

Биологические мембраны: Журнал мембранной и клеточной биологии, 2021, T. 38, № 4, стр. 284-291
Сравнение эффективности подавления KCl-индуцированных кальциевых ответов гиппокампальных нейронов различными блокаторами потенциал-зависимых Ca2+-каналов L-типа
Д. П. Ларюшкин a, b, *, С. А. Майоров a, С. Г. Гайдин a, В. П. Зинченко a, А. М. Косенков a
a Институт биофизики клетки РАН, ФИЦ ПНЦБИ РАН
142290 Московская обл., Пущино, Россия
b Институт теоретической и экспериментальной биофизики РАН
142290 Московская обл., Пущино, Россия
* E-mail: mr.ldp@yandex.ru
Поступила в редакцию 05.02.2021
После доработки 17.03.2021
Принята к публикации 19.03.2021
Аннотация
Потенциал-зависимые кальциевые каналы L-типа экспрессируются различными клетками млекопитающих. В нейронах они, как правило, расположены постсинаптически и отвечают за передачу возбуждающего сигнала, а также вносят вклад в активацию Ca2+-зависимых сигнальных каскадов. В настоящий момент одним из основных инструментов для изучения функций этих каналов является применение различных синтетических блокаторов. Целью настоящей работы было сравнение четырех блокаторов из трех фармакологических групп и определение концентрации, необходимой для полного подавления в нейронах гиппокампа притока Ca2+ через каналы L-типа в ответ на деполяризацию, индуцируемую добавлением KCl. Как показали эксперименты, повышение внутриклеточной концентрации Ca2+ в ответ на деполяризацию хоть и определяется в первую очередь притоком Ca2+ через каналы L-типа, тем не менее также обусловлено активацией других потенциал-зависимых кальциевых каналов. В связи этим нифедипин и исрадипин, обладающие, по всей видимости, большей селективностью по отношению к L-каналам, снижают амплитуду кальциевого ответа примерно в 2 раза. В свою очередь, дилтиазем и верапамил за счет неспецифического блокирования других, не относящихся к L-типу кальциевых каналов, полностью блокировали приток Ca2+, индуцированный аппликацией KCl. Следует отметить, что концентрация блокаторов, необходимая для полного подавления притока Ca2+ через потенциал-зависимые каналы, в обоих случаях составила 300 мкМ, при этом IC50 в случае дилтиазема и верапамила составили 54.2 и 56.4 мкМ, соответственно. Таким образом, вещества из группы дигидропиридинов предпочтительнее использовать в случае, если требуется наибольшая селективность по отношению к L-каналам. В свою очередь, высокие концентрации верапамила и дилтиазема могут применяться, если необходимо заблокировать большую часть потенциал-зависимых кальциевых каналов.
ВВЕДЕНИЕ
Потенциал-зависимые кальциевые каналы (ПЗКК/CaV) представляют собой семейство трансмембранных белков, избирательно проницаемых для ионов кальция. ПЗКК разделены на несколько типов, отличающихся фармакологическим профилем и биофизическими свойствами. На сегодняшний день в клетках млекопитающих охарактеризованы десять представителей семейства CaV-каналов, относящихся к четырем типам. Так, L-тип представлен каналами CaV1.1–CaV1.4, P-/Q-тип представлен CaV2.1 каналами, N-тип представлен каналами CaV2.2, R-тип – каналами CaV2.3, а T-тип включает каналы CaV3.1–CaV3.3. Типичная структура кальциевого канала L-типа представляет собой гетеротетрамер, состоящий из порообразующей трансмембранной α1-субъединицы, внутриклеточной β-субъединицы и внеклеточной α2δ-субъединицы. Основные биофизические и фармакологические свойства канала определяет α1-субъединица, структурная топология которой высококонсервативна [1].
Каналы L-типа широко распространены в различных клетках млекопитающих. Транскриптомный анализ показал наличие всех известных представителей каналов L-типа в лимфоцитах [2]. CaV1.1 в основном присутствует в клетках скелетных мышц, где играет ключевую роль в процессе сокращения, но также этот канал коэкспрессируется с рианодиновыми рецепторами в ГАМКергических нейронах [3]. CaV1.2 и CaV1.3 экспрессируются в клетках надпочечников, сердечных и нейрональных клетках [4], тогда как CaV1.4 в основном представлен в нейронах сетчатки [5]. В ряде исследований показано, что фармакологическая модуляция каналов L-типа является перспективным подходом к терапии различных паталогических состояний [6–10]. Изменения в функционировании CaV1.1 каналов коррелируют с гипокалиемическим периодическим параличом, а дисфункция CaV1.2 и CaV1.3 каналов приводит к таким патологиям, как синдром Тимоти, сердечная аритмия [6], биполярное расстройство и аутизм [7, 8].
На сегодняшний день для модуляции активности ПЗКК L-типа идентифицировано несколько низкомолекулярных соединений, которые разделены на четыре группы: фенилалкиламины, бензодиазепины, дигидропиридины и дифенилпиперазины [9]. Предполагается, что сайты связывания блокаторов находятся глубоко внутри молекулы канала и становятся доступными для связывания только в результате конформационных изменений, происходящих при активации и/или инактивации ионных каналов. Эти процессы зависят от потенциала, и при значительной продолжительной деполяризации каналы переходят в инактивированное состояние. Механизм действия этих веществ основан на стабилизации канала в инактивированном состоянии. Показано, что сайты связывания блокаторов в основном расположены близко к поре, в спиралях S5 и S6 [10]. Однако предполагается, что верапамил-подобные фенилалкиламины могут связываться непосредственно в порах канала [6].
Дигидропиридины
1,4-Дигидропиридин (ДГП) и его производные являются эффективными блокаторами кальциевых каналов L-типа. Эта группа препаратов используется при терапии сердечно-сосудистых заболеваний, таких как стенокардия и гипотензия [10, 11]. Производные дигидропиридина, нифедипин и исрадипин, также используются для лечения заболеваний сердца. Нимодипин, другое производное соединение ДГП, модулирует ПЗКК в нейронах мозга, улучшая результаты лечения аутизма и нейродегенерации [8]. ДГП связываются с инактивированными каналами L‑типа и стабилизируют их, нарушая тем самым приток кальция через этот тип каналов [10, 12]. Поскольку большинство дигидропиридинов являются липофильными молекулами, они также могут связываться с внешней поверхностью канала, обращенной к липидным молекулам, и взаимодействовать с S6 спиралью из DIII и DIV участков канала [13, 14]. Показано, что нифедипин, препарат группы дигидропиридинов, является неселективным блокатором ПЗКК, поскольку также блокирует следующие калиевые каналы: KV1.1, KV1.2, KV1.5, KV2.1, KV1.7, KV3.1 [15, 16]. При этом нами не было обнаружено публикаций, посвященных селективности исрадипина.
Фенилалкиламины
Согласно данным рентгеноструктурного анализа, фенилалкиламин и его производные связываются с центральной полостью поры канала на внутриклеточной стороне “фильтра селективности” [6], что приводит к его блокаде и препятствует прохождению ионов кальция. Типичным представителем фенилалкиламинов, широко применяемым при лечении гипертонии, является верапамил. Аналогично другим ДГП, верапамил связывается с инактивированными каналами L-типа [10, 12], но, связавшись с каналом, верапамил приводит к его необратимому блокированию после реполяризации [13]. Помимо L-каналов этот препарат также блокирует KV1.7, KV1.8, KV3.2, NaV2.1 каналы [17].
Бензодиазепины
Клинически одобренным и наиболее часто используемым для лечения различного рода аритмий блокатором ПЗКК из класса бензодиазепинов является дилтиазем. Дилтиазем проявляет умеренную селективность в отношении ПЗКК клеток гладких мышц сосудов, но не в отношении ПЗКК клеток сердца. Подобно блокаторам из других фармакологических групп препараты группы бензодиазепинов имеют тенденцию связываться с ПЗКК в инактивированном состоянии с тем же сайтом, что и ДГП. Данные экспериментов по фотоаффинному мечению указывают на то, что сайты связывания дилтиазема расположены в S6 спирали DIII и DIV участков канала [10]. Также сообщалось, что этот препарат блокирует каналы KV1.1, KV1.2, KV1.5, KV1.7, KV3.1 и канал 5-HT3 рецептора [16, 18].
Несмотря на то, что в настоящий момент синтезировано большое количество блокаторов ПЗКК L-типа, их эффективность, как правило, проверена с использованием систем, в которых были экспрессированы отдельные субъединицы. При этом практически отсутствуют работы, в которых бы доказывалась и сравнивалась эффективность соединений в условиях, приближенных к физиологическим. В настоящей работе мы изучали эффекты блокаторов ПЗКК L-типа, нифидипина и исрадипина, относящихся к классу дигидропиридинов, а также верапамила и дилтиазема, относящихся к классам фенилалкиламинов и бензодиазепинов, соответственно. Целью работы было сравнение действия этих соединений и определение концентраций блокаторов, необходимых для полного подавления притока Ca2+ через ПЗКК L-типа в нейроглиальной культуре клеток гиппокампа.
МАТЕРИАЛЫ И МЕТОДЫ
Приготовление культуры нейроглиальных клеток гиппокампа. В экспериментах использовали нейроглиальную культуру клеток гиппокампа, выделенного из головного мозга новорожденных крыс линии Sprague–Dawley. Извлеченный гиппокамп помещали в холодный раствор Версена и измельчали при помощи ножниц. Затем фрагменты ткани обрабатывали в течение 10 мин при 37°С 1% раствором трипсина. После этого ткань отмывали холодной средой Neurobasal-A для инактивации трипсина и аккуратно пипетировали. Полученную суспензию клеток переносили на круглые покровные стекла диаметром 25 мм, предварительно покрытые полиэтиленимином и помещенные в чашки Петри (диаметром 35 мм). Затем стекла помещали на 30 мин в CO2-инкубатор для прикрепления клеток, после чего заливали 2 мл среды для культивирования. В качестве среды для культивирования использовали Neurobasal-A, содержащую бессывороточную добавку B-27, 0.5 мМ глутамина и пенициллин–стрептомицин (1 : 100). Эксперименты проводили на 14–17 день культивирования.
Регистрация изменений концентрации внутриклеточного ионизированного кальция ([Са2+]i). Перед экспериментом клетки загружали в течение 40 мин при 28°C флуоресцентным двухволновым Са2+-чувствительным зондом Fura-2 AM (3 мкМ) в растворе HBSS (Hank`s balanced salt solution), содержащем (мМ): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 СаCl2, 10 глюкозы и 10 HEPES; pH 7.35. Все эксперименты с использованием данного флуоресцентного зонда проводили в растворе того же состава. Для регистрации изменений [Са2+]i использовали инвертированный моторизованный флуоресцентный микроскоп Leica DMI6000B (Leica Microsystems, Германия), оснащенный высокоскоростной монохромной CCD-камерой HAMAMATSU C9100, системой высокоскоростной смены возбуждающих светофильтров Leica Ultra-Fast Filter Wheels (время переключения 10–30 мс). Для съемки использовали объектив Leica HC PL APO 20×/0.7 IMM. В качестве источника возбуждения флуоресценции использовали осветитель Leica EL 6000 с ртутной лампой высокого давления HBO 103 W/2. Для возбуждения и регистрации флуоресценции Fura-2 использовали набор светофильтров FU2 с фильтрами возбуждения BP 340/30 и BP 387/15, светоделителем FT 410 и фильтром эмиссии BP 510/84. Полученные в двух различных каналах временные серии изображений обрабатывали в программе ImageJ. Изменения [Са2+]i (кальциевых ответов) в одиночных клетках определяли из отношения интенсивности флуоресценции Fura-2 при возбуждении светом с длиной волны 340 нм к интенсивности при возбуждении 380 нм. Аппликация KCl (35 мМ) производилась в присутствии антагонистов NMDA-рецепторов (D-AP5, 10 мкМ), АМРА/каинатных-рецепторов (NBQX, 10 мкМ) и блокаторов ПЗКК L-типа (нифедипина, исрадипина, верапамила и дилтиазема). Реагенты добавляли путем полной замены среды c помощью специальной перфузионной системы. Скорость записи всех серий изображений составляла 1 кадр в секунду. Все эксперименты выполнены при температуре 28–30°C.
Статистический анализ и обработка данных. Для графической обработки данных и статистического анализа использовался пакет программ OriginLab Pro 2016 (OriginLab, Northampton, США) и Prism GraphPad 8 (GraphPad Software, США). Статистическая обработка данных производилась с помощью однофакторного дисперсионного анализа с последующим множественным сравнением по методу Тьюки.
Реагенты. В работе использовали: HEPES (Диаэм, Россия), Fura-2 AM (Molecular Probes, США), бессывороточная добавка B-27, Neurobasal-A, трипсин 2.5%, глутамин (Life Technologies, США), D-АР5, NBQX динатриевая соль (Hello Bio, Великобритания), дилтиазем, нифедипин, исрадипин, верапамил, пенициллин–стрептомицин полиэтиленимин (Sigma-Aldrich, США).
РЕЗУЛЬТАТЫ
В рамках этой работы проведено сравнение эффективной дозы полумаксимального ингибирования (IC50) различных блокаторов потенциал-зависимых кальциевых каналов L-типа в нейроглиальной культуре клеток гиппокампа крысы. Для активации потенциал-зависимых каналов была использована деполяризация, индуцируемая аппликацией 35 мМ KCl. Поскольку в таких условиях в ответ на деполяризацию может происходить секреция глутамата, все эксперименты проводились в присутствии антагонистов NMDA-, АМРА- и каинатных рецепторов, чтобы исключить приток Ca2+ через эти каналы.
Из рис. 1а и 1б видно, что верапамил и дилтиазем дозозависимо снижают амплитуду изменения [Ca2+]i в ответ на аппликацию KCl во всех нейронах. При этом в обоих случаях максимальное подавление наблюдается при концентрации 300 мкМ. Значительное уменьшение амплитуды кальциевого ответа в случае верапамила отмечается уже при концентрации 1 мкМ, в то время как дилтиазем заметно снижает амплитуду ответа только в концентрации 10 мкМ. Несмотря на это, IC50 этих блокаторов имеют близкие значения, равные 56.4 мкМ для верапамила и 54.2 мкМ для дилтиазема.
Рис. 1.
Влияние ингибиторов ПЗКК L-типа на кальциевый ответ нейронов, индуцируемый деполяризацией. а, б, в, г – аппликация KCl (35 мМ) в присутствии антагонистов NMDA-рецепторов (D-AP5, 10 мкМ), АМРА/каинатных рецепторов (NBQX, 10 мкМ) и различных блокаторов ПЗКК L-типа. а', б ', в', г' – диаграммы, отражающие изменение амплитуды кальциевого ответа на аппликацию KCl в зависимости от концентрации блокаторов L-каналов. Каждая точка является усредненным ответом по 100 нейронам в отдельном эксперименте. ns – отсутствие статистически достоверного различия. а'', б '' – кривые доза–эффект для верапамила и дилтиазема.
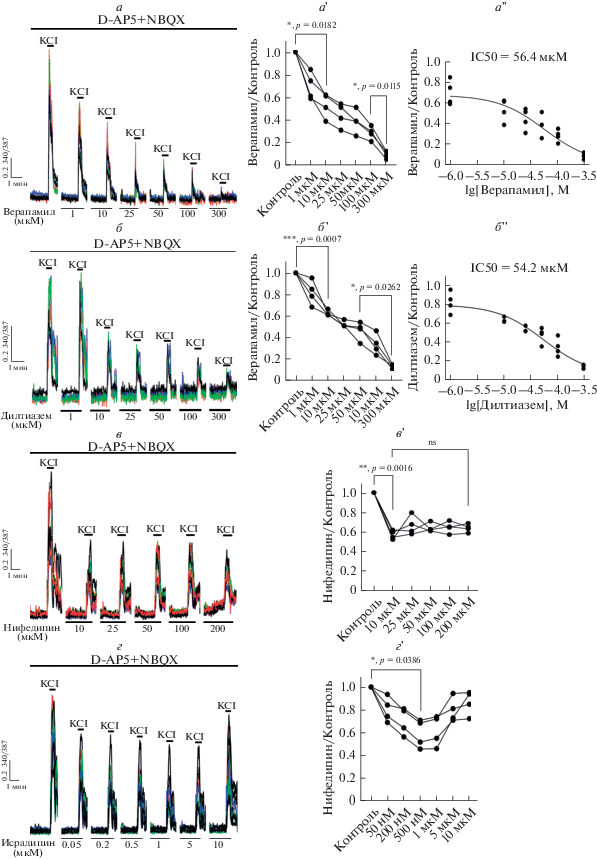
В свою очередь, нифедипин и исрадипин не способны полностью подавлять приток Ca2+, вызванный деполяризацией (рис. 1в и 1г). Нифедипин в концентрации 10 мкМ в среднем в 2 раза снижает амплитуду кальциевого ответа, однако дальнейшее повышение концентрации не приводит к значимым изменениям. Поскольку у исрадипина IC50 для субъединиц CaV1.2 и CaV1.3 находится в наномолярном диапазоне, мы использовали более низкие концентрации. Как и в случае с нифедипином, исрадипин в концентрации 50 нМ снижает амплитуду кальциевого ответа, однако более высокие концентрации уже не влияют на амплитуду ответа. Кроме того, стоит отметить, что высокие концентрации нифедипина приводят к подъему базового уровня [Ca2+]i в течение первых секунд воздействия.
На рис. 2а показано, что серия из нескольких аппликаций KCl не приводит к значительным изменениям в амплитуде кальциевого ответа. Таким образом, подавление кальциевого ответа в экспериментах на рис. 1 обусловлено именно воздействием блокаторов, а не связано с какими-либо клеточными процессами, инициируемыми деполяризацией.
Рис. 2.
Вклад кальциевых депо и других ПЗКК в кальциевый ответ нейронов. а – Кальциевый ответ нейронов на пять последовательных аппликаций KCl (35 мМ) в присутствии D-AP5 (10 мкМ) и NBQX (10 мкМ). б – Кальциевый ответ нейронов на аппликацию KCl (35 мМ) в присутствии D-AP5 (10 мкМ) и NBQX (10 мкМ), а также нифедипина (50 мкМ), блокатора ПЗКК Т-типа ML218 (10 мкМ) и блокатора ПЗКК N-, P/Q-типа ω-конотоксина MVIIC (1 мкМ). в – Кальциевый ответ клеток гиппокампа на аппликацию KCl (35 мМ) и ингибитора Са2+-АТP-азы ЭР тапсигаргина (2 мкМ). Цветом выделены сигналы клеток, отвечающих на добавку KCl (нейроны), невозбудимые клетки (предположительно астроциты) выделены черным цветом. Во всех экспериментах пауза между добавками веществ составляла 15 мин.
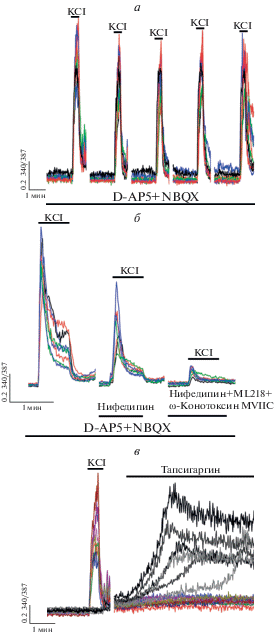
Поскольку в наших экспериментах нифедипин и исрадипин даже в высоких концентрациях оказались не способны полностью подавить кальциевый ответ, мы провели дополнительные эксперименты, в которых проверили вклад других ПЗКК и кальциевого депо – эндоплазматического ретикулума (ЭР). Эксперименты показали, что добавление нифедипина в комбинации с блокаторами ПЗКК Т-, N- и P/Q-типа приводит к практически полному подавлению кальциевого ответа при аппликации KCl (рис. 2б). Таким образом, несмотря на то, что ПЗКК L-типа являются основным источником притока Ca2+ при деполяризации, другие ПЗКК также вносят значительный вклад в данный процесс. На основании данного эксперимента можно предположить, что нифедипин и исрадипин селективно блокируют каналы L-типа. Однако поскольку Т-, N- и P/Q-каналы в сумме вносят значительный вклад в кальциевый приток при деполяризации, использование дигидропиридинов не приводит к полному подавлению кальциевого ответа при добавке KCl. В свою очередь, верапамил и дилтиазем полностью подавляют кальциевый приток, по всей видимости, за счет неселективного действия на другие ПЗКК.
Вклад депонированного кальция при деполяризации должен быть минимален, поскольку, как показано на рис. 2в, опустошение ЭР путем добавления ингибитора Са2+-АТP-азы ЭР тапсигаргина не приводит к детектируемому подъему [Ca2+]i в теле нейронов. В астроцитах в ответ на добавку ингибитора отмечается постепенное увеличение [Ca2+]i с последующим выходом на плато. Такая форма кальциевого ответа говорит о первоначальной мобилизации кальция из ЭР в ответ на ингибирование АТP-азы и последующей активации депо-управляемых кальциевых каналов.
Таким образом, из приведенных выше экспериментов можно сделать ряд выводов: 1) деполяризация, индуцируемая KCl, приводит к повышению [Ca2+]i, которое, в первую очередь, обусловлено притоком Ca2+ через ПЗКК L-типа, а также притоком через T-, N- и P/Q-каналы; 2) верапамил и дилтиазем полностью подавляют кальциевый ответ, индуцируемый деполяризацией, по всей видимости, за счет неселективного действия на другие ПЗКК при высоких концентрациях; 3) нифедипин и исрадипин более селективно по сравнению с верапамилом и дилтиаземом блокируют ПЗКК L-типа.
ОБСУЖДЕНИЕ
Потенциал-зависимые кальциевые каналы L-типа экспрессируются множеством типов клеток млекопитающих, включая нейроны различных отделов мозга и мышечные клетки. Как правило, в клетках мозга такие каналы состоят из CaV1.2 и CaV1.3 порообразующих субъединиц [4], которые определяют фармакологические свойства и проводимость канала. Кроме того, свойства и функции CaV1 каналов во многом зависят от взаимодействия с различными белками, такими как AKAP [19], денсин [20], STIM1 [21]. В нейронах эти каналы располагаются преимущественно в постсинаптических терминалях и влияют на передачу возбуждающего сигнала, участвуя в активации кальций-зависимых сигнальных каскадов, передающих сигнал в ядро и влияющих тем самым на транскрипцию генов, вовлеченных в регуляцию развития нейронов, процессов обучения и памяти [20, 22–24]. Несмотря на то, что для изучения ПЗКК активно используются молекулярно-генетические методы, использование различных блокаторов остается в настоящий момент одним из основных инструментов для исследования их функций. Многие из этих соединений уже с успехом используются в медицине в качестве одобренных лекарственных препаратов для терапии сердечно-сосудистых и нейродегенеративных заболеваний. Тем не менее функции ПЗКК на уровне отдельных нейронов, в частности ПЗКК L-типа, остаются до конца не изученными.
В этой работе мы сравнили действие блокаторов ПЗКК из трех групп – фенилалкиламины, бензодиазепины и дигидропиридины. Как показали эксперименты, соединения из этих групп значительно различаются по действию на кальциевый ответ нейронов, индуцируемый деполяризацией. Во всех экспериментах для деполяризации мы использовали добавку 35 мМ KCl. При добавлении избыточного количества K+ во внеклеточную среду меняется градиент концентрации этого иона, в результате чего происходит деполяризация возбудимых клеток. В ответ на деполяризацию происходит активация потенциал-зависимых каналов, в том числе и ПЗКК, что вызывает мощный приток Ca2+ в нейроны. Несмотря на то, что, согласно литературным данным, вещества как из групп фенилалкиламинов и бензодиазепинов, так и из группы дигидропиридинов способны блокировать не только ПЗКК, но и другие каналы, в данных условиях такие побочные действия не будут значительно влиять на кальциевый ответ при добавке KCl. В каждом эксперименте добавки производились на фоне антагонистов NMDA-, АМРА-/каинатных рецепторов, что исключает дополнительный приток Ca2+ через ионотропные рецепторы глутамата. Таким образом, подобная схема экспериментов позволяет оценить влияние блокаторов преимущественно на ПЗКК нейронов в условиях, приближенных к физиологическим.
Верапамил и дилтиазем способны полностью подавлять кальциевый ответ, индуцируемый деполяризацией. Однако максимальный эффект достигается при довольно высокой концентрации блокаторов. В свою очередь, соединения из группы дигидропиридинов не способны полностью подавлять кальциевый ответ. Нифедипин и исрадипин уже в минимальной концентрации снижают амплитуду кальциевого сигнала примерно в 2 раза, при этом дальнейшее повышение концентрации не приводит к значительным изменениям. Такая разница в действии блокаторов может объясняться различной селективностью в отношении к ПЗКК. Как было показано на рис. 2б, кальциевый приток в тело нейронов при деполяризации определяется не только ПЗКК L-типа. Таким образом, полное подавление кальциевого ответа при добавлении верапамила и дилтиазема достигается за счет неспецифического блокирования других, не относящихся к L-типу ПЗКК. Указанный эффект имеет место при высоких концентрациях блокаторов. В свою очередь, нифедипин и исрадипин, по всей видимости, более селективны по отношению к L-каналам по сравнению с другими ПЗКК. Однако поскольку широко представленные во взрослом гиппокампе CaV1.3 каналы [25] имеют слабую чувствительность к дигидропиридинам [26], нельзя утверждать, что нифедипин и исрадипин полностью блокируют все ПЗКК L-типа.
Таким образом, нельзя с уверенностью ответить на вопрос, какие именно блокаторы ПЗКК эффективнее. Скорее всего, выбор того или иного соединения будет зависеть от цели исследования. В случае, если важна селективность в отношении каналов L-типа, лучше использовать дигидропиридины или другие соединения, но в минимальных концентрациях. Однако, если необходимо заблокировать большинство ПЗКК, можно использовать верапамил или дилтиазем в высоких дозировках.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-04-00138.
Список литературы
Tuluc P., Yarov-Yarovoy V., Benedetti B., Flucher B.E. 2016. Molecular interactions in the voltage sensor controlling gating properties of CaV calcium channels. Structure. 24 (2), 261–271.
Gomes B., Savignac M., Moreau M., Leclerc C., Lory P., Guery J.-C., Pelletier L. 2004. Lymphocyte calcium signaling involves dihydropyridine-sensitive L-type calcium channels: Facts and controversies. Crit. Rev. Immunol. 24 (6), 425–447.
Hofmann F., Flockerzi V., Kahl S., Wegener J.W. 2014. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 94 (1), 303–326.
Striessnig J., Pinggera A., Kaur G., Bock G., Tuluc P. 2014. L-type Ca2+ channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 3 (2), 15–38.
Baumann L., Gerstner A., Zong X., Biel M., Wahl-Schott C. 2004. Functional characterization of the L-type Ca2+ channel Cav1.4alpha1 from mouse retina. Invest. Ophthalmol. Vis. Sci. 45 (2), 708–713.
Tang L., Gamal El-Din T.M., Swanson T.M., Pryde D.C., Scheuer T., Zheng N., Catterall W.A. 2016. Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature. 537 (7618), 117–121.
Berger S.M., Bartsch D. 2014. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 357 (2), 463–476.
Ortner N.J., Striessnig J. 2016. L-type calcium channels as drug targets in CNS disorders. Channels (Austin). 10 (1), 7–13.
Prakash P., Hancock J.F., Gorfe A.A. 2015. Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins. 83 (5), 898–909.
Hockerman G.H., Peterson B.Z., Johnson B.D., Catterall W.A. 1997. Molecular determinants of drug binding and action on L-type calcium channels. Annual Rev. Pharmacol. Toxicol. 37, 361–396.
Shaldam M.A., Elhamamsy M.H., Esmat E.A., El-Moselhy T.F. 2014. 1,4-Dihydropyridine calcium channel blockers: Homology modeling of the receptor and assessment of structure activity relationship. ISRN Med. Chem. 2014, 1–14.
Striessnig J., Ortner N.J., Pinggera A. 2015. Pharmacology of L-type calcium channels: Novel drugs for old targets? Curr. Mol. Pharmacol. 8 (2), 110–122.
Lipscombe D., Helton T.D., Xu W. 2004. L-type calcium channels: The low down. J. Neurophysiol. 92 (5), 2633–2641.
Striessnig J., Grabner M., Mitterdorfer J., Hering S., Sinnegger M.J., Glossmann H. 1998. Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol. Sci. 19 (3), 108–115.
Li X.-T., Li X.-Q., Hu X.-M., Qiu X.-Y. 2015. The inhibitory effects of Ca2+ channel blocker nifedipine on rat Kv2.1 potassium channels. PLoS One. 10 (4), e0124602.
Grissmer S., Nguyen A.N., Aiyar J., Hanson D.C., Mather R.J., Gutman G.A., Karmilowicz M.J., Auperin D.D., Chandy K.G. 1994. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45 (6), 1227–1234.
Madeja M., Müller V., Mußhoff U., Speckmann E.-J. 2000. Sensitivity of native and cloned hippocampal delayed-rectifier potassium channels to verapamil. Neuropharmacol. 39 (2), 202–210.
Gunthorpe M.J., Lummis S.C. 1999. Diltiazem causes open channel block of recombinant 5-HT3 receptors. J. Physiol. 519 Pt 3, 713–722.
Marshall M.R., Clark J.P., Westenbroek R., Yu F.H., Scheuer T., Catterall W.A. 2011. Functional roles of a C-terminal signaling complex of CaV1 channels and A-kinase anchoring protein 15 in brain neurons. J. Biol. Chem. 286 (14), 12627–12639.
Jenkins M.A., Christel C.J., Jiao Y., Abiria S., Kim K.Y., Usachev Y.M., Obermair G.J., Colbran R.J., Lee A. 2010. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30 (15), 5125–5135.
Wang Y., Deng X., Mancarella S., Hendron E., Eguchi S., Soboloff J., Tang X.D., Gill D.L. 2010. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 330 (6000), 105–109.
Wheeler D.G., Barrett C.F., Groth R.D., Safa P., Tsien R.W. 2008. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 183 (5), 849–863.
Zhang H., Fu Y., Altier C., Platzer J., Surmeier D.J., Bezprozvanny I. 2006. Ca1.2 and CaV1.3 neuronal L-type calcium channels: Differential targeting and signaling to pCREB. Eur. J. Neurosci. 23 (9), 2297–2310.
Kamijo S., Ishii Y., Horigane S.I., Suzuki K., Ohkura M., Nakai J., Fujii H., Takemoto-Kimura S., Bito H. 2018. A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J. Neurosci. 38 (24), 5551–5566.
Kim S.-H., Park Y.-R., Lee B., Choi B., Kim H., Kim C.-H. 2017. Reduction of Cav1.3 channels in dorsal hippocampus impairs the development of dentate gyrus newborn neurons and hippocampal-dependent memory tasks. PLoS One. 12 (7), e0181138.
Xu W., Lipscombe D. 2001. Neuronal Ca V 1.3α 1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21 (16), 5944–5951.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии