

Биологические мембраны: Журнал мембранной и клеточной биологии, 2021, T. 38, № 6, стр. 402-417
Роль p53-зависимых сигнальных путей в выживании и гибели нейронов и глиальных клеток при повреждении периферической нервной системы
С. В. Родькин a, *, В. А. Дзреян a, С. В. Демьяненко a, А. Б. Узденский a
a Южный федеральный университет, Академия биологии и биотехнологии,
лаборатория “Молекулярная нейробиология”
344090 Ростов-на-Дону, Россия
* E-mail: rodkin_stas@mail.ru
Поступила в редакцию 14.12.2020
После доработки 27.03.2021
Принята к публикации 29.03.2021
Аннотация
Нейротравма – одна из основных причин инвалидности и смертности людей. Тем не менее механизмы, которые опосредуют выживание и смерть клеток периферической нервной системы, до сих пор до конца не изучены. Белок p53, известный как опухолевый супрессор, является мастер-регулятором основных клеточных процессов, включая репарацию ДНК, прохождение клеточного цикла, метаболизм и апоптоз. Гиперэкспрессия p53, показанная в ряде экспериментальных моделей травмы периферических нервов, позволяет предположить важную роль этого белка в патогенезе нейротравм. В настоящем обзоре рассмотрены сигнальные и эпигенетические механизмы активации и регуляции p53, которые могут способствовать выживанию или гибели нейронов и глиальных клеток после травматического повреждения. Рассмотрены перспективы дальнейших исследований механизмов регуляции белка p53, в том числе с участием гистондеацетилаз, для разработки нейропротекторов.
ВВЕДЕНИЕ. ПРОБЛЕМЫ ТЕРАПИИ НЕЙРОТРАВМ
Травмы периферических нервов (ТПН) – одна из распространенных причин инвалидизации и смертности населения во всем мире [1–3]. Гетерогенность ТПН создает дополнительные трудности их исследования, а также сложности в диагностике и подборе правильного лечения [2].
Ряд работ повествуют о роли р53 как транскрипционного фактора, определяющего судьбу клетки и имеющего центральную значимость в патофизиологии инсульта и при травматических повреждениях головного и спинного мозга [4, 5], однако лишь в нескольких работах непосредственно исследовалась роль p53 в гибели и выживаемости нейронов периферической нервной системы. Гиперэкспрессия p53 в ряде экспериментальных моделей ТПН [6–12] позволяет предположить важную роль этого белка в их патогенезе.
Механизмы активации и регуляции p53 при нейротравмах на сегодняшний день изучены относительно слабо [13], но данная область исследований тем не менее представляется перспективной. Новые результаты позволят улучшить современное понимание структурных и функциональных изменений, вызываемых ТПН, а также будут способствовать разработке более эффективных подходов к терапевтическому вмешательству и восстановлению после нейротравм.
В связи с этим целью настоящего обзора явился анализ современных данных, указывающих на ключевую роль белка p53 в патогенезе ТПН. Опираясь в том числе и на результаты, полученные в нашей лаборатории, мы попытаемся разобрать вопросы, связанные с механизмами активации и сложной системой регуляции p53 в нейронах и глиальных клетках после аксотомии, а также затронем потенциальные терапевтические последствия воздействия на активность p53 в условиях нейротравмы.
СТРУКТУРА БЕЛКА p53
Белок р53 играет важную роль во внутриклеточной сигналиазации, регулируя клеточный цикл и транскрипцию множества генов, поэтому неудивительно, что р53 имеет сложную структурную организацию (рис. 1) [14]. Структура р53 имеет модульную доменную архитектонику, состоящую из ДНК-связывающих и тетрамеризационных доменов, включающих внутренне неупорядоченные участки на C- и N-концах (рис. 1). В активной форме р53 представляет собой тетрамер, каждый мономер которого состоит из доменов с различной степенью структурной стабильности, начиная от хорошо свернутого ДНК-связывающего домена (DNA-binding domain, DBD) и домена тетрамеризации (tetramerization domain, TET), до внутренне неупорядоченного трансактивирующего домена (transactivation domain, TAD) и крайнего С-концевого домена (C-terminal domain, CTD) [14–17].
Рис. 1.
Схема строения р53. Структура р53 включает в себя трансактивационный домен, состоящий из двух субдоменов TAD1 и TAD2, за которыми располагается область, насыщенная пролином (PRR), ДНК-связывающего центрального домена (DBD), образованного иммуноглобулин-подобным β-сэндвич-каркасом и расположенной на его конце ДНК-связывающей поверхностью, а также домена тетрамеризации и С-концевого домена CTD, регулирующим активность р53.
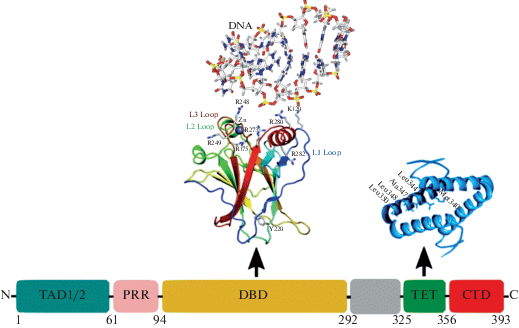
Основной ДНК-связывающий домен (DBD) состоит из центрального иммуноглобулин-подобного β-сэндвича и расположенной на его конце ДНК-связывающей поверхности, сформированной двумя большими петлями (L2 и L3) и мотивом “петля–лист–спираль” (loop-sheet-helix) [18]. Ион цинка, тетраэдрически координированный в позициях Cys176, His179, Cys238 и Cys242, стабилизирует области L2 и L3 [19]. Внутренняя термодинамическая стабильность DBD находится на относительно низком уровне, поскольку основной домен р53 имеет достаточно невысокую температуру плавления (44°C). Было показано, что низкая термодинамическая и кинетическая стабильность DBD обеспечивает быстрое изменение конформации р53 из свернутого в развернутое состояние и обратно, тем самым обеспечивая его дополнительную регуляцию в сочетании с убиквитин-протеасомной деградацией. Также это свойство DBD делает структуру р53 пластичной, обусловливая его более эффективное связывание с различными белками [20, 21].
Полноразмерный домен TAD p53 состоит из двух N-концевых субдоменов TAD1 (аминокислотные остатки 1–42) и TAD2 (аминокислотные остатки 43–73) и имеет суммарный заряд –17 из-за наличия в своей структуре большого количества кислых аминокислот (аспарагиновая и глутаминовая кислоты), перемежающихся с объемными гидрофобными участками. TAD входит в семейство “кислых” доменов активации (acidic activation domain, AAD) [21]. Сайт активации транскрипции располагается в пределах аминокислотных остатков 1–42. TAD не имеет третичной структуры, что характерно для большинства AAD, однако содержит α-спираль и два амфипатических витка [22]. В несвязанном состоянии TAD легко расщепляется протеазами, что свидетельствует о его слабой конформации. Возможно, низкая молекулярная организация TAD делает его структуру более пластичной, способствуя взаимодействию со множеством белков и облегчая его посттрансляционную модификацию различными ферментами. Основная функция TAD заключается в про-апоптотической транскрипции множества генов, однако было показано, что TAD также участвует и в процессах репарации ДНК [18–21].
На С-конце р53 между 311-м и 363-м аминокислотными остатками располагается домен TET, ответственный за тетрамеризацию, мономеры которого содержат β-цепь, связанную с α-спиралью одним аминокислотным остатком [22]. Исследования показали, что TET представляет собой димер димера. Два димера взаимодействуют между собой через свои α-спирали, а β-цепи располагаются на внешней стороне тетрамера, не принимая непосредственного участия в их ассоциации [23].
С-концевой домен (CTD) р53 представляет собой внутренне неупорядоченную область, меняющую свою конформацию в зависимости от ассоциации с белками-партнерами благодаря гибкой структуре, большой площади поверхности и экспонированию пептидных сайтов, обладающих функциями молекулярного распознавания. Недавние исследования показали, что CTD регулирует активность р53, в частности, обеспечивая отрицательный контроль над ДНК-связывающей способностью данного белка [23, 24].
РЕГУЛЯЦИЯ АКТИВНОСТИ БЕЛКА p53
Выделен целый ряд состояний, способных активировать р53: истощение запасов нуклеотидов, нарушения цитоскелета, нарушения биогенеза рибосом, гипоксия и ишемия, гипероксия, отсутствие или избыток некоторых факторов роста или цитокинов, нарушения клеточной адгезии и фокальных контактов, нарушение прикрепления клеток к субстрату (что сопровождается р53-зависимым аноикисом), действие монооксида азота (NO) и многое другое. Все эти состояния вызывают характерные для каждого из них модификации как самого белка р53, так и сигнальных систем, контролирующих его уровень и активность.
Ген р53 независимо от условий транскрибируется в клетке с постоянной интенсивностью, однако его транскрипты обладают исключительно коротким временем жизни, что достигается благодаря быстрой и непрерывной деградации в результате убиквитинирования белками MDM2 и MDMX. Дополнительный контроль функции р53 осуществляется посредством посттрансляционных модификаций (фосфорилирование, ацетилирование, метилирование и другие), а также посредством взаимодействия с белками-партнерами.
Деградация и стабилизация р53
E3-убиквитин-протеиновая лигаза MDM2 – основной отрицательный регулятор белка р53, катализирующий перенос активированного убиквитина с фермента группы Е2 на белок р53 [25]. При этом сам фермент MDM2 является продуктом гена, активируемого р53. Поскольку MDM2 является транскрипционной мишенью p53, MDM2-зависимая регуляция p53 существует в петле обратной связи, которая контролирует конститутивно низкую экспрессию p53 в нормальных условиях. Кроме того, MDM2 может быть важен для контроля кратковременного повышения уровня р53, необходимого, например, для восстановления ДНК после легкого генотоксического стресса без риска фатальных последствий чрезмерной активации р53. Все больше данных свидетельствуют о том, что MDM2 участвует в регуляции р53-зависимой гибели клеток в нейронах. В частности, повышение уровня MDM2 было обнаружено в ткани головного мозга при ишемии, где его отрицательная регуляция активности р53 важна для последующей репарации [26].
Процесс взаимодействия р53 с белком MDM2 тонко регулируется за счет множества механизмов, направленных как на регуляцию активности MDM2, так и на модификации его мишени – самого белка р53. Этот центральный регуляторный механизм дополнительно контролируется рядом других белков, таких как MDMX, опухолевым супрессором ARF и HAUSP [27].
MDMX (MDM4) также представляет собой белок, связывающий p53 и имеющий структурное сходство с MDM2, но, в отличие от MDM2, не обладающий убиквитинлигазной активностью. MDMX связывается с N-концевой областью р53, подавляя его транскрипционную активность, но не вызывает его разрушения. MDMX также способен гетероолигомеризоваться с MDM2, что приводит к ускорению разрушения MDMX и стабилизации MDM2. Таким образом, изменение соотношений этих двух белков может тонко регулировать количество и активность р53 [25–27].
Образование комплексов р53 с белками MDM2 и MDMX также тщательно регулируется. Например, рибосомные белки (L5, L11 и L23) связываются с MDM2, подавляют его активность в отношении р53 и одновременно стимулируют разрушение MDMX, что приводит к активации р53 в ответ на рибосомальный стресс [26].
Кроме того, важными отрицательными регуляторами MDM2 являются белок p19ARF и человеческий гомолог белка p14ARF, которые связываются с MDM2 и нейтрализуют его, тем самым повышая активность p53. В первичных нейронах накопление p19ARF и p53 происходило после NO-опосредованного апоптотического стресса через путь p21ras-MAPK [27].
Недавние исследования показали, что нарушение функции ядрышка может выступать в качестве важного датчика стресса, повышающего стабильность p53, вероятно, за счет повышения уровня p19ARF. Поскольку разрушение ядрышек достаточно, чтобы вызвать p53-зависимый апоптоз, предполагается, что именно оно выступает в качестве ключевого механизма интеграции всех известных стимулов активации p53, но это пока не до конца изучено в нейронах и остается лишь предположением [25–27].
Транскрипция ARF-белков положительно и отрицательно контролируется комплексами, содержащими транскрипционный фактор E2F1. В свою очередь, его экспрессия контролируется МАР-киназой р38 и фактором транскрипции с-Мус [25].
Модификация активности белка MDM2 также происходит и при его связывании с белком-активатором транскрипции р300/СВР, что приводит к переключению способности MDM2 с моноубиквитинирования на полиубиквитинирование р53, которое необходимо для его узнавания 26S-протеасомами и разрушения белка. Полиубиквитилирование p53, в отличие от моноубиквитирования, приводит к протеасомной деградации р53 в цитоплазме. Большая часть моноубиквитилированного p53 направляется в митохондрии [26–28].
Белок MDM2, так же как и р53, сам подвергается разрушению в 26S-протеасомах, но этот процесс может регулироваться за счет специальных ферментов, удаляющих убиквитиновые остатки. Установлено, что белок HAUSP, который также известен как убиквитин-специфическая протеаза 7 (USP7), является одним из таких ферментов. Он удаляет убиквитин с белка MDM2, в то время как другой белок Daxx образует комплекс с MDM2 и HAUSP и предотвращает самоубиквитинирование MDM2, что приводит к его стабилизации и ускорению разрушения р53. Кроме того, p53 может подвергаться деубиквитинированию с помощью белка HAUSP, который, оказывая противоположные эффекты на систему разрушения р53, способствует тонкой регуляции его активности [25].
Кроме того, обнаружено несколько других убиквитиновых лигаз E3, включая ARF-BP1/Mule, COP1, Pirh2 и MSL2, которые могут убиквитинировать p53, вызывая деградацию белка или изменение его субклеточной локализации. Однако биологическое значение этих результатов in vivo еще предстоит выяснить [27].
Белковая молекула р53 имеет неструктурированные участки в N- и С-концевых областях, что обусловливает ее разрушение по убиквитин-независимому пути в 20S-протеасомах. Данный процесс регулируется NAD(P)H-зависимой хиноновой оксидоредуктазой NQO1, которая при наличии NADH связывается с такими белками и предотвращает их разрушение в 20S-протеасомах [26].
Таким образом, многоуровневый подход к регулированию уровней p53 и MDM2 может обеспечить дополнительную защиту клетки от активации сигнального пути p53 [25–28].
Посттрансляционные модификации р53
Уровень белка р53 поддерживается на низком уровне в большинстве типов клеток, включая нейроны, благодаря тонким механизмам, обеспечивающим жесткую регуляцию его активности [25]. Такая динамическая регуляция определяет содержание белка в клетке, субклеточную локализацию, посттрансляционные модификации и партнеров по связыванию [29, 30]. Эти свойства впоследствии формируют необходимую скорость, интенсивность и продолжительность активации р53, а также его предпочтение отдельным транскрипционным мишеням, определяя, таким образом, возможные биологические ответы. Поэтому актуальным остается исследование механизмов регуляции белка p53 в процессах нейродегенерации и нейропротекции.
После повреждения ДНК или клеточного стресса р53 активируется посредством множества посттрансляционных модификаций, таких как фосфорилирование, ацетилирование и/или метилирование, а также введении остатков убиквитина и убиквитин-подобных белков SUMO и NEDD8 [30]. Такие модификации либо увеличивают стабильность р53, либо напрямую усиливают аффинность его связывания с ДНК. Перечисленные посттрансляционные модификации р53 могут варьироваться между популяциями нейронов, зависеть от типа и тяжести генотоксического стресса и могут изменяться в процессе развития и старения [25].
Описано более 20 сайтов в молекуле р53, подвергающихся ковалентным модификациям. Большая часть из них располагается в N- и С-концевых сегментах р53. Множество белков, взаимодействуя с р53, вносят ковалентные модификации в его структуру [25]. К таким белкам относится более 30 различных протеинкиназ, несколько протеинфосфатаз, несколько убиквитиновых лигаз и белков, регулирующих взаимодействие р53 с Е3-лигазами, деубиквитинирующие белки, белки, осуществляющие связывание с SUMO и NEDD8, несколько метилаз, ряд ацетилтрансфераз и деацетилирующих ферментов. Фосфорилирование p53 в области его N-конца способствует дегенерации двигательных нейронов [25].
Модификации в области N-конца р53 (например, фосфорилирование Ser15, Ser20, Thr18) также могут препятствовать его связыванию с MDM2 и другими Е3-лигазами, стабилизируя белок. Кроме того, за счет модификаций р53 происходит модуляция его способности взаимодействовать с коактиваторами и корепрессорами транскрипционного аппарата, а также изменение предпочтений в связывании с р53-респонсивными элементами тех или иных генов. Разнообразные модификации С-концевых участков р53 (фосфорилирование, ацетилирование, метилирование, пришивание убиквитин-подобных белков SUMO и NEDD8) приводят к нейтрализации ингибирующего действия С-концевого сегмента, к дальнейшей стабилизации р53, как позитивной, так и негативной модуляции его активности и к изменениям его внутриклеточной локализации [29, 30].
РАЗНООБРАЗИЕ КЛЕТОЧНЫХ ФУНКЦИЙ БЕЛКА p53
Белок p53 известен как опухолевый супрессор и “страж генома”. Он является регулятором важных функций клетки, включая репарацию ДНК, прохождение клеточного цикла, метаболизм и апоптоз [30–32]. В качестве фактора транскрипции p53 контролирует экспрессию множества генов, которые принимают участие в вышеперечисленных процессах [31, 33].
По последним данным, число генов, контролируемых белком p53, превышает 3000, хотя по более строгим оценкам число таких генов ограничено несколькими сотнями [33, 34]. Наряду с транскрипционно-зависимым путем регуляции, р53 принимает участие и в транскрипционно-независимых процессах, таких как регуляция функций митохондрий и запуск процессов апоптоза в клетках с митохондриальной дисфункцией [30–32].
Было продемонстрировано, что p53 в стрессовых условиях проникает в митохондрии и активирует экспрессию проапоптотических генов PUMA, BAX, APAF-1, NOXA, а также ингибирует экспрессию антиапоптотических генов семейства Bcl-2 (BCL2, BCL-X, BCL-in, MCL1). Эти проапоптотические белки вместе с p53 транспортируются в митохондрии, где они вызывают увеличение проницаемости внешней мембраны, что приводит к высвобождению цитохрома c и других апоптотических белков [35, 36].
Кроме того, в последние годы появилась информация о роли p53 в дифференцировке и пролиферации нервных стволовых клеток, а также в регуляции роста и регенерации аксонов [34]. Ингибирование p53 блокирует фактор роста нервов (NGF). В клетках, подвергшихся действию NGF, был обнаружен рост уровня p53 и снижение экспрессии Е3-убиквитин лигазы MDM2, которая маркирует p53 для его последующей протеасомной деградации. Кроме того, показано, что p53 активирует транскрипцию ряда генов, ответственных за перестройку цитоскелета. Как в нейронах коры головного мозга, так и в нейронах дорзальных ганглиев p53 активирует экспрессию cGMP-зависимой киназы cGKI, противодействующей коллапсу конуса роста. У мышей с нулевым уровнем p53 наблюдается нарушение регенерации аксонов после травмы лицевого нерва. Ряд других факторов транскрипции, включая NF-kB, которые важны для регенерации аксонов, могут потенциально пересекаться с р53-зависимыми сигнальными путями [34, 35].
В нормальных клетках экспрессия p53 обычно находится на низком уровне благодаря протеолизу, но в условиях стресса и при множественном повреждении ДНК экспрессия p53 повышается [4]. Ряд работ демонстрирует активацию p53 в различных экспериментальных моделях повреждения нервов [5, 35–37]. Кроме того, фармакологическое или генетическое ингибирование p53 чаще всего предотвращает гибель нейронов, а гиперактивация сигнального пути p53 обычно стимулирует нейродегенерацию. Это указывает на ключевую роль p53 в регуляции жизнеспособности нейронов после повреждения [30–32, 35, 36]. Однако роль р53 в гибели нейронов и глиальных клеток при повреждениях периферической нервной системы недостаточно ясна и нуждается в дальнейших исследованиях.
Мы сосредоточили свое внимание на изучении роли белка p53 в гибели и выживании нервных клеток после аксотомии – на экспериментальных моделях повреждения периферических аксонов у беспозвоночных и млекопитающих.
УЧАСТИЕ p53 В ГИБЕЛИ И ВЫЖИВАНИИ НЕРВНЫХ КЛЕТОК ПОСЛЕ АКСОТОМИИ
Нейротравма – одна из основных причин инвалидности и смертности людей в мире, особенно среди мужчин молодого и среднего возраста. Аксотомия относится к механическим повреждениям нервов и представляет собой полную перерезку нерва, инициирующую сложный каскад сигнальных и метаболических процессов, направленных на гибель или выживание нейрона [38–40].
Перерезка нерва (аксотомия) характеризуется тремя основными молекулярно-клеточными событиями: валлерова деградация отрезанного аксона, гибель поврежденного нейрона или его регенерация с отрастанием аксона и восстановлением нейронных связей [38]. Аксотомия вызывает нарушение целостности нейрональной мембраны, дестабилизацию клеточного гомеостаза, деструкцию микротрубочек, нарушение аксонального транспорта и повреждение внутриклеточных органелл [1]. При этом происходит активация и подавление множества белков в аксотомированных нейронах [1, 39]. Некоторые сигнальные белки ретроградно транспортируются к соме нейронов и стимулируют факторы транскрипции, чтобы инициировать экспрессию генов и синтез важных белков, которые затем обратно транспортируются к поврежденному месту аксона для восстановления его структуры и ускорения процесса регенерации [39]. В конечном итоге регенерирующие аксоны в периферической нервной системе могут снова восстановить контакт со своей мишенью, однако многие нейроны погибают [40]. К сожалению, клинически эффективных нейропротекторов, которые могут спасти поврежденные нейроны при аксотомии, пока не найдено [1–3, 40, 41].
Отсутствие решающих успехов в этом направлении говорит о недостаточном понимании молекулярно-клеточных механизмов повреждения нервов и последующих вторичных процессов, приводящих к клеточной смерти или к регенерации нерва и восстановлению утраченных функций. Поэтому актуальны исследования молекулярно-клеточных механизмов повреждения периферической нервной системы на модельных объектах. Трудностью таких исследований является большое разнообразие повреждений нервной системы [42]. Разработано несколько моделей нейротравм. Это модели растяжения, механического или гидростатического сдавливания, гидродинамического удара, удара падающего груза, рассечения нерва (аксотомии) [42–44]. Использование этих моделей помогает выяснить комплексные механизмы разных видов нейротравмы, ведущие к смерти нейронов и глиальных клеток [44].
В своих работах мы использовали три экспериментальные модели нейротравмы в биологических объектах разного уровня сложности (рис. 2): 1) аксотомированные рецепторы растяжения речного рака (РРР); 2) аксотомированные ганглии вентральной нервной цепочки (ВНЦ) рака и 3) аксотомированные ганглии корешков спинного мозга крысы (DRG), полученные при перерезке седалищного нерва [6, 7, 41, 45–50].
Рис. 2.
Экспериментальные модели аксотомии: рецептор растяжения и брюшная нервная цепочка рака Astacus leptodactylus, а также дорзальные ганглии крысы.
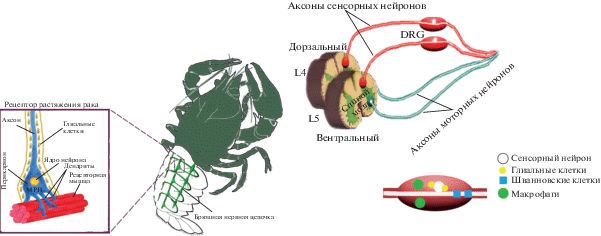
Рецептор растяжения речного рака является подходящим модельным объектом для изучения молекулярных механизмов нейрональных и глиальных ответов на аксотомию. Он состоит из одного механорецепторного нейрона (МРН), окруженного сателлитными глиальными клетками (СГК) (рис. 2).
На рис. 3 представлена схема участия изученных нами белков в некротической или апоптотической смерти удаленных глиальных клеток после аксотомии МРН.
Рис. 3.
Схема сигнальных путей, которые регулируют апоптоз и некроз глиальных клеток вокруг проксимального сегмента механорецепторного нейрона рака после аксотомии. Сплошные линии – собственные результаты, пунктирные линии – литературные данные. LIMK – актин-связывающая киназа LIM.
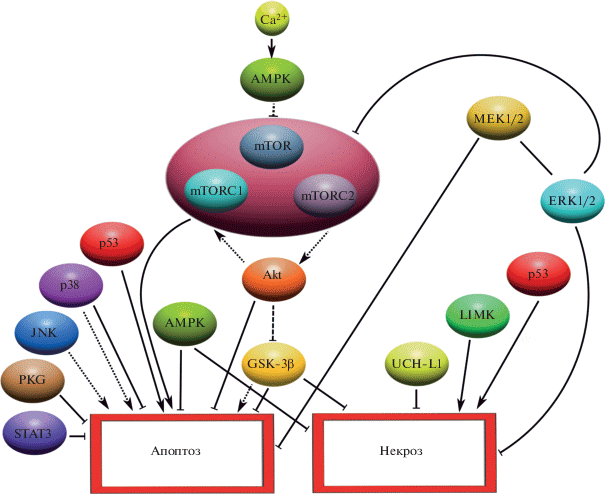
В своих работах мы использовали специфические ингибиторы для изучения роли таких белков, как: AMPK, mTOR , Akt, GSK-3β, MEK1/2, ERK1/2, p38 и JNK в индуцированном аксотомией некрозе и апоптозе СГК. СГК расположены на расстоянии нескольких миллиметров от места рассечения и окружают проксимальный сегмент аксона МРН [1]. Вначале рассмотрим литературные данные об участии данных систем в гибели клеток.
Увеличение внутриклеточного Ca2+ при аксотомии активирует протеинкиназу AMPК через сигнальный путь Ca2+/CaMKKβ, что приводит к ингибированию высококонсервативной серин/ треониновой протеинкиназы mTOR, входящей в качестве каталитической субъединицы в состав двух функционально различных гетерогенных комплексов mTORC1 и mTORC2 [51, 52]. Комплекс mTORC2 фосфорилирует киназу Akt – ключевой фермент сигнального пути PI3K/Akt/ mTOR, ответственного за выживание клетки [53–55]. С этим путем тесно связана гликогенсинтаза GSK-3β через Akt-опосредованное ингибирование. GSK-3β обладает противоположными эффектами, повышая или понижая порог инициации апоптотической клеточной смерти. Так, GSK-3β способствует гибели клетки, вызывая внутренний (митохондриальный) апоптоз, но ингибирует внешний, опосредованный рецепторами смерти, а также снижает риск некроза [56–67]. Akt ингибирует апоптоз на постмитохондриальном уровне без изменения экспрессии Bcl-2, Bcl-x или Bax через подавление каспазного каскада. В свою очередь, Akt активирует комплекс mTORC1, который активирует процессы апоптоза. Интересен также STAT3, сигнальный белок и активатор транскрипции, который обладает антиапоптотическим эффектом за счет регуляции экспрессии Bcl-xL, Bcl-2 и Mcl-1 [58]. Протеинкиназа G (PKG) регулирует апоптоз через фосфорилирование белка Bad. Протеинкиназа JNK и р38 практически во всех случаях связаны с индукцией апоптоза. Активация сигнального пути p38 приводит к множеству транскрипционных и трансляционных изменений, ведущих к запрограммированной гибели клетки [59]. Различные внутриклеточные и внеклеточные стрессовые факторы, включая аксотомию, приводят к стимуляции активности JNK. Данная киназа может перемещаться в кариоплазму, стимулируя множество проапоптотических факторов транскрипции [60, 61]. Особую роль в выживании и гибели клеток играет сигнальный путь MAPK RAS/RAF/MEK/ERK, активируемый широким спектром факторов роста и цитокинов.
Другой важный фермент, локализующийся преимущественно в нервной ткани, препятствующий некрозу нейроцитов и играющий центральную роль в поддержании жизнедеятельности и стабильности аксонов – убиквитин С-концевая гидролаза L1 (UCH-L1) [62]. Актин-связывающая киназа LIM, наоборот, увеличивает некроз глиальных клеток, вероятно, через сигнальный путь р21.
В наших исследованиях были выявлены некоторые несоответствия с данными литературы. Так, например, проапоптотическая MAP-киназа p38 в нашей работе проявляла антиапоптотические свойства в глиальных клетках речного рака. Ранее аналогичным образом была показана способность p38 защищать нейроны от NO-индуцированного апоптоза [63] и способствовать защите сердца от повреждения при реперфузии [64].
Обычно GSK-3β обладает проапоптотической активностью, а его ингибирование с помощью Akt подавляет апоптоз клеток. Однако после аксотомии МРН как Akt, так и GSK-3β могут участвовать в антиапоптотических процессах в СГК. Возможно, антиапоптотическая активность Akt связана с ингибированием проапоптотических белков Bad, Bim и каспазы-9, но не GSK-3β. Akt также может активировать mTORC1, который в наших экспериментах подавлял апоптоз [1].
Ингибирование AMP-активируемой протеинкиназы AMPK с помощью AICAR или дорсоморфина не влияло на некроз и апоптоз глиальных клеток после аксотомии. Скорее всего, уровень ATP в СГК не был настолько сильно нарушен, чтобы активировать AMPK после 8-часовой инкубации. Возможно, антиапоптотические процессы в сателлитных глиальных клетках после аксотомии МРН были связаны с каскадом Akt/mTORC1, а не с AMPK/mTOR [1].
Таким образом, белки MEK1/2, p38, Akt, GSK-3β и mTOR способствуют устойчивости глиальных клеток к апоптозу, индуцированному аксотомией. ERK1/2 и GSK-3β участвуют в защите СГК от вызванного аксотомией некроза. Поддержание выживания нейронов – одна из основных функций глиальных клеток нервной системы позвоночных. В РРР не только СГК способствуют выживанию нейронов, но и МРН поддерживает выживание глиальных клеток. Как именно поврежденный нейрон индуцирует защитные сигнальные пути в СГК, которые увеличивают их сопротивление, является проблемой для будущего исследования [1].
Также особый интерес заслуживает наше исследование по изучению локализации р53 в МРН рака и его роли в индуцированной аксотомией гибели СГК, удаленных от места рассечения аксона [6].
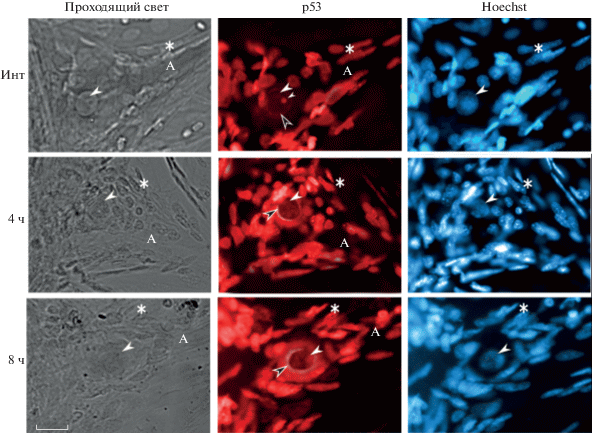
Иммунофлуоресцентная микроскопия показала, что в интактном МРН, аксоны которых были не повреждены и сохраняли связь с ганглиями брюшной нервной цепочки, р53 главным образом концентрировался в ядрышке и узком цитоплазматическом кольце шириной 2–4 мкм, расположенном вокруг ядра (рис. 4). Уровень р53 в перикарионе и глиальных ядрах, в которых не наблюдалось ядрышко, был заметно ниже, а в аксоне полностью отсутствовал. Перерезка аксона приводила к увеличению иммунофлуоресценции р53 через 4 и 8 ч в кариоплазме нейронов и глиальных клеток, в перикарионе МРН. При этом перинуклеарное кольцо не исчезало, а становилось более выраженным и интенсивным по сравнению с интактными нейроглиальными препаратами [6].
Рис. 4.
Локализация p53 (иммунофлуоресценция анти-р53 P5813, Sigma-Aldrich) в интактных (Инт) механорецепторных нейронах и через 4 и 8 ч после аксотомии, а также эти нейроглиальные препараты в проходящем свете и окрашенные Hoechst 33342, селективно флуорохромирующим клеточные ядра в синий цвет. Условные обозначения: А – аксон, звездочки – ядра глиальных клеток, большие белые стрелки – ядра нейронов, маленькая стрелка – ядрышко нейрона, черные стрелки – кариоплазматическое кольцо. Масштабный отрезок 20 мкм.
Проведенный ингибиторный анализ in vivo на нейронах РРР с использованием активаторов и ингибиторов p53 позволил сделать выводы о механизме участия р53 в апоптозе глиальных клеток [6]. Индуцированный аксотомией апоптоз отдаленных глиальных клеток усиливается в присутствии активаторов p53 WR-1065 и нутлина-3, но снижается пифитрином-α, который ингибирует транскрипционную активность p53 [6, 65]. Пифитрин-μ, который ингибирует действие p53 на митохондрии, увеличивал индуцированный аксотомией апоптоз удаленных глиальных клеток, но уменьшал их некроз. Следовательно, индуцированный аксотомией апоптоз удаленных глиальных клеток был связан с эффектом p53 на процессы транскрипции, тогда как глиальный некроз был скорее связан с независимым от транскрипции эффектом p53 на митохондрии. По-видимому, судьба удаленных глиальных клеток в аксотомированном РРР определяется балансом между различными модальностями активности p53 [6, 65].
Протеомные исследования показали, что через 1–3 ч после двусторонней аксотомии в вентральных ганглиях речного рака наряду с повышением белка p53 наблюдается гиперэкспрессия нескольких десятков важнейших белков, участвующих в ключевых клеточных процессах [50]. Это белки внутриклеточной сигнализации: p38, JNK, ERK1 и ERK2, RAF1, PKCα, MAPKAPK2, MKP-1, кальмодулин, APP; белки, участвующие в инициации, реализации и регуляции апоптоза: факторы транскрипции c-Myc, E2F1, p53; белки Bcl-10, GADD153, AIF, SMAC/DIABLO, каспазы 3, 6, 7 и 11; многофункциональные белки, способные инициировать апоптоз в определенных ситуациях: PAR4, NGFR, p75, NMDAR2a. Одновременно повышался уровень антиапоптотических белков: Bcl-x, p21, MDM2, ERK5, рецепторов эстрогенов. Повышался уровень некоторых белков, участвующих в перестройках актинового цитоскелета: кофилина, миозина II и Va, α-катенина, катенина p120CTN, p35. Вместе с тем другие, связанные с актином белки, такие как эзрин, дистрофин, тропомиозин, спектрин (α + β), фосфорилированный Pyk2 подавлялись. Различные цитокератины и βIV-тубулин, компоненты промежуточных филаментов и микротрубочек, также были подавлены, что могло быть результатом разрушения ткани. Подавление белков, участвующих в образовании клатриновых везикул (AP2α и AP2γ, адаптина (β1 + β2) и синтаксина), указывает на нарушение везикулярного транспорта, а ферментов синтеза катехоламинов (L-DOPA декарбоксилазы, тирозин- и триптофангидроксилаз) – на подавление синаптических процессов [50].
Другой объект нашего исследования – аксотомированные ганглии корешков спинного мозга крысы (DRG) (рис. 2) [7, 45]. Они состоят преимущественно из сенсорных нейронов, получающих информацию от седалищного нерва, иннервирующего задние конечности. После перерезки седалищного нерва нейроны DRG оказываются аксотомированными [44].
Перерезка седалищного нерва (sciatic nerve) на бедре у грызунов – одна из важных экспериментальных моделей нейротравмы. Популярность этой модели связана с доступностью седалищного нерва в средней части бедра животного для хирургического рассечения, а также меньшим неудобством и стрессом животного в сравнении с аксотомией нервов верхних конечностей [44].
В аксотомированных нейронах DRG апоптоз глиальных клеток наблюдался через 24 ч после перерезки седалищного нерва и усиливался на 7-й день, когда апоптоз некоторых нейронов только начинался [7]. Настоящие эксперименты показали, что глиальные клетки, удаленные от места перерезки нерва, были более уязвимы для аксотомии, чем нейроны DRG. Самым ранним проапоптотическим событием в поврежденной DRG была гиперэкспрессия фактора транскрипции E2F1 через 4 ч после перерезки седалищного нерва. Это предшествовало индукции p53 и активации каспазы-3 через 24 ч после аксотомии [7].
E2F1 является одним из важнейших регуляторов апоптоза [7]. Он индуцирует экспрессию ряда генов, регулирующих синтез и репарацию ДНК, клеточный цикл и апоптоз. В свою очередь, его экспрессия контролируется МАР-киназой р38 и фактором транскрипции с-Мус [66–68]. В нашей работе на аксотомированной ВНЦ и спинномозговых ганглиях крыс его уровень после аксотомии повышался уже через 1 и 4 ч, соответственно [69]. Появляется все больше свидетельств того, что факторы транскрипции p53 и E2F1 могут регулировать друг друга посредством взаимодействия, координируя как пролиферацию, так и гибель клеток и, в конечном итоге, поддерживая гемостаз в живых организмах [70]. При этом ингибирование пути р53/E2F1 предотвращает апоптоз нейронов [69]. Полученные нами данные указывают на возможное регулирование p53 со стороны E2F1 в аксотомированных нейронах DRG [7]. Однако это еще предстоит доказать в ходе будущих экспериментов.
Перерезка седалищного нерва через 24 ч вызывала снижение уровня р53 в ядерной фракции DRG ганглиев крысы и одновременно – повышение уровня p53 в цитоплазматической фракции [7]. Это указывает на перераспределение р53 между клеточными ядрами и цитоплазмой. В цитоплазме р53 непосредственно связывается с митохондриями и инициирует апоптоз [32, 70, 71]. Эти процессы совпадают по времени с усилением апоптоза глиальных клеток, но не нейронов в ганглиях [7].
РОЛЬ АЦЕТИЛИРОВАНИЯ/ ДЕАЦЕТИЛИРОВАНИЯ ГИСТОНОВ В РЕГУЛЯЦИИ p53 ПРИ АКСОТОМИИ
Ацетилирование p53 вызывает активацию многих генов, контролирующих остановку клеточного цикла и апоптоз [72–74]. p53 может быть ацетилирован с помощью гистонацетилтрансферазы p300/CBP по множественным остаткам лизина в пределах С-концевого регуляторного домена, что сильно коррелирует со стабилизацией и активацией белков [13]. Находящийся в комплексе с р300/СВР белок F также ацетилирует р53 в области сигнала ядерной локализации по лизину К320. Позже было показано, что p53 может in vivo ацетилироваться в ответ на множество разнообразных клеточных сигналов стресса. Белки p300/CBP и PCAF ацетилируют p53 по различным сайтам: p300/CBP – по K372, K373, K381 и K382 по C-концам, а PCAF – по K320 в линкерной области, соединяющей ДНК-связывающий и тетрамеризующий домены. Ацетилирование p53 по каждому из этих двух участков приводит к значительному повышению трансактивационной функции р53 [75]. В результате указанных процессов происходит стимуляция инициации транскрипции с р53-респонсивного промотора. Гиперэкспрессия PCAF способствовала регенерации сенсорных аксонов на расстоянии до 1 мм от места поражения в моделях повреждения спинного мозга. Две другие ацетилтрансферазы hMOF и TIP60 учувствуют в ацетилирование p53 по K120 в ДНК-связывающем домене. Интересно, что данная модификация не влияет на стабильность p53 и его способность связывать ДНК, однако ацетилирование p53 вне С-концевого домена имеет решающее значение для активации таких проапоптотических генов как PUMA и BAX [73].
Фосфорилирование p53, активирующее, как известно, его функции, может облегчать ацетилирование белка. Фосфорилированный p53 более эффективно взаимодействует с HAT (histone acetyltransferase), чем нефосфорилированный. Фосфорилирование p53 по N-концевым остаткам усиливает взаимодействие р53 с p300 [73–75].
Патологические воздействия, такие как нейротравмы, снижают ацетилирование гистонов и негистоновых белков. Вероятно, это происходит в результате активации гистондеацетилаз (HDACs), что нарушает белковый синтез [76–79]. В нашей лаборатории показано, что гистоновые деацетилазы класса I HDAC1, HDAC2 участвуют в регуляции выживаемости и гибели клеток пенумбры после ишемического инсульта в коре мозга крысы [79], а также в аксотомированных ганглиях брюшной нервной цепочки речного рака и спинномозговых ганглиях крыс [50].
Гистоновые деацетилазы помимо их функции в качестве эпигенетических регуляторов способны деацетилировать и централизованно регулировать активность некоторых факторов транскрипции, а также других белков в цитоплазме клеток [80]. Однако роль эпигенетических процессов, включая процессы деацетилирования негистоновых белков в регуляции гибели и выживаемости клеток после повреждения нервов, пока не изучена [76–80].
Белок p53 является негистоновым белком-мишенью HDAC (рис. 5). HDAC1, HDAC2, HDAC3 и SIRT1, член семейства HDAC класса III, могут действовать как деацетилазы p53 (рис. 5) [80].
Рис. 5.
Эффекты деацетилирования p53 c участием HDAC1, HDAC2, HDAC3 и SIRT1. Ac – ацильная группа; P – остаток фосфорной кислоты.
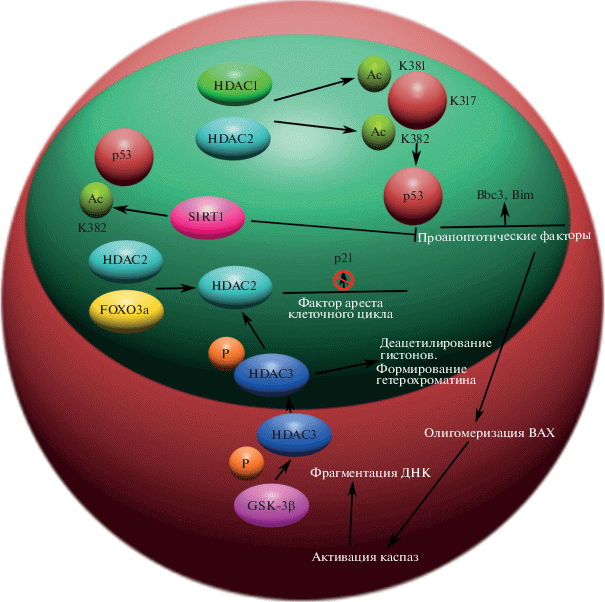
В нейронах деацетилирование p53, по-видимому, имеет решающее значение для экспрессии проапоптотических генов, в частности каспаз. HDAC1 и HDAC2 активируют р53 путем деацетилирования его остатков K381 и K382, что приводит к увеличению экспрессии генов, участвующих в апоптозе, включая Bbc3 (PUMA) и Bim. HDAC2 может выполнять дополнительную функцию по подавлению других генов-мишеней p53, таких как p21. В этом механизме HDAC2 действует с FOXO3a, который рекрутирует HDAC2 на сайт промотора p21 (рис. 5) [32, 35, 36].
Нами было показано, что перерезка седалищного нерва в первые 24 ч после аксотомии вызывает транслокацию HDAC1 из ядра в цитоплазму, где она может, по всей видимости, деацетилировать различные негистоновые белки, в том числе белок р53, гиперэкспрессия которого также наблюдалась в цитоплазматической фракции аксотомированных DRG крысы [24]. Деацетилирование р53 может привести к изменению его субклеточной локализации и, как следствие, его активности. Именно поэтому HDAC стали многообещающими мишенями для терапевтических вмешательств при патологических состояниях, включая нейродегенеративные патологии [80].
HDAC3, наряду с HDAC1 и HDAC2, играет первостепенную роль в деацетилировании гистонов и образовании гетерохроматина [81]. Гистондеацетилаза HDAC3 в нейронах локализована главным образом в цитоплазме и активируется путем фосфорилирования серин/треонин киназы GSK-3β: механизм, который обычно ингибируется стимуляцией факторами роста сигнального пути PI3K/Akt. Ряд исследований показывают ключевую нейротоксическую роль HDAC3 [9, 35, 37], однако специфичность активации HDAC3 при индукции гибели нейронов и влияние HDAC3 на уровень апоптоза и экспрессию p53 после аксотомии до конца не изучены [81].
Ряд работ демонстрирует защитный эффект ингибиторов HDAC на различных экспериментальных моделях повреждения нервов [80]. В нашей модели перерезки седалищного нерва было показано, что вальпроат натрия, неспецифический ингибитор HDAC I класса, защищает клетки DRG крысы от апоптоза и практически полностью отменяет вызванные аксотомией изменения экспрессии и субклеточной локализации p53. Вероятно, защитный эффект вальпроата натрия связан с регуляцией экспрессии и внутриклеточной локализации p53 путем подавления деацетилирования p53 с помощью HDAC1 [49].
Таким образом, стратегии, направленные на усиление ацетилирования p53 по лизинам К381 и К382, могут способствовать разработке соединений, которые будут проявлять нейропротекторные эффекты в периферической нервной системе при нейротравмах [35, 36, 73].
В отличие от HDAC I класса, которые продемонстрировали нейротоксический эффект в аксотомированных нейронах [80, 81], сиртуин 1 (SIRT1), член семейства никотинамидаденозиндинуклеотид-зависимых деацетилаз, ингибирует транскрипцию p53, деацетилируя его по лизину K382 на С-конце. Сиртуины выполняют различные функции в центральной нервной системе [80, 82–86]. Доказательства положительного воздействия SIRT1, полученные на моделях животных и исследованиях на людях, предполагают, что активация SIRT1 может быть потенциальным терапевтическим средством лечения нейродегенеративных заболеваний [82–86]. Однако в ряде работ на моделях нейродегенеративных заболеваний на животных также были сообщения о том, что активация SIRT1 не оказывает нейропротекторного действия [86]. Эти противоречивые результаты могут быть вызваны различными факторами. Кроме того, не изучена роль SIRT1 в повреждениях периферических нервов, что тоже будет интересно, учитывая возможное влияние этого белка на активность p53. Подавление p53 путем активации SIRT1 может быть потенциальной терапевтической стратегией при лечении заболеваний, связанных с повреждением нервов [82–86].
Приведенные нами данные подтверждают важность дальнейшего исследования механизмов регуляции белка p53 с участием HDACs в процессах нейродегенерации и нейропротекции. Использования низкомолекулярных ингибиторов активности HDAC в качестве терапевтических инструментов для защиты нейронов при повреждениях также предстоит изучить в дальнейшем.
ПЕРСПЕКТИВЫ ДАЛЬНЕЙШИХ ИССЛЕДОВАНИЙ
Таким образом, гиперэкспрессия белка p53, описанная в ряде модельных объектов повреждения периферических нервов [6, 8–12, 27], указывает на ключевую роль этого белка в регуляции жизнеспособности нейронов после нейротравмы. Накопленные данные свидетельствуют о том, что механизмы, регулирующие активность p53, и механизмы, с помощью которых p53 влияет на выживание и гибель нейронов, являются сложными и контролируются множеством сигнальных путей [87]. Сложная система регуляции активности p53 создает трудности для определения места и роли этого белка в патогенезе нейротравмы [7, 38]. По-видимому, судьба поврежденных нервных клеток определяется балансом между различными модальностями активности p53 [6].
Неясно, как перерезка аксона вызывает апоптоз глиальных клеток, удаленных на несколько миллиметров от места перерезки. Какие молекулы служат проапоптотическими сигналами нейроглии, и как они стимулируют р53, регулирующего гибель этих клеток? Можно предположить участие Ca2+ или NO в качестве низкомолекулярных мессенджеров в глиальном апоптозе. Сразу после аксотомии Са2+ может проникать в поврежденные глиальные клетки в месте пересечения и распространяться по глиальному синцитию. Са2+ способен актировать нейрональную NO-синтазу, которая продуцирует NO, а он в свою очередь легко переносится между клетками и вызывает их апоптоз. Альтернативно, NO может проникать в разрезанный аксон и индуцировать ретроградные сигнальные каскады, которые переносят некоторые проапоптотические молекулы в глиальные клетки. Известно, что в астроцитах NO индуцирует p53-опосредованную транслокацию Bax в митохондрии, которая вызывает апоптоз. Дальнейшие исследования должны проверить эти гипотезы [1, 6].
Открытым остается вопрос об эпигенетической регуляции p53 в нервных клетках при аксотомии [76, 80]. Не изучено влияние аксотомии на субклеточную локализацию p53. Это имеет важное значение, поскольку при действии патологических факторах HDACs I класса могут перемещаться из ядра в цитоплазму и ацетилировать там различные цитоплазматические белки, включая транскрипционный фактор р53 [80]. Деацетилирование р53 способствует изменению его субклеточной локализации, как следствие этого, его активности. Это и предстоит изучить в дальнейшей работе.
Полученные знания лягут в основу теоретической базы, что поможет лучше понять фундаментальные механизмы выживания и гибели нейронов и глиальных клеток при повреждении нервов. Белок p53 можно рассматривать как многообещающую молекулярную мишень для разработки потенциальных нейропротективных агентов, а его ингибирование является важной терапевтической стратегией, направленной на защиту и повышение выживаемости нейронов при нейротравмах.
Конфликт интересов. Авторы заявляют, что у них нет конфликта интересов.
Источники финансирования. Работа выполнена при финансовой поддержке гранта Министерства науки и высшего образования РФ № 0852-2020-0028 и стипендии Президента Российской Федерации для молодых ученых.
Соблюдение этических норм. Для данной статьи авторами не проводились эксперименты с участием людей или животных в качестве объектов.
Список литературы
Berezhnaya E., Bibov M., Komandirov M., Negin-skaya M., Rudkovskii M., Uzdensky A. 2017. Involvement of MAPK, Akt/GSK-3β and AMPK/mTOR signaling pathways in protection of remote glial cells from axotomy-induced necrosis and apoptosis in the isolated crayfish stretch receptor. Mol. Cell. Neurosci. 83, 1–5.
Rishal I., Fainzilber M. 2014. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 15, 32–42.
Navarro X., Vivo M., Valero-Cabre A. 2007. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 82, 163–201.
Demir Ö., Barros E., Offutt T., Rosenfeld M., Amaro R. 2021. An integrated view of p53 dynamics, function, and reactivation. Curr. Opin. Struct. Biol. 67, 187–194.
Talebi M., Talebi M., Kakouri E., Farkhondeh T., Pourbagher-Shahri A., Tarantilis P., Samarghandian S. 2021. Tantalizing role of p53 molecular pathways and its coherent medications in neurodegenerative diseases. Int. J. Biol. Macromol. 172, 93–103.
Rodkin S., Khaitin A., Pitinova M., Dzreyan V., Guzenko V., Rudkovskii M., Sharifulina S., Uzdensky A. 2020. The localization of p53 in the crayfish mechanoreceptor neurons and its role in axotomy-induced death of satellite glial cells remote from the axon transection site. J. Mol. Neurosci. 70, 532–541.
Dzreyan V., Rodkin S., Nikul V., Pitinova M., Uzdensky A. 2021. The expression of E2F1, p53, and caspase 3 in the rat dorsal root ganglia after sciatic nerve transection. J. Mol. Neurosci. 71, 826–835.
Patel N., Joseph C., Corcoran G., Ray S. 2010. Silymarin modulates doxorubicin-induced oxidative stress, Bcl-xL and p53 expression while preventing apoptotic and necrotic cell death in the liver. Toxicol. Appl. Pharmacol. 245, 143–152.
Akhter R., Sanphui P., Biswas S. 2014. The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J. Biol. Chem. 289, 10812–10822.
Checler F., Alves da Costa C. 2014. p53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther. 142, 99–113.
Culmsee C., Mattson M. 2005. p53 in neuronal apoptosis. Biochem. Biophys. Res. Communs. 331, 761–777.
Wan C., Ma X., Shi S., Zhao J., Nie X., Han J., Xiao J., Wang X., Jiang S., Jiang J. 2014. Pivotal roles of p53 transcription-dependent and -independent pathways in manganese-induced mitochondrial dysfunction and neuronal apoptosis. Toxicol. Appl. Pharmacol. 281, 294–302.
Chumakov P. 2007. Versatile functions of p53 protein in multicellular organisms. Biochemistry (Mosc.). 72, 1399–1421.
Fadda E., Nixon M. 2017. The transient manifold structure of the p53 extreme C-terminal domain: Insight into disorder, recognition, and binding promiscuity by molecular dynamics simulations. Phys. Chem. Chem. Phys. 19, 21287–21296.
Lee H., Mok K., Muhandiram R., Park K., Suk J., Kim D., Chang J., Sung Y., Choi K., Han K. 2000. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 275, 29 426–29 432.
Lee C., Martinez-Yamout M., Dyson H., Wright P. 2010. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry. 49, 9964–9971.
Chène P. 2001. The role of tetramerization in p53 function. Oncogene. 20, 2611–2617.
Wawryk-Gawda E., Chylińska-Wrzos P., Lis-Sochocka M., Chłapek K., Bulak K., Jędrych M., Jodłowska-Jędrych B. 2014. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma. 251, 525–533.
Wang Y., Rosengarth A., Luecke H. 2007. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. D, Biol. Crystallogr. 63, 276–281.
Brandt T., Kaar J., Fersht A., Veprintsev D. 2012. Stability of p53 homologs. PLoS One. 7, e47889.
Khoo K., Mayer S., Fersht A. 2009. Effects of stability on the biological function of p53. J. Biol. Chem. 284, 30974–30980.
Raj N., Attardi L. 2017. The transactivation domains of the p53 protein. Cold Spring Harb. Perspect. Med. 7, a026047.
Kannappan R., Mattapally S., Wagle P., Zhang J. 2018. Transactivation domain of p53 regulates DNA repair and integrity in human iPS cells. Am. J. Physiol. Heart Circ. Physiol. 315, 512–521.
Kumar A., Kumar P., Kumari S., Uversky V., Giri R. 2020. Folding and structural polymorphism of p53 C‑terminal domain: One peptide with many conformations. Arch. Biochem. Biophys. 684, 108342.
Brooks C., Gu W. 2010. New insights into p53 activation. Cell Research. 20, 614–621.
Karakostis K., López I., Peña-Balderas A., Fåhareus R., Olivares-Illana V. 2020. Molecular and biochemical techniques for deciphering p53-MDM2 regulatory mechanisms. Biomolecules. 11, 36.
Chao C. 2015. Mechanisms of p53 degradation. Clin. Chim. Acta. 438, 139–147.
Sullivan K., Galbraith M., Andrysik Z., Espinosa J. 2018. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143.
Liu Y., Tavana O., Gu W. 2019. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 11, 564–577.
Simabuco F., Morale M., Pavan I., Morelli A., Silva F., Tamura R. 2018. p53 and metabolism: From mechanism to therapeutics. Oncotarget. 9, 23780–23823.
Dai Q., Luo T., Luo S., Wang J., Wang S., Bai Y., Yang Y., Wang Y. 2016. p53 and mitochondrial dysfunction: novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 48. 337–347.
Wang D., Kinoshita C., Kinoshita Y., Morrison R. 2014. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta. 1842, 1186–1197.
Lahalle A., Lacroix M., De Blasio C., Cissé M., Linares L., Le Cam L. 2021. The p53 pathway and metabolism: The tree that hides the forest. Cancers (Basel). 13, 133.
Moxley A., Reisman D. 2021. Context is key: Understanding the regulation, functional control, and activities of the p53 tumour suppressor. Cell Biochem. Funct. 39, 235–247.
Schmitt H., Schlamp C., Nickells R. 2016. Role of HDACs in optic nerve damage-induced nuclear atrophy of retinal ganglion cells. Neurosci. Lett. 625, 11–15.
Lebrun-Julien F., Suter U. 2015. Combined HDAC1 and HDAC2 depletion promotes retinal ganglion cell survival after injury through reduction of p53 target gene expression. ASN Neuro. 7, 1759091415593066.
Bardai F., D’Mello S. 2011. Selective toxicity by HDAC3 in neurons: Regulation by Akt and GSK3beta. J. Neurosci. 31, 1746–1751.
Patodia S., Raivich G. 2012. Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 5, 8.
Dubový P., Klusáková I., Hradilová-Svíženská I., Joukal M. 2018. Expression of regeneration-associated proteins in primary sensory neurons and regenerating axons after nerve injury – an overview. Anat. Rec. (Hoboken). 301, 1618–1627.
Hill C., Coleman M., Menon D. 2016. Traumatic axonal injury: Mechanisms and translational opportunities. Trends Neurosci. 39, 311–324.
Khaitin A., Rudkovskii M., Uzdensky A. 2018. Ca2+ mediates axotomy-induced necrosis and apoptosis of satellite glial cells remote from the transection site in the isolated crayfish mechanoreceptor. Mol. Cell Neurosci. 88, 7–15.
McKay Hart A., Brannstrom T., Wiberg M., Terenghi G. 2002. Primary sensory neurons and satellite cells after peripheral axotomy in the adult rat Time course of cell death and elimination. Exp. Brain Res. 142, 308–318.
Melle C., Ernst G., Grosheva M., Angelov D., Irintchev A., Guntinas-Lichius O., von Eggeling F. 2009. Proteomic analysis of microdissected facial nuclei of the rat following facial nerve injury. J. Neurosci. Methods. 185, 23–28.
Savastano L., Laurito S., Fitt M., Rasmussen J., Gonzalez Polo V., Patterson S. 2014. Sciatic nerve injury: A simple and subtle model for investigating many aspects of nervous system damage and recovery. J. Neurosci. Meth. 227, 166–180.
Dzreyan V., Rodkin S., Pitinova M., Uzdensky A. 2021. HDAC1 expression, histone deacetylation, and protective role of sodium valproate in the rat dorsal root ganglia after sciatic nerve transection. Mol. Neurobiol. 58, 217–228.
Uzdensky A. 2018. Axotomy induces damage to glial cells remote from the transection site in the peripheral nervous system. Neural. Regen. Res. 13, 639–640.
Khaitin A., Rudkovskii M., Uzdensky A. 2015. The method of isolation of the crayfish abdominal stretch receptor maintaining a connection of the sensory neuron to the ventral nerve cord ganglion. Invertebr. Neurosci. 15, 176.
Uzdensky A., Berezhnaya E., Khaitin A., Kovaleva V., Komandirov M., Neginskaya M., Rudkovskii M., Sharifulina S. 2015. Protection of the crayfish mechanoreceptor neuron and glial cells from photooxidative injury by modulators of diverse signal transduction pathways. Mol. Neurobiol. 52, 811–825.
Casas C., Isus L., Herrando-Grabulosa M., Mancuso F., Borrás E., Sabidó E., Forés J., Aloy P. 2015. Network-based proteomic approaches reveal the neurodegenerative, neuroprotective and pain-related mechanisms involved after retrograde axonal damage. Sci. Rep. 5, 9185.
Demyanenko S., Dzreyan V., Uzdensky A. 2019. Axotomy-induced changes of the protein profile in the crayfish ventral cord ganglia. J. Mol. Neurosci. 68, 667– 678.
Sun B., Ou H., Ren F., Huan Y., Zhong T., Gao M., Cai H. 2018. Propofol inhibited autophagy through Ca2+/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol. Med. 24, 58.
Shi W., Xu D., Gu J., Xue C., Yang B., Fu L., Song S., Liu D., Zhou W., Lv J., Sun K., Chen M., Mei C. 2018. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ-AMPK-mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 449, 219–226.
Zhou H., Li X., Meinkoth J., Pittman R. 2000. Akt regulates cell survival and apoptosis at a postmitochondrial level. J. Cell Biol. 151, 483–494
Kakiuchi Y., Yurube T., Kakutani K., Takada T., Ito M., Takeoka Y., Kanda Y., Miyazaki S., Kuroda R., Nishida K. 2019. Pharmacological inhibition of mTORC1 but not mTORC2 protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism through Akt and autophagy induction. Osteoarthritis Cartilage. 27, 965–976
Liu Y., Xu H., An M. 2017. mTORC1 regulates apoptosis and cell proliferation in pterygium via targeting autophagy and FGFR3. Sci. Rep. 7, 7339.
Hermida M., Dinesh Kumar J., Leslie N. 2017. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 65, 5–15.
Beurel E., Jope R. 2006. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 79, 173–189.
Al Zaid Siddiquee K., Turkson J. 2008. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 18, 254–267.
Kralova J., Dvorak M., Koc M., Kral V. 2008. p38 MAPK plays an essential role in apoptosis induced by photoactivation of a novel ethylene glycol porphyrin derivative. Oncogene. 27, 3010–3020.
Sui X., Kong N., Ye L., Han W., Zhou J., Zhang Q., He C., Pan H. 2014. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 344, 174–179
Buitrago C., Ronda A., de Boland A., Boland R. 2006. MAP kinases p38 and JNK are activated by the steroid hormone 1alpha, 25(OH)2-vitamin D3 in the C2C12 muscle cell line. J. Cell. Biochem. 97, 698–708.
Bishop P., Rocca D., Henley J. 2016. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem J. 473, 2453–2462.
Chong Z., Lin S., Kang J., Maiese K. 2003. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell. Mol. Neurobiol. 23, 561–578.
Bassi R., Heads R., Marber M., Clark J. 2008. Targeting p38-MAPK in the ischaemicheart: Kill or cure? Curr. Opin. Pharmacol. 8,141–146.
Yang L., Greig N., Tweedie D., Jung Y., Chiang Y., Hoffer B., Miller J., Chang K., Wang J. 2020. The p53 inactivators pifithrin-μ and pifithrin-α mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy. Exp. Neurol. 324, 113135
Polager S., Ginsberg D. 2009. p53 and E2f: Partners in life and death. Nat. Rev. Cancer. 9, 738–748.
Shafiei-Roudbari S., Malekinejad H., Janbaz-Aciabar H., Razi M. 2017. Crosstalk between E2F1 and P53 transcription factors in doxorubicin-induced DNA damage: Evidence for preventive/protective effects of silymarin. J. Pharm. Pharmacol. 69, 1116–1124.
Liu Y., Zacksenhaus E. 2000. E2F1 mediates ectopic proliferation and stage-specific p53-dependent apoptosis but not aberrant differentiation in the ocular lens of Rb deficient fetuses. Oncogene. 19, 6065–6073.
Ma L., Yu H., Gan S., Gong R., Mou K., Xue J., Sun S. 2017. p53-Mediated oligodendrocyte apoptosis initiates demyelination after compressed spinal cord injury by enhancing ER-mitochondria interaction and E2F1 expression. Neurosci. Lett. 644, 55–61.
Fouad S., Hauton D., D’Angiolella V. 2021. E2F1: Cause and consequence of DNA replication stress. Front. Mol. Biosci. 7, 599332.
Kuusk A., Boyd H., Chen H., Ottmann C. 2020. Small-molecule modulation of p53 protein-protein interactions. Biol. Chem. 401, 921–931.
Brochier C., Dennis G., Rivieccio M., McLaughlin K., Coppola G., Ratan R., Langley B. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 33, 8621–8632.
Brooks C., Gu W. 2011. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2, 456–462.
Marcel V., Nguyen Van Long F., Diaz J. 2018. 40 years of research put p53 in translation. Cancers (Basel). 10, 152.
Kruse J., Gu W. 2008. SnapShot: p53 posttranslational modifications. Cell. 133, 930–930.e1.
Saha A., Tiwari S., Dharmarajan S., Otteson D., Belecky-Adams T. 2018. Class I histone deacetylases in retinal progenitors and differentiating ganglion cells. Gene Expr. Patterns. 30, 37–48.
Baltan S., Bachleda A., Morrison R., Murphy S. Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. 2011. Transl. Stroke Res. 2, 411–423.
Chen Y., Zang X., Pan J., Zhu X., Chen F., Chen Z., Xu Y. 2012. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin. Exp. Pharmacol. Physiol. 39, 751–758.
Demyanenko S., Dzreyan V., Uzdensky A. 2020. Expression of histone deacetylases HDAC1 and HDAC2 and their role in apoptosis in the penumbra induced by photothrombotic stroke. Mol. Neurobiol. 57, 226–238.
Thomas E., D’Mello S. 2018. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 145, 96–110.
Schmitt H., Pelzel H., Schlamp C., Nickells R. 2014. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol. Neurodegener. 9, 39.
Chen C., Zhou M., Ge Y., Wang X. 2020. SIRT1 and aging related signaling pathways. Mech. Ageing. Dev. 187, 111215.
Gomes B., Silva J., Romeiro C., Dos Santos S., Rodrigues C., Gonçalves P., Sakai J., Mendes P., Varela E., Monteiro M. 2018. Neuroprotective mechanisms of resveratrol in Alzheimer’s disease: Role of SIRT1. Oxid. Med. Cell. Longev. 2018, 8152373.
Fujita Y., Yamashita T. 2018. Sirtuins in neuroendocrine regulation and neurological diseases. Front. Neurosci. 12, 778.
Yang X., Si P., Qin H., Yin L., Yan L., Zhang C. 2017. The neuroprotective effects of SIRT1 on NMDA-induced excitotoxicity. Oxid. Med. Cell. Longev. 2017, 2823454.
Zhao X., Liu Y., Zhu G., Liang Y., Liu B., Wu Y., Han M., Sun W., Han Y., Chen G., Jiang J. 2019. SIRT1 downregulation mediated Manganese-induced neuronal apoptosis through activation of FOXO3a-Bim/PUMA axis. Sci. Total Environ. 646, 1047–1055.
Culmsee C., Mark P. Mattson. 2005. p53 in neuronal apoptosis. Biochem. Biophys. Res. Commun. 331, 761–777.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии