of PAR1 signaling by ADP acting via the P2Y12 receptor during platelet subpopulation formation. Biochim. Biophys. Acta. 1850 (12), 2518–2529.
Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2016. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 14 (9), 1867–1881.
Belyaev A.V., Dunster J.L., Gibbins J.M., Panteleev M.A., Volpert V. 2018. Modeling thrombosis in silico: Frontiers, challenges, unresolved problems and milestones. Phys. Life Rev. doi 10.1016/j.plrev.2018.02.005
Panteleev M.A., Saenko E.L., Ananyeva N.M., Ataullakhanov F.I. 2004. Kinetics of Factor X activation by the membrane-bound complex of Factor IXa and Factor VIIIa. Biochem. J. 381 (Pt 3), 779–794.
Zakharova N.V., Artemenko E.O., Podoplelova N.A., Sveshnikova A.N., Demina I.A., Ataullakhanov F.I., Panteleev M.A. 2015. Platelet surface-associated activation and secretion-mediated inhibition of coagulation factor XII. PLoS One. 10 (2), e0116665.
Topalov N.N., Yakimenko A.O., Canault M., Artemenko E.O., Zakharova N.V., Abaeva A.A., Loosveld M., Ataullakhanov F.I., Nurden A.T., Alessi M.-C., Panteleev M.A. 2012. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin alpha(IIb)beta(3). Arterioscler. Thromb. Vasc. Biol. 32 (10), 2475–2483.
Dale G.L., Friese P., Batar P., Hamilton S.F., Reed G.L., Jackson K.W., Clemetson K.J., Alberio L. 2002. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 415 (6868), 175–179.
Munnix I.C.A., Kuijpers M.J.E., Auger J., Thomassen C.M.L.G.D., Panizzi P., van Zandvoort M.A.M., Rosing J., Bock P.E., Watson S.P., Heemskerk J.W.M. 2007. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: Regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 27 (11), 2484–2490.
Mattheij N.J.A., Gilio K., van Kruchten R., Jobe S.M., Wieschhaus A.J., Chishti A.H., Collins P., Heemskerk J.W.M., Cosemans J.M.E.M. 2013. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. J. Biol. Chem. 288 (19), 13 325–13 336.
Artemenko E.O., Yakimenko A.O., Pichugin A.V., Ataullakhanov F.I., Panteleev M.A. 2016. Calpain-controlled detachment of major glycoproteins from the cytoskeleton regulates adhesive properties of activated phosphatidylserine-positive platelets. Biochem. J. 473 (4), 435–448.
Podoplelova N.A., Sveshnikova A.N., Kotova Y.N., Eckly A., Receveur N., Nechipurenko D.Y., Obydennyi S.I., Kireev I.I., Gachet C., Ataullakhanov F.I., Mangin P.H., Panteleev M.A. 2016. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 128 (13), 1745–1755.
Podoplelova N.A., Sveshnikova A.N., Kurasawa J.H., Sarafanov A.G., Chambost H., Vasil’ev S.A., Demina I.A., Ataullakhanov F.I., Alessi M.-C., Panteleev M.A. 2016. Hysteresis-like binding of coagulation factors X/Xa to procoagulant activated platelets and phospholipids results from multistep association and membrane-dependent multimerization. Biochim. Biophys. Acta. 1858 (6), 1216–1227.
Yakimenko A.O., Verholomova F.Y., Kotova Y.N., Ataullakhanov F.I., Panteleev M.A. 2012. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys. J. 102 (10), 2261–2269.
Abaeva A.A., Canault M., Kotova Y.N., Obydennyy S.I., Yakimenko A.O., Podoplelova N.A., Kolyadko V.N., Chambost H., Mazurov A.V., Ataullakhanov F.I., Nurden A.T., Alessi M.-C., Panteleev M.A. 2013. Procoagulant platelets form an alpha-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J. Biol. Chem. 288 (41), 29 621–29 632.
Yuki K., Bu W., Shimaoka M., Eckenhoff R. 2013. Volatile anesthetics, not intravenous anesthetic propofol bind to and attenuate the activation of platelet receptor integrin αIIbβ3. 8 (4), e60415.
Assinger A., Volf I., Schmid D. 2015. A Novel, rapid method to quantify intraplatelet calcium dynamics by ratiometric flow cytometry. PLoS One. 10 (4), 1–15.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии