

Биологические мембраны: Журнал мембранной и клеточной биологии, 2020, T. 37, № 4, стр. 271-281
Низкомолекулярные лиганды рецептора лютеинизирующего гормона с активностью антагонистов
К. В. Деркач a, Д. В. Дарьин b, А. О. Шпаков a, *
a Институт эволюционной физиологии и биохимии
им. И.М. Сеченова РАН
194223 Санкт-Петербург, Россия
b Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
* E-mail: alex_shpakov@list.ru
Поступила в редакцию 29.11.2019
После доработки 05.12.2019
Принята к публикации 22.12.2019
Аннотация
В настоящее время ведется поиск селективных антагонистов рецептора лютеинизирующего гормона (ЛГ), необходимых для подавления стероидогенеза при гормонозависимых опухолях и предотвращения синдрома гиперстимуляции яичников. Одним из подходов для решения этой проблемы является создание низкомолекулярных антагонистов аллостерического сайта этого рецептора, локализованного в его трансмембранном домене. Цель работы состояла в разработке гетероциклических соединений, производных тиено-[2,3-d]пиримидина (TP31), пиримидо[4,5,6-de][1,6]нафтиридина (PP10) и пиридо[3,4-d]пиримидина (PP17), и в изучении их способности влиять на функциональную активность рецептора ЛГ в условиях in vitro и in vivo. Показано, что соединение TP31 в микромолярных концентрациях подавляет стимулирующие эффекты хорионического гонадотропина человека (ХГЧ) и TP03, аллостерического агониста рецептора ЛГ, на активность аденилатциклазы в тестикулярных мембранах крысы, причем его действие в наибольшей степени проявлялось в отношении стимулирующих эффектов TP03. Это обусловлено большей селективностью антагониста TP31 в отношении cAMP-зависимых сигнальных каскадов, преимущественно активируемых TP03 и реализуемых через Gs-белки. Соединение PP17 в одинаковой степени ингибировало стимулирующие эффекты ХГЧ и TP03 на активность аденилатциклазы, но было менее активным в сравнении с TP31. При интратестикулярном (10 мг/кг) и внутрибрюшинном (45 мг/кг) введении самцам крыс соединения TP31 и PP17 снижали базовый уровень тестостерона в крови, а также подавляли продукцию тестостерона, стимулированную ХГЧ (100 МЕ/крысу), причем ингибирующий эффект TP31 был выражен намного сильнее. В сравнении с TP31 и PP17, соединение PP10 характеризовалось слабо выраженной антагонистической активностью в условиях in vitro и in vivo. Полученные данные указывают на то, что TP31, наиболее активный функциональный антагонист среди изученных соединений, связываясь с аллостерическим сайтом рецептора ЛГ, делает его малодоступным для аллостерических агонистов и нарушает передачу гормонального сигнала через рецептор ЛГ, что указывает на перспективность разработки ингибиторов ЛГ-зависимых путей и стероидогенеза на его основе.
ВВЕДЕНИЕ
Основными регуляторами стероидогенеза у мужчин и женщин являются лютеинизирующий гормон (ЛГ), который вырабатывается гонадотрофами гипофиза, и его структурный и функциональный гомолог – хорионический гонадотропин человека (ХГЧ), гипофизарная форма которого, как и ЛГ, вырабатывается гонадотрофами, а плацентарная форма продуцируется у женщин в первом триместре беременности. Все формы гонадотропинов специфично взаимодействуют с эктодоменом рецептора ЛГ, активируют его и запускают широкий спектр сигнальных каскадов в клетках Лейдига семенников или в клетках гранулезы и теки яичников, стимулируя процесс стероидогенеза [1–3]. Фармакологические препараты ЛГ и ХГЧ способны вызывать побочные эффекты, что обусловлено их низкой селективностью по отношению к внутриклеточным эффекторным системам, развитием к ним резистентности клеток-мишеней, необходимостью парентерального введения, иммуногенностью и гетерогенностью. Вследствие этого ведется разработка низкомолекулярных аллостерических агонистов рецептора ЛГ, способных стимулировать стероидогенез, не вызывая побочных эффектов, характерных для гонадотропинов [4, 5]. Наиболее эффективными среди них являются производные тиено-[2,3-d]пиримидина, в том числе ранее разработанные нами соединения TP01, TP03, TP04 и TP23 которые при различных способах введения самцам крыс стимулируют тестикулярный стероидогенез и повышают уровень тестостерона в крови животных [6–10].
Наряду с активаторами рецептора ЛГ, в клинике востребованы и его антагонисты, снижающие функциональную активность ЛГ-зависимых сигнальных каскадов. Они нацелены на предотвращение гиперактивации этих каскадов при развитии синдрома гиперстимуляции яичников [11, 12], а также могут использоваться для лечения гормонозависимых опухолей, ингибируя ЛГ-зависимую сигнализацию и снижая продукцию стероидных гормонов [13, 14]. Для этого обычно используют антагонисты гонадолиберина, стероидные и нестероидные антиандрогенные препараты, что, однако, сопряжено с развитием острого дефицита стероидных гормонов и тяжелыми репродуктивными расстройствами [13–15]. Альтернативой может стать разработка низкомолекулярных аллостерических регуляторов рецептора ЛГ с активностью антагонистов и(или) инверсионных агонистов. В 2009 году было получено соединение LUF5771, производное терфенила, наделенное активностью инверсионного агониста рецептора ЛГ, но оно до сих пор не используется в клинических условиях [16, 17]. Необходимо отметить, что, в отличие от рецептора ЛГ, разработано значительное число низкомолекулярных антагонистов и инверсионных агонистов для рецепторов тиреотропного (ТТГ) и фолликулостимулирующего гормонов (ФСГ), сходных по структурно-функциональной организации с рецептором ЛГ [5, 18–20].
Целью работы были разработка и изучение механизмов действия различных по структуре гетероциклических соединений, производных тиено-[2,3-d]пиримидина (TP31), пиримидо[4,5,6-de] [1, 6]нафтиридина (PP10) и пиридо[3,4-d]пиримидина (PP17), с активностью антагонистов рецептора ЛГ и зависимых от него сигнальных каскадов в условиях in vitro и in vivo. Антагонистическую активность оценивали по влиянию исследуемых соединений на стимулированную ХГЧ активность аденилатциклазы (АЦ) в тестикулярных мембранах крысы, а также на базовый и стимулированный ХГЧ уровень тестостерона в крови самцов крыс. Для исследования также использовали разработанное и изученное ранее тиено-[2,3-d]пиримидиновое производное TP03 с активностью полного аллостерического агониста рецептора ЛГ [7, 9, 10].
МАТЕРИАЛЫ И МЕТОДЫ
Для экспериментов были взяты самцы крыс Wistar (возраст 2.5–3 мес.), которых содержали в стандартных условиях вивария и на стандартном рационе. Все процедуры проводили в соответствии с правилами, разработанными Комитетом по биоэтике ИЭФБ РАН (15.02.2018 г.), и с правилами и требованиями, изложенными в документах “European Communities Council Directive 1986” (86/609/EEC) и “Guide for the Care and Use of Laboratory Animals”.
В биохимических экспериментах использовали креатинфосфат, креатинфосфокиназу из мышц кролика, cAMP, ATP, β,γ-имидогуанозин-5'-трифосфат (GppNHp) и другие реактивы производства фирмы Sigma (США). ХГЧ был производства Московского эндокринологического завода (Россия). Для определения активности АЦ использовали [α-32P]ATP (150 ГБк/ммоль) производства “Изотоп” (Россия). Для разделения смеси меченых нуклеотидов использовали активированную нейтральную окись алюминия (Sigma). Структуру синтезируемых соединений подтверждали с помощью 1H-ЯМР и масс-спектрометрии высокого разрешения. Для регистрации спектров 1H-ЯМР использовали спектрометр Bruker Avance III 400 (на ядрах 1H с частотой 400.13 MГц). Химические сдвиги указывали в миллионных долях (δ, м.д.), в качестве внутреннего стандарта использовали остаточный сигнал ДМСО-d6 (2.50 м.д.), константы спин-спинового взаимодействия (J) измеряли в приближении первого порядка и приводили в Гц. Масс-спектры высокого разрешения регистрировали с помощью спектрометра Bruker micrOTOF (electrospray ionization – time of flight, ESI-TOF) (Германия).
Соединение TP31, 5-амино-N-(трет-бутил)-2-(метилтио)-4-[3-(2-(этиламино)никотинамидо)-фенил]тиено[2,3-d]пиримидин-6-карбоксамид (рис. 1), синтезировали по той же схеме, что и соединение TP03, как описано ранее [7]. Для синтеза использовали реакцию ацилирования 5-амино-4-(3-аминофенил)-N-(трет-бутил)-2-(метилсульфанил)тиено[2,3-d]пиримидин-6-карбоксамида в присутствии 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксида гексафторфосфата (HATU) и N,N-диизопропилэтиламина. Целевой продукт очищали с помощью колоночной хроматографии (элюент: хлороформ−ацетон 10:1), перекристаллизовывали из метанола, выход составил 44%, т. пл. 149–150°С. Спектр ЯМР 1Н (δ, м. д. (J, Гц), ДМСО-d6) содержал следующие сигналы: 1.16 (3Н, т, CH2CH3, J 7.1); 1.37 (9Н, с, t-Bu); 2.62 (3H, c, SMe); 3.44 (2H, к, CH2CH3,J 7.1); 6.11 (2H, c, NH2); 6.64 (1H, дд, J 4.8, 7.6); 6.97 (1H, c, NH-t-Bu); 7.39 (1H, д, J 7.6); 7.58 (1H, т, J 7.9); 7.89–7.99 (2H, м); 8.02 (1H, c); 8.10 (1H, дд, J 1.7, 7.6); 8.23 (1H, дд, J 1.6, 4.8); 10.47 (1H, c). По данным масс-спектрометрии высокого разрешения, молекулярная масса ТР31 составила 536.1899 (вычислено для [M + H+] – 536.1897; C26H29N7O2S2).
Соединение PP10, этиловый эфир 8-амино-5-метил-2-(метилсульфанил)-6-(метиксикарбонил)-7Н-пиримидо[4,5,6-de] [1, 6]нафтиридин-9-карбоновой кислоты (рис. 1), было получено, как описано ранее [21]. Целевой продукт очищали с помощью колоночной хроматографии, перекристаллизацию проводили в смеси гексан–этилацетат 1 : 1. Выход реакции составил 63%, т. пл. 187-190°С (с разложением). Спектр ЯМР 1Н (δ, м. д. (J, Гц), ДМСО-d6) содержал следующие сигналы: 1.32 (т, 3H, J = 7.1, CH2CH3), 2.33 (с, 3H, SCH3), 2.54 (с, 3H, 7-Me), 3.85 (с, 3H, CO2CH3), 4.28 (к, 2H, J = 7.1, CH2CH3), 7.74 (2H, ш.с., NH2). По данным масс-спектрометрии высокого разрешения, молекулярная масса PР10 составила 376.1081 (вычислено для [M + Na+] – 376.1074; C16H18N5O4S).
Соединение PP17, этиловый эфир 7-амино-4-(3-(этоксикарбонил)пиперидин-1-ил)-2-(метилсульфанил)пиридо[4,3-d]пиримидин-8-карбоновой кислоты (рис. 1), было получено нуклеофильным замещением активного атома хлора циклическим амином (этилнипекотатом) из этилового эфира 7-амино-2-метилсульфанил-4-хлорпиридо[4,3-d]-пиримидин-8-карбоновой кислоты (соединение 1), синтез которого описан ранее [22].
В соответствии с разработанной методикой, к раствору этилового эфира нипекотиновой кислоты (1.59 ммоль) и N,N-диизопропилэтиламина (2.0 ммоль) в сухом N,N-диметилформамиде (2 мл) добавляли соединение 1 (1.33 ммоль). Раствор перемешивали 15 мин при комнатной температуре, добавляли к нему охлажденную воду (12 мл, 4°С), продукт экстрагировали хлористым метиленом (дважды по 10 мл), органическую фазу промывали водой (дважды по 8 мл), высушивали над прокаленным CaCl2, растворитель отгоняли в вакууме, остаток растирали с гексаном (10 мл), выдержали в течение 1 ч (+4°С), полученные кристаллы отфильтровали и высушивали. Выход реакции составил 66%, т. пл. 125–127°С. Спектр ЯМР 1Н (δ, м. д. (J, Гц), дейтерированный хлороформ) содержал следующие сигналы: 8.76 (c, 1H), 6.69 (ш.с., 2H), 4.47 (кв, J = 7.1, 2H), 4.42–4.35 (м, 1H), 4.17 (кв, J = 7.1, 2H), 4.14–4.07 (м, 1H), 3.46 (дд, J = 13.3, 10.0, 1H), 3.39–3.30 (м, 1H), 2.78–2.69 (м, 1H), 2.62 (с, 3H), 2.21–2.13 (м, 1H), 1.94–1.66 (м, 4H), 1.46 (т, J = 7.1, 3H), 1.27 (т, J = = 7.1, 3H). По данным масс-спектрометрии высокого разрешения, молекулярная масса PР17 составила 420.1712 (вычислено для [M + Na+] – 420.1700; C19H26N5O4S).
Для определения активности соединений в условиях in vitro из тканей семенников крыс Wistar (n = 6) выделяли фракции плазматических мембран. Животных наркотизировали, декапитировали, иссекали у них ткани семенников, которые затем промывали в охлажденном до 4°C 40 мМ Трис-HCl-буфере (pH 7.4), содержащем 5 мМ MgCl2, 10% сахарозу и ингибиторы протеаз (буфере А), измельчали и гомогенизировали в 10 объемах того же буфера. Гомогенат центрифугировали (1500 g, 10 мин), супернатант отделяли и повторно центрифугировали (20 000 g, 30 мин). Осажденные мембраны ресуспендировали в буфере А без сахарозы (содержание белка 1–2 мг/мл), замораживали и хранили при –80°C, используя в дальнейшем для определения активности АЦ.
Активность АЦ (ATP-пирофосфатлиазы циклизующей, НФ 4.6.1.1) во фракциях тестикулярных мембран определяли, как описано ранее [7], для чего мембраны (50–80 мкг белка) инкубировали в течение 12 мин при 37°С в смеси, содержащей 50 мМ Трис-HCl (pH 7.5), 5 мМ MgCl2, 0.1 мМ cAMP, 1 мМ ATP, 37 КБк [α-32P]ATP, 20 мМ креатинфосфата, 0.2 мг/мл креатинфосфокиназы. Реакцию проводили в течение 12 мин при 37°C, начиная добавлением фракции мембран и останавливая добавлением в пробу 100 мкл 0.5 M HCl. Пробы кипятили в течение 6 мин, кислоту нейтрализовали 100 мкл 1.5 M имидазола. Образовавшийся [32P]cAMP отделяли от других меченых нуклеотидов на колонках с нейтральным оксидом алюминия (градация II по Брокману), используя в качестве элюента 8 мл 10 мМ имидазол-HCl буфера (pH 7.4). Радиоактивность измеряли на счетчике LKB 1209/1215 RackBeta (LKB, Швеция). Активность АЦ оценивали по количеству cAMP, который получался в ходе реакции, и выражали в пмоль cAMP/мин/мг белка. Гидрофобные соединения TP03, TP31, PP10 и PP17 растворяли в диметилсульфоксиде (ДМСО). В контрольные пробы вместо соединений в том же объеме добавляли растворитель, рассматривая активность АЦ в них как базовую.
При изучении активности TP31, PP10 и PP17 в условиях in vivo их растворяли в ДМСО и вводили самцам крыс интратестикулярно в дозе 10 мг/кг или внутрибрюшинно в дозе 45 мг/кг. Контрольным крысам вместо препаратов интратестикулярно или внутрибрюшинно вводили ДМСО в том же объеме. Инъекции проводили в 11.00, концентрацию тестостерона оценивали до введения (10.00) и через 1, 3 и 5 ч (12.00, 14.00, 16.00) после введения препаратов. Для изучения влияния соединений на стимулирующий продукцию тестостерона эффект ХГЧ их вводили внутрибрюшинно в дозе 45 мг/кг за 20 мин до введения гонадотропина (в 10.40 и 11.00, соответственно). ХГЧ вводили подкожно в дозе 100 МЕ/крысу. В каждой группе было по 5 животных. Образцы крови для определения уровня тестостерона получали из хвостовой вены, используя местный наркоз с помощью анестезии 2% раствором лидокаина (в расчете 2–4 мг/кг). Концентрацию тестостерона определяли с помощью наборов Тестостерон-ИФА (Алкор-Био, Россия), используя спектрофотометр Anthos Absorbance Reader 2020 (Anthos Labtec Instruments, Австрия).
Статистический анализ осуществляли, используя программу Microsoft Office Excel 2007. Нормальность распределения проверяли с помощью критерия Шапиро–Уилка. Для сравнения двух выборок с нормальным распределением использовали t-критерий Стьюдента, для сравнения трех групп – дисперсионный анализ с поправкой Бонферрони. Статистически значимыми считали отличия при уровне значимости p < 0.05, данные представляли как среднее ± стандартная ошибка среднего (mean ± SEM).
РЕЗУЛЬТАТЫ
Среди серии синтезированных нами гетероциклических соединений были идентифицированы три, которые характеризовались способностью ингибировать стимулирующие эффекты ортостерических (ХГЧ) и аллостерических (TP03) агонистов рецептора ЛГ на активность аденилатциклазной системы в тестикулярных мембранах. Два из них, производное тиено-[2,3-d]пиримидина TP31 и производное пиридо[3,4-d]пиримидина PP17, были синтезированы и охарактеризованы нами впервые. В свою очередь, соединение PP10 является единственным известным в настоящее время представителем класса пери-конденсированных гетероциклов – пиримидо[4,5,6-de] [1, 6]нафтиридинов, а его синтез включает довольно редкую циклизацию путем окислительного нуклеофильного замещения в ароматическом ядре, впервые описанную нами ранее для такого рода субстратов [21].
Соединения TP31, PP10 и PP17 в концентрациях 10–6 и 10–4 М существенно не влияли на базовую активность АЦ в тестикулярных мембранах, в то время как ХГЧ (10–9 М) и соединение TP03 (10–5 М) ее повышали, причем АЦ эффект ХГЧ существенно превышал таковой TP03 (табл. 1). При совместном действии ХГЧ и TP03 стимулирующее влияние гонадотропина на активность АЦ не только сохранялось, но и в небольшой степени усиливалось. Активность фермента при совместной обработке ХГЧ и TP03 составила 151.0 ± ± 2.5 пмоль cAMP/мин/мг белка и была достоверно выше, чем при добавлении одного ХГЧ (p < 0.05). В тестикулярных мембранах, которые преинкубировали (5 мин, +4°C) с 10–4 М TP31, PP10 и PP17, стимулирующий АЦ эффект ХГЧ ослаблялся, причем TP31 подавлял его в значительной степени. В концентрации 10–6 М только соединение TP31 достоверно снижало эффект ХГЧ (табл. 1). Преинкубация мембран с PP10 в обеих концентрациях достоверно снижала, хотя и в небольшой степени, стимулирующее воздействие TP03 на активность АЦ, в то время как в случае преинкубации с соединениями TP31 и PP17 эффективной была только концентрация 10–4 М (табл. 1). В ней TP31 снижал стимулирующий эффект TP03, оцениваемый по приросту активности АЦ над ее базовым уровнем, на 84%, в то время как PP17 – на 48%. Таким образом, среди исследуемых соединений тиено-[2,3-d]пиримидин TP31 оказывает наиболее выраженное ингибирующее влияние на стимулированную ХГЧ и TP03 активность АЦ, причем его влияние в наибольшей степени выражено в отношении аллостерического агониста TP03. Ингибирующее влияние PP17 менее выражено и неселективно в отношении TP03, в то время как PP10 характеризовалось сравнительно низкой антагонистической активностью.
Таблица 1.
Влияние преинкубации фракций тестикулярных мембран с соединениями TP31, PP10 и PP17 на вызываемую ХГЧ и TP03 стимуляцию активности аденилатциклазы
| Преинкубация | Активность АЦ, пмоль cAMP/мин/мг белка, M ± SEM | ||
|---|---|---|---|
| контроль | ХГЧ, 10–9 М | TP03, 10–5 М | |
| ДМСО | 23.7 ± 1.1 | 135.2 ± 4.3a | 64.7 ± 2.9a |
| TP31, 10–6 М | 24.9 ± 0.9 | 110.2 ± 4.1a, b | 57.3 ± 2.9a |
| TP31, 10–4 М | 23.1 ± 1.0 | 68.5 ± 3.2a, b | 29.7 ± 1.8b |
| PP10, 10–6 М | 22.7 ± 0.8 | 124.2 ± 2.3a | 55.3 ± 1.1a, b |
| PP10, 10–4 М | 20.5 ± 0.6 | 105.0 ± 2.4a, b | 52.3 ± 1.6a, b |
| PP17, 10–6 М | 22.4 ± 0.5 | 122.3 ± 2.1a | 57.0 ± 1.9a |
| PP17, 10–4 М | 21.5 ± 1.0 | 88.7 ± 3.1a, b | 42.8 ± 1.5a, b |
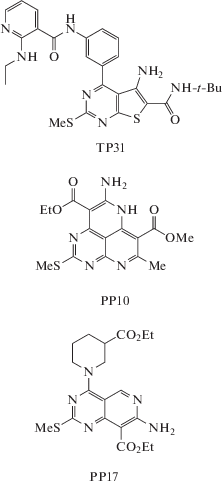
Интратестикулярное введение крысам TP31, PP10 и PP17 в дозе 10 мг/кг приводило к снижению уровня тестостерона в крови, в наибольшей степени в случае TP31 (рис. 2). Ингибирующий эффект нарастал во времени, и через 5 ч во всех исследуемых группах уровень тестостерона был достоверно снижен в сравнении с его начальным уровнем. Значения AUC12.00–16.00, представляющие собой интегрированную площадь под кривой “концентрация тестостерона (нМ) – время (12.00–16.00, ч)”, для крыс, обработанных TP31 и PP17, были ниже в сравнении с контролем (p < 0.05) (табл. 2).
Рис. 2.
Влияние интратестикулярного введения соединений TP31, PP10 и PP17 самцам крыс на уровень тестостерона в плазме крови. Соединения TP31, PP10 и PP17 вводили в 11.00. a – различия с начальным уровнем тестостерона статистически значимы при p < 0.05. Значения представлены как M ± SEM, n = 5.
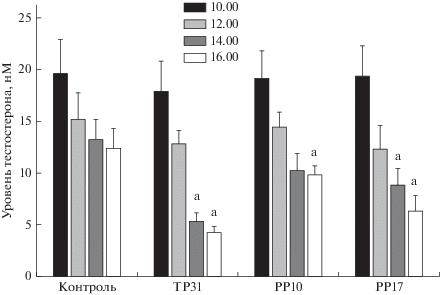
Таблица 2.
Рассчитанные значения интегрированной площади под кривыми “Концентрация тестостерона (нМ) – время (12.00–16.00)” при интратестикулярном и внутрибрюшинном введении самцам крыс соединений TP31, PP10 и PP17, в том числе в условиях стимуляции стероидогенеза с помощью ХГЧ
| Соединение | AUC12.00–16.00, усл. ед. | ||
|---|---|---|---|
| интратестикулярное введение | внутрибрюшинное введение | внутрибрюшинное введение совместно с ХГЧ | |
| ДМСО | 54.3 ± 6.9 | 76.8 ± 4.9 | 393.6 ± 22.8 |
| TP31 | 27.9 ± 2.4a | 31.0 ± 2.6a | 196.9 ± 13.4a |
| PP10 | 45.0 ± 3.9 | 50.6 ± 6.9 | 310.3 ± 22.4 |
| PP17 | 35.2 ± 6.1 | 42.4 ± 4.9a | 260.0 ± 15.6a, b |
Внутрибрюшинное введение крысам TP31 и PP17 в дозе 45 мг/кг снижало уровни тестостерона через 3 и 5 ч, в то время как в случае PP10 отмечали тенденцию к снижению, но различия с начальным уровнем тестостерона не были статистически значимыми на всем протяжении эксперимента (рис. 3). Введение TP31 и PP17 за 20 мин до инъекции ХГЧ в значительной степени снижало стимулированный гонадотропином уровень тестостерона, причем ингибирующее влияние TP31, как и при его воздействии на базовые уровни тестостерона, было выражено сильнее. Так, индуцированный гонадотропином прирост уровня гормона в группе, обработанной TP31, через 3 и 5 ч снижался в среднем в 4 раза, а значение AUC12.00–16.00 для этой группы было снижено в 2 раза (рис. 3, табл. 2). Более того, значение AUC12.00–16.00 для группы TP31 + ХГЧ было достоверно ниже в сравнении с таковым для группы PP17 + ХГЧ (табл. 2). Соединение PP10 при введении самцам крыс в дозе 45 мг/кг слабо влияло на стимулированную ХГЧ продукцию тестостерона (рис. 3, табл. 2). Таким образом, соединение TP31 при различных способах введения самцам крыс снижало как базовую, находящуюся под контролем эндогенного ЛГ, так и стимулированную экзогенным ХГЧ продукцию тестостерона и превосходило по этому показателю пиридо[3,4-d]пиримидиновое производное PP17, в то время как пиримидо[4,5,6-de] [1, 6]нафтиридиновое производное PP10 в условиях in vivo было малоактивным.
Рис. 3.
Влияние внутрибрюшинного введения самцам крыс соединений TP31, PP10 и PP17 на уровень тестостерона в крови животных (а) и на его стимуляцию хорионическим гонадотропином (б). В опытах без введения ХГЧ соединения TP31, PP10, PP17 вводили в 11.00. В опытах с введением ХГЧ соединения TP31, PP10 и PP17 вводили в 10.40, в то время как подкожную инъекцию гонадотропина (100 МЕ/крысу) осуществляли в 11.00. a – различия по сравнению с стартовыми концентрациями тестостерона в группах крыс (10.00) статистически значимы при p < 0.05; b – различия по сравнению с группой ДМСО + ХГЧ статистически значимы при p < 0.05; c – различия между группами TP31 + ХГЧ и PP10 + ХГЧ статистически значимы при p < 0.05. Значения представлены как M ± SEM, n = 5.
ОБСУЖДЕНИЕ
Несмотря на острую необходимость разработки селективных регуляторов рецептора ЛГ с активностью антагонистов, такие работы единичны. Среди таких регуляторов разработанное голландскими учеными соединение LUF5771, производное терфенила, которое подавляло стимуляцию рецептора ЛГ/ХГЧ гонадотропинами и низкомолекулярными аллостерическими агонистами [16, 17]. В концентрации 10 мкМ оно в 3 раза ускоряло скорость диссоциации производного тиено-[2,3-d]пиримидина Org 43 553 от рецептора ЛГ/ХГЧ и в 2–3 раза снижало стимулирующие эффекты ХГЧ и Org 43 553 на сигнальные каскады, зависимые от гонадотропинов [16]. Значительно большее число низкомолекулярных соединений с активностью антагонистов и инверсионных агонистов, относящихся к различным классам органических соединений, включая тиено-[2,3-d]пиримидины, разработано для рецепторов ФСГ и ТТГ. Как и в случае LUF5771, их ингибирующий эффект реализуется вследствие их специфичного взаимодействия с аллостерическим сайтом рецепторов ФСГ и ТТГ, который сформирован высококонсервативными участками трансмембранного домена и, как следствие, структурно близок аллостерическому сайту рецептора ЛГ [5, 18–20].
На основе данных литературы и собственных результатов по изучению тиено-[2,3-d]пиримидиновых производных нами была разработана, а затем и изучена активность серии гетероциклических соединений по их способности ингибировать стимулирующие эффекты ХГЧ и аллостерического агониста TP03 на активность аденилатциклазной сигнальной системы в тестикулярных мембранах. В клетках Лейдига эта система включает три основных компонента – рецептор ЛГ, гетеротримерный Gs-белок и фермент АЦ, и отвечает за большинство физиологических и биохимических эффектов ЛГ и ХГЧ, в том числе за стимуляцию ими стероидогенеза [1]. В результате проведенного скрининга были идентифицированы три соединения с различной гетероциклической структурой, в том числе впервые синтезированные нами TP31 и PP10, которые снижали стимулированную ХГЧ и TP03 активность АЦ в тестикулярных мембранах. Наибольшей активностью обладало производное тиено-[2,3-d]пиримидина TP31, которое в концентрации 100 мкМ практически полностью подавляло стимулирующий эффект TP03 на активность АЦ и в значительной степени снижало стимулирующий АЦ эффект ХГЧ (табл. 1).
Поскольку соединение TP31 в наибольшей степени предотвращает стимулирующее влияние TP03 на аденилатциклазную систему, можно предположить, что оно взаимодействует с аллостерическим сайтом рецептора ЛГ, делая его недоступным для TP03. Сходный механизм, состоящий в блокировании аллостерического сайта, лежит в основе действия большого числа созданных в настоящее время низкомолекулярных антагонистов рецепторов ФСГ и ТТГ, родственных рецептору ЛГ. Среди них содержащие гидрофобные бифенильные радикалы производные аминоалкиламидов, пиперазинов, пиррол[2,1-c]-бензодиазепинов и индола с активностью аллостерических антагонистов и(или) негативных аллостерических модуляторов рецептора ФСГ [4, 5, 20, 23], а также различные гетероциклические соединения NCGC00229600, NCGC00242595, NCGC00242364 и S37a с активностью нейтральных аллостерических антагонистов рецептора ТТГ [5, 18, 19, 24, 25].
Соединение PP17 уступало TP31 по способности снижать стимулирующие АЦ эффекты ХГЧ и TP03. В свою очередь, ингибирующее влияние PP10 на эти эффекты ХГЧ и TP03 было выражено слабо и практически не зависело от концентрации этого соединения, что свидетельствует об особенностях фармакологического профиля PP10 и может указывать на аллостерическую природу регуляции им рецептора ЛГ. Так, например, ADX61623, аллостерический регулятор рецептора ФСГ, в клетках с экспрессированным в них рецептором ФСГ подавлял индуцированную гонадотропином продукцию cAMP и прогестерона, но в высоких концентрациях, напротив, усиливал стероидогенез и повышал продукцию эстрадиола [26].
В условиях in vivo соединения TP31 и PP17 снижали как базовую, так и стимулированную ХГЧ продукцию тестостерона. Соединение TP31 было одинаково эффективным как при интратестикулярном, так и при внутрибрюшинном способах введения, в то время как ингибирующие эффекты PP17 при его внутрибрюшинном введении ослаблялись, что, вероятно, обусловлено частичной деградацией этого соединения в кровотоке и(или) сниженной его биодоступностью для клеток Лейдига. Снижение базового уровня тестостерона в условиях in vivo при отсутствии влияния TP31 на базовую активность АЦ в условиях in vitro указывает на то, что при введении самцам крыс TP31 подавляет стероидогенез, вызываемый эндогенным ЛГ. При этом соединение TP31 лишь частично снижает стероидогенный эффект ХГЧ, что может быть обусловлено сохранением части рецепторов ЛГ в активной, свободной от TP31 форме, способной активироваться гонадотропином. Поскольку регуляторы, наделенные активностью аллостерических антагонистов и негативных аллостерических модуляторов, с относительно низкой аффинностью связываются с аллостерическим сайтом рецептора, то даже при длительном воздействии их субмаксимальных концентраций, вследствие установления динамического равновесия между свободными и связанными формами рецепторов, часть рецепторов сохраняет способность к активации агонистами. Это является важным преимуществом аллостерических регуляторов, поскольку они не вызывают тотального подавления активности рецепторов, что характерно для антагонистов высокоаффинного ортостерического сайта [27, 28]. Имеются все основания полагать, что TP31 лишь частично ингибирует функции рецепторов ЛГ и, как следствие, не вызывает острого андрогенного дефицита. Это является их важным преимуществом, поскольку сильно выраженное подавление функций гипоталамо-гипофизарно-гонадной оси и контролируемого ею стероидогенеза – крайне нежелательный побочный эффект при использовании антиандрогенных препаратов и высоких доз аналогов гонадолиберина [13–15].
Сравнение структуры TP31 c ранее разработанным тиено-[2,3-d]пиримидиновыми производными с активностью агонистов показывает, что основной причиной появления у TP31 свойств функционального антагониста является введение во второе положение гетероциклического кольца остатка никотиновой кислоты дополнительной этиламинной группы. Это меняет объем и заряд варьируемого заместителя в 5-амино-4-(3-аминофенил)-N-(трет-бутил)-2-(метилсульфанил)тиено[2,3-d]пиримидин-6-карбоксамиде, используемом в качестве каркасной молекулы. У соединений TP01, TP03, TP04 и TP23 с активностью полных агонистов рецептора ЛГ в качестве такого заместителя выступают незамещенные остатки изоникотиновой и никотиновой кислот (TP01 и TP03), а также 1-метил-1Н-пиразольная (TP04) и 2-хлорникотиновая группы (TP23) [6–8]. Важно отметить, что даже небольшие изменения структуры каркасной молекулы приводят к полной потере полученными в результате таких модификаций тиено-[2,3-d] пиримидиновыми производными активности агонистов в отношении рецептора ЛГ, в то время как на антагонистическую активность такие модификации не влияют. На это указывает то, что соединение PP17, которое имеет отличающуюся от тиено-[2,3-d]пиримидина гетероциклическую систему и в котором отсутствуют трет-бутиламидная группа и варьируемый заместитель, имеет активность функционального антагониста рецептора ЛГ. В то же время оно с меньшей эффективностью, в сравнении с TP31, подавляет стимулирующие эффекты аллостерических агонистов этого рецептора, что позволяет предположить негативное влияние перечисленных выше структурных изменений в молекуле PP17 на его способность взаимодействовать с аллостерическим сайтом рецептора ЛГ.
Таким образом, нами впервые показано, что соединения TP31 и PP17, производные тиено-[2,3-d]пиримидина и пиридо[3,4-d]пиримидина, действуют как функциональные антагонисты рецептора ЛГ, препятствуя его стимуляции гонадотропином и аллостерическим агонистом TP03. По антагонистическому действию TP31 превосходит PP17, что обусловлено его более эффективным связыванием с аллостерическим сайтом рецептора. Причиной этого является большее соответствие тиено-[2,3-d]пиримидиновых производных пространственной структуре этого сайта. В сравнении с TP31 и PP17, соединение PP10, производное пиримидо[4,5,6-de] [1, 6]нафтиридина, характеризуется низкой антагонистической активностью в отношении рецептора ЛГ и зависимого от него стероидогенеза. Наиболее активное соединение TP31 может быть использовано в качестве прототипа для создания препаратов, предназначенных для подавления стероидогенной функции при лечении рака предстательной железы и некоторых других гормонозависимых опухолей, а также для коррекции и предотвращения синдрома гиперстимуляции яичников.
Работа поддержана Российским научным фондом (проект № 19-75-20122). ЯМР-исследования проведены с использованием оборудования ресурсного центра СПбГУ “Магнитно-резонансные методы исследования”, масс-спектры высокого разрешения получены на оборудовании ресурсного центра СПбГУ “Методы анализа состава вещества”.
Список литературы
Riccetti L., De Pascali F., Gilioli L., Potì F., Giva L.B., Marino M., Tagliavini S., Trenti T., Fanelli F., Mezzullo M., Pagotto U., Simoni M., Casarini L. 2017. Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod. Biol. Endocrinol. 15 (1), 2. https://doi.org/10.1186/s12958-016-0224-3
Riccetti L., Yvinec R., Klett D., Gallay N., Combarnous Y., Reiter E., Simoni M., Casarini L., Ayoub M.A. 2017. Human luteinizing hormone and chorionic gonadotropin display biased agonism at the LH and LH/CG receptors. Sci. Rep. 7 (1), 940. https://doi.org/10.1038/s41598-017-01078-8
Шпаков А.О. 2018. Гонадотропины – от теории к клинической практике. СПб.: ПОЛИТЕХ-ПРЕСС. 347 с.
van de Lagemaat R., Timmers C.M., Kelder J., van Koppen C., Mosselman S., Hanssen R.G. 2009. Induction of ovulation by a potent, orally active, low molecular weight agonist (Org 43553) of the luteinizing hormone receptor. Hum. Reprod. 24 (3), 640–648. https://doi.org/10.1093/humrep/den412
Nataraja S.G., Yu H.N., Palmer S.S. 2015. Discovery and development of small molecule allosteric modulators of glycoprotein hormone receptors. Front. Endocrinol. (Lausanne). 6, 142. https://doi.org/10.3389/fendo.2015.00142
Shpakov A.O., Dar’in D.V., Derkach K.V., Lobanov P.S. 2014. The stimulating influence of thienopyrimidine compounds on the adenylyl cyclase systems in the rat testes. Dokl. Biochem. Biophys. 456 (1), 104–107. https://doi.org/10.1134/S1607672914030065
Derkach K.V., Dar’in D.V., Bakhtyukov A.A., Loba-nov P.S., Shpakov A.O. 2016. In vitro and in vivo studies of functional activity of new low molecular weight agonists of the luteinizing hormone receptor. Biochemistry (Moscow). Suppl. Ser. A: Membr. Cell. Biol. 10 (4), 294–300. https://doi.org/10.1134/S1990747816030132
Derkach K.V., Legkodukh A.S., Dar’in D.V., Shpakov A.O. 2017. The stimulating effect of thienopyrimidines structurally similar to Org 43553 on adenylate cyclase activity in the testes and on testosterone production in male rats. Cell Tissue Biol. 11 (1), 73–80. https://doi.org/10.1134/S199 0519X17010035
Bakhtyukov A.A., Derkach K.V., Dar’in D.V., Shpakov A.O. 2019. Conservation of steroidogenic effect of the low-molecular-weight agonist of luteinizing hormone receptor in the course of its long-term administration to male rats. Dokl. Biochem. Biophys. 484 (1), 78–81. https://doi.org/10.1134/S1607672919 010216
Bakhtyukov A.A., Derkach K.V., Dar’in D.V., Stepochkina A.M., Shpakov A.O. 2019. A low molecular weight agonist of the luteinizing hormone receptor stimulates adenylyl cyclase in the testicular membranes and steroidogenesis in the testes od rats with type 1 diabetes. Biochemistry (Moscow). Suppl. Ser A: Membr. Cell. Biol. 13 (4), 301–309. https://doi.org/10.1134/S1990747819040032
Xing W., Lin H., Li Y., Yang D., Wang W., Zhang Q. 2015. Is the GnRH antagonist protocol effective at preventing OHSS for potentially high responders undergoing IVF/ICSI? PLoS One. 10 (10), e0140286. https://doi.org/10.1371/journal.pone.0140286
Morgante G., Massaro M.G., Di Sabatino A., Cappelli V., De Leo V. 2018. Therapeutic approach for metabolic disorders and infertility in women with PCOS. Gynecol. Endocrinol. 34 (1), 4–9. https://doi.org/10.1080/09513590.2017.1370644
Engel J.B., Schally A.V. 2007. Drug insight: Clinical use of agonists and antagonists of luteinizing-hormone-releasing hormone. Nat. Clin. Pract. Endocrinol. Metab. 3 (2), 157–167. https://doi.org/10.1038/ncpendmet0399
Godbole A.M., Njar V.C. 2011. New insights into the androgen-targeted therapies and epigenetic therapies in prostate cancer. Prostate Cancer. 2011, 918707. https://doi.org/10.1155/2011/918707
Lizneva D., Gavrilova-Jordan L., Walker W., Azziz R. 2016. Androgen excess: Investigations and management. Best Pract. Res. Clin. Obstet. Gynaecol. 37, 98–118. https://doi.org/10.1016/j.bpobgyn.2016.05.003
Heitman L.H., Narlawar R., de Vries H., Willemsen M.N., Wolfram D., Brussee J., Ijzerman A.P. 2009. Substituted terphenyl compounds as the first class of low molecular weight allosteric inhibitors of the luteinizing hormone receptor. J. Med. Chem. 52 (7), 2036–2042. https://doi.org/10.1021/jm801561h
Heitman L.H., Kleinau G., Brussee J., Krause G., Ijzerman A.P. 2012. Determination of different putative allosteric binding pockets at the lutropin receptor by using diverse drug-like low molecular weight ligands. Mol. Cell. Endocrinol. 351 (2), 326–336. https://doi.org/10.1016/j.mce.2012.01.010
Turcu A.F., Kumar S., Neumann S., Coenen M., Iyer S., Chiriboga P., Gershengorn M.C., Bahn R.S. 2013. A small molecule antagonist inhibits thyrotropin receptor antibody-induced orbital fibroblast functions involved in the pathogenesis of Graves ophthalmopathy. J. Clin. Endocrinol. Metab. 98 (5), 2153–2159. https://doi.org/10.1210/jc.2013-1149
Neumann S., Nir E.A., Eliseeva E., Huang W., Marugan J., Xiao J., Dulcey A.E., Gershengorn M.C. 2014. A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology. 155 (1), 310–314. https://doi.org/10.1210/en.2013-1835
Anderson R.C., Newton C.L., Millar R.P. 2019. Small molecule follicle-stimulating hormone receptor agonists and antagonists. Front. Endocrinol. (Lausanne). 9, 757. https://doi.org/10.3389/fendo.2018.00757
Bakulina O.Yu., Ivanov A.Yu., Dar’in D.V., Lobanov P.S. 2014. New transformations of 2-methylsulfanyl-4,6-dichloropyrimidine-5-carbaldehyde involving enamines: Synthesis of condensed azines. Mendeleev Commun. 24, 163–164. https://doi.org/10.1016/j.mencom.2014.04.013
Ryazanov S.G., Selivanov S.I., Dar’in D.V., Loba-nov P.S., Potekhin A.A. 2008. Chemoselective cyclocondensation of α-acylacetamidines with 2-methylsulfanyl-4,6-dichloropyrimidine-5-carbaldehyde. Russ. J. Org. Chem. 44 (2), 288–291.
Heitman L.H., Ijzerman A.P. 2008. G protein-coupled receptors of the hypothalamic-pituitary-gonadal axis: A case for Gnrh, LH, FSH, and GPR54 receptor ligands. Med. Res. Rev. 28 (6), 975–1011. https://doi.org/10.1002/med.20129
Neumann S., Eliseeva E., McCoy J.G., Napolitano G., Giuliani C., Monaco F., Huang W., Gershengorn M.C. 2011. A new small-molecule antagonist inhibits Graves’ disease antibody activation of the TSH receptor. J. Clin. Endocrinol. Metab. 96 (2), 548–554. https://doi.org/10.1210/jc.2010-1935
Marcinkowski P., Hoyer I., Specker E., Furkert J., Rutz C., Neuenschwander M., Sobottka S., Sun H., Nazare M., Berchner-Pfannschmidt U., von Kries J.P., Eckstein A., Schülein R., Krause G. 2019. A new highly thyrotropin receptor-selective small-molecule antagonist with potential for the treatment of Graves’ orbitopathy. Thyroid. 29 (1), 111–123. https://doi.org/10.1089/thy.2018.0349
Ayoub M.A., Yvinec R., Jégot G., Dias J.A., Poli S.M., Poupon A., Crépieux P, Reiter E. 2016. Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implications in steroidogenesis. Mol. Cell. Endocrinol. 436, 10–22. https://doi.org/10.1016/j.mce.2016.07.013
Lindsley C.W., Emmitte K.A., Hopkins C.R., Bridges T.M., Gregory K.J., Niswender C.M., Conn P.J. 2016. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 116 (11), 6707–6741. https://doi.org/10.1021/acs.chemrev.5b00656
Foster D.J., Conn P.J. 2017. Allosteric modulation of GPCRs: New insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron. 94 (3), 431–446. https://doi.org/10.1016/j.neuron.2017.03.016
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии